

Abstract
Gain-of-function mutations in the pore-forming subunit of IKs channels, KCNQ1, lead to short QT syndrome (SQTS) and lethal arrhythmias. However, how mutant IKs channels cause SQTS and the possibility of IKs-specific pharmacological treatment remain unclear. V141M KCNQ1 is a SQTS associated mutation. We studied its effect on IKs gating properties and changes in the action potentials (AP) of human ventricular myocytes. Xenopus oocytes were used to study the gating mechanisms of expressed V141M KCNQ1/KCNE1 channels. Computational models were used to simulate human APs in endocardial, mid-myocardial, and epicardial ventricular myocytes with and without β-adrenergic stimulation. V141M KCNQ1 caused a gain-of-function in IKs characterized by increased current density, faster activation, and slower deactivation leading to IKs accumulation. V141M KCNQ1 also caused a leftward shift of the conductance-voltage curve compared to wild type (WT) IKs (V1/2 = 33.6 ± 4.0 mV for WT, and 24.0 ± 1.3 mV for heterozygous V141M). A Markov model of heterozygous V141M mutant IKs was developed and incorporated into the O’Hara–Rudy model. Compared to the WT, AP simulations demonstrated marked rate-dependent shortening of AP duration (APD) for V141M, predicting a SQTS phenotype. Transmural electrical heterogeneity was enhanced in heterozygous V141M AP simulations, especially under β-adrenergic stimulation. Computational simulations identified specific IK1 blockade as a beneficial pharmacologic target for reducing the transmural APD heterogeneity associated with V141M KCNQ1 mutation. V141M KCNQ1 mutation shortens ventricular APs and enhances transmural APD heterogeneity under β-adrenergic stimulation. Computational simulations identified IK1 blockers as a potential antiarrhythmic drug of choice for SQTS.
Keywords: Arrhythmia, Anti-arrhythmic, IKs, KCNQ1, Short QT syndrome
1 Introduction
Short QT Syndrome (SQTS) is an inherited channelopathy associated with marked shortening of QT intervals on the ECG and sudden cardiac death in individuals with structurally normal hearts [1–4]. Hereditary SQTS is genetically heterogeneous with six currently identified forms, SQT1 to SQT6, based on the chronology of their discovery [5–11]. The mutation V141M KCNQ1 causes a gain-of-function of the slow delayed rectifier potassium current (IKs) and is one of the mutations that give rise to SQT2 [12, 13].
Shortening of the QT interval is a normal physiological response to an increase in heart rate. In patients with SQTS, the QT interval is relatively normal at fast rates, but abnormally short at slow heart rates [14]. Transmural dispersion of repolarization (TDR) has been suggested to correlate to ventricular arrhythmias in an experimental SQTS model that was generated by application of an IK,ATP opener, pinacidil, and in patients with SQTS [15, 16]. Reducing TDR has been suggested as an important antiarrhythmic property of quinidine in a SQTS model [17] and in patients exhibiting the SQT1 variant [14]. In other forms of SQTS, data regarding pharmacological therapy remain too limited to permit specific suggestions or recommendations.
This study evaluated the effect of V141M KCNQ1 mutation on TDR and the drug effects on TDR of different anti-arrhythmic agents. First, we determined the kinetic changes relative to the wild type of V141M mutant KCNQ1 + KCNE1 channel expressed in Xenopus oocytes. Then we used computer simulations in the O’Hara–Rudy (ORd) human ventricular cell model to study the electrophysiological consequences of V141M KCNQ1 in ventricular myocytes in terms of its effects on IKs kinetics, action potential (AP) with and without isoproterenol (ISO) challenge, AP rate adaptation, and transmural AP duration (APD) heterogeneity. We also examined the effects of various anti-arrhythmic drugs on V141M KCNQ1-augmented transmural APD heterogeneity and identified a potential target for suppressing TDR and consequently reducing arrhythmic risk in SQT2.
2 Methods
2.1 Mutagenesis and Oocyte Preparation
Xenopus oocytes that expressed human wild type (WT) KCNQ1 or V141M KCNQ1 and WT KCNE1 were used to record WT IKCNQ1+KCNE1 (WT IKs) and V141M IKCNQ1+KCNE1 (V141M IKs). KCNQ1 (provided by S. Goldstein, University of Chicago, Chicago, IL) and KCNE1 (provided by S. Nakanishi, Osaka Bioscience Institute, Osaka, Japan) were subcloned into the HindIII/XbaI cloning sites of pcDNA3.1+ vectors (Invitrogen, Grand Island, NY). The V141M KCNQ1 mutation was generated by using overlap extension amplification with high-fidelity polymerase chain reaction (PCR), verified by DNA sequencing (IDT technology, Coralville, IW). Messenger RNA was transcribed in vitro using the mMessage mMachine T7 polymerase kit (Applied Biosystems, Oyster Bay, NY). The follicle layer of oocytes was digested using type 1A collagenase (Sigma-Aldrich, Saint Louis, MO). Stage IV–V Xenopus oocytes were selected and injected with 4.6 ng mRNA per oocyte. For implementing a heterozygous genotype, we expressed heterozygous V141M IKs channels in oocytes that RNAs of WT KCNQ1 and V141M KCNQ1 at 1:1 ratio were injected. Total KCNQ1 and KCNE1 injection ratios for WT and V141M were the same at 6:1. Injected oocytes were incubated in ND96 solution (in mM: 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, and 5 Hepes, pH 7.60) at 18 °C for 3–5 days before recording.
2.2 Electrophysiology
Whole cell current recordings were obtained with the two-microelectrode voltage clamp technique. Microelectrodes were pulled from glass capillary tubes and filled with 3 M KCl. Oocytes were constantly superfused with ND96 at room temperature (~22 °C). The membrane potential was clamped using a GENECLAMP 500B amplifier (Axon Instruments, New York, NY). Data acquisition was controlled using PULSE/PULSEFIT software (HEKA, Farmingdale, NY).
2.3 Data Analysis
The electrophysiology data was analysed with Igor 4.09 (WaveMetrics, Lake Oswego, OR), and plotted with Prism 6 software (GraphPad, La Jolla, CA). Statistical evaluation was performed using the SAS program (JMP software, Version 9, Cary, NC). Where not otherwise specified, numerical variants were mean ± SD. Two-way ANOVA analysis was used, followed by Ryan-Einot-Gabriel-Welsch Multiple Range Test. A value of p < 0.05 was considered statistically significant.
