

Abstract
Obtaining well-diffracting crystals of macromolecules remains a significant barrier to structure determination. Here we propose and test a new approach to crystallization, in which the crystallization target is fused to a polymerizing protein module, so that polymer formation drives crystallization of the target. We test the approach using a polymerization module called 2TEL, which consists of two tandem sterile alpha motif (SAM) domains from the protein translocation Ets leukemia (TEL). The 2TEL module is engineered to polymerize as the pH is lowered, which allows the subtle modulation of polymerization needed for crystal formation. We show that the 2TEL module can drive the crystallization of 11 soluble proteins, including three that resisted prior crystallization attempts. In addition, the 2TEL module crystallizes in the presence of various detergents, suggesting that it might facilitate membrane protein crystallization. The crystal structures of two fusion proteins show that the TELSAM polymer is responsible for the majority of contacts in the crystal lattice. The results suggest that biological polymers could be designed as crystallization modules.
Keywords: crystallization module, protein crystallization, membrane protein, protein polymer, sterile alpha motif domains
Despite myriad technological advancements, obtaining well-diffracting crystals remains a major hurdle in structure determination. For example, a search of the TargetDB (Chen et al. 2004) database shows that diffraction-quality crystals were obtained for only 12% of the ∼28,000 soluble proteins that were expressed by structural genomics centers worldwide. Current crystallization methods are also ineffective for most membrane proteins (Ostermeier et al. 1995), which comprise a quarter of all the sequenced genomes (Wallin and von Heijne 1998). It is therefore important to develop methods which can effectively lower the crystallization barrier of membrane proteins and the so-called “high-hanging fruits” of structural genomics (Janda et al. 2004).
Prior studies suggest that the primary determinant of crystallization is the intrinsic crystallizability of the protein itself (Kendrew et al. 1954; McPherson 1982). Proteins with a high crystallization propensity crystallize in many conditions (Segelke 2001), whereas some recalcitrant proteins appear to require modifications to make them crystallizable (D'Arcy 1994; Dale et al. 2003; Derewenda 2004a; Keenan et al. 2005). Several methods are available to increase crystallization propensity, including limited proteolysis to remove disordered regions (Tooney and Cohen 1972; Cohen and Tooney 1974; Kwong et al. 1999), mutation of residues on the protein's surface that might be involved in crystal contacts (Lawson et al. 1991; D'Arcy et al. 1999; Czepas et al. 2004; Derewenda 2004a,b; Janda et al. 2004; Derewenda and Vekilov 2006), deglycosylation to diminish heterogeneity (Kwong et al. 1999), and antibody co-crystallization to rigidify the target and add potential crystal contacts (Iwata et al. 1995; Kleymann et al. 1995; Ostermeier et al. 1995; Ostermeier and Michel 1997; Hunte et al. 2000; Zhou et al. 2001; Hunte and Michel 2002; Jiang et al. 2003; Lee et al. 2005). Although useful, these methods must be applied in an ad hoc fashion for each target. Furthermore, since every variant generated using these approaches is effectively a new protein, crystallization conditions must be re-established by performing new rounds of crystallization screening.
In addition to increasing crystallization propensity, an ideal method would increase predictability of crystallization conditions, thereby removing the need to extensively screen conditions. One existing approach that could satisfy these requirements is the use of fusion modules in which difficult targets are linked to a highly crystallizable protein module in an effort to impart the crystallizability of the module to the target (Prive et al. 1994; Kuge et al. 1997; Stoll et al. 1998; Byrne et al. 2000; Liu et al. 2001; Zhan et al. 2001; Smyth et al. 2003). For example, well-diffracting crystals could be obtained when recalcitrant target proteins or peptides were fused to easily crystallized proteins such as hen egg white (HEW) lysozyme (Donahue et al. 1994), glutathione S-transferase (GST) (Zhan et al. 2001), or maltose-binding protein (MBP) (Smyth et al. 2003). The shortcoming of this method is that so far it has only been used to crystallize small proteins or peptides (Ware et al. 1999; Zhan et al. 2001; Smyth et al. 2003), most likely because the proteins that served as modules could form only weak crystal contacts that tend to be easily disrupted by the addition of larger target proteins. We hypothesize that a fusion module with more potent crystal contacts would be more able to impart its crystallization properties to the target.
The sterile alpha motif (SAM) domain from the translocation Ets leukemia protein (TEL) is a small, 78-residue protein that forms a left-handed helical polymer with six subunits per turn (Fig. 1; Kim et al. 2001). A Val to Glu mutation (E80 TELSAM) in the middle of the polymer interface results in a variant which was soluble at high pH but which polymerizes at low pH due to protonation of the Glu residue, providing a pH control for E80-TELSAM polymerization propensity (Kim et al. 2001). Numerous conditions with pH <7 also induce this protein to crystallize as needles or rods.
Figure 1.

E80 TELSAM forms a helical polymer in a pH-dependent fashion. (A) Recombinant WT-TELSAM protein aggregates when expressed, but E80 TELSAM only aggregates when the pH is below 7. (B) E80 TELSAM forms a helical polymer when crystallized. Alternating units are shaded differently to distinguish each TELSAM monomer. Six monomers form one helical turn in the polymer with a pitch of 53 Å.
Because the crystal contacts made by successive monomers correspond to a high-affinity and stable polymeric interface, TELSAM is an excellent candidate to serve as a crystallization module because it would be less subject to disruption by fusion partners than modules that provide only weak crystal contacts. Here, we constructed a crystallization module using two tandem TELSAM domains (2TEL) and fused this module to 12 soluble proteins. Eleven of these fusion proteins, including three which resisted conventional crystallization attempts, crystallized as needles or rods at low pH. In addition, the 2TEL module could crystallize in the presence of a high concentration of detergents commonly used for membrane protein crystallization. While the 2TEL module is clearly effective at driving crystal formation, the majority of the fusion protein crystals tend to be highly mosaic and only diffract to modest resolution. Structure determination of 2TEL fused to T4 lysozyme suggests a possible route to improving the crystallization module by shoring up weak intermodule crystal contacts.
