

Abstract
Early exposure to inflammatory signals may have lasting impact on immune function. Present throughout embryogenesis, macrophages are key cells providing innate immune protection to the developing fetus and newborn. Here we have used an established model of macrophage development to test how early inflammatory signals can impact cellular differentiation and function. Bone marrow derived macrophages were treated with E. coli lipopolysaccharide (LPS) two days after initial isolation and culture. LPS treatment during this early stage of differentiation decreased CSF1R expression and increased expression of the mature macrophage marker F4/80. These early changes in macrophage differentiation were similarly measured in cells from mice lacking IKKβ but the changes in CSF1R expression after LPS treatment was blocked with MAPK inhibition. LPS-induced changes in macrophage marker expression persisted following LPS removal, suggesting early inflammatory activation could induce lasting developmental impacts. Early LPS exposure inhibited macrophage phagocytosis of labeled E. coli while LPS had no effect on fully differentiated macrophages. Our data demonstrated that early inflammatory exposure to a microbial stimulus induced lasting phenotypic changes in macrophages.
Keywords: innate immunity, lipopolysaccharide, phagocytosis, macrophage progenitors
Introduction
During development, the fetal innate immune system removes damaged or dead cells and maintains tolerance to maternal antigens [1,2]. However at birth, newborns rapidly encounter environmental microbes and pathogens. During this transition period, newborns are particularly susceptible to disease, particularly pneumonia and sepsis [3,4]. Opportunistic bacteria like Group B Streptococcus and E. coli take advantage of the newborn transition period and cause devastating infections [5]. Infants born preterm are even more vulnerable to infections and inflammatory diseases [6]. While maturation of immunity does eventually occur in both term and preterm infants, the mechanisms regulating innate immue development are still being uncovered. In addition, early activation of developing immune cells may cause lasting consequences on healthy responses later in life.
Early lung infections are major drivers of pediatric disease. Respiratory viral infections in the first year of life increase the risk of asthma [7,8]. In preterm infants, infection and inflammation increase the risk of the chronic developmental lung disease bronchopulmonary dysplasia (BPD) [9–12]. We are now learning more about the role of innate immune development and pediatric lung disease. Macrophages are the primary immune cells in the lung and are responsible for protecting newborns from inhaled pathogens [13,14]. During development, macrophages arising from distinct sources differentiate into various lung macrophage populations. The earliest yolk sac derived macrophages migrate to multiple tissues, including the lung [15]. A second wave of macrophages from the fetal liver initially reside in the lung interstitium, but then migrate into the alveolar space following birth [16,17]. Differentiation of these immature cells into alveolar macrophages requires GM-CSF and PPARγ activation [18]. Differentiated alveolar macrophages express Siglec-F, CD11c, F4/80, and the macrophage scavenger receptors CD204 and CD206 [19,20].
Mature alveolar macrophages form the first line of defense against inhaled particles and pathogens. Efficient at phagocytosis, alveolar macrophages kill pathogens and recruit additional inflammatory cells when needed. During development, activation of fetal lung macrophages increased expression of mature alveolar macrophage markers. In addition to causing fetal lung inflammation, expressing a constitutively active IKKβ or a gain of function Nlrp3 allele in developing fetal macrophages increased expression of F4/80, CD204, and CD206 [21,22]. These data suggested that activation could promote and potentially accelerate lung macrophage differentiation. However, the signaling mechanisms involved in this maturation process were unclear.
Here we have used an experimental model of macrophage differentiation to test how inflammatory activation modulates fetal macrophage development. Culturing hematopoietic precursors from bone marrow with L929-conditioned media generates bone marrow-derived macrophages (BMDM) [23]. We used E. coli endotoxin lipopolysaccharide (LPS) as a stimulator of innate immune signaling to test if TLR-mediated inflammatory pathways in immature bone marrow precursors could influence macrophage differentiation. Our data presented here show not only that LPS alters macrophage marker expression, but also investigates the signaling mechanisms responsible for these changes. These results support our hypothesis that activation of developing macrophages alters differentiation and subsequent cell function.
Materials and Methods
Animals
C57BL/6 (The Jackson Laboratory; Bar Harbor, ME) or LysM-Cre:Ikkbflox/flox (LysM-ΔIKKβ) mice and their littermate controls were bred under standard conditions at the University of California, San Diego animal facilities. All animals experiments and protocols have been reviewed and approved by the University of California, San Diego Institutional Animal Care and Use Committee.
