

Abstract
Acetaminophen (APAP) overdose is the leading cause of Acute Liver Failure (ALF) with limited treatment options. It is known that liver regeneration following APAP induced ALF is a deciding factor in the final outcome. Previous studies from our laboratory using incremental dose model involving a regenerating (300 mg/kg, APAP300) and a non-regenerating (600 mg/kg, APAP600) dose of APAP in mice have revealed several pro- regenerative pathways that regulate regeneration after APAP overdose. Here we report that DNA damage and repair mechanisms regulate initiation liver regeneration following APAP overdose. Mice treated with non-regenerating APAP600 dose showed prolonged expression of pH2AX, a marker of the DNA double strand break (DSB) than APAP300. In regenerating APAP300 dose treated mice H2AX was rapidly dephosphorylated at Tyr142 indicating timely DNA repair. Expression of several DNA repair proteins was substantially lower in APAP600. Poly (ADP) ribose polymerase (PARP) activation, involved in DNA repair, was significantly higher in APAP300 group compared to APAP600 group. Activation of p53, the major cell cycle checkpoint protein, was significantly higher in APAP600 as demonstrated by substantially higher expression of its target genes. Taken together, these data show that massive DNA double damage occurs in high dose APAP toxicity and lack of prompt DSB repair after APAP overdose leads to prolonged growth arrest, proliferative senescence resulting in inhibited liver regeneration.
Keywords: Liver regeneration, DDR, PARP, p53, PARP, pH2AX, proliferation, injury
Introduction
Acetaminophen (APAP) is widely used analgesic and antipyretic drug present in several over the counter and prescription medications. It is safe at therapeutic doses of ≤ 4 grams per day however; overdose of APAP can cause acute liver injury (ALI), which can progress to acute liver failure (ALF)1. Overdose of APAP is the cause of almost 50% of ALF cases in the US with close to 35% mortality2,3 Despite being the major cause of ALF in Western world, therapeutic options for APAP induced ALF are extremely limited. Several studies in patients and rodents have demonstrated that stimulation of liver regeneration improves survival and prognosis after APAP overdose4–9. Although these studies highlight enhancing liver regeneration in the APAP-induced ALF patients as a plausible therapeutic option, the clinical application is delayed because the mechanisms of liver regeneration that drive liver regeneration after APAP overdose are not entirely known. Especially, the role of DNA damage response (DDR) in regulation of liver regeneration after APAP induced ALI has not been investigated.
DDR involves proteins that sense DNA damage and trigger a repair response to protect the cell. Sensor proteins in DDR sense the damage and send the signal to mediator and effector proteins via activation of apical kinases. Mediator proteins recruit DNA repair effector proteins at the damaged DNA site, which then carry out the repair process10,11. One of the major effector proteins in DDR is p53, which activates cell cycle checkpoints and induce cell cycle arrest till damage is repaired. However, if damage is beyond repair it can activate the cell death pathway12. Previous studies have shown that APAP injury results in nuclear DNA fragmentation preventing the cell proliferation by cell cycle arrest5,13,14.
We investigated the role of DDR in liver regeneration after APAP toxicity using a recently developed incremental dose model in our laboratory that includes comparing signaling between a regenerating (300 mg/kg, APAP300) and a non-regenerating (600 mg/kg, APAP600) dose in mice5. Our studies indicate that APAP overdose results in dose dependent DNA damage but at higher doses the DNA repair mechanisms fail resulting in initiation of cellular senescence and inhibition of liver regeneration. These studies have revealed a novel mechanism that connects cellular injury to initiation of liver regeneration after APAP overdose.
Methods
Animals, Treatment and Tissue Harvesting
All animal experiments were performed in compliance with protocols approved by the Institutional Animal Care and Use Committee at University of Kansas Medical Center. The details of incremental dose model have been previously published. Briefly, two to three month old male C57BL/6 mice were fasted overnight and injecting with either 300 or 600 mg/kg APAP intraperitoneally (i.p. dissolved in warm saline). Mice (n = 5 to 7) were sacrificed at 0, 3, 6, 12, 24, 48, 72 and 96 hr after APAP treatment and blood and livers were collected. Parts of liver tissue were processed separately to obtain paraffin sections, frozen sections, RNA samples, nuclear, cytoplasmic and RIPA total protein extracts as described previously15. Liver injury was assessed by serum alanine aminotransferase (ALT) activity. Liver regeneration was assessed using PCNA analysis as described before.
