

Summary
Daclizumab is a humanized monoclonal antibody (mAb) that binds to the interleukin-2 (IL-2) receptor α-chain (IL-2Rα; CD25) and blocks its interaction with IL-2, thus preventing the formation of high affinity IL-2 receptor (IL-2R). Because activated T cells upregulate high affinity IL-2R and because IL-2 is used to grow activated T cells in vitro, daclizumab was envisioned to selectively inhibit activated T cells. Under this assumption, daclizumab was successfully developed as add-on therapy to standard immunosuppressive regimen for prevention of acute allograft rejection in renal transplantation. After its regulatory approval for transplantation, daclizumab was tested in inflammatory uveitis and multiple sclerosis (MS) and in both diseases daclizumab reproducibly inhibited target organ inflammation. Mechanistic studies in MS showed that the mechanism of action (MOA) of daclizumab is surprisingly broad and that the drug has many unanticipated effects on the innate immunity. Specifically, daclizumab modulates the development of innate lymphoid cells (ILCs) by driving differentiation of ILC precursors away from pro-inflammatory lymphoid tissue inducer cells (LTi) towards immunoregulatory CD56bright NK cells, leading to their expansion and activation. Activated CD56bright NK cells migrate to the intrathecal compartment in MS and regulate autoreactive T cells via cytotoxicity. Finally, daclizumab also severely restricts initial steps of activation of T cells, by blocking trans-presentation of IL-2 by mature dendritic cells (mDCs) to antigen-specific T cells.
In conclusion, daclizumab therapy, which has proven its clinical efficacy in Phase III trials for relapsing-remitting MS (RRMS), has a unique MOA that does not limit migration of immune cells into the target tissue, but rather provides complex immunomodulatory effects with resultant inhibition of central nervous system (CNS) inflammation in MS.
Keywords: multiple sclerosis, autoimmunity, biological therapy, IL-2, CD25
Introduction
Interleukin 2 (IL-2) has been called “T cell growth factor” (Ruscetti, Morgan et al. 1977, Mizel and Farrar 1979), because since its discovery it has been used for T cell expansion in vitro. Upon activation of T cell receptor (TCR), T cells upregulate on their surface heterotrimeric “high affinity” IL-2 receptor (IL-2R; Figure 1). When the surface expression of IL-2R peaks (i.e. ~48–72h post-stimulation), activated naïve T cells also start producing large quantities of IL-2. Consequently, it was believed that this autocrine IL-2 signal is required for clonal expansion of activated T cells. As the induction of IL-2R on activated T cells is proportional to the strength of the TCR signal (Feinerman, Jentsch et al. 2010), T cells that receive higher antigen-specific stimulus compete better for subsequent IL-2 signal. This observation further reinforced the idea that IL-2 promotes T cell immunity. As a consequence, it was presumed that blockade of IL-2 signaling, for example by CD25-blocking therapies, would inhibit T cell effector functions (Waldmann, Kozak et al. 1987, Waldmann and O’Shea 1998, Waldmann 2002). The first such biological agent targeting IL-2 signaling pathway was daclizumab (Queen, Schneider et al. 1989).
Figure 1:
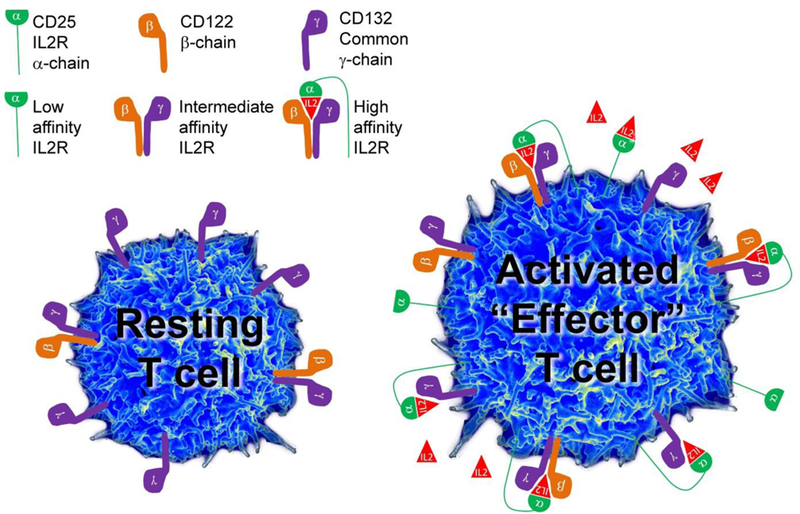
Schematics of the three IL-2 receptors (IL-2R) and their expression on the surface of resting, versus activated (effector) T cells.
IL-2 receptors and their expression on resting and activated “effector” T cells
The IL-2R system consists three chains, different combinations of which constitute 3 receptors that differ in their binding affinity to IL-2 and in their distribution on resting versus activated immune cells. The high affinity IL-2R consists of all three chains: two signaling molecules, γ-chain (CD132) and β-chain (CD122), and the non-signaling α-chain (CD25; Figure 1) (Wang, Rickert et al. 2005). The γ-chain is also called common γ-chain (γc), because it is utilized by several cytokines (IL-2, IL-4, IL-7, IL-15 and IL-21), while the β-chain is shared by two closely-related cytokines, IL-2 and IL-15 (Waldmann, Tagaya et al. 1998). Due to competition for limited quantities of different cytokines, this sharing of signaling chains has important functional consequences for cellular functions.
Constitutive expression of γc on resting T cells underlies their responsiveness to cytokines that mediate T cell homeostasis and survival, such as IL-7. Resting human T cells also express low levels of IL-2Rβ-chain, allowing them to receive IL-15 signal, and, under conditions of IL-2 abundance, also IL-2 signal. However, only a subgroup of CD4+ T cells called T-regulatory cells (T-regs), which are dependent on the transcriptional factor FoxP3, express high levels of CD25 in the resting state. Therefore, only resting T-regs are capable of binding low concentrations of IL-2, which is required for their in vivo survival, their homeostatic proliferation (because they have low levels of IL-7 receptor alpha chain) and their immunoregulatory functions (Setoguchi, Hori et al. 2005). Because of their expression of high-affinity IL-2R, T-regs can successfully “steal” the limited amounts of IL-2 secreted by weakly stimulated T cells, which represents one of the several mechanisms of T-reg-mediated immunoregulation of effector T cells (Feinerman, Jentsch et al. 2010). Expression of β- and γ-chain of IL-2R, which together form the “intermediate affinity IL-2R (Figure 1)” is sufficient to mediate IL-2 signaling when IL-2 concentrations are relatively high (Kd = 1nM). However, T cells that express CD25 can respond to 10–100-fold lower concentrations of IL-2 (Kd = 10pM; (Rickert, Wang et al. 2005)), explaining why T-regs win the tug-of-war with weakly stimulated effector T cells for the limited concentrations of IL-2 (Feinerman, Jentsch et al. 2010). The expression of IL-2Rβ-chain on resting T cells is low in comparison to γ-chain (Figure 1). Because relatively high concentrations of IL-2 are required to trigger intermediate affinity IL-2R, in resting state the γ-chain is preferentially paired with IL-7Rα-chain, mediating survival and homeostatic proliferation of resting naïve and memory T cells.
CD25 itself has low affinity for IL-2 (Kd = 10 nM) and therefore this non-signaling chain is called the “low affinity IL-2R” (Figure 1) (Rickert, Wang et al. 2005), even though in reality this receptor cannot transmit any signal. In fact, the only known role of CD25 is facilitation of IL-2 capture and therefore assembly of high affinity IL-2R. This can occur “in-cis”, i.e. when CD25 is expressed on the same cell that expresses intermediate affinity IL-2R, or “in-trans”, when resting T cells expressed only intermediate affinity IL-2R and CD25 is expressed on activated dendritic cell (DC). In this case, activated DC uses its CD25 to capture IL-2 that it released to the immune synapse with T cell during antigen (Ag) presentation, to “feed” resting T cells the IL-2 by complimenting intermediate affinity IL-2R on the T cell “in-trans” (Figure 2).
Figure 2:
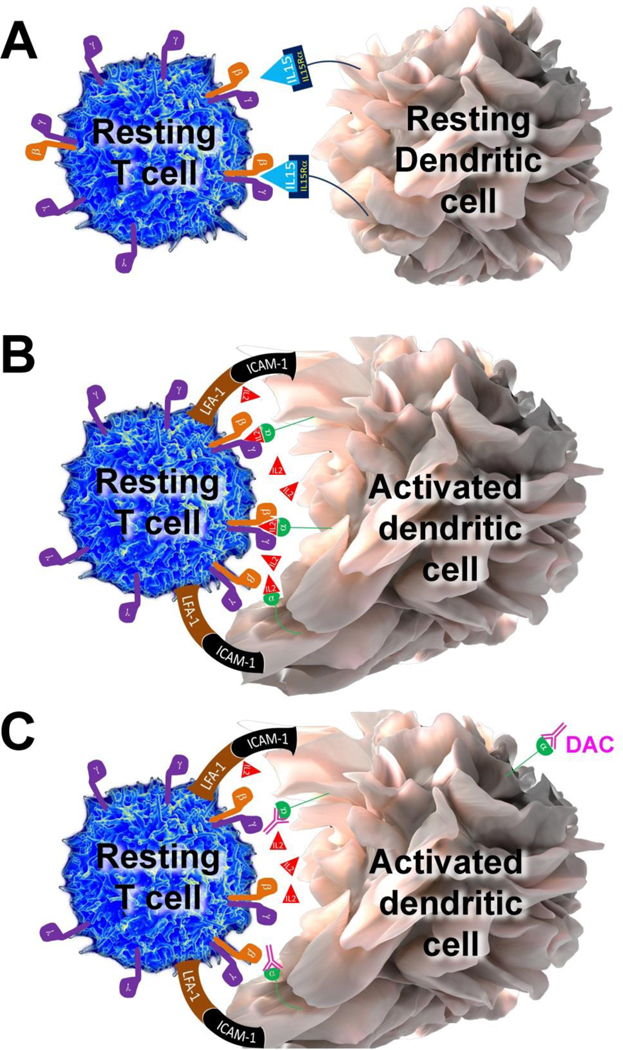
Trans-presentation of IL-15 and IL-2
A. Because of very high affinity between IL-15Rα chain for IL-15, IL-15Rα chain can easily capture and “hold” released IL-15 for long time-periods. This IL-15Rα/IL-15 complex can be then easily trans-presented to intermediate affinity IL-2/IL-15 receptor, expressed on T cells or NK cells.
B. In contrast, CD25 (IL-2Rα chain) has only low affinity for IL-2, so it is highly unlikely that CD25 would be able to effectively capture small concentrations of IL-2 and create stable CD25/IL-2 complexes for trans-presentation, if IL-2 can easily diffuse to the environment. However, if instead the IL-2 is released into the synaptic cleft, which represent an enclosed space between peripheral supramolecular activation cluster formed by adhesion molecules such as lymphocyte function associated antigen-1 (LFA-1) and intercellular adhesion molecule-1 (ICAM-1), then its diffusion is limited and high IL-2 concentrations can be achieved. Under those circumstances, CD25 expressed on the surface of dendritic cell (DC), which formed stable immune synapse (IS) with antigen-specific T cell, can effectively capture released IL-2 and trans-present it to the T cell at the time when T cell does not yet express CD25. This cytokine signal (Signal 3), delivered concomitantly with the Signal 1, provided by T cell receptor (TCR) specifically recognizing peptide loaded on the major histocompatibility complex (MHC) and the co-stimulatory Signal 2 (e.g. provided by interaction of CD28 with CD80 or CD86) seems to be necessary for efficient activation of human T cells and their differentiation to cytokine-producing effector cells.
