

Abstract
Cytoscape is an open source software project for integrating biomolecular interaction networks with high-throughput expression data and other molecular states into a unified conceptual framework. Although applicable to any system of molecular components and interactions, Cytoscape is most powerful when used in conjunction with large databases of protein-protein, protein-DNA, and genetic interactions that are increasingly available for humans and model organisms. Cytoscape's software Core provides basic functionality to layout and query the network; to visually integrate the network with expression profiles, phenotypes, and other molecular states; and to link the network to databases of functional annotations. The Core is extensible through a straightforward plug-in architecture, allowing rapid development of additional computational analyses and features. Several case studies of Cytoscape plug-ins are surveyed, including a search for interaction pathways correlating with changes in gene expression, a study of protein complexes involved in cellular recovery to DNA damage, inference of a combined physical/functional interaction network for Halobacterium, and an interface to detailed stochastic/kinetic gene regulatory models.
Computer-aided models of biological networks are a cornerstone of systems biology. A variety of modeling environments have been developed to simulate biochemical reactions and gene transcription kinetics (Endy and Brent 2001), cellular physiology (Loew and Schaff 2001), and metabolic control (Mendes 1997). Such models promise to transform biological research by providing a framework to (1) systematically interrogate and experimentally verify knowledge of a pathway; (2) manage the immense complexity of hundreds or potentially thousands of cellular components and interactions; and (3) reveal emergent properties and unanticipated consequences of different pathway configurations.
Typically, models are directed toward a cellular process or disease pathway of interest (Gilman and Arkin 2002) and are built by formulating existing literature as a system of differential and/or stochastic equations. However, pathway-specific models are now being supplemented with global data gathered for an entire cell or organism, by use of two complementary approaches. First, recent technological developments have made it feasible to measure pathway structure systematically, using high-throughput screens for protein-protein (Ito et al. 2001; von Mering et al. 2002), protein-DNA (Lee et al. 2002), and genetic interactions (Tong et al. 2001). To complement these data, a second set of high-throughput methods are available to characterize the molecular and cellular states induced by pathway interactions under different experimental conditions. For instance, global changes in gene expression are measured with DNA microarrays (DeRisi et al. 1997), whereas changes in protein abundance (Gygi et al. 1999), protein phosphorylation state (Zhou et al. 2001), and metabolite concentrations (Griffin et al. 2001) may be quantified with mass spectrometry, NMR, and other advanced techniques. High-throughput data pertaining to molecular interactions and states are well matched, in that both data types are global (providing information for all components or interactions in an organism); high-level (outlining relationships among pathway components without detailed information on reaction rates, binding constants, or diffusion coefficients); and coarse-grained (yielding qualitative data, such as the presence or absence of an interaction or the direction of an expression change, more readily than precise quantitative readouts).
Motivated by the explosion in experimental technologies for characterizing molecular interactions and states, researchers have turned to a variety of software tools to process and analyze the resulting large-scale data. For molecular interactions, general-purpose graph viewers such as Pajek (Batagelj and Mrvar 1998), Graphlet (www.infosun.fmi.uni-passau.de/Graphlet/), and daVinci (www.informatik.uni-bremen.de/daVinci/) are available to organize and display the data as a two-dimensional network; specialized tools such as Osprey (http://biodata.mshri.on.ca) and PIMrider (pim.hybrigenics.com) provide these capabilities and also link the network to molecular interaction and functional databases such as BIND (Bader et al. 2001), DIP (Xenarios and Eisenberg 2001), or TRANSFAC (Wingender et al. 2001). Similarly, for gene expression profiles and other molecular states, numerous programs such as GeneCluster (Tamayo et al. 1999), Tree-View (Eisen et al. 1998), and GeneSpring (www.silicongenetics.com) are available for clustering, classification, and visualization. However, a pressing need remains for software that is able to integrate both molecular interactions and state measurements together in a common framework, and to then bridge these data with a wide assortment of model parameters and other biological attributes. Moreover, a flexible and open system will be required to facilitate general and extensible computations on the interaction network (Karp 2001). It is through these computations that high-level interaction data may ultimately interface with, and drive development of, low-level physico-chemical models.