2.4 Computational Model Simulations
Simulations of the ventricular myocyte AP were based on a modified ORd model of the human ventricular cardiomyocyte, in which the signaling cascade from ISO application to PKA phosphorylation of target proteins was incorporated [18–20]. Parameters affected by PKA phosphorylation were computed by the Heijman et al. model of the β-adrenergic signaling pathway [21]. In the simulations, 1 μmol/L ISO was applied starting from steady state after pacing at a cycle length of 1000 ms for 1000 beats. The original ORd model used data from measurements in isolated undiseased human ventricular myocytes at 37 °C to formulate IKs [20, 22]. In this model, the Ca2+ dependence of IKs was also taken into consideration. The transition rates in the IKs Markov model used here were corrected based on recordings obtained at room temperature (provided in the Supplemental Information). Simulation of IKs activation was constrained with the G–V curves and the deactivation was constrained with experimental values of Tau (see Fig. 1). The scaling factors of IKs conductance without any drug effects for Endo/Epi and Mid cells were taken from the ORd human model in which GKs for Endo and Epi were both 1.4 and for Mid was 1.0 [20].
Fig. 1.
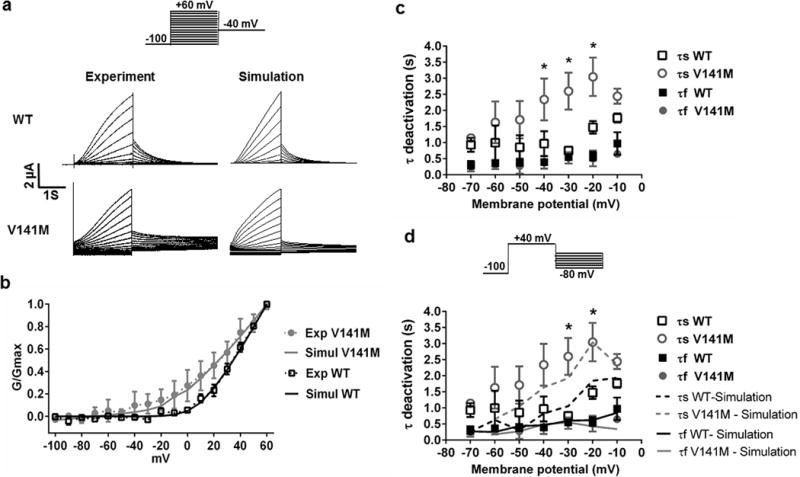
V141M KCNQ1 affects activation kinetics and voltage dependence of IKs. a Currents of WT KCNQ1/KCNE1 channels (WT IKs) and heterozygous V141M KCNQ1/KCNE1 channels (V141M IKs) expressed in Xenopus oocytes (left panels). The currents were elicited by test pulses to +60 mV from a holding potential of −100 mV. Tail currents were measured at −40 mV for WT and V141M IKs. The WT and V141M IKs were simulated using a Markov model (right panels; see supplementary materials). b V141M KCNQ1 mutation caused a leftward shift of the conductance–voltage curve (V1/2 = 33.6 ± 4.0 mV for WT, 24.0 ± 1.3 mV for heterozygous V141M; n = 5 for each). Simulation results (solid lines) are consistent with the experimental data (dashed lines). c Voltage dependence of activation and deactivation time constants (τ) of WT and heterozygous V141M IKs (n = 5 for each). τ activation was obtained from fitting the activating current traces with a double exponential function. V141M KCNQ1 causes faster activation of IKs (*p < 0.05 for τ1 and #p < 0.05 for τ2; V141M vs WT; n = 3 for each). d V141M KCNQ1 causes slower deactivation of IKs (*p value <0.05 for τs; V141M (n = 5) versus WT (n = 3). τ deactivation was obtained from fitting the deactivating current traces with a double exponential function. Lines indicate specific τ values for simulations
AP durations (APD) at 95% repolarization (APD95) for endocardial (Endo), mid-myocardial (Mid), and epicardial (Epi) myocytes were measured. APD95 was measured as the interval between the time of maximum AP upstroke velocity and the time at which the membrane voltage returned to 95% of its resting value. The largest difference among Endo, Mid, and Epi APD95 was normalized by the APD95 of Mid and the value in % was used to represent TDR.
To determine how V141M KCNQ1 affects repolarization in the context of heterogeneous heart tissue, simulations were performed on a 1-dimensional transmural fiber model. The fiber was composed of 60 Epi, 45 Mid, and 60 Endo myocytes. Gap junction conductance was homogenous throughout the fiber at 1.73 μs, except for a fivefold decrease at the Mid-to-Epi transition region. A 0.5-ms current stimulus was applied to Endo cell 1 to initiate Endo-to-Epi AP propagation. The resulting conduction velocity was 44 cm/s. The QT interval on the pseudo-ECG generated by the fiber was computed as the interval between the maximum negative derivative on the QRS and the maximum positive derivative on the T-wave.
We simulated antiarrhythmic drug effects by introducing conductance changes for specific ionic currents. Using conductance scaling, Benson et al. reproduced AP changes caused by sotalol and amiodarone in canine left ventricle transmural strips [22, 23]. To simulate amiodarone application of 30–40 mg/kg/day, maximal conductance of the late sodium current (INaL) was scaled by 0.2 in Mid, and maximal conductance of IKs was scaled by 0.2 in Endo and 0.7 in Epi [23]. To simulate 100 μM/L sotalol, we scaled the rapid delayed rectifier potassium current (IKr) maximal conductance by 0.5 in Endo, 0.3 in Mid, and 0.8 in Epi [23]. To simulate 6–10 μM/L quinidine, we scaled maximal conductance of INaL by 0.6, of ICaL by 0.75, of IKr by 0.6, of the inward rectifier current (IK1) by 0.6 [24–26], and of the transient outward current (Ito) by 0.6 in Mid, by 0.46 in Endo, and by 0.6 in Epi based on experimental results from guinea-pig, rabbit, or canine ventricular myocyte studies [24–27].