Bone marrow-derived macrophages
Bone marrow-derived macrophages (BMDM) were isolated and grown as previously described [23]. Femurs were isolated from 6–8 week old male C57BL/6, LysM-ΔIKKβ, and littermate control mice. Bone marrow cells were flushed out of the femurs using 10 ml of RPMI medium into 100 mm petri dishes with a 27.5-gauge needle. Single cell suspensions of bone marrow precursors were centrifuged, resuspended in L929-conditioned medium and cultured in non-tissue culture-treated 100 mm petri dishes or 48-well plates.
Lung Macrophages
To study the response of LysM-ΔIKKβ cells to LPS stimulation, lung single cell suspensions from 6 week-old LysM-ΔIKKβ mice and littermate controls (Ikkbflox/flox) were cultured overnight in L929-conditioned medium in non-tissue culture-treated 48-well plates. Medium containing non-adherent cells was removed the following day and adherent macrophage populations were stimulated with gel-purified E. coli lipopolysaccharide (LPS; 250 ng/ml; strain O55:B5 Sigma L6529; St. Louis, MO).
Immune stimulation
Bone marrow-derived macrophages were treated with LPS (250 ng/ml) for 16 h. For multiple exposure experiments, macrophages were treated with LPS for 16 h after which medium was removed and the cells were cultured in LPS-free L929-conditioned media for 24 h. These cells were then treated a second time with LPS or PAM3CSK4 (PAM3; 1μg/ml; Invivogen; San Diego, CA) for 3 h. A cocktail of MAPK pathway inhibitors was used at the following final concentrations: SB2035804 (p38 inhibitor) – 100 nM, PD98059 (ERK 1 and 2 inhibitor) – 10 μM and SP600125 (JNK 1 and 2 inhibitor) – 25 μM (Cell Signaling Technology; Boston, MA). The inhibitor cocktail was added to the culture for one hour prior to LPS treatment.
Flow cytometry
After LPS stimulation, macrophages were collected and centrifuged. Media was removed and the cell pellet was resuspended in flow cytometry staining buffer (PBS, 1% BSA, 5% FBS). The cell suspensions were then treated with TruStain CD16/32 (Fc block) at a 1:50 dilution (Biolegend; San Diego, CA). Cells were incubated on ice for 20 minutes. The cells were then stained with the various fluorophore conjugated antibodies at appropriate concentrations and incubated for 30 minutes on ice. Antibodies used were: CD45-FITC, CSF1R-PE, F4/80-APC and CD11b-Brilliant Violet. Live/Dead discrimination was done using the Zombie NIR™ Fixable Viability Kit from Biolegend. Cells were washed in flow cytometry buffer and then analyzed on a FACS Canto II flow cytometry instrument (Becton Dickinson; Franklin Lakes, NJ). Flow cytometry data was analyzed using FlowJo software.
ELISA
ELISA on macrophage supernatants were performed using Ready-Set-Go® kits (Thermo Fisher Scientific; Waltham, MA) for murine IL-6 and IL-10 according to manufacturer instructions.
Quantitative Real Time PCR
BMDM messenger total RNA was extracted using TRIzol (Thermo Fisher Scientific; Waltham, MA) and phenol extraction. 1 ng of RNA was then reverse transcribed to cDNA using Invitrogen SuperScript® III First Strand Synthesis System (Thermo Fisher Scientific; Waltham, MA) according to manufacturer instructions. Using the SYBR Green Mix (Bio-Rad; Hercules, CA) and specific primer pairs, quantitative real time polymerase chain reaction (Q-RTPCR) was performed using a CFX96 thermocycler (Bio-Rad; Hercules, CA). GAPDH was used as an internal reference gene for all experiments. Fold change was calculated using the 2-ΔΔCT method.