C. When CD25 on the DC is blocked by daclizumab, then primed T cell cannot receive IL-2 signal (Signal 3) concomitantly with its Signal 1 and 2, resulting in sub-optimal stimulation of T cell. The functional consequences are inhibition of antigen-specific T cell activation, formation of antigen-specific effector and memory T cells.
Upon activation, T cells strongly upregulate both IL-2Rβ-chain and CD25. This allows T cell to form large number of high-affinity IL-2R on its surface and thus respond to small quantities of IL-2. Such large numbers of high affinity IL-2R sequester most of the γ-chain, making effector T cell relatively insensitive to IL-7 and other γ-chain-signaling cytokines. This situation is further exacerbated by down-modulation of IL-7Rα-chain upon T cell activation. A T cell that receives strong IL-2 signal, but lacks IL-7 signal can proliferate and exert many effector functions, but is destined to die when the IL-2 levels become scarce (i.e. towards the end of the immune response). This phenomenon is often called activation-induced cell death (AICD). In contrast, T cells that become activated at the time when IL-2 concentrations are scarce do not undergo the same level of upregulation of IL-2R and down-modulation of IL-7R and they will preferentially survive at the end of the immune response as “memory” T cells.
Development of daclizumab as a therapeutic
Daclizumab is a humanized monoclonal antibody (mAb) of IgG1 subtype (Queen, Schneider et al. 1989) that blocks the interaction of CD25 with IL-2 (via the so called “Tac” epitope). As a consequence, daclizumab blocks low affinity and high affinity IL-2R, whereas it has no effect on IL-2 signaling through the intermediate affinity IL-2R. As we’ll see later, this property of daclizumab has important functional consequences for different types of immune cells.
Because CD25 is upregulated on T cell activation, it was believed that daclizumab would selectively inhibit activated (effector) T cells. Definitely, daclizumab (or its original murine anti-Tac mAb) inhibits high-affinity T cell signaling to IL-2 (Waldmann, Goldman et al. 1988, Goebel, Stevens et al. 2000, Martin, Perry et al. 2010). Because CD25 is also highly upregulated on Human T-lymphotropic virus-1 (HTLV-1)-associated adult T-cell leukemia/lymphoma cells, daclizumab was originally developed as treatment of this condition, where it was somewhat effective, but not curative. Subsequent demonstration that addition of daclizumab to standard immunosuppressive regimens (i.e. cyclosporine and steroids) provided clinical benefit for prevention of rejection of allogeneic renal transplants (Waldmann and O’Shea 1998) was fully in agreement with this putative MOA on effector T cells.
Based on these data, after its regulatory approval as adjunctive therapy for preventing rejection of allogeneic solid organ transplants (i.e. Zenapax®; Hoffmann-La Roche), the efficacy of daclizumab was tested in another HTLV-1 associated condition, tropical spastic paraparesis (HAM-TSP) with no definite clinical benefit (Lehky, Levin et al. 1998). However, mechanistic studies demonstrated decrease in HTLV-1 pro-viral load and inhibited spontaneous lymphoproliferation of activated T cells (Lehky, Levin et al. 1998). This prompted an idea that daclizumab may also inhibit other T-cell mediated inflammatory conditions, such as non-infectious inflammatory uveitis (Nussenblatt, Fortin et al. 1999, Nussenblatt, Thompson et al. 2003). This clinical application demonstrated clear clinical benefit of long-term daclizumab treatment.
Both these observations were consistent with the presumed inhibitory effect of daclizumab on effector T cells and prompted its testing in multiple sclerosis (MS). Initial trial (Clinicaltrials.gov identifier: NCT00001934) demonstrated that addition of daclizumab to patients who had inadequate clinical and radiological response to interferon-β (IFN-β) resulted in >80% inhibition of contrast-enhancing lesions (CEL) on brain MRI and stabilization of clinical disease activity (Bielekova, Richert et al. 2004, Rose, Watt et al. 2004). Follow-up studies (Rose, Burns et al. 2007, Bielekova, Howard et al. 2009) demonstrated that in the majority of MS patients, IFN-β could be withdrawn after 6 months of IFN-β/daclizumab combination therapy without decline in clinical efficacy of long-term daclizumab monotherapy. However, one of these studies (Bielekova, Howard et al. 2009) also demonstrated that IFN-β and daclizumab have synergistic effects and optimal response in 1/3 of patients required either continuation of IFN-β/daclizumab combination therapy or higher dose of daclizumab monotherapy (i.e. 2mg/kg intravenously every 4 weeks instead of traditional 1mg/kg intravenously every 4 weeks). This observation was puzzling, because already the standard dose of 1mg/kg led to 100% saturation of CD25 Tac epitope on peripheral blood mononuclear cells (PBMC) (Bielekova, Richert et al. 2004), and suggested for the first time that blockade of CD25 in tissues may be important for daclizumab’ s therapeutic effect in MS.
Because all aforementioned studies focused on a subgroup of MS patients who had high break-through activity on IFN-β, the question remained whether daclizumab monotherapy in treatment-naïve MS subjects would be equally effective. This question was answered in a new Phase II clinical trial (Clinicaltrials.gov identifier: NCT00071838), where comparable levels of MRI efficacy and stabilization/improvement of clinical outcomes was observed (Bielekova, Richert et al. 2011).
However, because all of these studies were open label, the efficacy of daclizumab on MS disease activity had to be reproduced in placebo-controlled studies. This was accomplished in two multicenter Phase II trials sponsored by the pharmaceutical industry: specifically in the CHOICE study (Clinicaltrials.gov identifier: NCT00109161)(Wynn, Kaufman et al. 2010), which investigated IFN-β/daclizumab combination therapy and in the Phase IIb SELECT trial (Clinicaltrials.gov identifier: NCT00870740)(Gold, Giovannoni et al. 2013), which investigated daclizumab monotherapy. The latter study utilized a new preparation of daclizumab called daclizumab high yield process (DAC HYP), which has identical amino-acid sequence with the original Zenapax® preparation, but a different production process resulted in altered glycosylation pattern of the molecule, which affect binding of daclizumab to Fc receptors. As reviewed elsewhere (Bielekova and Becker 2010), binding of mAb to Fc receptors can significantly modify the outcome of mAb therapy by promoting or inhibiting complement-dependent cytotoxicity (CDC) and antibody-dependent cellular cytotoxicity (ADCC). Furthermore, DAC HYP was developed for subcutaneous administration (once every 4 weeks) in contrast to intravenous administration of Zenapax®. The efficacy and safety of long-term DAC HYP monotherapy was examined in the Phase III extension trial called SELECTION (Giovannoni, Gold et al. 2014). Finally, efficacy of DAC HYP was also tested against active comparator (i.e. interferon β−1a) in DECIDE (Clinicaltrials.gov identifier: NCT01064401) trial (Kappos, Wiendl et al. 2015).
We will summarize safety and efficacy data on daclizumab that emerged collectively from all studies after a detailed explanation of MOA of daclizumab.
Mechanism of action (MOA) of daclizumab on the human immune system
Regarding the MOA of daclizumab, studies in MS significantly expanded previous knowledge and demonstrated that the original assumptions, which represented the rationale for development of daclizumab as a selective immuno-suppressive agent against effector T cells, were not entirely correct.
First, the notion that IL-2 simply stimulates T cell immunity in vivo was challenged by the observations that mice with genetic deletions of IL-2 or its signaling chains (CD25, CD122) have apparently normal immune responses to selected pathogens, but instead succumb to severe lymphoproliferation and autoimmunity (Schorle, Holtschke et al. 1991, Kundig, Schorle et al. 1993, Suzuki, Kundig et al. 1995, Willerford, Chen et al. 1995, Wakabayashi, Lian et al. 2006). Subsequent studies revealed a non-redundant role of IL-2 signaling in the biology of FoxP3+ T-regs (Shevach, McHugh et al. 2001, Almeida, Legrand et al. 2002, Malek 2003, Setoguchi, Hori et al. 2005, Turka and Walsh 2008) and an important role of high affinity IL-2R signaling in apoptosis of effector T cells (i.e. cytokine-withdrawal cell death and activation-induced cell death [AICD]) (Lenardo 1991, Van Parijs, Refaeli et al. 1999). These studies provided a mechanistic explanation for the lymphoproliferation and autoimmunity observed in IL-2 signaling deficient mice and highlighted a crucial role of IL-2 in immunoregulation. Intriguingly, this important contribution of IL-2 to regulation of the autoimmune responses has been confirmed by genetic linkage of IL-2 and/or its signaling components (e.g. CD25, CD122) with several human autoimmune diseases, including MS (Hafler, Compston et al. 2007, Lowe, Cooper et al. 2007, Maier, Lowe et al. 2009).
However, in contrast to mice, humans with genetic deletion of CD25 have in addition to lymphoproliferation and autoimmunity also severe immunodeficiency, which is usually the first presentation of CD25 genetic defect (Sharfe, Dadi et al. 1997, Roifman 2000, Caudy, Reddy et al. 2007). This observation indicates that the role of IL-2 in humans consists of both immune-stimulatory and immune-regulatory properties.
Although in our original MOA studies we attempted to confirm direct inhibitory effect of daclizumab on activated T cells, we observed no inhibition of T cell proliferation or their production of cytokines when T cells were isolated and polyclonally stimulated in the presence of in vivo-achievable concentrations of daclizumab (Bielekova, Catalfamo et al. 2006). In contrast, we observed that numbers of T-regs, their in vivo proliferation and in vitro suppressive functions towards effector T cells are all significantly inhibited by daclizumab therapy (Oh, Blevins et al. 2009, Martin, Perry et al. 2010). Furthermore, daclizumab inhibited apoptosis of effector T cells in vivo (Baan, Balk et al. 2003) and in vitro (Wuest, Edwan et al. 2011), consistent with elimination of pro-apoptotic AICD effects of high affinity IL-2 signaling (Lenardo 1991, Van Parijs, Refaeli et al. 1999). Collectively, these mechanistic studies were exceedingly perplexing, because they predicted that the net effect of daclizumab therapy should be activation of T cell immunity, as was observed in CD25 KO animals and humans with genetic deletion of CD25. However, clinical trials clearly demonstrated that MS disease activity is inhibited by daclizumab therapy. This apparent discrepancy suggested that daclizumab must have additional effects on the human immune system.