3 Results
3.1 V141M Mutation Accelerates IKs Activation, Decelerates Deactivation, and Causes a Negative Shift in IKs Voltage-Dependence
The KCNQ1/KCNE1 channels that were expressed in Xenopus oocytes were activated every 100 s from a holding potential of −100 mV with 3 s depolarizing pulses ranging from −100 to +60 mV (Fig. 1a). We had noticed much larger instantaneous currents on shorter sweep intervals and therefore used a long sweep interval of 100 s. After each test pulse, the membrane was repolarized to −40 mV to record tail currents. Measured from the conductance–voltage (G–V) relation curves, heterozygous V141M KCNQ1/KCNE1 channels were activated at more negative potentials compared to the WT (V1/2 = 33.6 ± 4.0 mV for WT, 24.0 ± 1.3 mV for V141M; n = 5 for each) (Fig. 1b). The rates of IKs activation were fitted with a double exponential function. The V141M caused faster activation of IKs at depolarizing potentials ranging from 0 to +50 mV (Fig. 1c). The same oocytes used for the voltage-dependence of IKs conductance were subjected to another pulse protocol where a depolarizing pulse was fixed at +40 mV followed by various repolarizing pulses ranging from −70 to −10 mV. The time required for tail currents to decay at different repolarizing potentials was also quantified by fitting the traces to a double exponential function. The heterozygous V141M KCNQ1 expression caused slower deactivation for IKs (Fig. 1d).
3.2 Simulated Voltage-Dependent Activation of WT IKs and Heterozygous V141M IKs is Consistent with Experimental Results
WT IKs and heterozygous V141M IKs were simulated for the voltage protocol employed in the experiment (Fig. 1a). The acceleration in IKs activation caused by the mutation was simulated by adjusting transition rates in the IKs Markov model (see Supplementary Material). The conductance-voltage relations show good agreement between the experimental and simulation results (Fig. 1b).
3.3 Simulations of Changes to Action Potentials, IKs, and ICaL in Endo, Mid, and Epi Myocytes
The Markov models for WT and V141M IKs were introduced in the ORd model to simulate the AP and corresponding time course of IKs and L-type calcium current (ICaL) in Endo, Mid, and Epi myocytes. Further, AP simulations were performed to evaluate the effects of β-adrenergic stimulation by using the Heijman et al. model of the β-adrenergic cascade [21] at 1 μmol/L ISO application. With V141M mutation, APD95 was shortened for all three myocyte types. The APD shortening resulted from the rapid activation of IKs and larger IKs peak amplitude during the AP (Fig. 2; Table 1). The rapidly repolarizing potential causes augmentation of ICaL amplitude, especially in the presence of ISO (Fig. 2). This, in turn, resulted in a “bump” during phase-3 repolarization of the AP which was most noticeable in Mid. The magnitude of V141M-induced APD shortening was further enhanced by the presence of ISO in all three myocyte types (Table 1). Consistent with the reported clinical presentation of SQTS [28], the rate-adaptation of APD was blunted by the V141M mutation (Fig. 3).
Fig. 2.
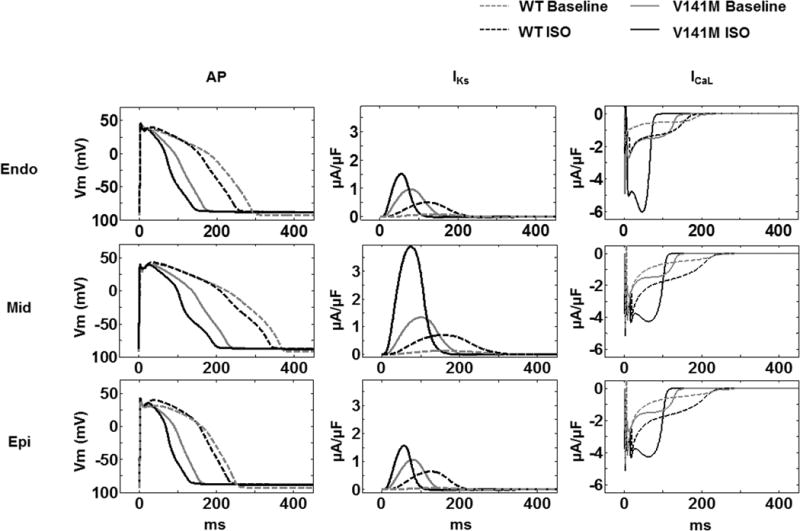
Simulated WT and heterozygous V141M KCNQ1 action potentials (AP) and the IKs and ICaL during the AP in endocardial (Endo), epicardial (Epi) and midmyocardial (Mid) myocytes with and without isoproterenol (ISO). A modified O’Hara-Rudy (ORd) human ventricular myocyte model was used in the simulations. The cycle length was 1000 ms. Baseline APs (in grey) and APs with ISO challenge (in black) were simulated for WT (dash lines) and V141M KCNQ1 (solid lines) respectively. APs were shortened significantly by the V141M KCNQ1 mutation with and without ISO. In all V141M cases, IKs and ICaL increased relative to WT during the AP
Table 1.
Computed APD95 and IKs for WT and V141M KCNQ1 using the ORd human ventricular model for Endo, Mid, and Epi myocytes
| Myocytes | Condition | ORd human ventricular myocyte model [20]
|
|||
|---|---|---|---|---|---|
| APD95 (ms) | Peak IKs amplitude (pA/pF) | APD95 shortening (ms/%) (V141M V.S. WT) | APD95 shortening (ms/%) (ISO V.S. baseline) | ||
| Endo | WT | 278.3 | 0.086 | ||
| V141M | 168.1 | 0.973 | 110.2/39.6 | ||
| WT ISO | 249.9 | 0.507 | 28.4/10.2 | ||
| V141M ISO | 140.1 | 1.516 | 109.8/43.9 | 28.0/16.7 | |
| Mid | WT | 344 | 0.128 | ||
| V141M | 230.6 | 1.336 | 113.4/33.0 | ||
| WT ISO | 338.9 | 0.696 | 5.1/1.5 | ||
| V141M ISO | 195.7 | 3.898 | 143.2/42.3 | 34.9/15.1 | |
| Epi | WT | 238.2 | 0.0695 | ||
| V141M | 155.5 | 1.066 | 82.7/34.7 | ||
| WT ISO | 231.8 | 0.648 | 6.4/2.9 | ||
| V141M ISO | 128.4 | 1.565 | 103.4/44.6 | 27.1/17.4 | |
Scaling factors for IKs conductance: Endo 1.4, Epi 1.4, and Mid 1.0
Fig. 3.