Antibodies
The following antibodies were used for analyses: (#4588) KU70, (#9532) PARP, (#9718) p-H2AX Ser139, (#2524) p53, (#9284) p-p53 S15 from Cell Signaling Technologies (Danvers, MA), (SC9051) DNAPkc, (SC1485) KU80, (SC166488) XLF, (SC8285) XRCC4, (SC28232) DNA Lig4, (SC642) BRCA1 from Santacruz Biotech. (#07-1590) p-H2Ax Tyr 142 from EMD Millipore Corporation (Billerica, MA), (PP-H1415-00) HNF4α from Perseus proteomics, (nb100-904) 53BP1 from Novus Biolabs and (#1020) PAR from Tulips biolabs. All Alxa fluor secondary antibodies were purchased from Invitrogen, Thermo Fisher and HRP conjugated secondary antibodies were purchased from Cell Signaling Technologies (Danvers, MA).
Western Blotting
Protein estimation of RIPA and nuclear extracts was done by BCA method and western blot analysis was performed using pooled protein extracts as described before15.
Immunofluorescence staining
Fresh-frozen liver sections (5 μm thick) were used to detect pH2AX Ser 139 immunofluorescence as described before16.
Real Time PCR
Total RNA was isolated from APAP300 and APAP600 livers using Trizol method according to the manufacturer’s protocol (Sigma, St. Louis, MO) and converted to cDNA as previously described4. Gene expression of various genes was determined by comparing mRNA levels from APAP treated groups at different time point with 0 hr control group using Real Time PCR analysis. SYBR Green technology was used for Real time PCR analysis on the Applied Biosystems Prism 7300 Real-time PCR Instrument according to manufacturer’s protocol. 18s gene expression in the same samples was used for data normalization. Primers used for real time PCR are listed in Table 1.
Table 1.
Primers used in this study
| Gene | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| GADD45α | CCGAAAGGATGGACACGGTG | TTATCGGGGTCTACGTTGAGC |
| GADD45β | CAACGCGGTTCAGAAGATGC | GGTCCACATTCATCAGTTTGGC |
| GADD45γ | GGGAAAGCACTGCACGAACT | AGCACGCAAAAGGTCACATTG |
| BTG2 | ATGAGCCACGGGAAGAGAAC | GCCCTACTGAAAACCTTGAGTC |
| PAI-1 | TTCAGCCCTTGCTTGCCTC | ACACTTTTACTCCGAAGTCGGT |
| 18S | TTGACGGAAGGGCACCACCAG | GCACCACCACCCACGGAATCG |
Statistical Analysis
Data are shown as mean ± SEM. Student’s T-test was used for statistical analysis. Difference between groups was considered statistically significant at P<0.05 and indicated by * in graphs.
Results
Sustained liver injury and inhibited liver regeneration following higher dose of APAP
Liver injury after APAP300 and APAP600 treatment was assessed using serum ALT and histopathological analysis of liver tissue over 0 to 96 hr time course5. At both doses, serum ALT activity was increased and peaked at 12 hr after treatment. In APAP300 treated mice, serum ALT activity regressed after 24 hr and returned to normal by 72 and 96 hr. However, in APAP600 treated mice ALT activity remained higher up to 24 hr and later decreased but was persistently higher until 96hr after APAP treatment as compared to APAP300 (Fig. 1A). All mice receiving APAP300 dose recovered from injury, whereas mice with APAP600 treatment showed 25% lethality and remaining mice had sustained injury up to 96 hr5.
Figure 1.
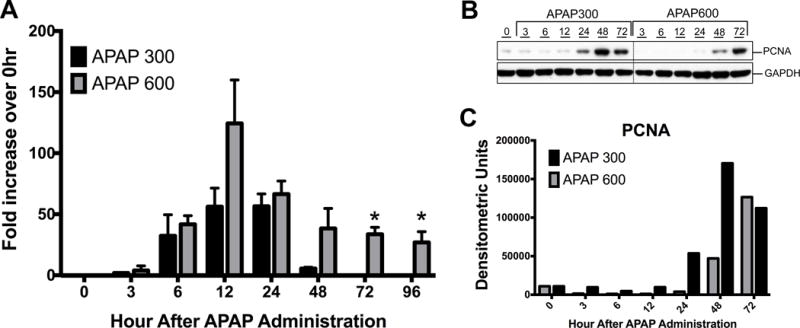
Sustained liver injury and inhibited liver regeneration following higher dose of APAP. (A) Liver injury analysis by serum ALT levels after APAP treatment. Shown as fold increase in ALT levels compare to 0hr. (B) Western blot analysis of PCNA in whole liver extract. (C) Densitometric analysis of PCNA western blot. * p<0.05 (APAP300 vs APAP600)
To determine the difference in liver regeneration after two doses of APAP, we determined expression of PCNA in mice liver5. Western blot analysis of PCNA revealed significantly delayed and reduced cell proliferation after APAP600 dose as compared to APAP300 dose (Fig. 1B and 1C). In APAP300 group, significant increase in PCNA was observed from 24 hr up to 72 hr. However in APAP600 group, PCNA expression was delayed to 48 hr and it was significantly lower than APAP300 group.