To address these needs, we have developed Cytoscape, a general-purpose modeling environment for integrating biomolecular interaction networks and states. We first provide an overview of Cytoscape's core functionality for representation and integration of biomolecular network models. We then describe three case studies of existing research projects in which the Cytoscape platform is applied to concrete biological problems or extended to implement new algorithms and network computations.
METHODS AND RESULTS
Cytoscape Core Functionality and Architecture
The central organizing metaphor of Cytoscape is a network graph, with molecular species represented as nodes and intermolecular interactions represented as links, that is, edges, between nodes. Cytoscape's Core software component provides basic functionality for integrating arbitrary data on the graph, a visual representation of the graph and integrated data, selection and filtering tools, and an interface to external methods implemented as plug-ins. Figure 1 illustrates key features through screenshots, whereas Figure 2 provides a schematic of their interrelationships.
Figure 2.

Schematic overview of the Cytoscape Core architecture. The Cytoscape window is the primary visual and programmatic interface to Cytoscape and contains the network graph and attribute data structures. Core methods that operate on these structures are graph editing, graph layout, attribute-to-visual mapping, and graph filtering. Annotations are available through a separate server.
Data Integration
Data are integrated with the graph model using Attributes. These are (name, value) pairs that map node or edge names to specific data values. For example, the node named “GAL4” may have an attribute named “expression ratio” whose value is 3.41. Attribute values may assume any type (e.g., text strings, discrete or continuous numbers, URLs, or lists) and are either loaded from a data repository or generated dynamically within a session. Graphical browsers allow the user to examine all attributes on the currently selected nodes and edges (Fig. 1c).
Figure 1.

Tour of Cytoscape core functionality. (a) Available network layout algorithms are accessed via the menu system; an example hierarchical layout is shown. (b) The data attribute-to-visual mapping control is used to integrate a variety of heterogenous data types on the network. Here, gene expression data are mapped to node color for the condition named “gal80R,” in which color is interpolated between green (negative values) and red (positive values) through gray as the midpoint. Node colors in a are derived using this mapping. (c) Attributes are displayed for selected nodes and edges in a browser window. As shown, multiple attributes and genes may be displayed in a customtabular format. (d) Annotations are transferred to node and edge attributes by choosing the desired ontology and hierarchical level from a list of those available.
Transfer of Annotations
Whereas an attribute is a single predicate of a node or edge, an Annotation represents a hierarchical classification (i.e., an ontology, formally, a directed acyclic graph) of progressively more specific descriptions of groups of nodes or edges. Annotations typically correspond to an existing repository of knowledge that is large, complex, and relatively static, such as the Gene Ontology database (GO Consortium 2001). For example, the term “hexose metabolism,” defined at level 6 of the GO Biological Process Ontology, spans several more specific processes at level 7, such as galactose metabolism and glucose metabolism. Cytoscape integrates annotations with other network data types by transferring the desired levels of annotation onto node or edge attributes. Using the Annotation controller (Fig. 1d), it is possible to have many levels of annotation all active and on display at the same time, each as a different attribute on the nodes or edges of interest.
Graph Layout
One of the most fundamental tools for interpreting molecular interaction data is visualization of nodes and edges as a two-dimensional network (Tollis et al. 1999). Cytoscape supports a variety of automated network layout algorithms, including spring-embedded layout, hierarchical layout, and circular layout. Among these, the spring embedder is the most widely used method for arranging general two-dimensional graphs (Eades 1984). It models a mechanical system in which edges of the graph correspond to springs, creating an attractive force between nodes that are far apart, and a repulsive force between nodes that are close together. Network layout for Figure 1a was performed using a hierarchical layout algorithm, and for Figures 3 and 4 using a spring embedder.
Figure 3.

Screening DNA-damage phenotypes against a scaffold of molecular interactions. A large molecular interaction network was integrated with 1615 yeast deletion phenotypes gathered systematically in response to MMS exposure. (a) Cytoscape's filter toolbox was first used to show only those proteins required for viable growth in MMS (i.e., MMS-essential proteins) and their immediate network neighbors. (b) The filtered network was then searched using the ActiveModules plug-in to identify interaction complexes containing significant numbers of MMS-essential proteins. One such region is shown, along with its corresponding p-value. Dark gray nodes represent MMS-essential proteins.