The first evidence that supported this hypothesis stemmed from a unexpected observation that emerged from ex vivo immunophenotyping studies performed in conjunction with the first trial of daclizumab in MS (Bielekova, Richert et al. 2004): we noticed remarkable expansion of lymphocytic cells that did not express TCR or B cell receptor (BCR), but instead had high expression of CD122 (IL-2Rβ) and intermediate expression of CD8α. Literature search suggested that these cells were natural killer (NK) cells, an important component of the innate immune response against viruses and tumors (Biron and Brossay 2001, Orange 2012). Consequently, in the follow-up studies we not only confirmed that expanding cells represent NK cells, but also observed that daclizumab therapy selectively expands only minor populations of blood NK cells, characterized by high expression of CD56 surface marker (Bielekova, Catalfamo et al. 2006). These CD56bright NK cells have been labeled as “immunoregulatory” (Cooper, Fehniger et al. 2001, Caligiuri 2008) for several reasons: 1. They are selectively expanded during pregnancy, especially in the first trimester and it is believed that they participate in mediating tolerance of the mother’s immune system to the genetically foreign fetus (Nishikawa, Saito et al. 1991, Guleria and Sayegh 2007); 2. CD56bright NK cells have, in comparison to the more prevalent CD56dim NK cells, lower levels of perforin and entirely lack granzyme B (GzB) – therefore, they were for a long time considered non-cytotoxic (Jacobs, Hintzen et al. 2001). 3. CD56bright NK cells are enriched in lymph nodes (Fehniger, Cooper et al. 2003) and can secrete large levels of cytokines early in the immune response (Saito, Nishikawa et al. 1993), so it was suggested that they may alter the phenotype of newly activated T cells during the T cell priming process. However, the direct evidence for their immunoregulatory role was lacking. The analysis of their function was limited due to their low frequencies in peripheral blood (i.e. 5–10% of NK cells, which represent around 1% of lymphocytes) and the inability to identify an analogous cell type in rodents, which lack the CD56 marker. Because daclizumab expanded CD56bright NK cells in peripheral blood up to 400–500% (Bielekova, Catalfamo et al. 2006, Bielekova, Howard et al. 2009, Gross, Schulte-Mecklenbeck et al. 2016), daclizumab-treated MS patients provided a unique opportunity to study the immunoregulatory functions of these cells in detail.
First, we observed that while daclizumab had no effect on expansion or cytokine production of polyclonally activated T cells in the absence of NK cells, when NK cells were present during T cell activation, survival of activated T cells was severely limited. Subsequent mechanistic studies demonstrated that this phenomenon was due to NK-cell mediated cytotoxicity towards activated autologous T cells (Bielekova, Catalfamo et al. 2006). This interpretation contradicted immunological dogma, that NK cells do not kill autologous cells with normal expression of MHC-I molecules, because killer inhibitory receptors (KIRs) expressed on the surface of NK cells provided inhibitory signal after their interaction with self-MHC-I molecules (Moretta, Biassoni et al. 2002). However, CD56bright NK cells lack inhibitory KIRs and instead utilize CD94/NKG2A heterodimer, which recognizes HLA-E expression on target cells as their main inhibitory receptor (Kaiser, Pizarro et al. 2008). Because HLA-E binds lead peptides from self-MHC-I molecules, which stabilize expression of HLA-E on cell surface, decline in MHC-I molecules (e.g. in cancer or virally-infected cells) results in downmodulation of HLA-E molecules. Therefore, this system monitors MHC-I expression on target cells indirectly (as compared to direct KIR/MHC-I interaction characteristic of CD56dim NK cells). We not only demonstrated that killing of activated autologous T cells is mediated predominantly by CD56bright NK cells (Bielekova, Catalfamo et al. 2006), but we also dissected the molecular mechanisms of this killing (Jiang, Chai et al. 2011).
Specifically, we demonstrated that CD56bright NK cells kill autologous activated T cells via perforin-mediate degranulation, which induces reactive oxygen species (ROS) and loss of mitochondrial transmembrane potential in the target cells. This represents characteristic signature of killing by two closely related granzymes, granzyme A (GzA) and K (GzK) (Bovenschen, Quadir et al. 2009). CD56bright NK cells are the only immune cells that express GzK constitutively (Bratke, Kuepper et al. 2005). While selective blockers of GzK currently do not exist, we observed that inhibition of GzK expression by siRNA technology significantly inhibited killing of syngeneic activated T cells by the NK-92 cell line, which originates from and retains functional characteristics of CD56bright NK cells (Gong, Maki et al. 1994, Jiang, Chai et al. 2011). Finally, going back to cryopreserved samples from daclizumab-treated MS patients, we observed that daclizumab therapy specifically enhanced expression of GzK, but not the two traditionally studied and more widely expressed granzymes, GzA and GzB (Jiang, Chai et al. 2011). The observation of a strong correlation between expansion of CD56bright NK cells and contraction of absolute numbers of CD4+ and CD8+ T cells induced by daclizumab therapy provided in vivo support for the NK-mediated cytotoxicity towards activated autologous T cells (Bielekova, Catalfamo et al. 2006). Although highly controversial at the time of its publication, the idea that CD56bright NK cells in humans (and NK cells in mice) kill autologous activated T cells that express MHC-I as a part of natural immunoregulatory process has now been reproduced by many investigators using different study systems (Nielsen, Odum et al. 2012, Waggoner, Cornberg et al. 2012, Gross, Schulte-Mecklenbeck et al. 2016, Laroni, Armentani et al. 2016).
We also observed that daclizumab therapy makes T cell targets more susceptible to NK-mediated killing (Bielekova, Catalfamo et al. 2006), for which we lacked mechanistic insight. These data were recently fully reproduced and expanded by demonstrating involvement of CD226 (DNAM-1)/CD155 receptor ligand interaction in the cytolysis of activated autologous T cells by CD56bright NK cells (Gross, Schulte-Mecklenbeck et al. 2016). DNAM-1 is activating receptor expressed on all blood NK cells, which interacts with two ligands: CD112 (nectin-2 or herpesvirus entry mediator 2) and CD155 (also known as poliovirus receptor). Analogous to CD25/IL-2 system, genetic polymorphism in CD226/DNAM-1 has been associated with susceptibility to several autoimmune diseases, including MS (Hafler, Maier et al. 2009). Gross et al demonstrated that DNAM-1 is downmodulated on CD56bright NK cells in the cerebrospinal fluid of both healthy donors (HDs) and MS patients, but the decrease is much more profound in MS. Similarly, the levels of its ligand, CD155, were slightly decreased on the surface of MS T cells in comparison to HDs. CD226/CD155 interaction stimulated killing of autologous activated T cells by CD56bright NK cells, and the expression defect in both of these molecules was at least partially corrected by daclizumab therapy (Gross, Schulte-Mecklenbeck et al. 2016). This explained the missing mechanisms for daclizumab-mediated restoration of susceptibility of effector T cells to cytolysis by immunoregulatory NK cells.
Thus, we conclude that CD56bright NK cells utilize CD226 as one of the activating receptors recognizing CD155 on the surface of antigen-activated T cells to mediate GzK (and possibly also related GzA)-dependent killing of autologous T cells. This immunoregulatory function is greatly enhanced by daclizumab therapy through activation and expansion of CD56bright NK cells and their upregulation of GzK expression on one hand, but also by upregulation of CD155 on effector T cells on the other hand. Because this type of cytotoxicity can be inhibited by strong, cell-permeable anti-oxidants, our data raise a voice of caution for indiscriminate use of antioxidants in subjects with inflammatory MS without proper evaluation in clinical trial setting.
The final question was where does the killing of activated autologous T cells happen? It was shown previously that CD56bright NK cells are enriched at inflammatory sites (Dalbeth, Gundle et al. 2004) but there were no studies performed that investigated access of these rare cells to the intrathecal compartment. Because in the MS disease process the ability of CD56bright NK cells to kill autologous activated T cells would be most relevant in the central nervous system (CNS), we asked if CD56bright NK cells could be detected in the cerebrospinal fluid (CSF) of MS patients and whether their numbers in the CSF are expanded after administration of daclizumab. Indeed, we found that CD56bright NK cells are enriched in the CSF as compared to blood and their CSF levels significantly increased 6.5 months after initiation of daclizumab treatment (Bielekova, Richert et al. 2011), an observation that was fully reproduced by independent investigators (Gross, Schulte-Mecklenbeck et al. 2016). The latter study also demonstrated presence of GzK-expressing CD56bright NK cells in MS lesions, where NK cells were in contact with T cells and polarized GzK-containing granules to such contact interphase, suggesting that afore-mentioned cytotoxicity occurs directly in the inflamed CNS tissue (Gross, Schulte-Mecklenbeck et al. 2016).
Intriguingly, we also observed that CD25 Tac epitope, the target of daclizumab therapy, was completely blocked not only in the blood, but also in the CSF after daclizumab treatment. Experimental evidence indicates that only 0.1% of blood concentrations of therapeutic mAb gain access to the CSF when the blood brain barrier (BBB) is intact (Rubenstein, Combs et al. 2003, Komori, Lin et al. 2016). This would correspond to a peak concentration of 10ng/ml of daclizumab in the CSF, which is approximately 100 fold lower concentration compared to that required for saturation of CD25 expression on all T cells. Therefore, if T cells were re-activated in the CNS, available concentrations of daclizumab would be insufficient to saturate de novo produced CD25. As a result, we had to conclude that the cells that were detected in the CSF acquired blockade of CD25 Tac epitope in the blood and they were not re-activated in the CNS compartment (Bielekova, Richert et al. 2011, Lin, Winokur et al. 2015). This means that either the inflammatory process was abrogated, or, alternatively, T cells that became activated in CNS tissue during daclizumab therapy were either killed by CD56bright NK cells or did not migrate out of CNS tissue back to the CSF. As further supporting evidence for inhibition of intrathecal inflammatory process in daclizumab-treated MS patients we observed that 6.5 months after initiation of daclizumab, CSF levels of cytokine IL-12p40 (Bielekova, Richert et al. 2011), and chemokine CXCL13 (Perry, Han et al. 2012), both of which are produced by activated macrophages, microglia and DCs, were decreased by 50–60%. Furthermore, daclizumab therapy also decreased IgG index, reflecting the intrathecal production of immunoglobulins (Perry, Han et al. 2012).
We also addressed the question why CD56bright NK cells are expanded and activated by daclizumab therapy. Interestingly, CD56bright NK cells, in contrast to CD56dim NK cells express some CD25 on their cell surface (Cooper, Fehniger et al. 2001, Bielekova, Catalfamo et al. 2006) and are selectively expanded in vivo in response to administration of limiting doses of IL-2 (Caligiuri, Zmuidzinas et al. 1990). This phenomenon has been interpreted as evidence that CD56bright NK cells receive IL-2 signal via high affinity IL-2R, while CD56dim NK cells, which lack CD25, can signal only via intermediate affinity IL-2R. However, because daclizumab therapy abrogates formation of high affinity IL-2R, if CD25 expression was the only difference between CD56bright and CD56dim NK cells, then daclizumab therapy should have inhibited, rather than expanded CD56bright NK cells. There is another striking difference between these NK cell subsets though, and that is the expression of IL-2Rβ (CD122), which is at least 10 fold higher on CD56bright as compared to CD56dim NK cells, and 100–1000 fold higher compared to resting T cells (Bielekova, Catalfamo et al. 2006). As a consequence, CD56bright NK cells have the highest expression of intermediate affinity IL-2R among all accessible human immune cells. We hypothesized, and experimentally confirmed, that this high expression of intermediate affinity IL-2R allows CD56bright NK cells to sustain their IL-2 signaling in the presence of daclizumab (Martin, Perry et al. 2010). Furthermore, as daclizumab limits consumption of IL-2 by T cells (which are dependent on high affinity IL-2R because of their limited expression of CD122), excess IL-2 can be utilized by CD56bright NK cells for signaling via the intermediate affinity IL-2R, which is not being inhibited by daclizumab. Thus, paradoxically, daclizumab therapy expands and activates CD56bright NK cells in an IL-2-dependent manner (Martin, Perry et al. 2010).