Prolonged DNA DSB and reduced repair protein expression after higher dose of APAP
To examine the mechanism underlying delayed and attenuated cell proliferation in higher dose mice with regard to DNA replication, we determined most lethal form of replication stress i.e. DNA double strand break (DSB). DSB was determined using western blot analysis and immunofluorescence detection of Ser 139 phosphorylation of histone 2AX (pH2AX), a hallmark of DSB. After both APAP300 and APAP600 doses, increased Ser 139 pH2AX expression was observed starting from 6hr (Fig. 2A-B). In APAP300 group pH2AX-Ser139 expression peaked at 12 hr, remained high at 24 hr and returned to control level at 48 and 72 hr. In APAP600 group pH2AX Ser 139 induction peaked at 12 hr and remained high up to 72 hr. Further, we studied tyrosine 142 phosphorylation (and dephosphorylation) on pH2Ax, a signal involved in recruitment of DNA DSB repair proteins. Previous studies have shown that pH2AX de-phosphorylation at Tyr142 is a critical in recruitment of repair proteins17,18. Western blot analysis of nuclear extracts showed an initial increase in pH2AX Tyr142 phosphorylation up to 24 hr and a significantly decreased at 48 and 72 hr in APAP300 group indicating initiation of DNA repair. In contrast, APAP600 group mice had higher levels of pH2AX Tyr142 throughout the time course (Fig. 2A–C). To determine which cells exhibit pH2AX after APAP overdose, we performed double immunofluorescence staining with pH2AX and hepatocyte marker HNF4α. Immunofluorescence staining revealed that hepatocytes immediately surrounding APAP induced necrotic zone were positive for DSB (Fig. 2C).
Figure 2.
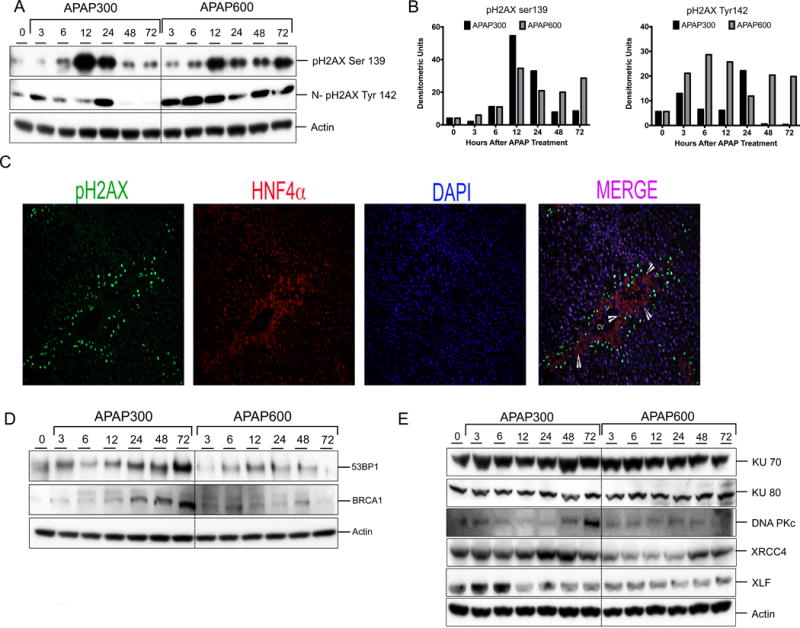
Prolonged DNA DSB and reduced repair protein expression after higher dose of APAP. (A) Western blot analysis of phos-H2AX Ser139 using total liver extract and phos-H2AX Tyr142 using nuclear extract. (B) Bar graphs showing densitometric analysis of pH2AX ser139 and Tyr142 Western blots (C) Representative immunofluorescence staining for pH2AX Ser139 (green), HNF4α (red) and DAPI (blue) for cell nuclei. Arrowheads are pointing to necrotic cells. (D) Western blot analysis of DNA repair mediator proteins 53BP1, BRCA1in total liver extract (E) Western blot analysis of DNA repair effector proteins KU70, KU80, DNAPkc, XRCC4, XLF, Lig4 using total liver extract.