Figure 4.
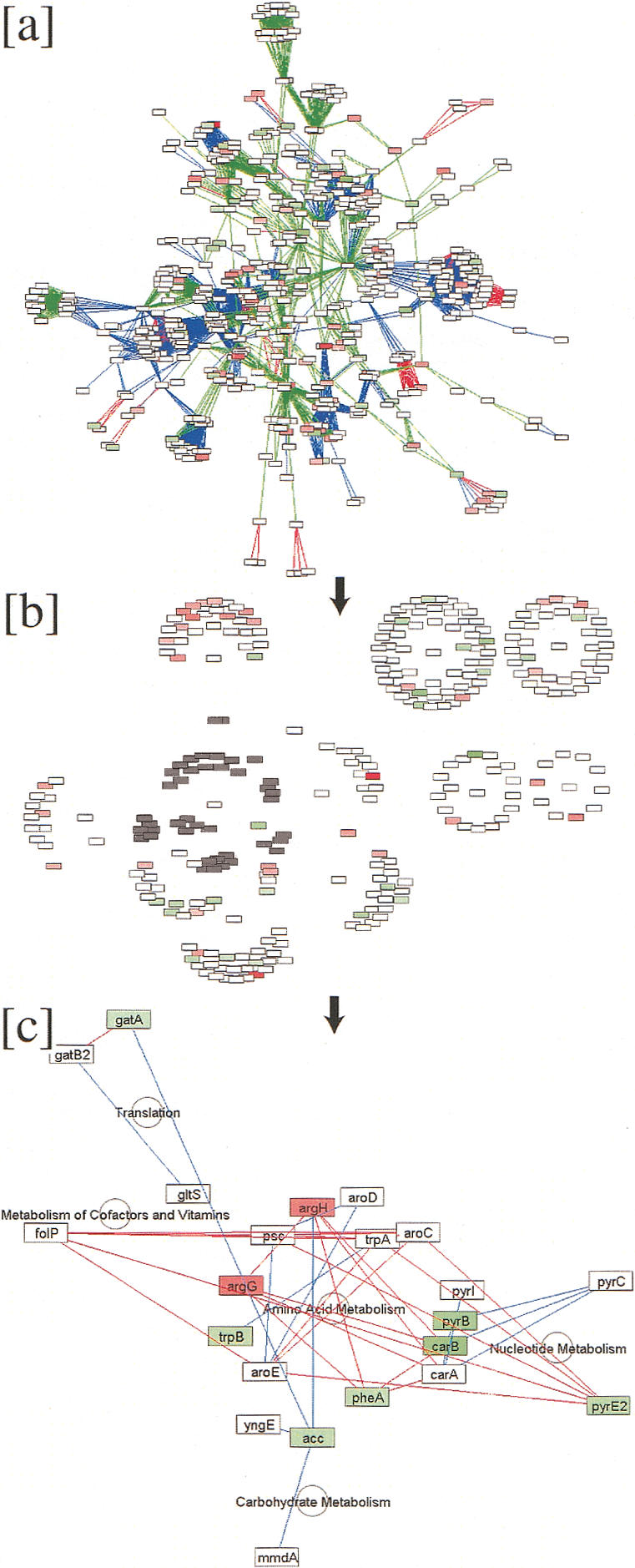
Function-guided layout of the Halobacterium inferred protein network facilitates simultaneous exploration of large discrete databases. (a) The largest connected component of the Halobacterium network is shown; red, green, and blue edges indicate phylogenetic interactions, protein-protein interactions inferred from yeast, and domain-fusion events, respectively (see text). Node colors indicate mRNA expression changes in a phototrophy deficient strain relative to the parent, in which red is induced and green is repressed. (b) Attribute-based layout was used to organize the network according to major functional classes. The cluster predominantly involved with amino-acid metabolism is selected for further exploration (shaded nodes), with edges hidden for clarity. (c) A highly connected subnetwork within the amino-acid metabolism cluster reveals the effect of suppression of phototrophy on amino-acid metabolismand highlights interactions with nucleotide metabolism and other pathways.