Phagocytosis Assay
Phagocytosis assays were performed using pHrodo Green E.coli Bioparticles from Thermo Fisher (Thermo Fisher Scientific; Waltham, MA). Following treatment with LPS, macrophages were briefly washed, resuspended in fresh culture medium, and plated into a 96-well microplate (Falcon; Corning, NY). The re-plated cells were incubated for 30 minutes at 37°C. The microplate was centrifuged at 1000 rpm for 10 minutes and culture medium was removed afterwards. The medium was replaced with 50µl pHrodo Green E. coli Bioparticles suspended in Live Cell Imaging Buffer (1 mg/ml final concentration; Thermo Fisher Scientific; Waltham, MA). The microplate was incubated for 30 minutes at 37°C without CO2 to avoid artificial phagosome acidification. Following incubation, the cells were washed, resuspended in 200 µl flow cytometry staining buffer and analyzed via flow cytometry.
Data/Statistical Analysis
Data were analyzed using either Student’s t test or one-way ANOVA within Prism.
Results
Our previous data showed that immune activation altered macrophage development [21,22]. To further investigate this process, we used a primary cell culture model of macrophage differentiation. Bone marrow precursors differentiate into macrophages when cultured with L929-conditioned medium for at least 7 d. To use this model in testing the effects of inflammatory activation on macrophage differentiation, we first measured the kinetics of macrophage marker expression during culture in conditioned media (Figure 1A). Freshly isolated bone marrow precursors failed to express significant levels of the macrophage markers CSF1R, F4/80, or CD11b. After 3 d of culture, we detected expression of each macrophage marker with small populations of F4/80-negative and CD11b-negative cells. Expression of CSF1R, F4/80 and CD11b was higher following 7 d of culture. To test the effects of inflammatory activation in naïve precursors before differentiation was complete, we focused on how LPS could impact macrophage marker expression in immaturely differentiated cells 2 d after isolation.
Figure 1. LPS treatment altered macrophage differentiation.

(A). Freshly isolated bone marrow precursors expressed the macrophage markers CSF1R, F4/80, and CD11b after 3 d culture in L929 conditioned media. (B). Adding LPS to the media (250 ng/ml for 16 h) on day 0 had no effect on CSF1R or F4/80 expression. However treating bone marrow precursors with LPS after 2 d (middle) or 7 d (right) decreased CSF1R (top panels) and increased F4/80 expression (bottom panels). Representative FACS data shown. (C). MFI data from multiple 2 d experiments showed that LPS reduced CSF1R (**P < 0.005, n = 11) and increased F4/80 (**P < 0.005, n = 11). (D). Each separate bone marrow isolette displayed similar trends in F4/80 and CSF1R expression following LPS treatment on day 2. (E). LPS increased mRNA expression of Adgre1 (gene encoding F4/80) and the scavenger receptor Msr1 (CD204) (***P < 0.001, n = 10).
Bone marrow precursors cultured in L929-conditioned media for 2 d were treated with LPS and assayed for macrophage marker expression 16 h later (Figure 1B,C). LPS treatment reduced CSF1R expression and increased F4/80 compared to control cells. These effects were not observed in freshly isolated and plated precursors treated with LPS for 16 h (left panels in Figure 1B), possibly due to lack of TLR4 expression or inadequate macrophage differentiation. Similar LPS-dependent changes were observed when cells cultured in conditioned media for 7 d were treated with LPS (far right panels in Figure 1B). LPS increased F4/80 and decreased CSF1R MFI across a number of independent experiments; each treated on day 2 and analyzed 16 h later (Figure 1C). The LPS-induced trends were consistent within each experiment (Figure 1D). LPS did not alter Csf1r mRNA levels as measured by real time PCR, suggesting a post-translational change in surface expression of CSF1R protein as has been reported (Figure 1E, [24]). Based on our previous experiments testing fetal inflammation on macrophage marker expression [21,22], we measured expression of both Adgre1 (the gene encoding F4/80) and two mannose/scavenger receptor genes Msr1 (CD204) and Mrc1 (CD206). LPS increased expression of Adgre1 and Msr1, but did not alter expression of Mrc1 (Figure 1E).
In macrophages, LPS activation of TLR4 signals through both IKKβ/NF-κB [25] and MAPK [26] pathways. To test the pathways connecting LPS/TLR4 signaling to macrophage differentiation, we measured the effects of LPS on macrophage differentiation using bone marrow from mutant mice lacking functional IKKβ in myeloid cell populations. LPS induced lower levels of Il6 in LysM-ΔIKKβ lung macrophages compared to littermate control cells, confirming the important role IKKβ in the LPS esponse (Figure 2A). We then isolated bone marrow precursors from LysM-Cre:Ikkbflox/flox (LysM-ΔIKKβ) mice and littermate controls, treating cells with LPS after 2 d of culture in L929-conditioned media. As seen in Figure 2B, LPS had similar effects on cells from both LysM-ΔIKKβ and Ikkbflox/flox littermate controls, reducing CSF1R expression and increasing F4/80 expression. The trends effects of LPS on CSF1R and F4/80 expression were consistent across three independent experiments (Figure 2C). Therefore the effects of LPS on macrophage differentiation did not appear to require IKKβ-dependent signaling.