We observed a strong correlation between in vivo expansion of CD56bright NK cells and inhibition of focal brain inflammatory activity measured by CEL (Bielekova, Catalfamo et al. 2006), suggesting that immunoregulation of T cell responses by CD56bright NK cells may represent a decisive MOA of daclizumab in MS. This observation indicated that the level of expansion of CD56bright NK cells and the decrease in ratios of T cells (as target cells) to CD56bright NK cells (as effector cells) could represent a useful biomarker indicative of therapeutic response to daclizumab therapy, as it differentiated full responders from partial responders in our MS cohort (Bielekova, Howard et al. 2009). These data have been reproduced in two independent multicenter Phase II trials of daclizumab in MS: CHOICE (Wynn, Kaufman et al. 2010) and SELECT trials (Elkins, Sheridan et al. 2012). However, even patients with the smallest levels of expansion of CD56bright NK cells had discernable efficacy on MRI and clinical markers, indicating that while expansion of CD56bright NK cells is an important mechanism, it cannot be the only mechanism responsible for drug’s efficacy (see below).
Since our original description of CD56bright NK cells as an immunoregulatory population relevant to MS disease process (Bielekova, Catalfamo et al. 2006), expansion of these cells was observed by other effective therapies for MS, such as IFN-β (Bielekova, Howard et al. 2009, Vandenbark, Huan et al. 2009) and rituximab (Reis, Athanazio et al. 2008). Furthermore, unbiased cytometric profiling in MS uncovered a deficiency in non-T, non-B cell lymphocytes that expressed CD8α surface marker as the only distinguishing feature between MS subjects and controls (De Jager, Rossin et al. 2008). It is probable that these cells represent a subpopulation of CD56bright NK cells. However, several follow-up studies did not identify numerical or proportional defects in NK or CD56bright NK cell numbers in MS (Gross, Schulte-Mecklenbeck et al. 2016, Laroni, Armentani et al. 2016). On the other hand functional deficiencies in NK cells, especially in their secretion of IFN-γ, which is mainly secreted by CD56bright NK cells, have been associated with MS and other autoimmune diseases by many studies performed over the past 30 years (Benczur, Petranyl et al. 1980, Kastrukoff, Morgan et al. 1998, French and Yokoyama 2004). It is likely that CD56bright NK cells work together with other regulatory cell populations, such as FoxP3+ T-regs or Tr1 regulatory cells, to maintain immune tolerance under physiological conditions. Whether any of these regulatory cells play a more important role in specific immune-mediated disorders remains an open question.
The strong correlation between daclizumab-driven expansion of CD56bright NK cells and inhibition of the MS disease process suggested that this is the main MOA responsible for beneficial effect of daclizumab in MS. However, consistent with later observations from clinical trials (Elkins, Sheridan et al. 2015) in our extensive experience with daclizumab therapy, we identified a patient who did not expand CD56bright NK cells at all, but still experienced more than 80% suppression of CEL and stabilization of clinical disability (Ohayon, Oh et al. 2013). This patient taught us there had to be another MOA, unrelated to CD56bright NK cells.
We started our search by focusing on myeloid DCs, because these cells upregulate CD25 upon their activation by microbial stimuli and are also endowed with the ability to synthesize and secrete IL-2 (Granucci, Vizzardelli et al. 2001). Because mature DCs (mDCs) are the most important antigen presenting cells (APC) that activate T cells in an antigen-specific manner, we asked if daclizumab affects this crucial function of mDCs. Indeed, we observed that peak in vivo achievable concentrations of daclizumab (10μg/ml) that had no discernable effect on proliferation of polyclonally activated T cells almost completely abolished expansion of antigen-specific T cells activated by mDCs (Wuest, Edwan et al. 2011). Utilizing selective pre-treatment of mDCs or T cells with daclizumab, siRNA technology and ultimately also T cells derived from a rare human subject with genetic deletion of CD25, we demonstrated that it is the blockade of CD25 on mDCs, and not on T cells that underlies this inhibition. We then hypothesized that mDCs are activated by IL-2 signal and that this activation is necessary for effective priming of antigen-specific T cells. However, this hypothesis turned out to be incorrect, because myeloid DCs, either in resting or activated state do not express CD122 and therefore cannot receive IL-2 (or IL-15) signal (Driesen, Popov et al. 2008, Wuest, Edwan et al. 2011).
As a result, we pursued the alternative hypothesis, that mDCs utilize their CD25 to present IL-2 to primed T cells “in trans”; in a manner analogous to what was previously described for IL-15 (Dubois, Mariner et al. 2002) (Figure 2A). In other words, mDCs utilize their CD25 in trans to complement intermediate affinity IL-2R on resting T cells, and thus allow T cells to receive high affinity IL-2R signal at a time when T cells express only intermediate affinity IL-2R. The only problem with this hypothesis was the discrepancy in affinities of IL-15Rα versus IL-2Rα (i.e. CD25) for their respective ligands: while IL-15Rα has a very strong affinity for IL-15, such that the majority (if not all) IL-15 is bound to IL-15Rα in vivo (Figure 2A), CD25 has a very low affinity for IL-2, which allows it to bind IL-2 only under circumstances of IL-2 abundance. Thus, it is unlikely that CD25 would be able to effectively capture small quantities of IL-2 secreted by mDC if IL-2 can freely diffuse to the environment. We conceptually solved this dilemma by hypothesizing that trans-presentation of IL-2 by mDCs occurs across the immune synapse (IS; Figure 2B). Thus, instead of indiscriminative release of IL-2 to the environment, we hypothesized that mDCs release their IL-2 into the small synaptic cleft formed between an mDC and an antigen-specific T cell and that this physical constraint limits the diffusion of IL-2 away from the primed T cell. Consequently, sufficiently high concentrations of IL-2 are reached in the IS to allow effective capture of IL-2 by CD25 expressed on mDC and subsequent trans-presentation of IL-2 to the primed, antigen-specific T cell.
This hypothesis was fully supported by subsequent mechanistic studies: First, we observed that trans-presentation of IL-2 by CD25-expressing mDC to CD25 negative T cells was inefficient when T cells did not carry a TCR specific for the antigen being presented by the mDC (i.e. when the stable IS was not formed between the mDC and T cell). This was true even when we added IL-2 exogenously to such mDC-T cell co-cultures. In contrast, when CD25-negative T cells were specific for the antigen presented by mDC, they promptly received strong IL-2 signal without any exogenous addition of IL-2, and this IL-2 signaling was significantly reduced if mDCs were pre-treated with daclizumab (Wuest, Edwan et al. 2011) (Figure 2C). We also observed that CD25, expressed solely on mDCs, co-localized to the IS with the antigen-specific T cell and that it was only the T cell, and not the mDC, that received the IL-2 signal as measured by phosphorylated STAT5 molecules. The reason mDC-mediated IL-2 signaling on T cells occurred strictly in an antigen-specific manner was the fact that only when mDCs were co-cultured with T cells that recognized their antigen-MHC-II complex would mDCs release their IL-2. Therefore, there appears to be a bilateral communication between mDC and T cell (likely triggered by cognate TCR/antigen-MHC interaction; called “Signal 1”) before the mDC decides to release its IL-2 to the synaptic cleft. This assures that limiting amounts of IL-2 produced at the beginning of the immune response are not wasted for “bystander” T cells, but are delivered to T cells that have received full TCR and co-stimulatory signals (Signal 2) and thus can mediate an effective immune response. In in vitro assays, Signal 3, provided by mDC-derived IL-2 was necessary for efficient expansion of antigen-specific T cells, and when it was abrogated (by blocking CD25 on mDCs), T cells proliferated poorly, even though daclizumab-pretreated mDCs expressed many MHC-peptide complexes and co-stimulatory molecules. This observation is consistent with immunodeficiency observed in children with genetic deletion of CD25. Of note, T cells could enter the proliferation cycle when supplemented in vitro with IL-7 as an alternative source of γc-signaling cytokine. However, T cells expanded this way did not express full effector functions, compared to T cells that received IL-2 signal during priming. A similar observation was made in the animal system, where IL-2 signal was necessary during the priming of antigen-specific CD8+ T cells for effective development of T cell memory (Williams, Tyznik et al. 2006). Again, these observations are compatible with the curious combination of immunodeficiency and lymphoproliferation that is the characteristic phenotype of individuals with genetic deletion of CD25.
Interestingly, for this MOA to occur, daclizumab must saturate CD25 in sites where the antigen-presentation happens, such as lymphoid organs (Savo, Book et al. 1999) and inflamed tissues. Because concentrations of intravenously administered mAb are expected to be higher in the blood than in tissues, this requirement may explain the previously-mentioned paradoxical observations that efficacy of daclizumab decreases 4–6 weeks after the last IV dose despite the fact that the CD25 epitope remains almost completely saturated (>95%) in the blood (Bielekova, Richert et al. 2004).
Final described MOA, represents inhibitory effect of daclizumab on innate lymphoid cells (ILCs), especially on development of their pro-inflammatory subtype called lymphoid tissue inducer (LTi) cells (Perry, Han et al. 2012). Like in the case of CD56bright NK cells, it was broader immunophenotyping associated with the NIH daclizumab trial (Clinicaltrials.gov identifier: NCT01143441) that alerted us to the link between LTi cells and daclizumab therapy (Perry, Han et al. 2012).
ILCs are heterogeneous group of lymphocytes that belong to innate, rather than adaptive immune systems. While NK cells are the most familiar subset of ILCs, this category also contains recently described cells with constitutive expression of retinoic acid receptor-related orphan receptor γt (ROR γt). Depending on the tissue from which these cells have been isolated, they express slightly different phenotype and have been labeled by different names (Sawa, Cherrier et al. 2010, Spits and Di Santo 2011), such as LTi cells, ILC22 (i.e. IL-22-producing ILCs, which also express NKp44) and ILC17 (IL-17 producing ILCs). It is clear that these different ILC categories are developmentally related, as they all originate from CD34+ hematopoietic precursors and are all dependent on transcriptional regulator Id2 (Yokota, Mansouri et al. 1999).
Although LTi cells play fundamental role in the formation of secondary lymphoid tissues during fetal development (Aloisi and Pujol-Borrell 2006), tertiary ectopic follicles associated with chronic inflammation can form in ROR γt-deficient animals (Lochner, Ohnmacht et al. 2011), where either activated T cells or B cells can acquire lymphoid tissue-inducing capacity. Therefore, the role of adult LTi cells remains unclear. Nevertheless, it has been hypothesized that through their constitutive expression of OX40 and CD30, adult LTi cells may play vital role in the evolution and maintenance of CD4+ T cell memory (Withers, Gaspal et al. 2012) and related B cell/antibody (Ab) responses, including formation of high-affinity class-switched IgG (Lane, Gaspal et al. 2012).