BRCA1 and 53BP1 are critical mediator proteins involved in DDR, which can interact with broken DNA ends and help binding of DNA repair effector proteins at the damaged DNA site19,20. A marked increase in 53BP1 and BRCA1 protein levels was seen from 12 hr up to 72 hr after APAP300 treatment as compare to 0 hr control. In contrast, 53BP1 and BRCA1 protein expression was down regulated after APAP600 treatment (Fig. 2D).
We have previously demonstrated that in APAP600 group cells were arrested at G0/G1 phase5. In G0/G1 phase of the cell cycle, DSB is repaired mainly executed by non-homologous end joining (NHEJ)21. Therefore, we studied NHEJ repair pathway proteins to further examine the difference in DNA repair between APAP300 and APAP600 groups. We determined expression of proteins involved in NHEJ repair including KU70, KU80, DNA Pkc, XRCC4, XLF, and DNA Lig4 using Western blot analysis (Fig. 2D). The data indicated significant upregulation of XRCC4, XLF, DNA Pkc and Lig4 in APAP300, all of which were downregulated in APAP600 group as compared to the 0 hr control (Fig. 2D). We did not observe any difference in KU70 and KU80 protein levels between APAP300 and APAP600 group.
These data suggest that after higher dose of APAP there is reduced DNA repair protein expression and inadequate chromatin modification resulting in impaired DSB repair.
Reduced PARP activation following higher dose of APAP
Another critical protein in DDR is poly (ADP) ribose polymerase (PARP-1) that can sense the DNA damage and mediate the stress response by poly-ADPribosylation of nuclear proteins. It results in chromatin remodeling, which favors DNA repair22. We did not observe any difference in total PARP1 protein expression between APAP300 and APAP600 group mice. However, nuclear PARP1 (N-PARP1) was significantly downregulated in APAP600 treated mice as compare to APAP300 treatment (Fig. 3A–B). Next we determined PARP activation by staining for PARylated proteins using immunohistochemistry (Fig. 3C). Following APAP300 dose PARP activation was observed in time dependent manner. No PAR staining was evident till 12 hr (data not shown), but significant nuclear PAR staining was evident from 12 to 72 hr after APAP300 treatment. PAR staining intensity significantly increased at 24 hr, sustained till 72 hr and disappeared by 96 hr after APAP300 treatment. On the contrary, in APAP600 treated mice, at 12 and 24 hr very few cells stained positive for PAR with low intensity. At 48 hr many PAR positive cells were observed however staining intensity was weak as compare to APAP300. PAR staining disappeared by 72 hr following APAP600 treatment. These data indicate that PARP activation is significantly higher and sustained following APAP300 treatment however it is delayed and weak following APAP600 treatment.
Figure 3.
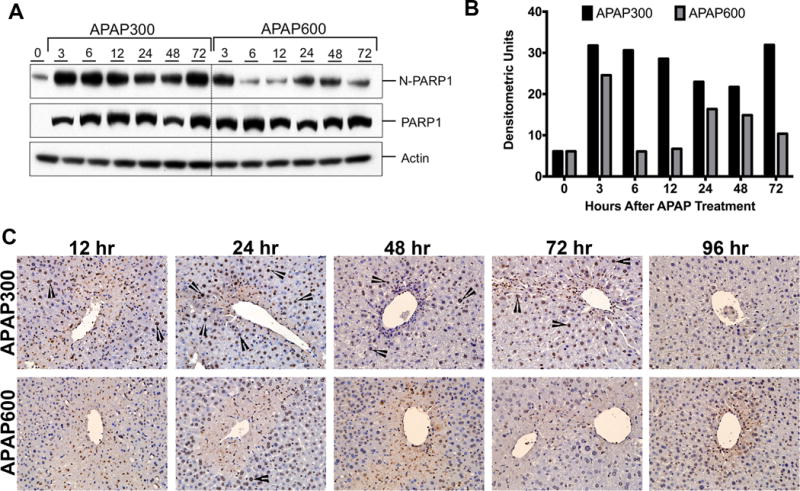
Delayed activation of PARP following APAP600 treatment. (A) Western blot analysis and (B) densitometric analysis of the PARP1 blots in total liver extract and nuclear extract (C) Immunohistochemical analysis of PARylated proteins. Arrowheads point to nuclear PAR staining.