Attribute-to-Visual Mapping
Whereas layout determines the location of the nodes and edges in the window, an attribute-to-visual mapping allows data attributes to control the appearance of their associated nodes and edges. Cytoscape supports a wide variety of visual properties, such as node color, shape, and size; node border color and thickness; and edge color, thickness, and style; a data attribute is mapped to a visual property using either a lookup table or interpolation, depending on whether the attribute is discrete valued or continuous. Figure 1b shows an example in which expression ratio attributes are mapped to node colors. By visually superimposing molecular states on the interaction pathways hypothesized to regulate those states, attribute-to-visual mappings directly connect observed data to an underlying model.
Graph Selection and Filtering
To reduce the complexity of a large molecular interaction network, it is necessary to selectively display subsets of nodes and edges. Nodes and edges may be selected according to a wide variety of criteria, including selection by name, by a list of names, or on the basis of an attribute. More complex network selection queries are supported by a filtering toolbox that includes a Minimum Neighbors filter, which selects nodes having a minimum number of neighbors within a specified distance in the network; a Local Distance filter, which selects nodes within a specified distance of a group of preselected nodes; a Differential Expression filter, which selects nodes according to their associated expression data; and a Combination filter, which selects nodes by arbitrary and/or combinations of other filters.
The Cytoscape Core is written in Java and has been released under an LGPL Open Source license; graph structures and some layout algorithms (hierarchical and circular) are implemented using the yFiles Graph Library (www.yworks.de).
Customizing Cytoscape Through Plug-ins
Plug-in modules provide a powerful means of extending the Core to implement new algorithms, additional network analyses, and/or biological semantics.6 Plug-ins are given access to the Core network model and can also control the network display. Although the Cytoscape Core is Open Source, plug-ins are separable software that may be protected under any license the plug-in authors desire.
To illustrate the power of this architecture to address different biological problems within the Cytoscape environment, we explore three case studies of existing plug-ins: a plug-in that examines the overlap between node attribute values and the structure of molecular interaction network to identify significant interaction pathways (Fig. 3, explored in Case Study 1); a plug-in that organizes the network layout according to putative functional attributes of genes (Fig. 4, explored in Case Study 2); and a plug-in that uses the Systems Biology Markup Language (Hucka et al. 2002) to enable lower-level stochastic simulation of network models (Fig. 5, also explored in greater detail at http://www.cytoscape.org/plugins/SBW/). The first plug-in has been instrumental in two previously published research projects (Begley et al. 2002; Ideker et al. 2002), whereas the second and third plug-ins are at earlier stages of research and development.
Figure 5.

Cytoscape and the Systems Biology Workbench stochastically simulating gene regulation. In collaboration with the ERATO Systems Biology Workbench (SBW) project (Hucka et al. 2002), we developed a plug-in that allows Cytoscape to read and simulate SBML-encoded biochemical models. A Cytoscape network view of the SBML model is shown (left), accompanied by a user interface to the simulator (top, right) and an x-y plot of stochastic simulation results (bottom, right). In the x-y plot, the top curve shows the concentration of the N protein, which regulates genes expressed early in λ phage life cycle. Details are available at http://www.cytoscape.org/plugins/SBW/.
Case Study 1: Using the ActiveModules Plug-in to Map Cellular Pathways Responding to Genetic Perturbations and Environmental Stimuli
Active modules are connected subnetworks within the molecular interaction network whose genes show significant coordinated changes in mRNA-expression (or other) state over particular experimental conditions. Determining active modules reduces network complexity by pinpointing just those regions whose states are perturbed by the conditions of interest, while removing false-positive interactions and interactions not involved in the perturbation response. The remaining subnetworks represent concrete hypotheses as to the underlying signaling and regulatory mechanisms in the cell. This approach has been implemented as the ActiveModules plug-in to Cytoscape (implemented in C++ and linked to Java through a JNI bridge) and is available at www.cytoscape.org.
The approach is described in full in Ideker et al. (2002), where it is applied to identify network modules associated with gene expression changes in galactose-induced yeast cultures. The ActiveModules plug-in has also been used to screen for pathways and protein complexes important for cellular recovery to DNA damage—see Figure 3 and Begley et al. (2002) for more details. Both applications demonstrate how Cytoscape's integrated modeling environment may be used to map transcriptional and signaling pathways in a systematic, top-down fashion.