Results
Design and crystal structure of the 2TEL crystallization module
We speculated that larger targets might require more space to pack in a fusion protein crystal than would be provided by the relatively small 78-residue TELSAM domain. We therefore, created a module with two TELSAM domains linked together (Fig. 2A) so that the fusion partner could be placed at every other TELSAM domain.
Figure 2.

The 2TEL module crystallized as a helical polymer. (A) The 2TEL module was composed of an E80 TELSAM connected to a WT TELSAM via a Gly4Ser linker. (B) 2TEL polymerization behavior. At high pH, deprotonated Glu80 prevents 2TEL polymerization. Conversely, protonation at low pH permits polymerization. (C) The crystal structure of the 2TEL module forms a double helix. One polymer is illustrated in a surface representation while the other is shown as a ribbon diagram.
Our initial TELSAM-based modules were all composed of an E80 TELSAM at the N-terminus and a WT TELSAM at the C-terminus (E80-WT) connected by integral repeats of the linker sequence Gly4Ser (Fig. 2A). These E80-WT modules were soluble at high pH but precipitated at low pH (data not shown), suggesting pH-dependent protonation of E80 and consequent polymerization (Fig. 2B). All four E80-WT modules crystallized in various conditions, indicating that linker addition did not lower the crystallization propensity of the protein. Furthermore, all the crystals had needlelike morphologies, suggesting that the TELSAM helical polymer was formed in the crystals. To confirm this hypothesis, we determined the structure of the E80-WT module with a single Gly4Ser linker using molecular replacement with the E80-TELSAM structure (Kim et al. 2001) as a search model. Figure 2C shows that the fused TELSAM domains formed a double-helical structure in which two TELSAM polymers were intertwined with one another. The double-helical structure is clearly not biologically relevant as this architecture cannot accommodate other domains of TEL (Chakrabarti and Nucifora 1999). Nevertheless, the crystal structure confirmed that our linker choices did not disrupt polymer formation, a necessary condition for TELSAM-aided crystallization. We use the single-linker module in all our future experiments and refer to it as the 2TEL module.
Crystals of the 2TEL module fused to soluble proteins of known structure
To test whether the 2TEL module could mediate crystallization of a target protein, we fused it to nine different soluble proteins whose crystal structures had been determined previously (Table 1). Initial target proteins ranged from much smaller (7 kDa) to slightly larger (26 kDa) than the 2TEL module (22 kDa). Because the nature of the linker peptide between 2TEL and the target protein might play a critical role in crystallization (Smyth et al. 2003; Qiao et al. 2004), each target protein was fused to 2TEL using seven linker sequences of various lengths and sequence composition (Table 2). In addition, we also fused each target protein either N- or C-terminal of the 2TEL module, since the order of proteins has been shown to affect fusion protein expression and crystallization. The combination of linker type and connection resulted in up to 14 possible constructs for each target protein. Over 90% of the fusion proteins were expressed and were purified with a single Ni-affinity chromatography step to yield >95% pure proteins as assessed by SDS PAGE (data not shown). Crystallization trials were carried out in-house using commercial screens or sent to the Hauptman-Woodward Institute (HWI) for high-throughput microbatch screening of 1536 conditions (Luft et al. 2003).
Table 1.
List of soluble proteins that were fused to the 2TEL module

Table 2.
Amino acid sequence of 2TEL linkers

Eight out of the nine target proteins could be crystallized as a fusion to 2TEL (Table 1), including the 26 kDa GST, the largest target protein ever crystallized using fusion protein co-crystallization methods. We confirmed that these were indeed crystals of the fusion proteins, rather than proteolyzed fragments by washing the crystals and analyzing them on denaturing polyacrylamide gels (data not shown). Two characteristics of the 2TEL fusion protein crystals suggest that their crystallization was mediated by 2TEL polymerization: Most crystals were formed at low pH, and they had needle- or hexagonal rodlike morphologies akin to the crystals of the 2TEL module by itself (Fig. 3A), but this could only be verified in two cases for which structures were solved, as discussed below.
Figure 3.
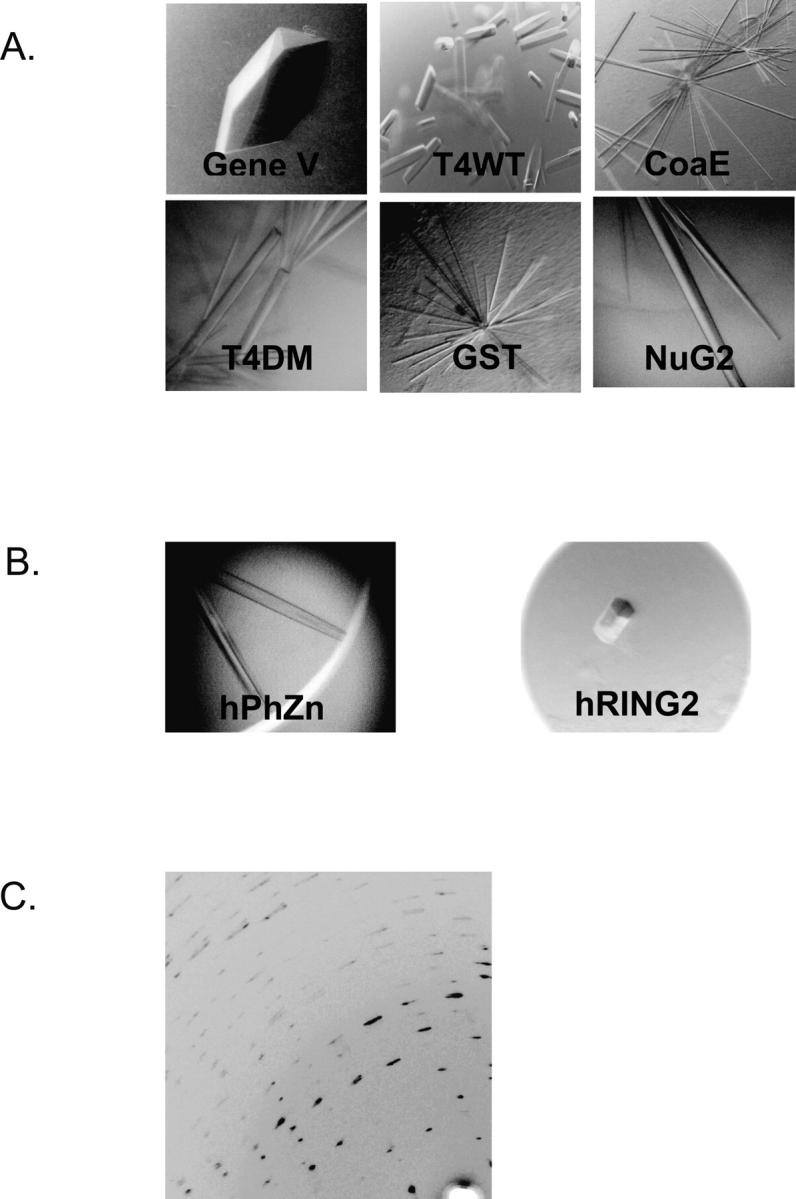
(A) Examples of crystals of 2TEL fused to various soluble proteins of known structures. (B) Crystals of 2TEL fused to novel soluble protein: 2TEL–hPhZn (left) and 2TEL-hRING2 (right). (C) Highly mosaic diffraction pattern of 2TEL-hRING2.