Figure 2. Effects of LPS on BMDM differentiation do not require IKKβ.
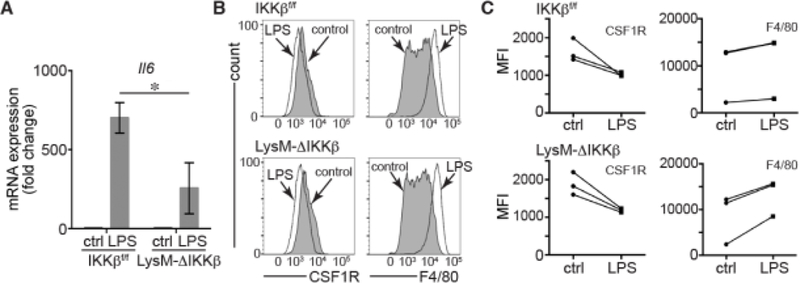
(A). Lung macrophages from LysM-ΔIKKβ mice had reduced Il6 mRNA expression compared to Ikkbflox/flox littermate controls (*P < 0.05, n = 6). Cells not treated with LPS labeled as “ctrl.” (B). LPS had similar effects on CSF1R and F4/80 expression in BMDM from LysM-ΔIKKβ and Ikkbflox/flox (littermate control) mice. Cells were treated with LPS on day 2 of culture and analyzed by FACS. (C). MFI from independent experiments showing consistent trends in CSF1R and F4/80 expression following LPS treatment in Ikkbflox/flox and LysM-ΔIKKβBMDM. Cells not treated with LPS labeled as “ctrl.”
We next tested if the effects of LPS on macrophage differentiation required MAPK signaling (Figure 3). Bone marrow precursors cultured for 2 d in conditioned media were treated with a MAPK inhibitor cocktail 1 h prior to LPS exposure. MAPK inhibition prevented the LPS-induced decrease in CSF1R expression (Figure 3A). However, the LPS effect on increased F4/80 expression alone did not appear to be impacted by MAPK inhibitor treatment. Across five independent experiments, MAPK inhibition prevented LPS from decreasing the percentage of CSF1R expressing cells within the F4/80 macrophage population (Figure 3B,C). These data suggested that at least some of the LPS effects on macrophage differentiation involve MAPK signaling but not IKKβ.
Figure 3. Effect of MAPK inhibition on LPS-induced changes in BMDM differentiation.

(A). FACS of BMDM treated with LPS or LPS following 1 h pretreatment with a MAPK inhibitor cocktail. MAPK inhibition prevented the LPS-induced reduction in CSF1R expression (*P < 0.05, n = 5). However F4/80 expression was not affected. (B,C). Representative FACS experiment showing effects of LPS +/−MAPK inhibition on F4/80 and CSF1R expression in BMDM. MAPK inhibition prevented the LPS-induced changes in the percentage of F4/80+ cells also expressing CSF1R. Each line in (C) represents an independent experiment.
To test if LPS-mediated changes in macrophage differentiation were transient or persistent, we treated 2 d bone marrow precursors with LPS for 16 h, removed the LPS by changing the media, and measured macrophage marker expression 24 h later (Figure 4A). FACS showed that the LPS-induced changes in F4/80 and CSF1R persisted after 24 h of LPS removal. We next tested if early LPS-induced changes could alter the macrophage response to a second round of TLR activation. For these experiments, we treated 2 d bone marrow precursors with LPS for 16 h, removed the media for 24 h, and then treated the cells a second time for 3 h with either LPS or the TLR2 agonist Pam3CysSerLys4 (PAM3) (Figure 4B). To assay TLR responsiveness, we measured Il6 mRNA by real time PCR. LPS-induced Il6 expression was similar in cells that had been treated with LPS on both day 2 and day 4, or only on day 4. Interestingly, treating cells on day 2 with LPS increased the induction of Il6 expression when challenged with PAM3 on day 4 compared to samples only treated with PAM3 on day 4 (Figure 4C). This result suggested early LPS might augment TLR2-mediated transcription. However, we did not measure similar changes in IL-6 or IL-10 protein released into the cell media (Figure 4D). These data suggest that early activation might affect transcriptional responses to additional stimuli but not necessarily significantly alter cytokine release.