Case Study 2: Using an Attribute-Based Layout Algorithm to Construct and Analyze a Combined Functional/Physical Network for Halobacterium
Halobacterium NRC-1 is an extremely halophilic archaeon with a fully sequenced genome. The hallmark of Halobacterium is its ability to effectively switch from aerobic to anaerobic growth. During the anaerobic period, it derives energy from two major sources, phototrophy—that is, energy from light (Oesterhelt and Stoeckenius 1973), and fermentation of arginine (Ruepp and Soppa 1996).
To better define the systems-level relationships between these important energy transduction pathways, Cytoscape was used to construct a global Halobacterium protein interaction network integrated with functional attributes and expression profiles (Fig. 4a; Baliga et al. 2002). Interactions in this network were inferred from three sources:
Domain-fusion interactions. A domain-fusion interaction (Enright et al. 1999) was inferred from the observation that the orthologs of two separate proteins in Halobacterium were covalently fused as domains within a single protein in the genome of another species. Domain-fusion data spanning 44 genomes were obtained from the Predictome Web site (http://predictome.bu.edu), resulting in 1460 interactions of this type among 526 halobacterial proteins.
Phylogenetic interactions. Proteins with the same pattern of presence/absence in the genomes of many sequenced organisms often have similar functions (Pellegrini et al. 1999); this phylogenetic interaction implies functional association but not necessarily physical interaction. Phylogenetic interaction data were also obtained from the Predictome Web site, connecting a total of 276 proteins with 486 interactions of this type.
Inferred protein-protein interactions. The yeast two-hybrid protein-protein interaction network was mapped onto halobacterial orthologs of yeast proteins as defined by the COG database (Tatusov et al. 2001) to infer 2169 putative protein-protein interactions among 406 Halobacterium proteins.
In total, 929 of the 2413 proteins encoded in the Halobacterium genome could be connected with 5022 interactions of these three types. This global network was then annotated with two sets of node attributes, representing functional classifications and gene expression ratios. Functional classification attributes were taken from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Kanehisa et al. 2002), whereas mRNA expression ratios were drawn from data measured in response to knockout of the bat gene, a transcriptional regulator of phototrophy (Baliga et al. 2002).
To better understand the relationship between network interactions and protein functions, we used an attribute-based layout algorithm to visually organize proteins in the network into tightly connected clusters on the basis of their functional attributes (Fig. 4b). This algorithm, originally implemented as a plug-in7, invokes the basic spring-embedder algorithm (see Cytoscape Core Functionality and Architecture), but uses additional attractive forces between nodes having the same value of a selected attribute. The overall effect is to partition the graph into high-level regions on the basis of attribute value, then to group nodes in each region on the basis of edge connectivity. For example, the nodes labeled gatA and gatB2 (Fig. 4c) are associated with the attribute “Translation,” and thus appear together as a module in the same region of the network. Because attributes can represent a wide variety of biological data, attribute-based layout also makes it possible to visually organize the network according to subcellular localization, gene expression ratio, or any other desired biological attribute. By superimposing expression ratios on the network, it becomes clear that the proteins associated with Amino Acid Metabolism not only have the largest number of network interconnections, but are also the most differentially expressed in a bat knockout.
Case Study 3: Stochastic Simulation of λ Phage Life Cycle Using the Systems Biology Workbench
In collaboration with members of the ERATO Systems Biology Workbench (SBW) project, we implemented a plug-in that allows Cytoscape users to read biochemical models encoded in the Systems Biology Markup Language (Hucka et al. 2002) and to run simulations through SBW. This makes low-level biochemical models and simulators accessible through Cytoscape. Figure 5 shows a model of gene regulation of λ phage (Gibson and Bruck 2001) displayed in Cytoscape, with controls and results plotted from the Gibson simulator. Details of the simulation are available at http://www.cytoscape.org/plugins/SBW/.