We observed that some target proteins could crystallize using several linker types, whereas others would only form crystals with one linker type, illustrating the importance of linker composition in the crystallization propensity of these fusion proteins. We also observed that some precipitants, for example 2–8 M ammonium nitrate or 15%–35% PEG 3350, were able to crystallize a diverse set of target proteins, suggesting that screens centered around these precipitants might be particularly effective for 2TEL fusions (see below).
Crystals of the 2TEL module fused to recalcitrant soluble proteins
We next tested whether the 2TEL module could drive the crystallization of more difficult targets. Our candidates for these recalcitrant proteins were the Zn-binding domain of the human polyhomeotic protein (hPhZn) (Plath et al. 2004; Zhang et al. 2004) and the C-terminal domain of the human RING2 protein (hRING2) (Schoorlemmer et al. 1997; Satijn and Otte 1999; Gorfinkiel et al. 2004). Both proteins qualify as hard targets because previous screening at the HWI had yielded no crystals (C. Kim, pers. comm.). Using linkers listed in Table 2, we fused 2TEL to hPhZn (2TEL–hPhZn) or hRING2 (2TEL–hRING2), and expressed and purified these fusion proteins. Purified 2TEL–hRING2 (with the Ala-Ala-Gly-Pro linker) could still bind to cbx7 (Schoorlemmer et al. 1997), the in vitro-binding partner of hRING2 (C. Kim, pers. comm.), which indicated that neither 2TEL nor the linker interfered with hRING2 biological activity.
High-throughput screening of both proteins at the HWI yielded needle- or hexagonal rodlike crystals which could be reproduced in-house (Fig. 3B). Interestingly, the 2TEL–hRING2 fusion yielded crystals in >100 conditions tested at the HWI. This was by far the largest number of successful crystallization conditions we have ever observed for any 2TEL fusion protein, which is remarkable considering that the hRING2 protein on its own refused to crystallize under these same crystallization conditions. These two examples clearly show the effectiveness of the 2TEL module in aiding the crystallization of soluble proteins which are difficult to crystallize using conventional means.
Optimization of our initial crystallization conditions yielded crystals of 2TEL–hRING2 which diffracted to 2.3 Å at the Advanced Light Source synchrotron facility. Unfortunately, these crystals had very high mosaicity, and the data could not be used for structure determination (Fig. 3C). High mosaicity indicates imperfect packing of blocks of molecules in the crystals (Snell et al. 1995), and we are currently testing various post-crystallization treatments such as crystal annealing and crosslinking (Heras and Martin 2005) to mitigate this problem.
A minimal crystallization screen yielded crystals of 2TEL fused to a novel soluble protein
It is possible that the 2TEL module could reduce the trial-and-error nature of crystallization because 2TEL polymerization provides a common driving force for crystallization. Thus, it may be useful to limit initial crystallization screening to conditions near those that were successful with other 2TEL fusions. We observed that ammonium nitrate at low pH successfully crystallized four out of the 11 proteins that we tested so far (T4WT, NuG2, hPhZn, and GST), and we therefore hypothesized that an ammonium nitrate-based minimal screen could be particularly effective with new 2TEL fusions.
As a test of this idea, we fused 2TEL to the soluble domain of the Rhomboid protease, a transmembrane serine protease (2TEL-RhoS) (Urban 2006). An NMR structure of the 8 kDa soluble domain (RhoS) structure has been determined (Del Rio et al. 2007), but no crystals have been obtained to our knowledge. Indeed, we were unable to obtain crystals of the domain alone in the 1536-condition HWI screen. Nevertheless, needlelike crystals of 2TEL–RhoS were obtained in a small, 24-condition screen using only 2–8 M ammonium nitrate as the precipitant, buffered with 0.1 M sodium acetate pH 3.6–5.6. Supplemental Figure S1A shows crystals that we obtained after a single round of optimization, which diffracted to 4 Å. This result suggests that the crystallization properties of 2TEL can indeed be imparted to the fusion partner, and prior crystallization information can be used to guide the design of a minimal crystallization screen for future 2TEL fusion proteins.
The 2TEL module crystallized in the presence of detergents
Membrane proteins present a particularly difficult crystallization challenge, so we performed a preliminary test of whether the 2TEL module has the potential to drive membrane protein crystallization.
Because detergents are commonly used for membrane protein crystallization, it is important to determine whether the 2TEL polymeric structure would be disrupted when 2TEL is crystallized at high concentration of these detergents. We selected five detergents and determined the concentration that was commonly used for membrane protein crystallization for each detergent using published literature (Table 3). Because most proteins in the literature were concentrated prior to crystallization trials, the actual detergent concentration in the protein solution could be up to 5–10 times the concentration reported. Thus, we screened for 2TEL crystals using the literature-reported value and 10 times the reported value (Table 3). The detergent concentrations ranged between 2 and 30 times the critical micelle concentration (CMC) of the detergents used (Niegowski et al. 2006).