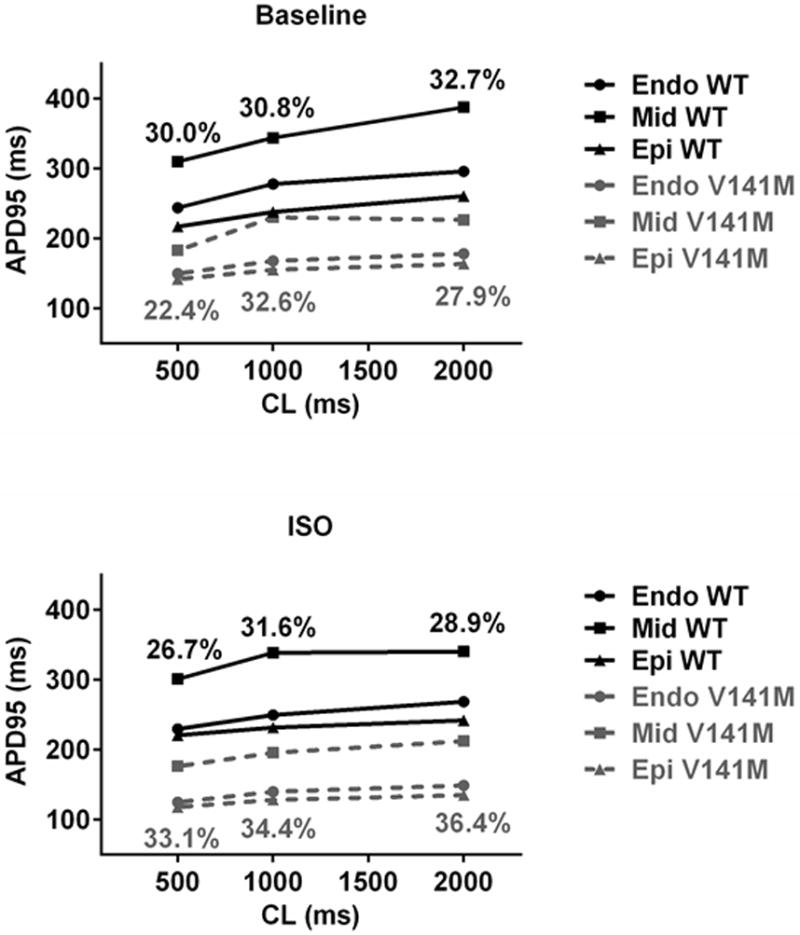
AP simulations show blunted APD rate adaptation and increased transmural heterogeneity of APD associated with the V141M KCNQ1 mutation. At fast cycle lengths of 500 ms and slow cycle lengths of 2000 ms, WT KCNQ1 APD increased 52.0, 77.6, and 43.5 ms in Endo, Mid and Epi, respectively, but V141M KCNQ1 mutation APD increased 28.3, 43.7, and 21.5 ms respectively (upper panel). At cycle lengths of 500 and 2000 ms, APD heterogeneity diminished from 30.0 to 26.7% and from 32.7 to 28.9% respectively in the presence of isoproterenol (ISO) challenge in WT. In contrast, the heterogeneity of APD was augmented with ISO in V141M KCNQ1. In the presence of ISO, APD heterogeneity increased from 22.4 to 33.1%, from 27.9 to 36.4%, and from 32.6 to 34.4%, at cycle lengths of 500, 2000, or 1000 ms, respectively. The heterogeneity increase was due to preferential abbreviation of epicardial (Epi) and endocardial (Endo) cells as compared with mid-myocardial (Mid) cells
3.4 V141M KCNQ1 Mutation Increases Transmural Heterogeneity of Repolarization
Increased transmural heterogeneity of repolarization has been suggested as a pro-arrhythmic substrate in SQTS [15]. We quantified the transmural heterogeneity of repolarization as described in the Methods section. At baseline, APD heterogeneity augmentation by the V141M mutation was most noticeable at a cycle length of 1000 ms (Fig. 3). However, in the presence of ISO, V141M-augmented APD heterogeneity was present at long and short cycle lengths as well. In WT, ISO induced less transmural APD heterogeneity at both rapid and slow cycle lengths. In contrast, with V141M KCNQ1 mutation, ISO induced more transmural heterogeneity at slow cycle lengths (Fig. 3).
3.5 Specific IK1 Blockade Reduces the Transmural APD Heterogenecity
The effects of different anti-arrhythmic drug actions and several specific channel blockers on the V141M-augmented transmural APD heterogeneity were examined by computer simulations. Amiodarone and sotalol are class III antiarrhythmic agents whose main electrophysiological effects include AP prolongation [23]. However, in our simulations of the cellular effects of 100 μM/L sotalol or amiodarone (30 mg/kg/day), both drugs failed to reduce the transmural heterogeneity of APD. The next anti-arrhythmic drug we tested was quinidine, which has multiple depressing effects on sodium, calcium and potassium currents and generally prolongs the AP [24–26, 29]. Quinidine has shown promise in treating SQTS patients [30]. However, it did not reduce the transmural heterogeneity of APD in our simulations for the V141M mutation. Specific IKs blockade also failed to reduce the transmural heterogeneity of APD at either 50 or 90% blockade. Finally, with 90% specific IK1 blockade, the transmural heterogeneity of V141M APD was reduced below that of WT. The APD heterogeneity was reduced by specific IK1 blockade at both long and short cycle lengths (Fig. 4).
Fig. 4.