Figure 4. LPS caused persistent changes in BMDM differentiation and response to TLR agonists.

(A). The LPS-induced changes in CSF1R and F4/80 expression were unchanged after LPS was removed for 24 h. (B). LPS-treated BMDM (16 h on day 2) were challenged with a second treatment of either LPS again or the TLR2 agonist Pam3CysSerLys4 (PAM3) on day 4. (C). LPS treatment on day 2 increased the relative expression of Il6 following PAM3 stimulation on day 4 (*P < 0.05, n = 6). (D) Release of IL-6 and IL-10 into the cell culture media after either LPS or PAM3 exposure on day 4 was not affected by LPS treatment on day 2.
We next tested the effects of LPS during differentiation on microbial and particle phagocytosis. Cultured BMDMs were incubated with pHrodo E. coli bioparticles, which emit a fluorescent signal when engulfed and exposed to the acidic environment of the phagosome. The majority of control cells had high levels of pHrodo fluorescence, consistent with efficient uptake and acidification (PEHI; Figure 5A). A smaller percentage of cells had lower intensity (PELO), potentially due to incomplete acidification within an early endosomal compartment or immature phagosome development. In LPS treated cells, the percentage of PEHI cells was lower, with more cells having low levels of pHrodo fluorescence or no fluorescent signal at all (Figure 5A–C). Mature macrophages (day 7) did not show a difference between the populations of cells that had low or high levels of pHrodo fluorescence (Figure 5A–D). Overall, these results suggest that treating immature, developing macrophages with LPS decreased phagocytic activity.
Figure 5. LPS treatment during BMDM differentiation reduced phagocytosis.

BMDM treated on day 2 or day 7 with LPS (250 ng/ml for 16 h) were then cultured with pHrodo-labeled E. coli bioparticles. Phagocytosis (endocytosis and intravesicular acidification) was measured by FACS. (A,B). Representative FACS plot (of nine total experiments) showing populations of low fluorescence (PELO) and high fluorescence (PEHI) within BMDM cultures. (C,D). Percentage of cells with low (PELO) or high (PEHI) pHrodo fluorescence. Nine experiments were performed on cells from three separate bone marrow isolates. Data were normalized to controls (control = 100%). In day 2 experiments (C), LPS increased the percentage of cells with low pHrodo E. coli fluorescence (****P < 0.001) and decreased cells with higher fluorescence intensity (****P < 0.001). BMDM treated with LPS on day 7 (D) had similar percentages of cells with low or high pHrodo intensity compared to controls.
Discussion
Our data showed that early activation altered differentiation of immature macrophages. Treating incompletely differentiated bone marrow derived precursors with LPS reduced CSF1R and increased F4/80 expression compared to untreated controls. The findings paralleled our previous data showing that transgenic macrophage activation in vivo appeared to accelerate fetal lung macrophage differentiation [21,22]. The changes in CSF1R expression in developing macrophages required MAPK signaling but not IKKβ. Interestingly, the differential expression of macrophage cell surface markers appeared to be persistent even after LPS was removed. Early activation also led to later functional differences in phagocytosis and transcriptional responses to secondary stimuli. These results therefore support the concept of early immune activation having lasting effects on macrophage biology.
We previously showed that macrophage activation in utero increases expression of mature alveolar macrophage markers. Transgenic expression of gain of function Ikkb or Nlrp3 alleles increased expression of F4/80 (Adgre1), CD204 (Msr1), and CD206 (Mrc1), each highly expressed by mature alveolar macrophages. These two mutant strains targeted innate immune activation upstream (Ikkb) and downstream (Nlrp3) of NF-κB transcriptional activity [21,22]. In macrophages, LPS activates both MAPK [26] and NF-κB [25]. Our data here showed that at least some components of LPS-mediated changes in BMDM differentiation required MAPK signaling but not IKKβ, the rate-limiting enzyme in NF-κB activation. Our findings could be related to MAPK-dependent ATF7 phosphorylation, which plays a key role in promoting LPS-mediated innate immune memory [27]. Consistent with the concept of “trained immunity” or innate immune memory [28–31], our findings suggest that immune activation of immature macrophages also causes changes in cell differentiation and potential training of the innate immune response.