Table 3.
Range of detergent concentration used for 2TEL crystallization

We crystallized the 2TEL module using known conditions (see Materials and Methods) and added each detergent in Table 3 to these conditions. As shown in Supplemental Figure S1B, we were able to obtain 2TEL crystals even at the highest concentration for four of the tested detergents. The only detergent for which we failed to obtain crystals was lauryldimethylamine N-oxide (LDAO) at the highest concentration, although we could still obtain 2TEL crystals in 13 mM LDAO, ∼10-fold greater than its CMC. These results indicate that the high concentration of detergents do not disrupt 2TEL polymerization, a result that supports the idea that 2TEL polymerization could be used as a means to drive the crystallization of membrane proteins.
Crystal structures of 2TEL fused to T4 lysozyme
To see how the 2TEL module aids the crystallization of target proteins, we determined the crystal structures of two 2TEL fusion proteins. The target proteins were two variants of T4 lysozyme which we call T4WT and T4DM (Heinz and Matthews 1994). Structure determinations of the fusion proteins are detailed in the Materials and Methods section, and data collection and refinement statistics are presented in Table 4.
Table 4.
Data collection and refinement statistics

The structures of the T4WT and T4DM fusions and how they were organized in their respective crystal lattices are shown in Figure 4. The TELSAM polymer is a major structural component in both of the fusion protein crystal lattices, forming a hexagonal tube into which the target proteins are placed. These structures highlighted two types of interactions that the 2TEL module made in both lattices. First, as we hypothesized, alternating WT- and E80-TELSAM monomers in the 2TEL module formed the familiar TELSAM helical polymeric structure (Kim et al. 2001). These interactions are responsible for the long axis of the needle- or rodlike crystals. The change in helical pitch from 53 Å for the T4DM fusion to 58.1 Å for the T4WT fusion indicates that the TELSAM polymer is adept at adjusting to accommodate the addition of a fused target protein. Second, we also observe that neighboring 2TEL polymers make lateral interactions (Fig. 4) which are likely responsible for the diameter of the needle- or rodlike crystals.
Figure 4.

Crystal structures and packing of the 2TEL module (sticks model) fused to T4 lysozyme (ribbon diagram) viewed down the helical polymer axis. Six helical polymers are included for each structure. Lysozyme molecules fused to 2TEL on neighboring hexagonal tubes are deleted for clarity. Lateral contacts (see text) between neighboring polymers are highlighted.
In contrast to the ordered structures formed as a result of numerous interactions made by the 2TEL polymer, both T4WT and T4DM made few contacts in the crystal lattice (see Materials and Methods), which is reflected in the much higher average B-factor values of the T4-lysozyme moieties (Table 4). These contacts are not the same as previously observed for both T4WT and T4DM when they were crystallized on their own (Heinz and Matthews 1994). Thus, the 2TEL module appears to be the primary contributor to crystal lattice stabilization. The 2TEL moiety packs in a similar fashion in both structures, so it is likely that changes in lysozyme packing in the two crystals is due to the different linker sequences used to connect lysozyme to 2TEL. Inspection of the 2TEL–T4WT structure showed that the rigid 16-residue helical linker placed the T4WT molecule into the groove of a neighboring 2TEL polymer, increasing the helical pitch of the 2TEL polymer to 58.1 Å. This arrangement prevents T4WT from making the canonical “back-to-back” crystal contacts observed when it is crystallized on its own. In contrast, the short three-residue linker connecting the C-terminus of 2TEL to the N-terminus of T4DM caused the back surface to be oriented toward the 2TEL polymer instead of a neighboring lysozyme molecule. This orientation also prevented T4DM from making its usual crystal contacts.
T4 lysozyme is composed of two domains and it has been shown that the N-terminal domain can adopt a wide range of hinge-bending motion relative to the C-terminal domain (Faber and Matthews 1990). In both the structures of 2TEL–T4WT and 2TEL–T4DM, only the C-terminal domain of T4 lysozyme makes any crystal contacts at all. The high average B factors for the N-terminal domain of T4 lysozyme indicate that it is relatively disordered in these crystals. On the other hand, it appears that hinge-bending movement allows the C-terminal domain to move into a position which can make productive crystal contacts with neighboring 2TEL or lysozyme molecules.
Discussion
We have tested our proposal that biological polymers such as the TELSAM domain could serve as effective crystallization modules. We have shown that the 2TEL module could direct the crystallization of 11 out of 12 soluble proteins, even proteins that did not crystallize on their own and proteins that were larger than the 2TEL module itself. Moreover, the method may be useful for membrane proteins as detergents do not significantly inhibit crystallization of the module itself.
There are a number of potential advantages of the polymer driven crystallization method: (1) In contrast to antibody co-crystallization methods (Hunte and Michel 2002), the target protein does not require specific interactions with the target protein, making the approach more general. (2) The polymer contacts are stronger than typical crystal contacts so that the properties of the polymer may dominate the crystallization properties of the fusion protein. This allows for the possibility that similar crystallization conditions could be used for many fusion partners. Indeed, we were able to design a minimal screen that produced crystals of 2TEL fused to a novel soluble protein (Supplemental Fig. S1A).
The major practical limitation of the current module is poor crystal quality. In general we see limited resolution, high mosaicity, and occasional crystal twinning. Moreover, in the two solved crystal structures, parts of the fusion partners are poorly ordered. Given the large number of targets, these defects are partly due to the lack of intensive efforts at crystal optimization. More fundamentally, however, the current 2TEL module is yet not optimized for crystallization of a fusion protein. The crystal structures of 2TEL–T4WT and 2TEL–T4DM show that the natural intra-polymer interfaces of TELSAM are strong and force crystallization in the direction of the polymer, as we hypothesized. Most of the 2TEL fusion proteins crystallized as thin needles, however, which limits the diffracting volume (Fig. 3A) and lowers crystal quality. Thin crystals are likely a consequence of poor lateral contacts made between polymers (Fig. 4). Thus, if we can improve these lateral contacts, we may be able to significantly enhance the utility of the 2TEL module. It may be possible to optimize the lateral contacts by reducing surface entropy (Derewenda 2004a; Derewenda and Vekilov 2006), by computational design approaches (Nauli et al. 2002) or by engineering in Tyr/Ser side chains, which were recently shown to be particularly well-suited for making tight and specific contacts (Fellouse et al. 2004; Koide et al. 2007). In a similar fashion, it may be possible to engineer the surface of TELSAM so that it has a higher probability of interacting favorably with fusion partners, improving their order. Future work on the development of the TELSAM polymer crystallization module will focus on re-engineering the polymer surface to favor protein–protein contacts.