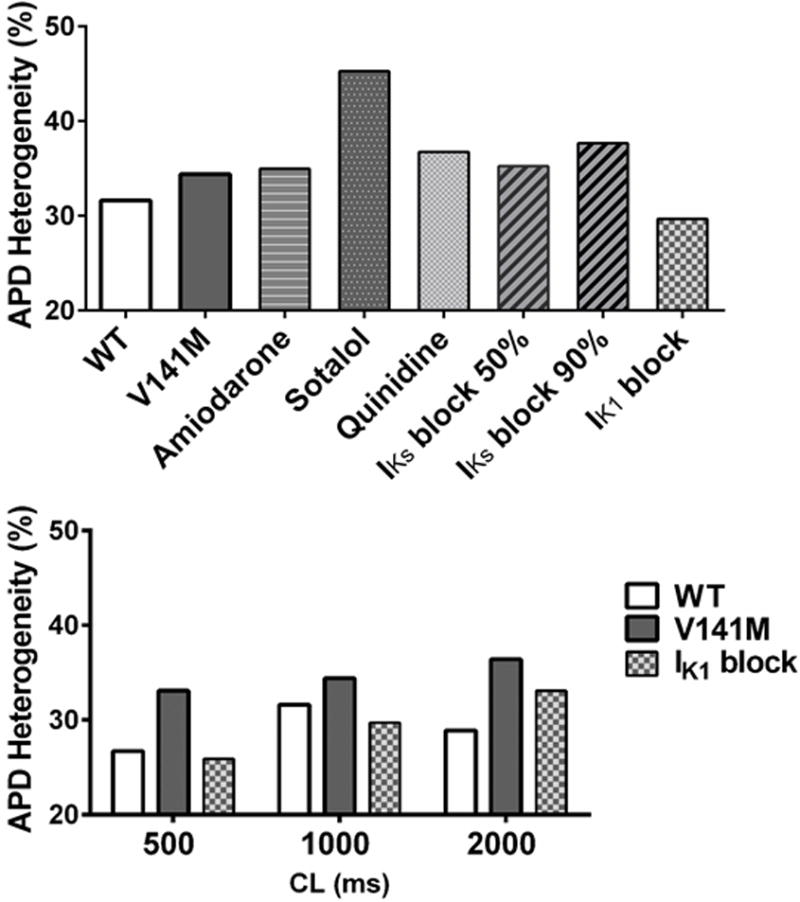
Effects of different anti-arrhythmic agents or channel blockers on the V141M KCNQ1-augmented transmural heterogeneity of APD. At a cycle length of 1000 ms with isoproterenol (ISO) (upper panel), amiodarone, sotalol, quinidine and IKs block (either at 50 or 90% block) all failed to reduce the mutation-augmented transmural heterogeneity of APD (upper panel). Only the IK1 block (with 90% block) improved APD heterogeneity at both long and short cycle lengths under ISO challenge (lower panel)
3.6 Specific IK1 Blockade Abolishes V141M Mutation-Induced QT Interval Shortening
We examined effects of the V141M KCNQ1 mutation on QT intervals of a pseudo-ECG that was computed for a 1D transmural fiber at different pacing cycle lengths (Fig. 5a). The phenotype of QT shortening was reproduced by introducing a V141M IKs Markov model into the Endo, Mid, and Epi myocytes of the fiber (Fig. 5b). The QT intervals for mutant V141M were shorter than WT at all (short and long) cycle lengths, especially with ISO challenge (Fig. 5c). V141M-induced QT shortening with ISO challenge was abolished by 90% specific IK1 blockade (Fig. 5b, c).
Fig. 5.
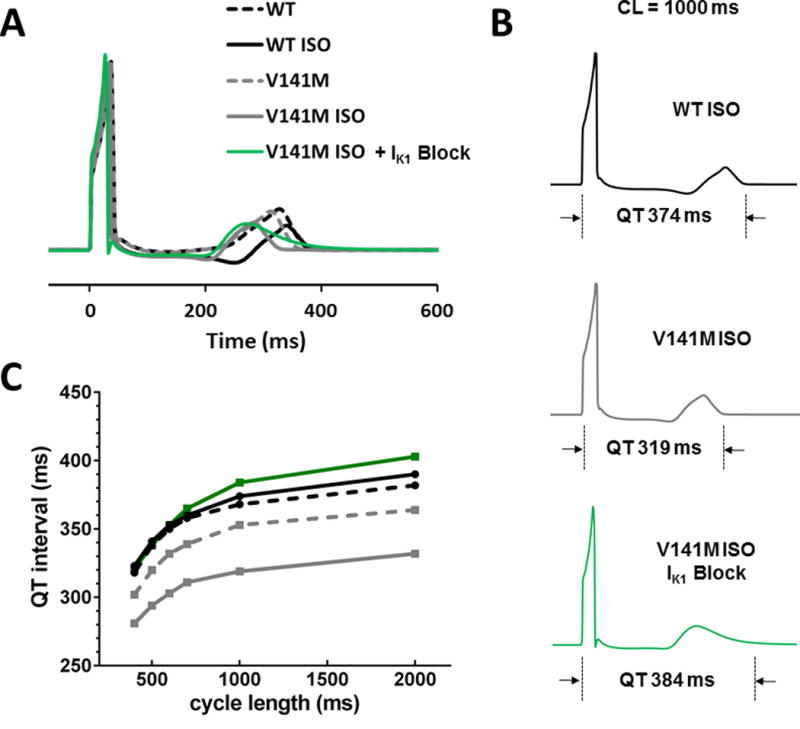
Effects of IK1 blockade on the pseudo-ECG and QT interval. a Pseudo-ECGs at a cycle length of 1000 ms for WT and V41M KCNQ1 mutation, at baseline and with isoproterenol, and with 90% IK1 block. b QT interval measurements for each pseudo-ECG are indicated. c QT intervals on the pseudo-ECG are plotted against the cycle length. In the case of V141M, the QT interval was abnormally shortened with ISO; the shortening of the QT interval was corrected by specific IK1 blockade. ECG electrocardiogram, WT wild type, V141M V141M KCNQ1 mutation
4 Discussion
The results of the present study show that: (1) the V141M KCNQ1 mutation causes gain-of-function of IKs mainly by accelerating channel activation and decelerating deactivation; (2) V141M KCNQ1 mutation-induced shortening of APs is more prominent in the presence of β-adrenergic stimulation; (3) V141M KCNQ1 augments transmural heterogeneity of repolarization, a property that has been suggested as an arrhythmogenic mechanism in SQTS; and (4) specific IK1 blockade has a beneficial effect of reducing the transmural APD heterogeneity and normalizing the short QT interval associated with the V141M KCNQ1 mutation.