Increased transcriptional activation of p53 at higher dose of APAP
p53 is the major effector protein of DDR pathway, which can activate cell cycle checkpoint and arrest the cell cycle till damage is repaired. Stabilization and activation of p53 protein has been shown to play an important role in many cellular processes such as cell cycle arrest, cell senescence, cell death, cell metabolism etc23,24. We determined p53 activation by measuring changes in p53 protein by Western blotting and quantifying mRNA for several p53 target genes after APAP overdose. Western blot analysis of APAP300 and APAP600 samples indicated marked increase in p53 stabilization after APAP treatment (Fig. 3A–B). Interestingly, p53 protein levels were significantly higher in APAP600 group as compare to APAP300 from 6hr up to 72hr. APAP600 treated mice exhibited 6-fold higher p53 expression at 6 hr after APAP treatment. Similarly, Ser 15 phosphorylation of p53, which indicates activation of p53, was significantly higher in APAP600 at all time points (Fig. 3A). Real time PCR analysis showed that expression of several p53 responsive genes involved in cell cycle inhibition (GADD45α, GADD45β, GADD45γ, BTG2) (Fig. 3C), and cell senescence (PAI1) (Fig. 3D) increased consistently with increased p53 activity. Previously we have demonstrated that p21 mRNA is significantly higher in the APAP600, the non-regenerating dose as compared to APAP300, the regenerating dose5. A marked increase in all cell cycle inhibitor and cell senescence gene expression was seen in both groups after APAP treatment. In APAP300 group, mRNA levels of all these genes were significantly reduced from 24hr to 96hr. However, these cell cycle inhibitor and senescence gene expressions was sustained and significantly higher in APAP600 group. These data indicate that sustained activation of p53 after APAP600 treatment results in cell cycle arrest, replicative senescence and may be cell death.
Discussion
APAP is a safe analgesic and antipyretic drug when taken at recommended daily dose. It is safely metabolized in liver and excreted in urine. However, overdose of APAP causes ALI and even ALF, which is a number one cause of ALF in USA and UK.2,25 The mechanism of APAP toxicity involves generation of ROS, release of endonucleases, extensive DNA fragmentation and subsequent cell necrosis26. In response to injury, healthy hepatocytes surrounding the necrotic zone divide rapidly and help repair the injured liver4,5,27. In previous study we have demonstrated that liver regeneration is stimulated rapidly following treatment with 300 mg/kg of APAP (regenerating dose) but it is significantly delayed and blunted after a 600 mg/kg of APAP (non-regenerating dose)5. The main reason behind this is the cells that surround the necrotic zone, which normally undergo proliferation to fuel liver regeneration are arrested in mice treated with the higher non-regenerating dose of APAP. Our previous studies have shown that the reason behind the delayed regeneration following APAP600 treatment is not lack of enough number of viable cells. Even after this high dose only about 40% hepatocytes at the maximum undergo necrosis leaving up to 60% hepatocytes intact to divide and replace the dead cells. However, these cells are extremely stressed due to ongoing injury and are incapable of entering cell cycle as shown by G0 to G1 arrest in the previous work. However, the mechanisms behind this cell cycle arrest in these stressed hepatocytes in the APAP600 treated mice are not completely known.
In the present study, we determined if this cell cycle arrest at non-regenerating doses is due to enhanced DNA damage and blunted DNA repair processes. Our data indicate that DNA damage occurs following both the regenerating (APAP300) and non-regenerating (APAP600) doses of APAP but DNA repair process is significantly inhibited following treatment with non-regenerating dose of APAP. Furthermore, immunofluorescence data revealed that the hepatocytes immediately next to the necrotic zone exhibit extensive DNA damage. These are the same hepatocytes that are required to proliferate in order to ensue liver regeneration. Whereas previous studies have shown that DNA damage is part of necrotic cell death after APAP, our data are the first to demonstrate that DNA damage and subsequent lower DNA repair inhibit liver regeneration, repair and recovery after APAP overdose.
Because we observed sustained DSB in non-regenerating animals, we further studied whether DSB repair is inactive and cells are arrested due to failure to replicate damaged DNA in APAP600. De-phosphorylation of p-H2AX at Tyr 142 is one of the chromatin modifications that facilitate DSB repair17,18. We observed that Tyr142 phosphorylation was maintained for significantly longer time following treatment with the non-regenerating APAP600 dose, which would delay recruitment of DNA repair proteins. Additionally, higher dose of APAP suppressed expression of mediator proteins 53BP1 and BRCA1 (Fig. 2C). Expression of several DSB repair effector proteins was suppressed at non-regenerating doses. Because DSB repair is the collective effort of various proteins, lack of several critical proteins will result in delayed or completely suppressed DAB repair in APAP600 treated mice. Furthermore, dephosphorylation of H2AX at Tyr 142, which is required for easy access of repair proteins to DSB sites was significantly lower in APAP600 treated mice. This may have made damage site inaccessible for the repair protein in non-regenerating dose treated mice. These results collectively show that DSB repair is deregulated in non-regenerating animals leading to sustain DSB.