Materials and Methods
Cloning, protein expression, and purification of 2TEL fusion proteins
All 2TEL-based proteins were cloned into a pBAD HisA vector (Invitrogen). Each component of the 2TEL crystallization module—E80 TELSAM, Gly4Ser linker, WT TELSAM, helical/flexible linker, and target protein—is bounded by unique restriction enzymes to facilitate the introduction of new fragments.
For expression, plasmids were transformed into TOP10 cells (Invitrogen) and plated on LB plates containing 100 μg/mL ampicillin (Sigma). One liter LB ampicillin (100 μg/mL) cultures were inoculated with a starter culture prepared from single colonies. Growth with shaking at 37°C was performed until OD600 reached 0.6. Each culture was then induced with 0.02% of l-arabinose (Sigma) and left to express the desired protein for 3–4 h. Cells were harvested and stored frozen in −20°C.
Each cell pellet was resuspended in 20 mM Tris, pH 8.6, 0.5 M NaCl. For target proteins with Cys residues, 14 mM β-mercaptoethanol was added to the resuspension buffer; 1 mM PMSF (Sigma) and 60 Kunitz units of DNase I (Sigma, D4263) were added to the resuspended cells. The cell suspension was pulsed–sonicated for 6 × 1 min using a Branson Sonifier macrotip at a setting of 5 at a half-duty cycle. Cell extract was centrifuged in a Sorvall GSA rotor at ∼4000g to separate soluble proteins from the insoluble fraction. The soluble fraction was loaded into a 5-mL HisTrap column (GE Healthcare). Ni-affinity purification was performed using an Äkta Prime system (GE Healthcare). Typically 2TEL fusion proteins eluted at 150 mM imidazole. Fractions were pooled and dialyzed overnight against 20 mM Tris, pH 8.6, 0.2 M NaCl (14 mM β-mercaptoethanol included as necessary). Dialyzed proteins were concentrated using Vivaspin 15-mL concentrators (ISC Bioscience) to a concentration suitable for crystallization, usually between 4 and 5 mg/mL for 2TEL fusion proteins.
Crystallization
High-throughput crystallization of all 2TEL fusion proteins was performed at the HWI at protein concentrations between 1 and 10 mg/mL. Initial crystals were reproduced and optimized in hanging-drop setups in-house. Most crystals were obtained at pH <5 and with needle- or rodlike morphologies characteristic of crystals built from TELSAM polymers. After several crystallization trials pointed out the importance of ammonium nitrate as the precipitating agent, we developed our own crystallization screen for 2TEL fusion proteins with the following composition: 2–8 M ammonium nitrate with sodium citrate between pH 3.5 and 6.6.
For crystallization in various detergents (Supplemental Fig. S1B), 2TEL was mixed in equal volume with the following precipitant: 0.52 M manganese chloride, 0.1 M sodium acetate, pH 5.6, and detergent at the final concentration indicated in the figure.
2TEL was crystallized in 0.3 M MnCl2 and 0.1 M sodium acetate, pH 5.0. 2TEL–hRING2 crystallized in 20% PEG3350, 0.1 M ammonium formate, and 20 mM calcium chloride. 2TEL–hPhZn crystallized in 1.4 M ammonium nitrate, 0.1 M sodium citrate, pH 4.7, and 33 mM l-cysteine. 2TEL–RhoS crystals were obtained in a screen using 2–8 M ammonium nitrate as a precipitant, at pH 3.6–5.6 with sodium acetate as a buffer. 2TEL–T4WT was crystallized in 4.8 M ammonium nitrate and sodium citrate, pH 6.2. 2TEL–T4DM was crystallized in 0.3 M MnCl2, 0.1 M sodium acetate, pH 6.0, and 20 mM HEDS.
X-ray data collection and structure determination
Diffraction data for 2TEL were collected at the Advanced Light Source (ALS) Beamline 8.2.1 and processed in space group P31 at 2.6 Å resolution. Molecular replacement was performed using PHASER with E80 TELSAM as the search model (PDB code: 1JI7). Refinement and molecular modeling were performed using CCP4i (Potterton et al. 2003) and COOT (Emsley and Cowtan 2004), respectively. The final model had an R = 21.1% and an R free = 24.1%.
Diffraction data for 2TEL–T4WT were collected at the Advanced Light Source (ALS) Beamline 8.2.1 and processed in space group P32 to 2.4 Å with two molecules in the asymmetric unit. The length of the unit cell c axis, which corresponds to the helical pitch of the polymer, was 58.1 Å, slightly larger than the 53 Å observed for the E80–TELSAM polymer (Kim et al. 2001). Molecular replacement using only E80 TELSAM as the search model was successful in obtaining initial phases. After a difference density calculation, we saw a large positive electron density that corresponded to the T4WT moiety as well as density corresponding to the 16-residue helical linker (data not shown). We repeated the molecular replacement search using coordinates for both E80 and T4WT (Heinz and Matthews 1994). It was clear as we refined this initial model that parts of the T4WT molecule (residues 1–55), which did not pack with nearby molecules, could not be modeled accurately due to disorder. In contrast, the 2TEL moiety could be easily refined due to the numerous interactions it made in the lattice. Thus instead of building the N-terminal domain of T4WT, we decided to manually dock it (using T4WT PDB coordinates) into the density. Our final model had R = 22% and R free = 25%.