Hong et al. reported that the V141M KCNQ1 mutation only altered the gating of IKs channels in the presence of KCNE1 auxiliary subunits [12]. They also showed that coexpression of V141M and WT KCNQ1 with KCNE1 produced a current with intermediate biophysical properties between homozygous V141M and WT IKs. Consistent with their results, our heterozygous V141M IKs also demonstrated an instantaneous current. Notably, the magnitude of the instantaneous current became smaller as we prolonged the sweep interval from 40 to 100 s. With the longer sweep interval, tail currents were suitable for an accurate fitting analysis of the G-V relationship.
The V141M KCNQ1 mutation has been linked to SQTS2, with presentation including extremely short QT interval, fetal bradycardia, and atrial fibrillation [12, 13]. Heterogeneous AP shortening was shown to result in augmented dispersion of APD across the ventricular wall with another SQTS2 mutation [31]. Our simulations showed that the APD shortening resulting from the V141M KCNQ1 mutation is also heterogeneous (Table 1) and leads to an increase in transmural APD heterogeneity and dispersion of repolarization.
In long QT syndrome 1, which is also caused by KCNQ1 mutation affecting IKs, a greater reactivity of the sympathetic control of the QT interval has been recognized as a protective factor [32]. Similarly, it is crucial to determine the reactivity of the sympathetic control of the QT interval to evaluate the arrhythmic risk related to V141M KCNQ1 mutation. To our knowledge, there is no published report showing how the V141M mutant responds to β-adrenergic stimulation. Therefore we assumed that β-adrenergic targets are the same as in WT IKs and respond similarly to adrenergic stimulation. Based on this assumption, β-adrenergic stimulation augments APD heterogeneity for V141M KCNQ1 mutation.
In the simulations, ICaL is severely reduced by including V141M in the model (Fig. 2). The literature reports a phenotype of concurrent sinus bradycardia in patients with V141M mutation [12, 13]. Reduced ICaL density was shown to contribute to slowing of diastolic depolarization of the sinoatrial AP, resulting in slowing of the intrinsic heart rate, even with preserved β-adrenergic response [33]. Reduced ICaL may also affect contractility, but clinical reports did not mention abnormal ventricular function [12].
Notably different from wild type, the degree of APD heterogeneity in V141M KCNQ1 is greater at a long cycle length. Reduced rate-adaptation of the QT interval has been observed in SQTS [14, 28]. SQTS2 patients with the V141M KCNQ1 mutation have been reported to present with bradycardia resulting from sinus and atrioventricular node dysfunction in utero and after birth [4, 12, 13]. Our simulations suggest that in SQTS2 with V141M KCNQ1 mutation, arrhythmia vulnerability is higher at a slow heart rate. In the simulations, both amiodarone and sotalol failed to reduce V141M-augmented APD heterogeneity. Amiodarone and sotalol can inhibit sinus and atrioventricular nodal function but can potentially worsen the bradycardia observed in SQTS2 patients. Therefore we suggest that these drugs should probably be avoided for SQTS2 patients with the V141M KCNQ1 mutation.
There has been growing evidence that quinidine is effective in treating SQTS [14, 30, 34]. However, quinidine can cause serious side effects such as thrombocytopenia and agranulocytosis [35]. In the simulations, quinidine did not reduce APD heterogeneity during β-adrenergic stimulation. An important finding of this study is that IK1 blockade markedly reduced APD heterogeneity in SQTS. Specific IK1 blockade was shown to reduce the incidence of ventricular fibrillation and ischemia–reperfusion ventricular arrhythmias in rats, rabbits, and primates by prolonging APD. Based on pioneering efforts using a transmural ventricular wedge preparation to relate the AP to electrocardiographic waveforms in experiments and in silico [36, 37], we applied the pseudo-ECG modeling approach to provide QT interval changes in KCNQ1 V141M mutation under variable conditions. The specific IK1 block was also effective in reducing TDR [38, 39]. Our simulations provide support for specific IK1 blockade as a potential antiarrhythmic strategy in patients with SQTS. The results are relevant to a better understanding of SQTS2, offering a clue to more feasible risk stratification, and helping to dissect the mechanisms underlying the efficacy of pharmacologic interventions.
In this study, we used the ORd model without considering the possibility of mutation effects on the membrane expression levels of IKs. In addition to affecting IKs kinetics, it is possible that the V141M KCNQ1 mutation also changes IKs densities in a transmurally heterogeneous fashion, adding to TDR. The purpose of the channel expression system is to determine the WT and V141M IKs channel kinetics in order to perform the subsequent human AP simulations. The oocyte system provide large IKs therefore the endogenous KCNQ1 current can be ignored. There are other systems would offer different advantages. The mammalian heterologous expression systems such as HEK293 cells provide feasibility for applying a physiological temperature (37 °C), and for inducing cAMP as a PKA activation. The human cardiomyocytes that are generated from inducible pluripotent stem cells would allow us to precisely determine the sensitivity to channel inhibitors or drugs for V141M mutation. However there remains problems to reproduce a homogenous population of ventricular myocytes with typical and same phenotype in dishes [40].
In conclusion, we demonstrated shortening of APD and QT in V141M KCNQ1 mutation due to accelerated activation and decelerated deactivation kinetics of IKs. The APD shortening is transmurally heterogeneous and results in increased transmural dispersion. This effect can contribute to arrhythmia vulnerability in SQTS2 patients with the V141M mutation. Simulated application of IK1 blocker improved transmural APD heterogeneity and QT interval widening, suggesting specific IK1 blockade as a potential antiarrhythmic strategy in SQTS.
Supplementary Material
Acknowledgments
We thank Dr. Jingyi Shi and Kelli Delaloye for KCNQ1 and KCNE1 subcloning. We thank Dr. Guohui Zhang for technical help with data analysis. We also thank Dr. Po-Yuan Chen for technical help with the computational simulation. The authors also thank the Statistical Analysis Laboratory, Department of Medical Research, Kaohsiung Medical University Hospital, Kaohsiung Medical University. This study was funded by Taiwan National Science Council Grant NSC 102-2314-B-037 -041 and KMUH97-7G31 (to HC Lee), NIH–NHLBI Grants R01-HL-049054 and R01-HL-033343 (to Y. Rudy), and NIH Grants R01-HL70393 and R01-NS060706 (to J. Cui).