We observed that untreated MS patients have significantly higher levels of c-kit+/RORγt-expressing LTi cells in the blood and CSF (Perry, Han et al. 2012, Lin, Winokur et al. 2015) in comparison to healthy controls. These observations were reproduced by independent investigators studying independent cohort of MS patients, although the elevations of LTi cells in the blood did not reach formal statistical significance (Degn, Modvig et al. 2015). Daclizumab therapy normalizes this CSF abnormality (Perry, Han et al. 2012, Lin, Winokur et al. 2015), by skewing development of CD34+ hematopoietic stem cells and c-kit+ undifferentiated ILC precursors away from LTi lineage and toward CD56bright NK cells, by enhancing IL-2 signaling through intermediate affinity IL-2R (Perry, Han et al. 2012).
Although we cannot directly visualize or quantify meningeal lymphoid follicles in living MS subjects (Magliozzi, Howell et al. 2007, Howell, Reeves et al. 2011), daclizumab therapy decreased intrathecal production of chemokine CXCL13 and of IgG, measured as IgG index (Perry, Han et al. 2012). Intriguingly, effect of daclizumab on IgG production was specific for the intrathecal compartment, because daclizumab did not lower blood levels of IgG, IgA or IgM (Bielekova, Richert et al. 2004, Bielekova, Catalfamo et al. 2006). Because daclizumab does not limit migration of immune cells to the intrathecal compartment (Bielekova, Richert et al. 2011, Lin, Winokur et al. 2015), decrease in intrathecal levels of CXCL13, which is highly expressed in tertiary lymphoid follicles (Magliozzi, Columba-Cabezas et al. 2004, Magliozzi, Howell et al. 2007), indirectly supports the notion that inhibition of LTi cells by daclizumab may have impeded the maintenance of meningeal lymphoid aggregates. Since formation of meningeal lymphoid follicles in MS has been associated with greater pathology of the underlying gray matter (Howell, Reeves et al. 2011) future studies should investigate whether long-term administration of daclizumab inhibits atrophy of the CNS gray matter that is in direct contact with CSF/meningeal compartment.
MOA of daclizumab: unanswered questions
When multiple different effects on the immune system are described, the question often asked is which of these MOA is the most important for the observed therapeutic effect. Because these various effects cannot be separated from each other in vivo, it is impossible to answer this question within the context of one therapy. Instead, if the same mechanism is being targeted by another successful therapy (e.g. expansion of CD56bright NK cells by IFN-β), then the likelihood that the described mechanism is important for studied disease process increases. Similarly, correcting abnormality that is associated with the disease state (e.g. ability of daclizumab to normalize increased numbers of circulating LTi cells in MS subjects), provides support to the notion that the observed MOA may be pathophysiologically relevant. Therefore, it is only the aggregate experience with different therapies that may elucidate most important pathophysiological drivers of the disease and consequently, lead to knowledge that is necessary for design of more targeted therapies.
At the same time, we should not expect a single MOA underlying each therapeutic modality. Even monoclonal antibodies, which target only one specific molecule exert multiple in vivo effects, often dependent on situational diversity. Such pleiotropy and functional redundancies make biological systems robust and resilient (Mesarovic, Sreenath et al. 2004). Indeed, example of IL-2 shows that a single cytokine can have profound and at times opposing effects on multiple cells of the immune system, based on the dynamics of expression of its signaling chains and competition for available cytokines. Thus, it is likely that all described MOA of daclizumab are important on the population level. That does not eliminate the possibility that occasional patients, likely due to their genetic background, may not be able to utilize some MOA (Ohayon, Oh et al. 2013, Elkins, Sheridan et al. 2015) and may therefore rely more prominently on remaining ones, or experience suboptimal therapeutic response.
Efficacy and safety of daclizumab therapy in MS
All open label baseline-versus-treatment studies demonstrated that intravenously administered daclizumab (1mg/kg every 4 weeks) inhibited CEL on brain MRI by more than 75%, regardless of whether daclizumab was administered as monotherapy or as add-on to IFN-β (Bielekova, Richert et al. 2004, Rose, Watt et al. 2004, Rose, Burns et al. 2007, Bielekova, Howard et al. 2009, Bielekova, Richert et al. 2011). This was associated with stabilization (Bielekova, Richert et al. 2004, Rose, Watt et al. 2004, Rose, Burns et al. 2007) or even improvements (Bielekova, Howard et al. 2009, Bielekova, Richert et al. 2011) of clinical outcomes. Limited selection of highly-active MS subjects and open-label design without placebo control were major drawbacks of these early studies (Liu, Wang et al. 2012, Schneider and Arbour 2012), although the consistent efficacy on objective outcome measures, such as CEL or MSFC provided strong impetus for commercial development of daclizumab for the treatment of inflammatory MS.
In the first industry-sponsored, placebo controlled trial (CHOICE study, N=230; Clinicaltrials.gov identifier: NCT00109161), addition of subcutaneously administered daclizumab (low dose: 1mg/kg every 4 weeks and high dose: 2mg/kg every 2 weeks) to IFN-β, decreased CEL lesion by 72% in the high dose arm (p=0.004) and by 25% (p=0.51) in the low dose arm (Wynn, Kaufman et al. 2010). The high dose arm yielded systemic levels of daclizumab comparable to the intravenous 1mg/kg dose, which was utilized in open-label trials. In the Phase IIb SELECT trial (N=600; Clinicaltrials.gov identifier: NCT00390221), two doses of DAC HYP monotherapy (150mg or 300mg every 4 weeks) administered subcutaneously for 1 year were compared to placebo. Both doses of daclizumab inhibited formation of CEL (by 68.8% and 79.2%; p<0.001) and new or enlarging T2 lesions (by 70.4% and 79.0%; p<0.001) on brain MRI (Gold, Giovannoni et al. 2012). This efficacy on MRI parameters of MS lesion formation was paralleled by significant inhibition of annualized relapse rate (by 54.3% and 50%; p<0.001) and by inhibition of disability progression (by 57%; p=0.021 for 150mg dose and by 43%; p=0.091 for 300mg dose).
SELECTION extension trial examined several important issues, such as rebound phenomenon after stopping of DAC HYP treatment, efficacy after re-initiation of therapy and long-term efficacy/safety. This trial confirmed no meaningful difference between 150mg and 300mg doses, lack of rebound of disease activity beyond baseline values and continuous efficacy of daclizumab (on relapses, MRI markers and disability) after treatment re-initiation or its continuous use (Giovannoni, Gold et al. 2014). This study also reproduced observations from the open-label studies that linked daclizumab administration to expansion of CD56bright regulatory natural killer (NK) cells.
Finally, DECIDE trial, demonstrated statistically-significant superiority of DAC HYP (150mg SQ q 4 weeks) against weekly IM administration of interferon-β1a in annualized relapse rate (45% inhibition) and the number of new or enlarging T2 lesions (54% reduction)(Kappos, Wiendl et al. 2015).
In general, all clinical trials in MS found daclizumab well-tolerated, with little induced immunogenicity. The overall incidence of adverse events (AE) and the rate of discontinuation of therapy were similar between placebo and daclizumab arms in the CHOICE (Wynn, Kaufman et al. 2010) and SELECT (Gold, Giovannoni et al. 2012) trials and between IFN-b and daclizumab in DECIDE trial (Kappos, Wiendl et al. 2015). The most common AEs observed in MS trials of daclizumab belong to four categories: 1. Skin rashes, 2. Lymphadenopathy, 3. Elevation of liver function tests (LFTs) and 4. Infections (Bielekova, Richert et al. 2004, Bielekova, Howard et al. 2009, Wynn, Kaufman et al. 2010, Bielekova, Richert et al. 2011, Gold, Giovannoni et al. 2012, Kappos, Wiendl et al. 2015).
We will discuss these categories of AE in view of the described MOA: Skin rashes were seen in 13% of daclizumab subjects and 8% of placebo subjects in the CHOICE study (Wynn, Kaufman et al. 2010). In NIH open label trials we observed frequent skin rashes, mostly of mild intensity that responded to emollients or topical steroids (Cortese, Ohayon et al. 2016). We now advise patients to routinely use sunscreen and skin moisturizers, because we have observed an increased frequency of skin rashes on sun-exposed areas and with dry, scaling skin. However, we have also observed few prolonged and more severe skin rashes, which required systemic steroids and/or discontinuation of therapy (Bielekova, Richert et al. 2004, Bielekova, Howard et al. 2009, Bielekova, Richert et al. 2011, Oh, Saidha et al. 2014). Serious cutaneous events were observed in 1% of DAC HYP-treated subjects in the SELECT trial (Gold, Giovannoni et al. 2012) and in 2% of DAC HYP-treated subjects in DECIDE trial (Kappos, Wiendl et al. 2015). It is not clear what underlies enhanced skin reactivity in daclizumab-treated subjects; one hypothesis implicates daclizumab-driven inhibition of FoxP3 T-regs (Oh, Blevins et al. 2009), while we observed in lesional skin biopsies increased representation of CD56+ NK cells (Cortese, Ohayon et al. 2016), suggesting that the same cell population that mediates beneficial effects of daclizumab on MS may be responsible for (at least some) cutaneous side-effects.
Mild generalized lymphadenopathy is often associated with daclizumab therapy (Bielekova, Richert et al. 2004, Bielekova, Howard et al. 2009) without any pathological consequences. We have evaluated several subjects with prominent or persistent lymphadenopathy using fine needle biopsy and found no pathological changes in flow cytometry or pathology profile ((Oh, Saidha et al. 2014) and unpublished observations).
Elevations of LFTs have been observed in daclizumab-treated cohorts; while in NIH trials we observed only transient elevation of LFTs which did not require discontinuation of therapy (Bielekova, Richert et al. 2004, Bielekova, Howard et al. 2009, Bielekova, Richert et al. 2011), serious elevations of LFTs (five times above upper limit of normal) were observed in 4% of DAC HYP-treated subjects in SELECT trial and in 6% of subjects in DECIDE trial (as compared to 3% of IFN-β treated subjects in the same study. Again, the mechanism behind this phenomenon is unclear, however, CD56bright NK cells have been reported to be a prominent immune cell population in the liver under physiological conditions (Moroso, Metselaar et al. 2010).
Although in our open label NIH studies we observed a slight increase in the frequency of mild infectious AE (mostly urinary and upper respiratory tract infections), the placebo-controlled CHOICE study did not demonstrate an increased incidence of infections in the daclizumab arms (Wynn, Kaufman et al. 2010). In contrast, the SELECT trial reported an increase in serious infections (2%) in the DAC HYP cohort and DECIDE trial confirmed these findings by observing 4% rate of serious infections in DAC HYP group in comparison to 2% rate of serious infections in IFN-β group. One DAC HYP-treated subject died due to a complications of psoas abscess (Gold, Giovannoni et al. 2012). However, thus far no opportunistic infections were observed. MOA studies indicate that daclizumab-treated subjects may be at risk of severe infectious because of the inhibitory effect of daclizumab on T cell priming by limiting IL-2 trans-presentation by DCs. This mechanism would be predicted to have stronger effect on sterile inflammation as opposed to infections, where robust activation of innate immunity may partially compensate for the deficiency of Signal 3; this hypothesis is indirectly confirmed by observations that rate of mild infections is comparable between DAC-HYP and placebo arms and that patients under daclizumab therapy mount protective immune responses to routine annual influenza vaccination (Lin, Winokur et al. 2016).