DISCUSSION
Cytoscape is a general-purpose, open-source software environment for the large scale integration of molecular interaction network data. Dynamic states on molecules and molecular interactions are handled as attributes on nodes and edges, whereas static hierarchical data, such as protein-functional ontologies, are supported by use of annotations. The Cytoscape Core handles basic features such as network layout and mapping of data attributes to visual display properties. Cytoscape plug-ins extend this core functionality and may be released under separate license agreements if desired. We have described several projects that Cytoscape has supported to-date:
Use of the ActiveModules plug-in to identify pathways and protein complexes activated by galactose gene knockouts and by DNA damage
Inference and attribute-based layout of a combined physical/functional interaction network for Halobacterium
Access to stochastic/kinetic simulation tools through SBML
An immediate future priority is to establish direct connections between Cytoscape and interaction databases such as DIP (Xenarios and Eisenberg 2001), expression databases such as GEO (www.ncbi.nlm.nih.gov/geo), and annotation ontologies such as GO (GO Consortium 2001). Currently, these data must be externally parsed into annotations or attributes. One solution to this problem is data federation, in which a relational database management system serves as middleware providing transparent access to a number of heterogenous data sources.
A second, longer-term direction is to further explore mechanisms for bridging high-level interaction networks with lower-level, physico-chemical models of specific biological processes. Whereas Cytoscape focuses on high-level representation of components and interactions, low-level models are addressed by ongoing software development projects such as Ecell (Tomita et al. 1999), VirtualCell (Loew and Schaff 2001), Gepasi (Mendes 1997), and the Systems Biology Workbench (Hucka et al. 2002). We have illustrated one strategy for transitioning from high-to low-level models, in which a large molecular interaction network is screened to identify subnetworks of interest using either gene expression or genomic phenotyping (Case Study 1). These top-down pathway mapping approaches greatly reduce the size and scope of the modeling problem to a single subnetwork, providing an entry point for lower-level modeling efforts. Subnetworks of interest may then be developed into lower-level models, as shown in Figure 5 for the Systems Biology Workbench.
Perhaps most importantly, Cytoscape's future directions will ultimately depend on the needs and efforts of an active research community. Whereas Cytoscape will continue to be supported and developed by our own research groups, it will also be driven by an active community of users and developers who contribute functionality and expertise through plug-ins, core improvements, and parallel versions.
Acknowledgments
While this work was in review, the Cytoscape core development team expanded to include the laboratory of Dr. Chris Sander at the Memorial Sloan-Kettering Cancer Research Center. We are particularly indebted to Gary Bader and Ethan Cerami in that lab for their recent efforts. Many thanks also go to Iliana Avila-Campillo, Andrew Finney, Mike Hucka, and Vesteinn Thorsson. T.I. is the David Baltimore Whitehead Fellow and was funded through a grant from Pfizer; A.M., P.S., and B.S. were funded through NIH Grant P20 GM64361; N.B. was funded through NSF Grant 0220153. Lastly, we gratefully acknowledge the MIT UROP office for support of J.W., N.A., and D.R.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1239303.
[The Cytoscape v1.1 Core runs on all major operating systems and is freely available for download from http://www.cytoscape.org/ as an open source Java application.]
Biological semantics vary widely within the biological community as well as fromproject to project. If biological semantics were in the Cytoscape Core, we would be faced with a difficult question; which semantics should we use? Cytoscape avoids this problem by leaving it to plug-in writers to adopt semantics adequate to the problem at hand. Of course, there is often substantial biological significance associated with data in the Core. For example, the core may represent a node (an abstract concept free of biological semantics) whose label is GCN4 (a text string with significance to yeast biologists as an important transcription factor) or we might use the Core to define node attributes entitled “expression ratio” or “cellular compartment.” In this way, great freedom and flexibility—the ability to accommodate new biological problems—is gained by not inscribing the notion of specific biological entities directly into the semantics of Cytoscape's Core.
Although attribute-based layout was initially implemented as a plug-in, its general applicability and tight integration with several of Cytoscape's core features (graph layout and node attribute mapping) ultimately led us to incorporate it into the Cytoscape Core platform. Thus, plug-ins also provide a general means of introducing and testing new features.