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s40846-017-0257-x) contains supplementary material, which is available to authorized users.
Compliance with Ethical Standards
Conflict of interest None.
References
- 1.Gussak I, Brugada P, Brugada J, Wright RS, Kopecky SL, Chaitman BR, et al. Idiopathic short QT interval: A new clinical syndrome? Cardiology. 2000;94(2):99–102. doi: 10.1159/000047299. [DOI] [PubMed] [Google Scholar]
- 2.Bjerregaard P, Gussak I. Short QT syndrome: Mechanisms, diagnosis and treatment. Nature Clinical Practice Cardiovascular Medicine. 2005;2(2):84–87. doi: 10.1038/ncpcardio0097. [DOI] [PubMed] [Google Scholar]
- 3.Schimpf R, Wolpert C, Gaita F, Giustetto C, Borggrefe M. Short QT syndrome. Cardiovascular Research. 2005;67(3):357–366. doi: 10.1016/j.cardiores.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 4.Villafañe J, Atallah J, Gollob MH, Maury P, Wolpert C, Gebauer R, et al. Long-term follow-up of a pediatric cohort with short QT syndrome. Journal of the American College of Cardiology. 2013;61(11):1183–1191. doi: 10.1016/j.jacc.2012.12.025. [DOI] [PubMed] [Google Scholar]
- 5.Brugada R, Hong K, Dumaine R, Cordeiro J, Gaita F, Borggrefe M, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004;109(1):30–35. doi: 10.1161/01.cir.0000109482.92774.3a. [DOI] [PubMed] [Google Scholar]
- 6.Hong K, Bjerregaard P, Gussak I, Brugada R. Short QT syndrome and atrial fibrillation caused by mutation in KCNH2. Journal of Cardiovascular Electrophysiology. 2005;16(4):394–396. doi: 10.1046/j.1540-8167.2005.40621.x. [DOI] [PubMed] [Google Scholar]
- 7.Priori SG, Pandit SV, Rivolta I, Berenfeld O, Ronchetti E, Dhamoon A, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circulation Research. 2005;96(7):800–807. doi: 10.1161/01.RES.0000162101.76263.8c. [DOI] [PubMed] [Google Scholar]
- 8.Bellocq C, van Ginneken AC, Bezzina CR, Alders M, Escande D, Mannens MM, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation. 2004;109(20):2394–2397. doi: 10.1161/01.cir.0000130409.72142.fe. [DOI] [PubMed] [Google Scholar]
- 9.Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115(4):442–449. doi: 10.1161/circulationaha.106.668392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Templin C, Ghadri JR, Rougier JS, Baumer A, Kaplan V, Albesa M, et al. Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6) European Heart Journal. 2011;32(9):1077–1088. doi: 10.1093/eurheartj/ehr076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abriel H, Zaklyazminskaya EV. Cardiac channelopathies: Genetic and molecular mechanisms. Gene. 2013;517(1):1–11. doi: 10.1016/j.gene.2012.12.061. [DOI] [PubMed] [Google Scholar]
- 12.Hong K, Piper DR, Diaz-Valdecantos A, Brugada J, Oliva A, Burashnikov E, et al. De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero. Cardiovascular Research. 2005;68(3):433–440. doi: 10.1016/j.cardiores.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 13.Maltret A, Wiener-Vacher S, Denis C, Extramiana F, Morisseau-Durand MP, Fressart V, et al. Type 2 short QT syndrome and vestibular dysfunction: Mirror of the Jervell and Lange-Nielsen syndrome? International Journal of Cardiology. 2014;171(2):291–293. doi: 10.1016/j.ijcard.2013.11.078. [DOI] [PubMed] [Google Scholar]
- 14.Wolpert C, Schimpf R, Giustetto C, Antzelevitch C, Cordeiro J, Dumaine R, et al. Further insights into the effect of quinidine in short QT syndrome caused by a mutation in HERG. Journal of Cardiovascular Electrophysiology. 2005;16(1):54–58. doi: 10.1046/j.1540-8167.2005.04470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Extramiana F, Antzelevitch C. Amplified transmural dispersion of repolarization as the basis for arrhythmogenesis in a canine ventricular-wedge model of short-QT syndrome. Circulation. 2004;110(24):3661–3666. doi: 10.1161/01.CIR.0000143078.48699.0C. [DOI] [PubMed] [Google Scholar]
- 16.Anttonen O, Vaananen H, Junttila J, Huikuri HV, Viitasalo M. Electrocardiographic transmural dispersion of repolarization in patients with inherited short QT syndrome. Annals of Noninvasive Electrocardiology. 2008;13(3):295–300. doi: 10.1111/j.1542-474X.2008.00234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Milberg P, Tegelkamp R, Osada N, Schimpf R, Wolpert C, Breithardt G, et al. Reduction of dispersion of repolarization and prolongation of postrepolarization refractoriness explain the antiarrhythmic effects of quinidine in a model of short QT syndrome. Journal of Cardiovascular Electrophysiology. 2007;18(6):658–664. doi: 10.1111/j.1540-8167.2007.00813.x. [DOI] [PubMed] [Google Scholar]
- 18.O’Hara T, Rudy Y. Arrhythmia formation in sub-clinical (“silent”) long QT syndrome requires multiple insults: Quantitative mechanistic study using the KCNQ1 mutation Q357R as example. Heart Rhythm. 2012;9(2):275–282. doi: 10.1016/j.hrthm.2011.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee HC, Rudy Y, Po-Yuan P, Sheu SH, Chang JG, Cui J. Modulation of KCNQ1 alternative splicing regulates cardiac IKs and action potential repolarization. Heart Rhythm. 2013;10(8):1220–1228. doi: 10.1016/j.hrthm.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Hara T, Virág L, Varró A, Rudy Y. Simulation of the undiseased human cardiac ventricular action potential: Model formulation and experimental validation. PLoS Computational Biology. 2011;7(5):e1002061. doi: 10.1371/journal.pcbi.1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heijman J, Volders PG, Westra RL, Rudy Y. Local control of beta-adrenergic stimulation: Effects on ventricular myocyte electrophysiology and Ca(2+)-transient. Journal of Molecular and Cellular Cardiology. 2011;50(5):863–871. doi: 10.1016/j.yjmcc.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sicouri S, Moro S, Litovsky S, Elizari MV, Antzelevitch C. Chronic amiodarone reduces transmural dispersion of repolarization in the canine heart. Journal of Cardiovascular Electrophysiology. 1997;8(11):1269–1279. doi: 10.1111/j.1540-8167.1997.tb01018.x. [DOI] [PubMed] [Google Scholar]