Further studies showed that APAP600 treated mice exhibited reduced nuclear PARP1 levels in spite of similar amounts of total PARP1 levels as compared to APAP300. Normally PARP1 is present in the nucleus where it is involved in protein PARylation28,29. Our data indicate that 6 hr following APAP600, PARP1 is rapidly removed from the nucleus while PARP levels are maintained in APAP300 treated mice. This was consistent with significantly decreased PARylation of nuclear proteins in APAP600. However, we did not see any significant difference in total PARP levels in either APAP300 or APAP600 treated mice over the time course. These data indicate that following high dose of APAP (APAP600), PARP may be actively exported out of the nucleus, reducing its nuclear activity. The mechanism of this enhanced nuclear transport is not clear. In agreement with previous study30, these data show that PARP activation is not associated with increased liver injury following APAP toxicity. However these data suggest that PARP activation is a critical step in DSB repair following APAP overdose. Further studies are required to delineate the mechanism of nuclear export of PARP1 following higher dose of APAP.
p53 is a primary effector protein that plays a critical role in cell cycle regulation during DDR. Under stress conditions p53 is stabilized and activated through various posttranslational modifications. One such modification is phosphorylation at Ser15 that leads to transcriptional activation of p53. Activated p53 regulates plethora of downstream gene expression involved in cell cycle inhibition and senescence24. Our data indicate significantly higher and sustained activation of p53 following APAP600 dose (Fig. 4A). The expression of p53 target genes (Cell cycle inhibitor- GADD45α, GADD45β, GADD45γ, cell senescence PAI1, p21) increased after both APAP300 and APAP600 but it was significantly higher in APAP600 dose at all time points. Previous studies indicate that moderate activation of p53 results in cell cycle arrest that permit cell to repair the DNA damage, however excessive and sustained activation of p53 results in replicative senescence and cell death23. Further, BIRC5, a negative target of p53 was repressed at all time points in non-regenerative dose consistent with higher p53 activation. These data suggest that moderate activation of p53 at regenerative dose results in transient cell cycle arrest whereas, sustained excessive activation of p53 at non-regenerative dose may cause prolonged growth arrest, replicative senescence or cell death. Further studies are required to demonstrate the exact role of p53 and some of these target genes in liver regeneration after APAP overdose.
Figure 4.
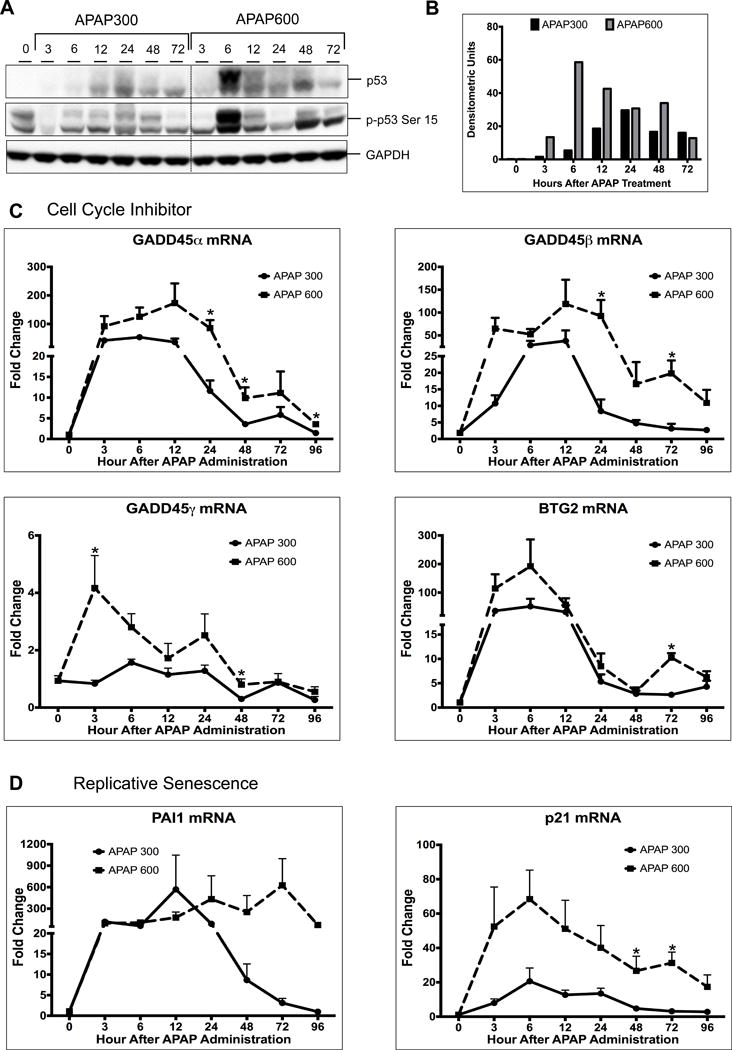
Activation of p53 is higher following APAP600 treatment. (A) Western blot analysis of total p53 and phospho-p53 Ser15 using total liver extract (B) Bar graph showing densitometric analysis of total p53 Western blots (C) Real time PCR analysis of p53 target genes regulating cell cycle inhibition Gadd45α, Gadd45β, Gadd45γ, and BTG2 (D) replicative senescence PAI1. *p<0.05 (APAP300 vs APAP600)
In conclusion, our study indicates that DNA damage and repair response plays a critical role in deciding whether liver regeneration will be ‘timely’ or ‘delayed’ following APAP overdose. At high doses of APAP, DSB repair is impaired resulting in inhibited liver regeneration. This study is the first to highlights the complex signaling pathway involved in DNA DSB repair in regulation of liver regeneration following APAP induced ALI. These data also indicate that improving DNA repair may have therapeutic benefit after APAP overdose.