Changes in CSF1R expression could lead to multiple, pleiotropic effects on macrophages, including cell proliferation and differentiation. CSF1R signaling in response to CSF1 generally suppresses the innate immune response in exchange for more trophic cellular effects [32]. Previous reports showed that LPS reduced CSF1R cell surface expression on mature treated macrophages, potentially leading to more vigorous immune responses [24]. Our data here demonstrate that LPS also lowers CSF1R expression in developing macrophages. Consistent with the idea of trained innate immunity, LPS treated cells expressed higher levels of Il6 transcripts when challenged later with the TLR2 ligand PAM3. While this increase did not apparently lead to detectable changes in secreted cytokine levels, the data do suggest differences in transcriptional responses in cells exposed early to immune activators.
Recent work has provided significant insight into the transcriptional mechanisms regulating macrophage differentiation. The master macrophage regulator PU.1 promotes the deposition of mono-methylated lysine 4 in histone 3 (H3K4me1) to create small open regions of accessible DNA of approximately 150–300 base pairs [33–35]. Macrophage transcription factors including C/EBPα, IRF4, IRF5 and PPARγ as well as stimulus-dependent factors like NF-κB are then able to bind to DNA driving the expression of specific genes. PU.1 appears to interact with both lineage-specific and stimulus-dependent transcription factors. For example, specific interactions between the amino-terminal of PU.1 and C/EBP drive transcription of GM-CSF receptor α [36] Pretreatment of human monocytes with β-glucan increased trimethylation of lysine 4 in histone 3 (H3K4me3) and acetylation of histone 3 at lysine 27 (H3K27ac) in differentiated macrophages [37]. Furthermore, in peritoneal macrophages, LPS treatment led to disruption of heterochromatin and a decrease in the level of repressive histone H3K9me2 marker [27]. Such genome-wide epigenetic changes could alter cellular behavior when responding later to subsequent immune challenges.
Early LPS exposure also reduced the efficient phagocytosis of E. coli particles. In our experiments, macrophage precursors cultured for 2 d and treated with LPS had phagocytosis patterns resembling cell cultured for 7 d. These data were again consistent with innate immune activation promoting certain aspects of macrophage maturation. As efficient phagocytosis of both microbes and particles is a key function of macrophages, these changes could represent significant alterations in immunity. Newborn infants (especially those born preterm) are often exposed to pathogenic microbes before macrophage differentiation completes [9,38]. Therefore these early exposures could lead to developmental changes and subsequent disease pathogenesis. Our data do not consistently point to simple training of macrophages to be more inflammatory or tolerant. This apparent complexity was reflected in our previous data showing that activation of fetal lung macrophages induced both inflammatory cytokine expression (“M1”) and increased levels of “M2” markers consistent with reduced inflammatory responsiveness [22]. Additional studies using complementary models of macrophage differentiation will further test how developmental changes influence tissue immunity.
Acknowledgments
This work was supported by the National Institutes of Health (HL086324, HL126703) and the Gerber Foundation (3830, 5346). We are thankful to our colleagues at the University of California, San Diego and Rady Children’s Hospital for their helpful advice and feedback. We thank Gilberto Hernandez for his technical support and Lori Broderick for her helpful feedback on the manuscript.
Footnotes
Conflict of Interest Disclosure
The authors declare no conflicts of interest.