References
- Bader, G.D., Donaldson, I., Wolting, C., Ouellette, B.F., Pawson, T., and Hogue, C.W. 2001. BIND—The biomolecular interaction network database. Nucleic Acids Res. 29: 242-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baliga, N.S., Pan, M., Goo, Y.A., Yi, E.C., Goodlett, D.R., Dimitrov, K., Shannon, P., Aebersold, R., Ng, W.V., and Hood, L. 2002. Coordinate regulation of energy transduction modules in Halobacterium sp. analyzed by a global systems approach. Proc. Natl. Acad. Sci. 99: 14913-14918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batagelj, V. and Mrvar, A. 1998. Pajek—Program for large network analysis. Connections 21: 47-57. [Google Scholar]
- Begley, T.J., Rosenbach, A.S., Ideker, T., and Samson, L.D. 2002. Damage recovery pathways in Saccharomyces cerevisiae revealed by genomic phenotyping and interactome mapping. Mol. Cancer Res. 1: 103-112. [PubMed] [Google Scholar]
- DeRisi, J.L., Iyer, V.R., and Brown, P.O. 1997. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278: 680-686. [DOI] [PubMed] [Google Scholar]
- Eades, P. 1984. A heuristic for graph drawing. Congressus Numerantium 42: 142-160. [Google Scholar]
- Eisen, M.B., Spellman, P.T., Brown, P.O., and Botstein, D. 1998. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. 95: 14863-14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endy, D. and Brent, R. 2001. Modelling cellular behaviour. Nature 409: 391-395. [DOI] [PubMed] [Google Scholar]
- Enright, A.J., Iliopoulos, I., Kyrpides, N.C., and Ouzounis, C.A. 1999. Protein interaction maps for complete genomes based on gene fusion events. Nature 402: 86-90. [DOI] [PubMed] [Google Scholar]
- Gibson, M.A. and Bruck, J. 2001. A probabilistic model of a prokaryotic gene and its regulation in computational modeling of genetic and biochemical networks (eds. J.M. Bouer and H. Boluri). MIT Press, Cambridge.
- Gilman, A. and Arkin, A.P. 2002. GENETIC “CODE”: Representations and dynamical models of genetic components and networks. Annu. Rev. Genomics Hum. Genet. 3: 341-369. [DOI] [PubMed] [Google Scholar]
- GO Consortium. 2001. Creating the gene ontology resource: Design and implementation. Genome Res. 11: 1425-1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin, J.L., Mann, C.J., Scott, J., Shoulders, C.C., and Nicholson, J.K. 2001. Choline containing metabolites during cell transfection: An insight into magnetic resonance spectroscopy detectable changes. FEBS Lett. 509: 263-266. [DOI] [PubMed] [Google Scholar]
- Gygi, S.P., Rist, B., Gerber, S.A., Turecek, F., Gelb, M.H., and Aebersold, R. 1999. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 17: 994-999. [DOI] [PubMed] [Google Scholar]
- Hucka, M., Finney, A., Sauro, H.M., Bolouri, H., Doyle, J., and Kitano, H. 2002. The ERATO Systems Biology Workbench: Enabling interaction and exchange between software tools for computational biology. Pac. Symp. Biocomput. 450-461. [DOI] [PubMed]
- Ideker, T., Ozier, O., Schwikowski, B., and Siegel, A.F. 2002. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics 18: S233-S240. [DOI] [PubMed] [Google Scholar]
- Ito, T., Chiba, T., and Yoshida, M. 2001. Exploring the protein interactome using comprehensive two-hybrid projects. Trends Biotechnol. 19: S23-S27. [DOI] [PubMed] [Google Scholar]
- Kanehisa, M., Goto, S., Kawashima, S., and Nakaya, A. 2002. The KEGG databases at GenomeNet. Nucleic Acids Res. 30: 42-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp, P.D. 2001. Pathway databases: A case study in computational symbolic theories. Science 293: 2040-2044. [DOI] [PubMed] [Google Scholar]
- Lee, T.I., Rinaldi, N.J., Robert, F., Odom, D.T., Bar-Joseph, Z., Gerber, G.K., Hannett, N.M., Harbison, C.T., Thompson, C.M., Simon, I., et al. 2002. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298: 799-804. [DOI] [PubMed] [Google Scholar]