Acknowledgments
Financial Support: These studies were supported by NIH-COBRE (P20 RR021940-03), NIEHS Toxicology Training Grant (T32ES007079-34) and NIDDK R0198414
References
- 1.Shiffman S, Rohay JM, Battista D, Kelly JP, Malone MK, Weinstein RB, Kaufman DW. Patterns of acetaminophen medication use associated with exceeding the recommended maximum daily dose. Pharmacoepidemiol Drug Saf. 2015;24(9):915–21. doi: 10.1002/pds.3830. [DOI] [PubMed] [Google Scholar]
- 2.Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, Hynan LS, Reisch JS, Schiodt FV, Ostapowicz G, Shakil AO, et al. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology. 2005;42(6):1364–72. doi: 10.1002/hep.20948. [DOI] [PubMed] [Google Scholar]
- 3.Ostapowicz G, Fontana RJ, Schiodt FV, Larson A, Davern TJ, Han SH, McCashland TM, Shakil AO, Hay JE, Hynan L, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med. 2002;137(12):947–54. doi: 10.7326/0003-4819-137-12-200212170-00007. [DOI] [PubMed] [Google Scholar]
- 4.Apte U, Singh S, Zeng G, Cieply B, Virji MA, Wu T, Monga SP. Beta-catenin activation promotes liver regeneration after acetaminophen-induced injury. Am J Pathol. 2009;175(3):1056–65. doi: 10.2353/ajpath.2009.080976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhushan B, Walesky C, Manley M, Gallagher T, Borude P, Edwards G, Monga SP, Apte U. Pro-regenerative signaling after acetaminophen-induced acute liver injury in mice identified using a novel incremental dose model. Am J Pathol. 2014;184(11):3013–25. doi: 10.1016/j.ajpath.2014.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Donahower BC, McCullough SS, Hennings L, Simpson PM, Stowe CD, Saad AG, Kurten RC, Hinson JA, James LP. Human recombinant vascular endothelial growth factor reduces necrosis and enhances hepatocyte regeneration in a mouse model of acetaminophen toxicity. J Pharmacol Exp Ther. 2010;334(1):33–43. doi: 10.1124/jpet.109.163840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu B, Colletti LM. Stem cell factor and c-kit are involved in hepatic recovery after acetaminophen-induced liver injury in mice. Am J Physiol Gastrointest Liver Physiol. 2008;295(1):G45–G53. doi: 10.1152/ajpgi.00024.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.James LP, Lamps LW, McCullough S, Hinson JA. Interleukin 6 and hepatocyte regeneration in acetaminophen toxicity in the mouse. Biochem Biophys Res Commun. 2003;309(4):857–63. doi: 10.1016/j.bbrc.2003.08.085. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt LE, Dalhoff K. Alpha-fetoprotein is a predictor of outcome in acetaminophen-induced liver injury. Hepatology. 2005;41(1):26–31. doi: 10.1002/hep.20511. [DOI] [PubMed] [Google Scholar]
- 10.Sulli G, Di Micco R, d’Adda di Fagagna F. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat Rev Cancer. 2012;12(10):709–20. doi: 10.1038/nrc3344. [DOI] [PubMed] [Google Scholar]
- 11.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408(6811):433–9. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 12.Carvajal LA, Manfredi JJ. Another fork in the road–life or death decisions by the tumour suppressor p53. EMBO Rep. 2013;14(5):414–21. doi: 10.1038/embor.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ray SD, Sorge CL, Raucy JL, Corcoran GB. Early loss of large genomic DNA in vivo with accumulation of Ca2+ in the nucleus during acetaminophen-induced liver injury. Toxicol Appl Pharmacol. 1990;106(2):346–51. doi: 10.1016/0041-008x(90)90254-r. [DOI] [PubMed] [Google Scholar]