References
- 1.Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, Schilling JD, Schwendener R, Sergin I, Razani B, Forsberg EC, Yokoyama WM, Unanue ER, Colonna M, Randolph GJ, Mann DL: Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014;40:91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McGovern N, Shin A, Low G, Low D, Duan K, Yao LJ, Msallam R, Low I, Shadan NB, Sumatoh HR, Soon E, Lum J, Mok E, Hubert S, See P, Kunxiang EH, Lee YH, Janela B, Choolani M, Mattar CNZ, Fan Y, Lim TKH, Chan DKH, Tan KK, Tam JKC, Schuster C, Elbe-Burger A, Wang XN, Bigley V, Collin M, Haniffa M, Schlitzer A, Poidinger M, Albani S, Larbi A, Newell EW, Chan JKY, Ginhoux F: Human fetal dendritic cells promote prenatal T-cell immune suppression through arginase-2. Nature 2017;546:662–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nissen MD: Congenital and neonatal pneumonia. Paediatr Respir Rev 2007;8:195–203. [DOI] [PubMed] [Google Scholar]
- 4.Hoffman JA, Mason EO, Schutze GE, Tan TQ, Barson WJ, Givner LB, Wald ER, Bradley JS, Yogev R, Kaplan SL: Streptococcus pneumoniae infections in the neonate. Pediatrics 2003;112:1095–1102. [DOI] [PubMed] [Google Scholar]
- 5.Stoll BJ, Hansen NI, Sanchez PJ, Faix RG, Poindexter BB, Van Meurs KP, Bizzarro MJ, Goldberg RN, Frantz ID 3rd, Hale EC, Shankaran S, Kennedy K, Carlo WA, Watterberg KL, Bell EF, Walsh MC, Schibler K, Laptook AR, Shane AL, Schrag SJ, Das A, Higgins RD, Eunice Kennedy Shriver National Institute of Child H, Human Development Neonatal Research N: Early onset neonatal sepsis: the burden of group B Streptococcal and E. coli disease continues. Pediatrics 2011;127:817–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collins A, Weitkamp JH, Wynn JL: Why are preterm newborns at increased risk of infection? Arch Dis Child Fetal Neonatal Ed 2018;103:F391–F394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jackson DJ: Early-life viral infections and the development of asthma: a target for asthma prevention? Curr Opin Allergy Clin Immunol 2014;14:131–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jackson DJ, Gangnon RE, Evans MD, Roberg KA, Anderson EL, Pappas TE, Printz MC, Lee WM, Shult PA, Reisdorf E, Carlson-Dakes KT, Salazar LP, DaSilva DF, Tisler CJ, Gern JE, Lemanske RF Jr.: Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am J Respir Crit Care Med 2008;178:667–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jobe AH: Mechanisms of Lung Injury and Bronchopulmonary Dysplasia. Am J Perinatol 2016;33:1076–1078. [DOI] [PubMed] [Google Scholar]
- 10.Jackson CM, Wells CB, Tabangin ME, Meinzen-Derr J, Jobe AH, Chougnet CA: Pro-inflammatory immune responses in leukocytes of premature infants exposed to maternal chorioamnionitis or funisitis. Pediatr Res 2017;81:384–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jobe AH: The new bronchopulmonary dysplasia. Curr Opin Pediatr 2011;23:167–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jobe AH: Blood cytokines and BPD. J Pediatr 2009;154:A2. [DOI] [PubMed] [Google Scholar]
- 13.Blackwell TS, Hipps AN, Yamamoto Y, Han W, Barham WJ, Ostrowski MC, Yull FE, Prince LS: NF-kappaB signaling in fetal lung macrophages disrupts airway morphogenesis. J Immunol 2011;187:2740–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hiraiwa K, van Eeden SF: Contribution of lung macrophages to the inflammatory responses induced by exposure to air pollutants. Mediators Inflamm 2013;2013:619523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, Rodewald HR: Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015;518:547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ginhoux F, Guilliams M: Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016;44:439–449. [DOI] [PubMed] [Google Scholar]
- 17.van de Laar L, Saelens W, De Prijck S, Martens L, Scott CL, Van Isterdael G, Hoffmann E, Beyaert R, Saeys Y, Lambrecht BN, Guilliams M: Yolk Sac Macrophages, Fetal Liver, and Adult Monocytes Can Colonize an Empty Niche and Develop into Functional Tissue-Resident Macrophages. Immunity 2016;44:755–768. [DOI] [PubMed] [Google Scholar]
- 18.Schneider C, Nobs SP, Kurrer M, Rehrauer H, Thiele C, Kopf M: Induction of the nuclear receptor PPAR-gamma by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol 2014;15:1026–1037. [DOI] [PubMed] [Google Scholar]