- 23.Benson AP, Aslanidi OV, Zhang H, Holden AV. The canine virtual ventricular wall: A platform for dissecting pharmacological effects on propagation and arrhythmogenesis. Progress in Biophysics and Molecular Biology. 2008;96(1–3):187–208. doi: 10.1016/j.pbiomolbio.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 24.Wu L, Guo D, Li H, Hackett J, Yan GX, Jiao Z, et al. Role of late sodium current in modulating the proarrhythmic and antiarrhythmic effects of quinidine. Heart Rhythm. 2008;5(12):1726–1734. doi: 10.1016/j.hrthm.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu MH, Su MJ, Lue HC. Age-related quinidine effects on ionic currents of rabbit cardiac myocytes. Journal of Molecular and Cellular Cardiology. 1994;26(9):1167–1177. doi: 10.1006/jmcc.1994.1135. [DOI] [PubMed] [Google Scholar]
- 26.Hiraoka M, Sawada K, Kawano S. Effects of quinidine on plateau currents of guinea-pig ventricular myocytes. Journal of Molecular and Cellular Cardiology. 1986;18(10):1097–1106. doi: 10.1016/s0022-2828(86)80296-6. [DOI] [PubMed] [Google Scholar]
- 27.Zhou P, Yang X, Li C, Gao Y, Hu D. Quinidine depresses the transmural electrical heterogeneity of transient outward potassium current of the right ventricular outflow tract free wall. Journal of Cardiovascular Disease Research. 2010;1(1):12–18. doi: 10.4103/0975-3583.59979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel C, Yan GX, Antzelevitch C. Short QT syndrome: From bench to bedside. Circulation: Arrhythmia and Electrophysiology. 2010;3(4):401–408. doi: 10.1161/circep.109.921056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yatani A, Wakamori M, Mikala G, Bahinski A. Block of transient outward-type cloned cardiac K + channel currents by quinidine. Circulation Research. 1993;73(2):351–359. doi: 10.1161/01.res.73.2.351. [DOI] [PubMed] [Google Scholar]
- 30.Kaufman ES. Quinidine in short QT syndrome: An old drug for a new disease. Journal of Cardiovascular Electrophysiology. 2007;18(6):665–666. doi: 10.1111/j.1540-8167.2007.00815.x. [DOI] [PubMed] [Google Scholar]
- 31.Zhang H, Kharche S, Holden AV, Hancox JC. Repolarisation and vulnerability to re-entry in the human heart with short QT syndrome arising from KCNQ1 mutation—A simulation study. Progress in Biophysics and Molecular Biology. 2008;96(1–3):112–131. doi: 10.1016/j.pbiomolbio.2007.07.020. [DOI] [PubMed] [Google Scholar]
- 32.Porta A, Girardengo G, Bari V, George AL, Jr, Brink PA, Goosen A, et al. Autonomic control of heart rate and QT interval variability influences arrhythmic risk in long QT syndrome type 1. Journal of the American College of Cardiology. 2015;65(4):367–374. doi: 10.1016/j.jacc.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Larson ED, St Clair JR, Sumner WA, Bannister RA, Proenza C. Depressed pacemaker activity of sinoatrial node myocytes contributes to the age-dependent decline in maximum heart rate. Proceedings of the National Academy of Sciences. 2013;110(44):18011–18016. doi: 10.1073/pnas.1308477110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaita F, Giustetto C, Bianchi F, Schimpf R, Haissaguerre M, Calo L, et al. Short QT syndrome: Pharmacological treatment. Journal of the American College of Cardiology. 2004;43(8):1494–1499. doi: 10.1016/j.jacc.2004.02.034. [DOI] [PubMed] [Google Scholar]
- 35.Perdomo J, Yan F, Ahmadi Z, Jiang XM, Stocker R, Chong BH. Quinine-induced thrombocytopenia: Drug-dependent GPIb/IX antibodies inhibit megakaryocyte and pro-platelet production in vitro. Blood. 2011;117(22):5975–5986. doi: 10.1182/blood-2010-10-314310. [DOI] [PubMed] [Google Scholar]
- 36.Gima K, Rudy Y. Ionic current basis of electrocardiographic waveforms: A model study. Circulation Research. 2002;90(8):889–896. doi: 10.1161/01.res.0000016960.61087.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Antzelevitch C, Shimizu W, Yan GX, Sicouri S, Weissenburger J, Nesterenko VV, et al. The M cell: Its contribution to the ECG and to normal and abnormal electrical function of the heart. Journal of Cardiovascular Electrophysiology. 1999;10(8):1124–1152. doi: 10.1111/j.1540-8167.1999.tb00287.x. [DOI] [PubMed] [Google Scholar]
- 38.Rees SA, Curtis MJ. Further investigations into the mechanism of antifibrillatory action of the specific IK1 blocker, RP58866, assessed using the rat dual coronary perfusion model. Journal of Molecular and Cellular Cardiology. 1995;27(12):2595–2606. doi: 10.1006/jmcc.1995.0046. [DOI] [PubMed] [Google Scholar]
- 39.Rees SA, Curtis MJ. Specific IK1 blockade: A new antiarrhythmic mechanism? Effect of RP58866 on ventricular arrhythmias in rat, rabbit, and primate. Circulation. 1993;87(6):1979–1989. doi: 10.1161/01.cir.87.6.1979. [DOI] [PubMed] [Google Scholar]
- 40.Sinnecker D, Goedel A, Laugwitz KL, Moretti A. Induced pluripotent stem cell-derived cardiomyocytes: A versatile tool for arrhythmia research. Circulation Research. 2013;112(6):961–968. doi: 10.1161/circresaha.112.268623. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.