- 14.Shen W, Kamendulis LM, Ray SD, Corcoran GB. Acetaminophen-induced cytotoxicity in cultured mouse hepatocytes: effects of Ca(2+)-endonuclease, DNA repair, and glutathione depletion inhibitors on DNA fragmentation and cell death. Toxicol Appl Pharmacol. 1992;112(1):32–40. doi: 10.1016/0041-008x(92)90276-x. [DOI] [PubMed] [Google Scholar]
- 15.Wolfe A, Thomas A, Edwards G, Jaseja R, Guo GL, Apte U. Increased activation of the Wnt/beta-catenin pathway in spontaneous hepatocellular carcinoma observed in farnesoid X receptor knockout mice. J Pharmacol Exp Ther. 2011;338(1):12–21. doi: 10.1124/jpet.111.179390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walesky C, Gunewardena S, Terwilliger EF, Edwards G, Borude P, Apte U. Hepatocyte-specific deletion of hepatocyte nuclear factor-4alpha in adult mice results in increased hepatocyte proliferation. Am J Physiol Gastrointest Liver Physiol. 2013;304(1):G26–37. doi: 10.1152/ajpgi.00064.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cook PJ, Ju BG, Telese F, Wang X, Glass CK, Rosenfeld MG. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature. 2009;458(7238):591–596. doi: 10.1038/nature07849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiao A, Li H, Shechter D, Ahn SH, Fabrizio LA, Erdjument-Bromage H, Ishibe-Murakami S, Wang B, Tempst P, Hofmann K, et al. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature. 2009;457(7225):57–62. doi: 10.1038/nature07668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol. 2014;15(1):7–18. doi: 10.1038/nrm3719. [DOI] [PubMed] [Google Scholar]
- 20.Jackson SP. Sensing and repairing DNA double-strand breaks. Carcinogenesis. 2002;23(5):687–96. doi: 10.1093/carcin/23.5.687. [DOI] [PubMed] [Google Scholar]
- 21.Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47(4):497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 22.Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell. 2010;39(1):8–24. doi: 10.1016/j.molcel.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137(3):413–31. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 24.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9(5):402–12. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 25.Bernal W, Wendon J. Acute liver failure. N Engl J Med. 2013;369(26):2525–34. doi: 10.1056/NEJMra1208937. [DOI] [PubMed] [Google Scholar]
- 26.Jaeschke H, Bajt ML. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol Sci. 2006;89(1):31–41. doi: 10.1093/toxsci/kfi336. [DOI] [PubMed] [Google Scholar]
- 27.Bajt ML, Knight TR, Farhood A, Jaeschke H. Scavenging peroxynitrite with glutathione promotes regeneration and enhances survival during acetaminophen-induced liver injury in mice. J Pharmacol Exp Ther. 2003;307(1):67–73. doi: 10.1124/jpet.103.052506. [DOI] [PubMed] [Google Scholar]
- 28.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7(7):517–28. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 29.Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol. 2012;13(7):411–24. doi: 10.1038/nrm3376. [DOI] [PubMed] [Google Scholar]
- 30.Cover C, Fickert P, Knight TR, Fuchsbichler A, Farhood A, Trauner M, Jaeschke H. Pathophysiological role of poly(ADP-ribose) polymerase (PARP) activation during acetaminophen-induced liver cell necrosis in mice. Toxicol Sci. 2005;84(1):201–8. doi: 10.1093/toxsci/kfi065. [DOI] [PubMed] [Google Scholar]