- 19.Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H: Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol 2013;49:503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S: Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013;38:79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stouch AN, McCoy AM, Greer RM, Lakhdari O, Yull FE, Blackwell TS, Hoffman HM, Prince LS: IL-1beta and Inflammasome Activity Link Inflammation to Abnormal Fetal Airway Development. J Immunol 2016;196:3411–3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stouch AN, Zaynagetdinov R, Barham WJ, Stinnett AM, Slaughter JC, Yull FE, Hoffman HM, Blackwell TS, Prince LS: IkappaB kinase activity drives fetal lung macrophage maturation along a non-M1/M2 paradigm. J Immunol 2014;193:1184–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weischenfeldt J, Porse B: Bone Marrow-Derived Macrophages (BMM): Isolation and Applications. CSH Protoc 2008;2008:pdb prot5080. [DOI] [PubMed]
- 24.Sester DP, Trieu A, Brion K, Schroder K, Ravasi T, Robinson JA, McDonald RC, Ripoll V, Wells CA, Suzuki H, Hayashizaki Y, Stacey KJ, Hume DA, Sweet MJ: LPS regulates a set of genes in primary murine macrophages by antagonising CSF-1 action. Immunobiology 2005;210:97–107. [DOI] [PubMed] [Google Scholar]
- 25.Xie QW, Kashiwabara Y, Nathan C: Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J Biol Chem 1994;269:4705–4708. [PubMed] [Google Scholar]
- 26.An H, Xu H, Yu Y, Zhang M, Qi R, Yan X, Liu S, Wang W, Guo Z, Qin Z, Cao X: Up-regulation of TLR9 gene expression by LPS in mouse macrophages via activation of NF-kappaB, ERK and p38 MAPK signal pathways. Immunol Lett 2002;81:165–169. [DOI] [PubMed] [Google Scholar]
- 27.Yoshida K, Maekawa T, Zhu Y, Renard-Guillet C, Chatton B, Inoue K, Uchiyama T, Ishibashi K, Yamada T, Ohno N, Shirahige K, Okada-Hatakeyama M, Ishii S: The transcription factor ATF7 mediates lipopolysaccharide-induced epigenetic changes in macrophages involved in innate immunological memory. Nat Immunol 2015;16:1034–1043. [DOI] [PubMed] [Google Scholar]
- 28.Bekkering S, Blok BA, Joosten LA, Riksen NP, van Crevel R, Netea MG: In Vitro Experimental Model of Trained Innate Immunity in Human Primary Monocytes. Clin Vaccine Immunol 2016;23:926–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Netea MG: Immunological memory in innate immunity. J Innate Immun 2014;6:117–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Netea MG: Training innate immunity: the changing concept of immunological memory in innate host defence. Eur J Clin Invest 2013;43:881–884. [DOI] [PubMed] [Google Scholar]
- 31.Bistoni F, Vecchiarelli A, Cenci E, Puccetti P, Marconi P, Cassone A: Evidence for macrophage-mediated protection against lethal Candida albicans infection. Infect Immun 1986;51:668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stanley ER, Chitu V: CSF-1 receptor signaling in myeloid cells. Cold Spring Harb Perspect Biol 2014;6. [DOI] [PMC free article] [PubMed]
- 33.Lawrence T, Natoli G: Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol 2011;11:750–761. [DOI] [PubMed] [Google Scholar]
- 34.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK: Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 2010;38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghisletti S, Barozzi I, Mietton F, Polletti S, De Santa F, Venturini E, Gregory L, Lonie L, Chew A, Wei CL, Ragoussis J, Natoli G: Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity 2010;32:317–328. [DOI] [PubMed] [Google Scholar]
- 36.Hohaus S, Petrovick MS, Voso MT, Sun Z, Zhang DE, Tenen DG: PU.1 (Spi-1) and C/EBP alpha regulate expression of the granulocyte-macrophage colony-stimulating factor receptor alpha gene. Mol Cell Biol 1995;15:5830–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, Cheng SC, Ratter J, Berentsen K, van der Ent MA, Sharifi N, Janssen-Megens EM, Ter Huurne M, Mandoli A, van Schaik T, Ng A, Burden F, Downes K, Frontini M, Kumar V, Giamarellos-Bourboulis EJ, Ouwehand WH, van der Meer JW, Joosten LA, Wijmenga C, Martens JH, Xavier RJ, Logie C, Netea MG, Stunnenberg HG: Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014;345:1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Speer CP: New insights into the pathogenesis of pulmonary inflammation in preterm infants. Biol Neonate 2001;79:205–209. [DOI] [PubMed] [Google Scholar]