Diffraction data for 2TEL–T4DM were collected at the ALS Beamline 8.2.1 and processed in space group P32 to 2.6 Å with two molecules in the asymmetric unit. The length of the unit cell c-axis, which corresponds to the helical pitch of the polymer, was 53.6 Å, similar to that of the E80-TELSAM polymer (Kim et al. 2001). Molecular replacement using only E80 TELSAM as the search model was successful in obtaining initial phases. Similarly to T4WT, the T4DM moiety makes few contacts with other molecules and is generally hard to model. As in the case of T4WT, we manually docked various domains of T4DM during refinement to yield our final model which has R = 21% and R free = 25%.
Data deposition
Coordinates for all structures have been deposited to the RCSB with the following accession numbers: 2QB1 (for 2TEL), 2QAR (for 2TEL–T4WT), and 2QB0 (for 2TEL–T4DM).
Electronic supplemental material
The Electronic supplemental material shows the crystals of 2TEL–RhoS (Supplemental Fig. S1A) and crystals of 2TEL in the presence of various detergents (Supplemental Fig. S1B).
Acknowledgments
We thank the Hauptman-Woodward Institute (Buffalo, New York) for the initial screening of crystallization conditions, Lingmin Loh for optimization of the 2TEL–lysozyme crystals, Michael Sawaya for help with structure refinement of 2TEL–lysozyme, Chongwoo Kim at the University of Texas, San Antonio, for his gift of the hPhZn and hRING2 plasmids, Brian Matthews at the University of Oregon, Eugene, for his gift of the T4-lysozyme plasmids, Don Gray at the University of Texas, Dallas, for his gift of the g5p plasmid, and David Baker at the University of Washington, Seattle, for his gift of the protein L and NuG2 plasmids. This work was supported by a Cancer Research Institute Postdoctoral Fellowship to S.N. and a National Institutes of Health Grant (R01 CA081000) to J.U.B.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: James U. Bowie, Department of Chemistry and Biochemistry, UCLA–DOE Institute for Genomics and Proteomics, Boyer Hall, 611 Charles E. Young Drive East, Los Angeles, CA 90095-1570, USA; e-mail: bowie@mbi.ucla.edu; fax: (310) 206-4749.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.073074207.
References
- Byrne B., Abramson, J., Jansson, M., Holmgren, E., and Iwata, S. 2000. Fusion protein approach to improve the crystal quality of cytochrome bo (3) ubiquinol oxidase from Escherichia coli . Biochim. Biophys. Acta 1459: 449–455. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S.R. and Nucifora, G. 1999. The leukemia-associated gene TEL encodes a transcription repressor which associates with SMRT and mSin3A. Biochem. Biophys. Res. Commun. 264: 871–877. [DOI] [PubMed] [Google Scholar]
- Chen L., Oughtred, R., Berman, H.M., and Westbrook, J. 2004. TargetDB: A target registration database for structural genomics projects. Bioinformatics 20: 2860–2862. [DOI] [PubMed] [Google Scholar]
- Cohen C. and Tooney, N.M. 1974. Crystallisation of a modified fibrinogen. Nature 251: 659–660. [DOI] [PubMed] [Google Scholar]
- Czepas J., Devedjiev, Y., Krowarsch, D., Derewenda, U., Otlewski, J., and Derewenda, Z.S. 2004. The impact of Lys → Arg surface mutations on the crystallization of the globular domain of RhoGDI. Acta Crystallogr. D Biol. Crystallogr. 60: 275–280. [DOI] [PubMed] [Google Scholar]
- D'Arcy A. 1994. Crystallizing proteins—A rational approach? Acta Crystallogr. D Biol. Crystallogr. 50: 469–471. [DOI] [PubMed] [Google Scholar]
- D'Arcy A., Stihle, M., Kostrewa, D., and Dale, G. 1999. Crystal engineering: A case study using the 24 kDa fragment of the DNA gyrase B subunit from Escherichia coli . Acta Crystallogr. D Biol. Crystallogr. 55: 1623–1625. [DOI] [PubMed] [Google Scholar]
- Dale G.E., Oefner, C., and D'Arcy, A. 2003. The protein as a variable in protein crystallization. J. Struct. Biol. 142: 88–97. [DOI] [PubMed] [Google Scholar]
- Del Rio A., Dutta, K., Chavez, J., Ubarretxena-Belandia, I., and Ghose, R. 2007. Solution structure and dynamics of the N-terminal cytosolic domain of rhomboid intramembrane protease from Pseudomonas aeruginosa: Insights into a functional role in intramembrane proteolysis. J. Mol. Biol. 365: 109–122. [DOI] [PubMed] [Google Scholar]
- Derewenda Z.S. 2004a. Rational protein crystallization by mutational surface engineering. Structure 12: 529–535. [DOI] [PubMed] [Google Scholar]
- Derewenda Z.S. 2004b. The use of recombinant methods and molecular engineering in protein crystallization. Methods 34: 354–363. [DOI] [PubMed] [Google Scholar]
- Derewenda Z.S. and Vekilov, P.G. 2006. Entropy and surface engineering in protein crystallization. Acta Crystallogr. D Biol. Crystallogr. 62: 116–124. [DOI] [PubMed] [Google Scholar]
- Donahue J.P., Patel, H., Anderson, W.F., and Hawiger, J. 1994. Three-dimensional structure of the platelet integrin recognition segment of the fibrinogen γ chain obtained by carrier protein-driven crystallization. Proc. Natl. Acad. Sci. 91: 12178–12182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P. and Cowtan, K. 2004. COOT: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60: 2126–2132. [DOI] [PubMed] [Google Scholar]
- Faber H.R. and Matthews, B.W. 1990. A mutant T4 lysozyme displays five different conformations. Nature 348: 263–266. [DOI] [PubMed] [Google Scholar]
- Fellouse F.A., Wiesmann, C., and Sidhu, S.S. 2004. Synthetic antibodies from a four-amino-acid code: A dominant role for tyrosine in antigen recognition. Proc. Natl. Acad. Sci. 101: 12467–12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorfinkiel N., Fanti, L., Melgar, T., Garcia, E., Pimpinelli, S., Guerrero, I., and Vidal, M. 2004. The Drosophila Polycomb group gene Sex combs extra encodes the ortholog of mammalian Ring1 proteins. Mech. Dev. 121: 449–462. [DOI] [PubMed] [Google Scholar]