- Loew, L.M. and Schaff, J.C. 2001. The Virtual Cell: A software environment for computational cell biology. Trends Biotechnol. 19: 401-406. [DOI] [PubMed] [Google Scholar]
- Mendes, P. 1997. Biochemistry by numbers: Simulation of biochemical pathways with Gepasi 3. Trends Biochem. Sci. 22: 361-363. [DOI] [PubMed] [Google Scholar]
- Oesterhelt, D. and Stoeckenius, W. 1973. Functions of a new photoreceptor membrane. Proc. Natl. Acad. Sci. 70: 2853-2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini, M., Marcotte, E.M., Thompson, M.J., Eisenberg, D., and Yeates, T.O. 1999. Assigning protein functions by comparative genome analysis: Protein phylogenetic profiles. Proc. Natl. Acad. Sci. 96: 4285-4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruepp, A. and Soppa, J. 1996. Fermentative arginine degradation in Halobacterium salinarium (formerly Halobacterium halobium): Genes, gene products, and transcripts of the arcRACB gene cluster. J. Bacteriol. 178: 4942-4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamayo, P., Slonim, D., Mesirov, J., Zhu, Q., Kitareewan, S., Dmitrovsky, E., Lander, E.S., and Golub, T.R. 1999. Interpreting patterns of gene expression with self-organizing maps: Methods and application to hematopoietic differentiation. Proc. Natl. Acad. Sci. 96: 2907-2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov, R.L., Natale, D.A., Garkavtsev, I.V., Tatusova, T.A., Shankavaram, U.T., Rao, B.S., Kiryutin, B., Galperin, M.Y., Fedorova, N.D., and Koonin, E.V. 2001. The COG database: New developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 29: 22-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollis, I.G., Battista, G.D., Eades, P., and Tamassia, R. 1999. Graph drawing—Algorithms for the visualization of graphs. Prentice Hall, Upper Saddle River, NJ.
- Tomita, M., Hashimoto, K., Takahashi, K., Shimizu, T.S., Matsuzaki, Y., Miyoshi, F., Saito, K., Tanida, S., Yugi, K., Venter, J.C., et al. 1999. E-CELL: Software environment for whole-cell simulation. Bioinformatics 15: 72-84. [DOI] [PubMed] [Google Scholar]
- Tong, A.H., Evangelista, M., Parsons, A.B., Xu, H., Bader, G.D., Page, N., Robinson, M., Raghibizadeh, S., Hogue, C.W., Bussey, H., et al. 2001. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294: 2364-2368. [DOI] [PubMed] [Google Scholar]
- von Mering, C., Krause, R., Snel, B., Cornell, M., Oliver, S.G., Fields, S., and Bork, P. 2002. Comparative assessment of large-scale data sets of protein-protein interactions. Nature 417: 399-403. [DOI] [PubMed] [Google Scholar]
- Wingender, E., Chen, X., Fricke, E., Geffers, R., Hehl, R., Liebich, I., Krull, M., Matys, V., Michael, H., Ohnhauser, R., et al. 2001. The TRANSFAC system on gene expression regulation. Nucleic Acids Res. 29: 281-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xenarios, I. and Eisenberg, D. 2001. Protein interaction databases. Curr. Opin. Biotechnol. 12: 334-339. [DOI] [PubMed] [Google Scholar]
- Zhou, H., Watts, J.D., and Aebersold, R. 2001. A systematic approach to the analysis of protein phosphorylation. Nat. Biotechnol. 19: 375-378. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://biodata.mshri.on.ca/; Osprey Network Visualization System
- http://pim.hybrigenics.com/; PIMRider
- http://predictome.bu.edu/; Predictome Project
- http://www.cytoscape.org/; Cytoscape v1.1 Home Page
- http://www.cytoscape.org/plugins/SBW/; Supplementary data on model exchange via SBML
- http://www.informatik.uni-bremen.de/daVinci/; daVinci V2.1
- http://www.infosun.fmi.uni-passau.de/Graphlet/; Graphlet Toolkit 5.0.1
- http://www.silicongenetics.com/; GeneSpring 5.0
- http://www.yworks.de/; yFiles Graph Library
- http://www.ncbi.nlm.nih.gov/geo; GEO