Will daclizumab cause progressive multifocal encephalopathy? Because daclizumab does not limit access of immune cells to the intrathecal compartment and expanded CD56bright NK cells may provide effective immunity against (at least herpes) viruses (Orange 2012) we would predict risk of PML lower in daclizumab-treated patients as compared to those treatments that directly limit access of immune system to CNS. However, only broad post-marketing experience will determine the actual risk of PML with long-term daclizumab administration.
Finally, none of the clinical trials demonstrated increased risk of cancer with daclizumab therapy and again, from the mechanistic standpoint we would predict that activation of (CD56bright) NK cells may enhance NK-mediated immuno-surveillance against cancer, even if T cell immunity is diminished in parallel. This conclusion is supported by in vivo observations from experience in transplantation, where transplant patients that received daclizumab (in addition to standard immunosuppressive therapy) had in fact lower levels of secondary cancers (Webster, Playford et al. 2004) than those patients that received identical immunosuppression without daclizumab.
Conclusions
At the time we initiated studies of daclizumab in MS, the model of its MOA was based on the idea that IL-2 is a crucial T cell growth factor and by blocking high affinity IL-2 signaling on T cells, daclizumab would inhibit effector functions of activated T cells. Instead, in vivo observations supplemented by mechanistic in vitro studies revealed unexpected effects of daclizumab on cells belonging to the innate immune system, CD56bright NK cells, mDCs and ILCs. These effects were not previously identified in animals with genetically deleted IL-2 or its signaling components, signifying that carefully conducted mechanistic studies linked to human interventional trials have the potential to discover novel biological mechanisms (Bielekova, Vodovotz et al. 2014). Such new insights may be also relevant for the pathophysiology of the targeted disease. The MOA of daclizumab also reminds us that adaptive immune responses, which are dysregulated in autoimmunity, are tightly controlled by cells of the innate immune system. Thus, both of these systems need to be studied in an integrated manner before we can fully understand mechanisms that lead to the breakdown of tolerance and development of immune-mediated pathology.
Acknowledgment:
The work was supported by the Intramural research program of the NINDS. B.B. is a co-inventor on NIH patents related to daclizumab therapy and as such has received patent royalty payments.
Bibliography:
- Almeida AR, Legrand N, Papiernik M and Freitas AA (2002). “Homeostasis of peripheral CD4+ T cells: IL-2R alpha and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers.” J Immunol 169(9): 4850–4860. [DOI] [PubMed] [Google Scholar]
- Aloisi F and Pujol-Borrell R (2006). “Lymphoid neogenesis in chronic inflammatory diseases.” Nat Rev Immunol 6(3): 205–217. [DOI] [PubMed] [Google Scholar]
- Baan CC, Balk AH, van Riemsdijk IC, Vantrimpont PJ, Maat AP, Niesters HG, Zondervan PE, van Gelder T and Weimar W (2003). “Anti-CD25 monoclonal antibody therapy affects the death signals of graft-infiltrating cells after clinical heart transplantation.” Transplantation 75(10): 1704–1710. [DOI] [PubMed] [Google Scholar]
- Benczur M, Petranyl GG, Palffy G, Varga M, Talas M, Kotsy B, Foldes I and Hollan SR (1980). “Dysfunction of natural killer cells in multiple sclerosis: a possible pathogenetic factor.” Clin Exp Immunol 39(3): 657–662. [PMC free article] [PubMed] [Google Scholar]
- Bielekova B and Becker B (2010). “Monoclonal antibodies in MS: Mechanism of action.” Neurology 74(Supplement 1): S31–S40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielekova B, Catalfamo M, Reichert-Scrivner S, Packer A, Cerna M, Waldmann TA, McFarland H, Henkart PA and Martin R (2006). “Regulatory CD56bright natural killer cells mediate immunomodulatory effects of IL-2R-alpha-targeted therapy (daclizumab) in multiple sclerosis.” PNAS 103(15): 5941–5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielekova B, Howard T, Packer AN, Richert N, Blevins G, Ohayon J, Waldmann TA, McFarland HF and Martin R (2009). “Effect of anti-CD25 antibody daclizumab in the inhibition of inflammation and stabilization of disease progression in multiple sclerosis.” Arch Neurol 66(4): 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielekova B, Richert N, Herman ML, Ohayon J, Waldmann TA, McFarland H, Martin R and Blevins G (2011). “Intrathecal effects of daclizumab treatment of multiple sclerosis.” Neurology 77(21): 1877–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielekova B, Richert N, Howard T, Blevins G, Markovic-Plese S, McCartin J, Wurfel J, Ohayon J, Waldmann TA, McFarland HF and Martin R (2004). “Humanized anti-CD25 (daclizumab) inhibits disease activity in multiple sclerosis patients failing to respond to interferon-beta.” Proc Natl Acad Sci U S A 101(23): 8705–8708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielekova B, Vodovotz Y, An G and Hallenbeck J (2014). “How implementation of systems biology into clinical trials accelerates understanding of diseases.” Front Neurol 5: 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron CA and Brossay L (2001). “NK cells and NKT cells in innate defense against viral infections.” Curr Opin Immunol 13(4): 458–464. [DOI] [PubMed] [Google Scholar]
- Bovenschen N, Quadir R, van den Berg AL, Brenkman AB, Vandenberghe I, Devreese B, Joore J and Kummer JA (2009). “Granzyme K displays highly restricted substrate specificity that only partially overlaps with granzyme A.” J Biol Chem 284(6): 3504–3512. [DOI] [PubMed] [Google Scholar]
- Bratke K, Kuepper M, Bade B, Virchow JC Jr. and Luttmann W (2005). “Differential expression of human granzymes A, B, and K in natural killer cells and during CD8+ T cell differentiation in peripheral blood.” Eur J Immunol 35(9): 2608–2616. [DOI] [PubMed] [Google Scholar]
- Caligiuri MA (2008). “Human natural killer cells.” Blood 112(3): 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caligiuri MA, Zmuidzinas A, Manley TJ, Levine H, Smith KA and Ritz J (1990). “Functional consequences of interleukin 2 receptor expression on resting human lymphocytes. Identification of a novel natural killer cell subset with high affinity receptors.” J Exp Med 171(5): 1509–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudy AA, Reddy ST, Chatila T, Atkinson JP and Verbsky JW (2007). “CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes.” J Allergy Clin Immunol 119(2): 482–487. [DOI] [PubMed] [Google Scholar]
- Cooper MA, Fehniger TA and Caligiuri MA (2001). “The biology of human natural killer-cell subsets.” Trends Immunol 22(11): 633–640. [DOI] [PubMed] [Google Scholar]
- Cortese I, Ohayon J, Fenton K, Lee CC, Raffeld M, Cowen EW, DiGiovanna JJ and Bielekova B (2016). “Cutaneous adverse events in multiple sclerosis patients treated with daclizumab.” Neurology [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalbeth N, Gundle R, Davies RJ, Lee YC, McMichael AJ and Callan MF (2004). “CD56bright NK cells are enriched at inflammatory sites and can engage with monocytes in a reciprocal program of activation.” J Immunol 173(10): 6418–6426. [DOI] [PubMed] [Google Scholar]
- De Jager PL, Rossin E, Pyne S, Tamayo P, Ottoboni L, Viglietta V, Weiner M, Soler D, Izmailova E, Faron-Yowe L, O’Brien C, Freeman S, Granados S, Parker A, Roubenoff R, Mesirov JP, Khoury SJ, Hafler DA and Weiner HL (2008). “Cytometric profiling in multiple sclerosis uncovers patient population structure and a reduction of CD8low cells.” Brain 131(Pt 7): 1701–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degn M, Modvig S, Dyring-Andersen B, Bonefeld CM, Frederiksen JL, Geisler C and von Essen MR (2015). “Increased prevalence of lymphoid tissue inducer cells in the cerebrospinal fluid of patients with early multiple sclerosis.” Mult Scler [DOI] [PubMed] [Google Scholar]
- Driesen J, Popov A and Schultze JL (2008). “CD25 as an immune regulatory molecule expressed on myeloid dendritic cells.” Immunobiology 213(9–10): 849–858. [DOI] [PubMed] [Google Scholar]
- Dubois S, Mariner J, Waldmann TA and Tagaya Y (2002). “IL-15Ralpha recycles and presents IL-15 In trans to neighboring cells.” Immunity 17(5): 537–547. [DOI] [PubMed] [Google Scholar]
- Elkins J, Sheridan J, Amaravadi L, Riester K, Selmaj K, Bielekova B, Parr E and Giovannoni G (2015). “CD56(bright) natural killer cells and response to daclizumab HYP in relapsing-remitting MS.” Neurol Neuroimmunol Neuroinflamm 2(2): e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins J, Sheridan J, Armaravadi L, Riester K and O’Neil G (2012). “CD56bright natural killer cell expansion predicts response to daclizumab HYP treatment in RRMS: Results of the SELECT trial.” Neurology 78 Meeting abstracts: S31.004. [Google Scholar]
- Fehniger TA, Cooper MA, Nuovo GJ, Cella M, Facchetti F, Colonna M and Caligiuri MA (2003). “CD56bright natural killer cells are present in human lymph nodes and are activated by T cell-derived IL-2: a potential new link between adaptive and innate immunity.” Blood 101(8): 3052–3057. [DOI] [PubMed] [Google Scholar]
- Feinerman O, Jentsch G, Tkach KE, Coward JW, Hathorn MM, Sneddon MW, Emonet T, Smith KA and Altan-Bonnet G (2010). “Single-cell quantification of IL-2 response by effector and regulatory T cells reveals critical plasticity in immune response.” Mol Syst Biol 6: 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French AR and Yokoyama WM (2004). “Natural killer cells and autoimmunity.” Arthritis Res Ther 6(1): 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannoni G, Gold R, Selmaj K, Havrdova E, Montalban X, Radue EW, Stefoski D, McNeill M, Amaravadi L, Sweetser M, Elkins J, O’Neill G and Investigators SS (2014). “Daclizumab high-yield process in relapsing-remitting multiple sclerosis (SELECTION): a multicentre, randomised, double-blind extension trial.” Lancet Neurol 13(5): 472–481. [DOI] [PubMed] [Google Scholar]