- Guan Y., Zhang, H., Konings, R.N., Hilbers, C.W., Terwilliger, T.C., and Wang, A.H. 1994. Crystal structures of Y41H and Y41F mutants of gene V protein from Ff phage suggest possible protein–protein interactions in the GVP–ssDNA complex. Biochemistry 33: 7768–7778. [PubMed] [Google Scholar]
- Guddat L.W., Vos, S., Martin, J.L., Keough, D.T., and de Jersey, J. 2002. Crystal structures of free, IMP-, and GMP-bound Escherichia coli hypoxanthine phosphoribosyltransferase. Protein Sci. 11: 1626–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz D.W. and Matthews, B.W. 1994. Rapid crystallization of T4 lysozyme by intermolecular disulfide cross-linking. Protein Eng. 7: 301–307. [DOI] [PubMed] [Google Scholar]
- Henriksen A., Aghajari, N., Jensen, K.F., and Gajhede, M. 1996. A flexible loop at the dimer interface is a part of the active site of the adjacent monomer of Escherichia coli orotate phosphoribosyltransferase. Biochemistry 35: 3803–3809. [DOI] [PubMed] [Google Scholar]
- Heras B. and Martin, J.L. 2005. Post-crystallization treatments for improving diffraction quality of protein crystals. Acta Crystallogr. D Biol. Crystallogr. 61: 1173–1180. [DOI] [PubMed] [Google Scholar]
- Hunte C. and Michel, H. 2002. Crystallisation of membrane proteins mediated by antibody fragments. Curr. Opin. Struct. Biol. 12: 503–508. [DOI] [PubMed] [Google Scholar]
- Hunte C., Koepke, J., Lange, C., Rossmanith, T., and Michel, H. 2000. Structure at 2.3 Å resolution of the cytochrome bc(1) complex from the yeast Saccharomyces cerevisiae co-crystallized with an antibody Fv fragment. Structure 8: 669–684. [DOI] [PubMed] [Google Scholar]
- Iwata S., Ostermeier, C., Ludwig, B., and Michel, H. 1995. Structure at 2.8 Å resolution of cytochrome c oxidase from Paracoccus denitrificans . Nature 376: 660–669. [DOI] [PubMed] [Google Scholar]
- Janda I., Devedjiev, Y., Cooper, D., Chruszcz, M., Derewenda, U., Gabrys, A., Minor, W., Joachimiak, A., and Derewenda, Z.S. 2004. Harvesting the high-hanging fruit: The structure of the YdeN gene product from Bacillus subtilis at 1.8 angstroms resolution. Acta Crystallogr. D Biol. Crystallogr. 60: 1101–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y., Lee, A., Chen, J., Ruta, V., Cadene, M., Chait, B.T., and MacKinnon, R. 2003. X-ray structure of a voltage-dependent K+ channel. Nature 423: 33–41. [DOI] [PubMed] [Google Scholar]
- Keenan R.J., Siehl, D.L., Gorton, R., and Castle, L.A. 2005. DNA shuffling as a tool for protein crystallization. Proc. Natl. Acad. Sci. 102: 8887–8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendrew J.C., Parrish, R.G., Marrack, J.R., and Orlans, E.S. 1954. The species specificity of myoglobin. Nature 174: 946–949. [DOI] [PubMed] [Google Scholar]
- Kim C.A., Phillips, M.L., Kim, W., Gingery, M., Tran, H.H., Robinson, M.A., Faham, S., and Bowie, J.U. 2001. Polymerization of the SAM domain of TEL in leukemogenesis and transcriptional repression. EMBO J. 20: 4173–4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleymann G., Ostermeier, C., Ludwig, B., Skerra, A., and Michel, H. 1995. Engineered Fv fragments as a tool for the one-step purification of integral multisubunit membrane protein complexes. Biotechnology (N.Y.) 13: 155–160. [DOI] [PubMed] [Google Scholar]
- Koide A., Gilbreth, R.N., Esaki, K., Tereshko, V., and Koide, S. 2007. High-affinity single-domain binding proteins with a binary-code interface. Proc. Natl. Acad. Sci. 104: 6632–6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuge M., Fujii, Y., Shimizu, T., Hirose, F., Matsukage, A., and Hakoshima, T. 1997. Use of a fusion protein to obtain crystals suitable for X-ray analysis: Crystallization of a GST-fused protein containing the DNA-binding domain of DNA replication-related element-binding factor, DREF. Protein Sci. 6: 1783–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman B., O'Neill, J.W., Kim, D.E., Zhang, K.Y., and Baker, D. 2002. Accurate computer-based design of a new backbone conformation in the second turn of protein L. J. Mol. Biol. 315: 471–477. [DOI] [PubMed] [Google Scholar]
- Kwong P.D., Wyatt, R., Desjardins, E., Robinson, J., Culp, J.S., Hellmig, B.D., Sweet, R.W., Sodroski, J., and Hendrickson, W.A. 1999. Probability analysis of variational crystallization and its application to gp120, the exterior envelope glycoprotein of type 1 human immunodeficiency virus (HIV-1). J. Biol. Chem. 274: 4115–4123. [DOI] [PubMed] [Google Scholar]
- Lawson D.M., Artymiuk, P.J., Yewdall, S.J., Smith, J.M., Livingstone, J.C., Treffry, A., Luzzago, A., Levi, S., Arosio, P., Cesareni, G., et al. 1991. Solving the structure of human H ferritin by genetically engineering intermolecular crystal contacts. Nature 349: 541–544. [DOI] [PubMed] [Google Scholar]
- Lee S.Y., Lee, A., Chen, J., and MacKinnon, R. 2005. Structure of the KvAP voltage-dependent K+ channel and its dependence on the lipid membrane. Proc. Natl. Acad. Sci. 102: 15441–15446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Manna, A., Li, R., Martin, W.E., Murphy, R.C., Cheung, A.L., and Zhang, G. 2001. Crystal structure of the SarR protein from Staphylococcus aureus . Proc. Natl. Acad. Sci. 98: 6877–6882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luft J.R., Collins, R.J., Fehrman, N.A., Lauricella, A.M., Veatch, C.K., and DeTitta, G.T. 2003. A deliberate approach to screening for initial crystallization conditions of biological macromolecules. J. Struct. Biol. 142: 170–179. [DOI] [PubMed] [Google Scholar]
- McPherson A. 1982. Preparation and analysis of protein crystals. Wiley, Chichester, UK.
- McTigue M.A., Williams, D.R., and Tainer, J.A. 1995. Crystal structures of a schistosomal drug and vaccine target: Glutathione S-transferase from Schistosoma japonica and its complex with the leading antischistosomal drug praziquantel. J. Mol. Biol. 246: 21–27. [DOI] [PubMed] [Google Scholar]
- Minor W., Cymborowski, M., Otwinowski, Z., and Chruszcz, M. 2006. HKL-3000: The integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62: 859–866. [DOI] [PubMed] [Google Scholar]
- Nauli S., Kuhlman, B., Le Trong, I., Stenkamp, R.E., Teller, D., and Baker, D. 2002. Crystal structures and increased stabilization of the protein G variants with switched folding pathways NuG1 and NuG2. Protein Sci. 11: 2924–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niegowski D., Hedren, M., Nordlund, P., and Eshaghi, S. 2006. A simple strategy towards membrane protein purification and crystallization. Int. J. Biol. Macromol. 39: 83–87. [DOI] [PubMed] [Google Scholar]
- Ostermeier C. and Michel, H. 1997. Crystallization of membrane proteins. Curr. Opin. Struct. Biol. 7: 697–701. [DOI] [PubMed] [Google Scholar]
- Ostermeier C., Iwata, S., Ludwig, B., and Michel, H. 1995. Fv fragment-mediated crystallization of the membrane protein bacterial cytochrome c oxidase. Nat. Struct. Biol. 2: 842–846. [DOI] [PubMed] [Google Scholar]
- O'Toole N., Barbosa, J.A., Li, Y., Hung, L.W., Matte, A., and Cygler, M. 2003. Crystal structure of a trimeric form of dephosphocoenzyme A kinase from Escherichia coli . Protein Sci. 12: 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plath K., Talbot, D., Hamer, K.M., Otte, A.P., Yang, T.P., Jaenisch, R., and Panning, B. 2004. Developmentally regulated alterations in Polycomb repressive complex 1 proteins on the inactive X chromosome. J. Cell Biol. 167: 1025–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potterton E., Briggs, P., Turkenburg, M., and Dodson, E. 2003. A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr. 59: 1131–1137. [DOI] [PubMed] [Google Scholar]
- Prive G.G., Verner, G.E., Weitzman, C., Zen, K.H., Eisenberg, D., and Kaback, H.R. 1994. Fusion proteins as tools for crystallization: The lactose permease from Escherichia coli . Acta Crystallogr. D Biol. Crystallogr. 50: 375–379. [DOI] [PubMed] [Google Scholar]
- Qiao F., Song, H., Kim, C.A., Sawaya, M.R., Hunter, J.B., Gingery, M., Rebay, I., Courey, A.J., and Bowie, J.U. 2004. Derepression by depolymerization; structural insights into the regulation of Yan by Mae. Cell 118: 163–173. [DOI] [PubMed] [Google Scholar]
- Satijn D.P. and Otte, A.P. 1999. RING1 interacts with multiple Polycomb-group proteins and displays tumorigenic activity. Mol. Cell. Biol. 19: 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoorlemmer J., Marcos-Gutierrez, C., Were, F., Martinez, R., Garcia, E., Satijn, D.P., Otte, A.P., and Vidal, M. 1997. Ring1A is a transcriptional repressor that interacts with the Polycomb-M33 protein and is expressed at rhombomere boundaries in the mouse hindbrain. EMBO J. 16: 5930–5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segelke B.W. 2001. Efficiency analysis of sampling protocols used in protein crystallization screening. J. Cryst. Growth 232: 553–562. [Google Scholar]
- Smyth D.R., Mrozkiewicz, M.K., McGrath, W.J., Listwan, P., and Kobe, B. 2003. Crystal structures of fusion proteins with large-affinity tags. Protein Sci. 12: 1313–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell E.H., Weisgerber, S., Helliwell, J.R., Holzer, K., and Schroer, K. 1995. Improvements in lysozyme protein crystal perfection through microgravity growth. Acta Crystallogr. D Biol. Crystallogr. 51: 1099–1102. [DOI] [PubMed] [Google Scholar]
- Stoll V.S., Manohar, A.V., Gillon, W., MacFarlane, E.L., Hynes, R.C., and Pai, E.F. 1998. A thioredoxin fusion protein of VanH, a d-lactate dehydrogenase from Enterococcus faecium: Cloning, expression, purification, kinetic analysis, and crystallization. Protein Sci. 7: 1147–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooney N.M. and Cohen, C. 1972. Microcrystals of a modified fibrinogen. Nature 237: 23–25. [DOI] [PubMed] [Google Scholar]
- Urban S. 2006. Rhomboid proteins: Conserved membrane proteases with divergent biological functions. Genes & Dev. 20: 3054–3068. [DOI] [PubMed] [Google Scholar]
- Wallin E. and von Heijne, G. 1998. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 7: 1029–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware S., Donahue, J.P., Hawiger, J., and Anderson, W.F. 1999. Structure of the fibrinogen γ-chain integrin binding and factor XIIIa cross-linking sites obtained through carrier protein driven crystallization. Protein Sci. 8: 2663–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Y., Song, X., and Zhou, G.W. 2001. Structural analysis of regulatory protein domains using GST-fusion proteins. Gene 281: 1–9. [DOI] [PubMed] [Google Scholar]
- Zhang H., Christoforou, A., Aravind, L., Emmons, S.W., van den Heuvel, S., and Haber, D.A. 2004. The C. elegans Polycomb gene SOP-2 encodes an RNA-binding protein. Mol. Cell 14: 841–847. [DOI] [PubMed] [Google Scholar]
- Zhou Y., Morais-Cabral, J.H., Kaufman, A., and MacKinnon, R. 2001. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature 414: 43–48. [DOI] [PubMed] [Google Scholar]