- Goebel J, Stevens E, Forrest K and Roszman TL (2000). “Daclizumab (Zenapax) inhibits early interleukin-2 receptor signal transduction events.” Transpl Immunol 8(3): 153–159. [DOI] [PubMed] [Google Scholar]
- Gold R, Giovannoni G, Selmaj K, Havrdova E, Moltalban X, Radue EW, Stefoski D, Robinson R, Riester K, Elkins J and O’Neill G (2012). “A randomized, double-blind, placebo-controlled study to evaluate the safety and efficacy of daclizumab HYP monotherapy in relapsing-remitting multiple sclerosis: Primary results of the SELECT trial.” Neurology 78 Meeting Abstracts: S01.005. [Google Scholar]
- Gold R, Giovannoni G, Selmaj K, Havrdova E, Montalban X, Radue EW, Stefoski D, Robinson R, Riester K, Rana J, Elkins J, O’Neill G and S. s. i. for the (2013). “Daclizumab high-yield process in relapsing-remitting multiple sclerosis (SELECT): a randomised, double-blind, placebo-controlled trial.” Lancet [DOI] [PubMed] [Google Scholar]
- Gong JH, Maki G and Klingemann HG (1994). “Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells.” Leukemia 8(4): 652–658. [PubMed] [Google Scholar]
- Granucci F, Vizzardelli C, Pavelka N, Feau S, Persico M, Virzi E, Rescigno M, Moro G and Ricciardi-Castagnoli P (2001). “Inducible IL-2 production by dendritic cells revealed by global gene expression analysis.” Nat Immunol 2(9): 882–888. [DOI] [PubMed] [Google Scholar]
- Gross CC, Schulte-Mecklenbeck A, Runzi A, Kuhlmann T, Posevitz-Fejfar A, Schwab N, Schneider-Hohendorf T, Herich S, Held K, Konjevic M, Hartwig M, Dornmair K, Hohlfeld R, Ziemssen T, Klotz L, Meuth SG and Wiendl H (2016). “Impaired NK-mediated regulation of T-cell activity in multiple sclerosis is reconstituted by IL-2 receptor modulation.” Proc Natl Acad Sci U S A [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guleria I and Sayegh MH (2007). “Maternal acceptance of the fetus: true human tolerance.” J Immunol 178(6): 3345–3351. [DOI] [PubMed] [Google Scholar]
- Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, De Jager PL, de Bakker PI, Gabriel SB, Mirel DB, Ivinson AJ, Pericak-Vance MA, Gregory SG, Rioux JD, McCauley JL, Haines JL, Barcellos LF, Cree B, Oksenberg JR and Hauser SL (2007). “Risk alleles for multiple sclerosis identified by a genomewide study.” N Engl J Med 357(9): 851–862. [DOI] [PubMed] [Google Scholar]
- Hafler JP, Maier LM, Cooper JD, Plagnol V, Hinks A, Simmonds MJ, Stevens HE, Walker NM, Healy B, Howson JM, Maisuria M, Duley S, Coleman G, Gough SC, C. International Multiple Sclerosis Genetics, Worthington J, Kuchroo VK, Wicker LS and Todd JA (2009). “CD226 Gly307Ser association with multiple autoimmune diseases.” Genes Immun 10(1): 5–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell OW, Reeves CA, Nicholas R, Carassiti D, Radotra B, Gentleman SM, Serafini B, Aloisi F, Roncaroli F, Magliozzi R and Reynolds R (2011). “Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis.” Brain 134(9): 2755–2771. [DOI] [PubMed] [Google Scholar]
- Jacobs R, Hintzen G, Kemper A, Beul K, Kempf S, Behrens G, Sykora KW and Schmidt RE (2001). “CD56bright cells differ in their KIR repertoire and cytotoxic features from CD56dim NK cells.” Eur J Immunol 31(10): 3121–3127. [DOI] [PubMed] [Google Scholar]
- Jiang W, Chai NR, Maric D and Bielekova B (2011). “Unexpected Role for Granzyme K in CD56bright NK Cell-Mediated Immunoregulation of Multiple Sclerosis.” J Immunol 187(2): 781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser BK, Pizarro JC, Kerns J and Strong RK (2008). “Structural basis for NKG2A/CD94 recognition of HLA-E.” Proc Natl Acad Sci U S A 105(18): 6696–6701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappos L, Wiendl H, Selmaj K, Arnold DL, Havrdova E, Boyko A, Kaufman M, Rose J, Greenberg S, Sweetser M, Riester K, O’Neill G and Elkins J (2015). “Daclizumab HYP versus Interferon Beta-1a in Relapsing Multiple Sclerosis.” N Engl J Med 373(15): 1418–1428. [DOI] [PubMed] [Google Scholar]
- Kastrukoff LF, Morgan NG, Zecchini D, White R, Petkau AJ, Satoh J and Paty DW (1998). “A role for natural killer cells in the immunopathogenesis of multiple sclerosis.” J Neuroimmunol 86(2): 123–133. [DOI] [PubMed] [Google Scholar]
- Komori M, Lin YC, Cortese I, Blake A, Ohayon J, Cherup J, Maric D, Kosa P, Wu T and Bielekova B (2016). “Insufficient disease inhibition by intrathecal rituximab in progressive multiple sclerosis.” Ann Clin Transl Neurol 3(3): 166–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundig TM, Schorle H, Bachmann MF, Hengartner H, Zinkernagel RM and Horak I (1993). “Immune responses in interleukin-2-deficient mice.” Science 262(5136): 1059–1061. [DOI] [PubMed] [Google Scholar]
- Lane PJ, Gaspal FM, McConnell FM, Kim MY, Anderson G and Withers DR (2012). “Lymphoid tissue inducer cells: innate cells critical for CD4(+) T cell memory responses?” Ann N Y Acad Sci 1247(1): 1–15. [DOI] [PubMed] [Google Scholar]
- Laroni A, Armentani E, Kerlero de Rosbo N, Ivaldi F, Marcenaro E, Sivori S, Gandhi R, Weiner HL, Moretta A, Mancardi GL and Uccelli A (2016). “Dysregulation of regulatory CD56 NK cells/T cells interactions in multiple sclerosis.” J Autoimmun [DOI] [PubMed] [Google Scholar]
- Lehky TJ, Levin MC, Kubota R, Bamford RN, Flerlage AN, Soldan SS, Leist TP, Xavier A, White JD, Brown M, Fleisher TA, Top LE, Light S, McFarland HF, Waldmann TA and Jacobson S (1998). “Reduction in HTLV-I proviral load and spontaneous lymphoproliferation in HTLV-I-associated myelopathy/tropical spastic paraparesis patients treated with humanized anti-Tac.” Ann Neurol 44(6): 942–947. [DOI] [PubMed] [Google Scholar]
- Lenardo MJ (1991). “Interleukin-2 programs mouse T lymphocytes for apoptosis.” Nature 353: 858–861. [DOI] [PubMed] [Google Scholar]
- Lin YC, Winokur P, Blake A, Wu T, Manischewitz J, King LR, Romm E, Golding H and Bielekova B (2016). “Patients with MS under daclizumab therapy mount normal immune responses to influenza vaccination.” Neurol Neuroimmunol Neuroinflamm 3(1): e196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Winokur P, Blake A, Wu T, Romm E and Bielekova B (2015). “Daclizumab reverses intrathecal immune cell abnormalities in multiple sclerosis.” Ann Clin Transl Neurol 2(5): 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Wang L, Zhan SY and Xia Y (2012). “Daclizumab for relapsing remitting multiple sclerosis.” Cochrane Database Syst Rev 4: CD008127. [DOI] [PubMed] [Google Scholar]
- Lochner M, Ohnmacht C, Presley L, Bruhns P, Si-Tahar M, Sawa S and Eberl G (2011). “Microbiota-induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of RORgamma t and LTi cells.” J Exp Med 208(1): 125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe CE, Cooper JD, Brusko T, Walker NM, Smyth DJ, Bailey R, Bourget K, Plagnol V, Field S, Atkinson M, Clayton DG, Wicker LS and Todd JA (2007). “Large-scale genetic fine mapping and genotype-phenotype associations implicate polymorphism in the IL2RA region in type 1 diabetes.” Nat Genet 39(9): 1074–1082. [DOI] [PubMed] [Google Scholar]
- Magliozzi R, Columba-Cabezas S, Serafini B and Aloisi F (2004). “Intracerebral expression of CXCL13 and BAFF is accompanied by formation of lymphoid follicle-like structures in the meninges of mice with relapsing experimental autoimmune encephalomyelitis.” J Neuroimmunol 148(1–2): 11–23. [DOI] [PubMed] [Google Scholar]
- Magliozzi R, Howell O, Vora A, Serafini B, Nicholas R, Puopolo M, Reynolds R and Aloisi F (2007). “Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology.” Brain 130(Pt 4): 1089–1104. [DOI] [PubMed] [Google Scholar]
- Maier LM, Lowe CE, Cooper J, Downes K, Anderson DE, Severson C, Clark PM, Healy B, Walker N, Aubin C, Oksenberg JR, Hauser SL, Compston A, Sawcer S, C. International Multiple Sclerosis Genetics, De Jager PL, Wicker LS, Todd JA and Hafler DA (2009). “IL2RA genetic heterogeneity in multiple sclerosis and type 1 diabetes susceptibility and soluble interleukin-2 receptor production.” PLoS Genet 5(1): e1000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malek TR (2003). “The main function of IL-2 is to promote the development of T regulatory cells.” J Leukoc Biol 74(6): 961–965. [DOI] [PubMed] [Google Scholar]
- Martin JF, Perry JS, Jakhete NR, Wang X and Bielekova B (2010). “An IL-2 paradox: blocking CD25 on T cells induces IL-2-driven activation of CD56(bright) NK cells.” J Immunol 185(2): 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesarovic MD, Sreenath SN and Keene JD (2004). “Search for organising principles: understanding in systems biology.” Syst Biol (Stevenage) 1(1): 19–27. [DOI] [PubMed] [Google Scholar]
- Mizel SB and Farrar JJ (1979). “Revised nomenclature for antigen-nonspecific T-cell proliferation and helper factors.” Cell Immunol 48(2): 433–436. [DOI] [PubMed] [Google Scholar]
- Moretta L, Biassoni R, Bottino C, Cantoni C, Pende D, Mingari MC and Moretta A (2002). “Human NK cells and their receptors.” Microbes Infect 4(15): 1539–1544. [DOI] [PubMed] [Google Scholar]
- Moroso V, Metselaar HJ, Mancham S, Tilanus HW, Eissens D, van der Meer A, van der Laan LJ, Kuipers EJ, Joosten I and Kwekkeboom J (2010). “Liver grafts contain a unique subset of natural killer cells that are transferred into the recipient after liver transplantation.” Liver Transpl 16(7): 895–908. [DOI] [PubMed] [Google Scholar]
- Nielsen N, Odum N, Urso B, Lanier LL and Spee P (2012). “Cytotoxicity of CD56(bright) NK cells towards autologous activated CD4+ T cells is mediated through NKG2D, LFA-1 and TRAIL and dampened via CD94/NKG2A.” PLoS One 7(2): e31959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa K, Saito S, Morii T, Hamada K, Ako H, Narita N, Ichijo M, Kurahayashi M and Sugamura K (1991). “Accumulation of CD16-CD56+ natural killer cells with high affinity interleukin 2 receptors in human early pregnancy decidua.” Int Immunol 3(8): 743–750. [DOI] [PubMed] [Google Scholar]
- Nussenblatt RB, Fortin E, Schiffman R, Rizzo L, Smith J, Van Veldhuisen P, Sran P, Yaffe A, Goldman CK, Waldmann TA and Whitcup SM (1999). “Treatment of noninfectious intermediate and posterior uveitis with the humanized anti-Tac mAb: a phase I/II clinical trial.” Proc Natl Acad Sci U S A 96(13): 7462–7466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussenblatt RB, Thompson DJ, Li Z, Peterson JS, Robinson RR, Shames RS, Nagarajan S, Tang MT, Mailman M, Velez G, Roy C, Levy-Clarke GA, Suhler EB, Djalilian A, Sen HN, Al-Khatib S, Ursea R, Srivastava S, Bamji A, Mellow S, Sran P, Waldmann TA and Buggage RR (2003). “Humanized anti-interleukin-2 (IL-2) receptor alpha therapy: long-term results in uveitis patients and preliminary safety and activity data for establishing parameters for subcutaneous administration.” J Autoimmun 21(3): 283–293. [DOI] [PubMed] [Google Scholar]
- Oh J, Saidha S, Cortese I, Ohayon J, Bielekova B, Calabresi PA and Newsome SD (2014). “Daclizumab-induced adverse events in multiple organ systems in multiple sclerosis.” Neurology [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh U, Blevins G, Griffith C, Richert N, Maric D, Lee CR, McFarland H and Jacobson S (2009). “Regulatory T cells are reduced during anti-CD25 antibody treatment of multiple sclerosis.” Arch Neurol 66(4): 471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohayon J, Oh U, Richert N, Martin J, Vortmeyer A, McFarland H and Bielekova B (2013). “CNS vasculitis in a patient with MS on daclizumab monotherapy.” Neurology 80(5): 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orange JS (2012). “Unraveling human natural killer cell deficiency.” J Clin Invest 122(3): 798–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry JS, Han S, Xu Q, Herman ML, Kennedy LB, Csako G and Bielekova B (2012). “Inhibition of LTi Cell Development by CD25 Blockade Is Associated with Decreased Intrathecal Inflammation in Multiple Sclerosis.” Sci Transl Med 4(145): 145ra106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queen C, Schneider WP, Selick HE, Payne PW, Landolfi NF, Duncan JF, Avdalovic NM, Levitt M, Junghans RP and Waldmann TA (1989). “A humanized antibody that binds to the interleukin 2 receptor.” Proc Natl Acad Sci U S A 86(24): 10029–10033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis EA, Athanazio DA, Lima I, NO ES, Andrade JC, Jesus RN, Barbosa LM, Reis MG and Santiago MB (2008). “NK and NKT cell dynamics after rituximab therapy for systemic lupus erythematosus and rheumatoid arthritis.” Rheumatol Int 29(4): 469–475. [DOI] [PubMed] [Google Scholar]
- Rickert M, Wang X, Boulanger MJ, Goriatcheva N and Garcia KC (2005). “The structure of interleukin-2 complexed with its alpha receptor.” Science 308(5727): 1477–1480. [DOI] [PubMed] [Google Scholar]
- Roifman CM (2000). “Human IL-2 receptor alpha chain deficiency.” Pediatr Res 48(1): 6–11. [DOI] [PubMed] [Google Scholar]
- Rose JW, Burns JB, Bjorklund J, Klein J, Watt HE and Carlson NG (2007). “Daclizumab phase II trial in relapsing and remitting multiple sclerosis: MRI and clinical results.” Neurology 69(8): 785–789. [DOI] [PubMed] [Google Scholar]
- Rose JW, Watt HE, White AT and Carlson NG (2004). “Treatment of multiple sclerosis with an anti-interleukin-2 receptor monoclonal antibody.” Ann Neurol 56(6): 864–867. [DOI] [PubMed] [Google Scholar]
- Rubenstein JL, Combs D, Rosenberg J, Levy A, McDermott M, Damon L, Ignoffo R, Aldape K, Shen A, Lee D, Grillo-Lopez A and Shuman MA (2003). “Rituximab therapy for CNS lymphomas: targeting the leptomeningeal compartment.” Blood 101(2): 466–468. [DOI] [PubMed] [Google Scholar]
- Ruscetti FW, Morgan DA and Gallo RC (1977). “Functional and morphologic characterization of human T cells continuously grown in vitro.” J Immunol 119(1): 131–138. [PubMed] [Google Scholar]
- Saito S, Nishikawa K, Morii T, Enomoto M, Narita N, Motoyoshi K and Ichijo M (1993). “Cytokine production by CD16-CD56bright natural killer cells in the human early pregnancy decidua.” Int Immunol 5(5): 559–563. [DOI] [PubMed] [Google Scholar]
- Savo AM, Book BK, Henson S, Hakimi J and Pescovitz MD (1999). “Daclizumab rapidly saturates interleukin-2 receptor-alpha (CD25) on lymph node lymphocytes in children.” Transplant Proc 31(1–2): 1182–1183. [DOI] [PubMed] [Google Scholar]
- Sawa S, Cherrier M, Lochner M, Satoh-Takayama N, Fehling HJ, Langa F, Di Santo JP and Eberl G (2010). “Lineage relationship analysis of RORgammat+ innate lymphoid cells.” Science 330(6004): 665–669. [DOI] [PubMed] [Google Scholar]
- Schneider R and Arbour N (2012). “Journal club: Intrathecal effects of daclizumab treatment of multiple sclerosis.” Neurology 78(22): e131–133. [DOI] [PubMed] [Google Scholar]
- Schorle H, Holtschke T, Hunig T, Schimpl A and Horak I (1991). “Development and function of T cells in mice rendered interleukin-2 deficient by gene targeting.” Nature 352(6336): 621–624. [DOI] [PubMed] [Google Scholar]
- Setoguchi R, Hori S, Takahashi T and Sakaguchi S (2005). “Homeostatic maintenance of natural Foxp3+ CD25+ CD4+ regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization.” J Exp Med 201(5): 723–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharfe N, Dadi HK, Shahar M and Roifman CM (1997). “Human immune disorder arising from mutation of the alpha chain of the interleukin-2 receptor.” Proc Natl Acad Sci U S A 94(7): 3168–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevach EM, McHugh RS, Piccirillo CA and Thornton AM (2001). “Control of T-cell activation by CD4+ CD25+ suppressor T cells.” Immunol Rev 182: 58–67. [DOI] [PubMed] [Google Scholar]
- Spits H and Di Santo JP (2011). “The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling.” Nat Immunol 12(1): 21–27. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Kundig TM, Furlonger C, Wakeham A, Timms E, Matsuyama T, Schmits R, Simard JJ, Ohashi PS, Griesser H and et al. (1995). “Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta.” Science 268(5216): 1472–1476. [DOI] [PubMed] [Google Scholar]
- Turka LA and Walsh PT (2008). “IL-2 signaling and CD4+ CD25+ Foxp3+ regulatory T cells.” Front Biosci 13: 1440–1446. [DOI] [PubMed] [Google Scholar]
- Van Parijs L, Refaeli Y, Lord JD, Nelson BH, Abbas AK and Baltimore D (1999). “Uncoupling IL-2 signals that regulate T cell proliferation, survival, and Fas-mediated activation-induced cell death.” Immunity 11(3): 281–288. [DOI] [PubMed] [Google Scholar]
- Vandenbark AA, Huan J, Agotsch M, La Tocha D, Goelz S, Offner H, Lanker S and Bourdette D (2009). “Interferon-beta-1a treatment increases CD56bright natural killer cells and CD4+CD25+ Foxp3 expression in subjects with multiple sclerosis.” J Neuroimmunol 215(1–2): 125–128. [DOI] [PubMed] [Google Scholar]
- Waggoner SN, Cornberg M, Selin LK and Welsh RM (2012). “Natural killer cells act as rheostats modulating antiviral T cells.” Nature 481(7381): 394–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi K, Lian ZX, Moritoki Y, Lan RY, Tsuneyama K, Chuang YH, Yang GX, Ridgway W, Ueno Y, Ansari AA, Coppel RL, Mackay IR and Gershwin ME (2006). “IL-2 receptor alpha(−/−) mice and the development of primary biliary cirrhosis.” Hepatology 44(5): 1240–1249. [DOI] [PubMed] [Google Scholar]
- Waldmann T, Tagaya Y and Bamford R (1998). “Interleukin-2, interleukin-15, and their receptors.” Int Rev Immunol 16(3–4): 205–226. [DOI] [PubMed] [Google Scholar]
- Waldmann TA (2002). “The IL-2/IL-15 receptor systems: targets for immunotherapy.” J Clin Immunol 22(2): 51–56. [DOI] [PubMed] [Google Scholar]
- Waldmann TA, Goldman CK, Bongiovanni KF, Sharrow SO, Davey MP, Cease KB, Greenberg SJ and Longo DL (1988). “Therapy of patients with human T-cell lymphotrophic virus I-induced adult T-cell leukemia with anti-Tac, a monoclonal antibody to the receptor for interleukin-2.” Blood 72(5): 1805–1816. [PubMed] [Google Scholar]
- Waldmann TA, Kozak RW, Tsudo M, Oh-ishi T, Bongiovanni KF and Goldman CK (1987). “IL-2 receptors in adult T-cell leukemia: a target for immunotherapy.” Hamatol Bluttransfus 31: 110–115. [DOI] [PubMed] [Google Scholar]
- Waldmann TA and O’Shea J (1998). “The use of antibodies against the IL-2 receptor in transplantation.” Curr Opin Immunol 10(5): 507–512. [DOI] [PubMed] [Google Scholar]
- Wang X, Rickert M and Garcia KC (2005). “Structure of the quaternary complex of interleukin-2 with its alpha, beta, and gammac receptors.” Science 310(5751): 1159–1163. [DOI] [PubMed] [Google Scholar]
- Webster AC, Playford EG, Higgins G, Chapman JR and Craig JC (2004). “Interleukin 2 receptor antagonists for renal transplant recipients: a meta-analysis of randomized trials.” Transplantation 77(2): 166–176. [DOI] [PubMed] [Google Scholar]
- Willerford DM, Chen J, Ferry JA, Davidson L, Ma A and Alt FW (1995). “Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment.” Immunity 3(4): 521–530. [DOI] [PubMed] [Google Scholar]
- Williams MA, Tyznik AJ and Bevan MJ (2006). “Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells.” Nature 441(7095): 890–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withers DR, Gaspal FM, Mackley EC, Marriott CL, Ross EA, Desanti GE, Roberts NA, White AJ, Flores-Langarica A, McConnell FM, Anderson G and Lane PJ (2012). “Cutting Edge: Lymphoid Tissue Inducer Cells Maintain Memory CD4 T Cells within Secondary Lymphoid Tissue.” J Immunol 189(5): 2094–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuest SC, Edwan JH, Martin JF, Han S, Perry JS, Cartagena CM, Matsuura E, Maric D, Waldmann TA and Bielekova B (2011). “A role for interleukin-2 trans-presentation in dendritic cell-mediated T cell activation in humans, as revealed by daclizumab therapy.” Nat Med 17(5): 604–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn D, Kaufman M, Montalban X, Vollmer T, Simon J, Elkins J, O’Neill G, Neyer L, Sheridan J, Wang C, Fong A and Rose JW (2010). “Daclizumab in active relapsing multiple sclerosis (CHOICE study): a phase 2, randomised, double-blind, placebo-controlled, add-on trial with interferon beta.” Lancet Neurol 9(4): 381–390. [DOI] [PubMed] [Google Scholar]
- Yokota Y, Mansouri A, Mori S, Sugawara S, Adachi S, Nishikawa S and Gruss P (1999). “Development of peripheral lymphoid organs and natural killer cells depends on the helix-loop-helix inhibitor Id2.” Nature 397(6721): 702–706. [DOI] [PubMed] [Google Scholar]