

Abstract
Phenobarbital (PB) is a nongenotoxic tumor promoter in the liver. One mechanism by which PB may exert its tumor promoting activity is by inducing oxidative stress. We previously found that PB administration increased hepatic NF-κB DNA binding activity. In this study we examined the hypothesis that the effects of PB on cell proliferation and apoptosis are dependent on NF-κB. We used a mouse model that is deficient in the p50 subunit of NF-κB; previous studies had found that p50 -/- mice were less sensitive to the induction of hepatic cell proliferation by PCBs or peroxisome proliferators. Mice (p50 -/- and wild-type B6129) were fed a control diet or one containing 0.05% PB for 3, 10 or 34 days. At the end of the experiment, the mice were euthanized and livers removed and processed. PB increased cell proliferation at 3 and 10 days (but not at 34 days), but the deletion of the NF-κB p50 subunit did not inhibit these increases. p50 -/- mice had higher cell proliferation at the 3 day (only in mice fed PB) and 34 day time points. PB decreased hepatocyte apoptosis after 3 days, slightly decreased it after 10 days, and did not affect it after 34 days. The deletion of the NF-κB p50 subunit did not influence PB’s effect on apoptosis. In p50 -/- mice, apoptosis was increased after 3 or 10 days compared to wild-type mice, but no effect was seen after 34 days. The hepatic expression of the NF-κB-regulated gene TNF-α correlated more with the hepatic cell proliferation data than with hepatic apoptosis, and was not decreased by the deletion of the p50 subunit. These findings show that the p50 subunit of NF-κB is not required for the alteration of hepatocyte proliferation or apoptosis by PB up to 34 days after its administration.
Keywords: phenobarbital, NF-κB, cell proliferation, apoptosis
INTRODUCTION
Phenobarbital (PB) was identified as a hepatic tumor promoter by Peraino et al. (1971). It was later demonstrated to be one of the most efficacious tumor promoting agents in the liver (Pitot et al., 1987). PB is a non-genotoxic carcinogen but when administered chronically, it increases cell proliferation and produces hepatomegaly in rodents (Jirtle and Meyer, 1991). After continued treatment, cell proliferation levels return to normal and further mitogenic stimuli does not produce more cell proliferation (Jirtle et al., 1991; Jirtle and Meyer, 1991). Chronic administration of PB to rats and mice results in hepatocarcinogenesis (Butler and Hempsall, 1978; Feldman et al., 1981). PB is a prototype of a group of structurally unrelated chemicals that induce a specific subset of cytochrome P450 genes (Sueyoshi and Negishi, 2001). In various studies, depending on the time point, inhibition of cell proliferation, increased DNA synthesis and increased TGF-β concentration have all been shown in liver tissue (Barbason et al., 1983; Farber, 1987; Jirtle and Meyer, 1991; Manjeshwar et al., 1992) after PB administration. Some studies have shown that growth of preneoplastic lesions by PB may be attributed to inhibition of apoptosis, increased oxidative stress by reactive oxygen species and spontaneous aberrations in DNA replication and repair (Schulte-Hermann et al., 1990; Waxman and Azaroff, 1992; Butterworth et al., 1995). In addition to cytochrome P450s, PB induces several other enzymes such as NADPH cytochrome P450 reductase and transferases (Waxman and Azaroff, 1992; Frueh et al., 1997; Honkakoski and Negishi, 1998). Studies have shown that PB’s mechanism of action is mediated by the constitutive androstane receptor (CAR) (Sueyoshi and Negishi, 2001). In the nucleus this receptor binds to specific responsive elements of genes activated by PB, called the phenobarbital responsive enhancer module (PBREM). Many CYP2B genes are shown to be activated by PB by this CAR-PBREM pathway (Sueyoshi and Negishi, 2001). A recent microarray analysis in wild type and CAR null mice showed more than 70 genes, including a group of genes consisting of xenobiotic metabolizing enzymes, that were either induced or repressed in response to PB treatment (Ueda et al., 2002).
It has been postulated that PB acts through the same autocrine growth pathway as TGF-α by inhibiting apoptosis (Sandgren et al., 1995; Smith et al., 1995; Kaufmann et al., 1997; Santoni-Rugiu et al., 1998), which is thought to be caused by activation of reactive oxygen species (ROS). ROS may also be involved in the intracellular regulation of transcription factors like NF-κB and AP-1 through protein kinase-C activation (Feig et al., 1994; Brawn et al., 1995; Storz and Polla, 1996; Klaunig et al., 1998). It has been shown that PB administration increases the DNA binding of the NF-κB in rats (Li et al., 1996; Calfee-Mason et al., 2002). It is already shown that NF-κB is involved in the regulation of cell proliferation and apoptosis (Barkett and Gilmore, 1999; Hinz et al., 1999). The NF-κB complex is present as a dimer that includes two of the several subunits (p50, p52, p65 (RelA), c-rel, RelB) but the most common form of the complex is a p50-p65 heterodimer. When not activated, NF-κB is sequestered in cytosol by the inhibitory complex IκB. Upon activation, the IκB degrades and the NF-κB complex translocates to the nucleus to bind to the responsive elements of the induced genes (Li and Karin, 1999)
Because PB can activate NF-κB and because of the important role of NF-κB in the regulation of cell proliferation and apoptosis, we examined if PB’s effects on cell proliferation and apoptosis are dependent on NF-κB. We used a knockout mouse model in which the p50 subunit of the NF-κB complex was not present. The p50 -/- and wild type mice were fed PB in the diet (0.05%) for three different time periods (3 days, 10 days and 34 days) and the effects on hepatocyte proliferation, apoptosis, and expression of the NF-κB-regulated gene TNF-α were quantified.
MATERIAL AND METHODS
Chemicals
Phenobarbital and bromodeoxyuridine (BrdU) were obtained from Sigma Chemical Company (St. Louis, MO). The BrdU antibody was purchased from Becton Dickinson (San Jose, CA). The consensus NF-κB oligonucleotide (5-AGTTGAGGGGACTTTCCCAGGC-3) was obtained from Promega (Madison, WI). The Vectastain kit for immunostaining was obtained from Vector Laboratories (Burlingame, CA). Alzet 1003D osmotic pumps for BrdU infusion were purchased from Durect (Cupertino, CA). The antigen retrieval solution Citra was purchased from BioGenex (San Ramon, CA). All other chemicals unless noted were obtained from Sigma Chemicals (St. Louis, MO).
Experimental design
7-8 week old female B6129 mice and homozygous p50-/- mice were obtained from our breeding colony (originally purchased from Jackson Laboratory, Bar Harbor, ME). Experiments were conducted for three different time points: 3 days, 10 days, and 34 days. Each experiment consisted of four groups of animals: wild type control, wild type PB treated, p50-/- control, and p50-/- PB treated. Control animals were fed an unrefined diet (Harlan Teklad Global 18% Protein Rodent Diet-2918) while the animals in the PB treatment groups were fed 0.05% PB mixed in the powdered unrefined diet for 3, 10 and 34 days. For the 10- and 34-day time points, the animals were implanted s.c. with osmotic pumps containing BrdU (20 mg/ml) 3 days before euthanasia. For 3 day study, animals received a s.c. injection of BrdU (100 mg/kg) 2 hours before euthanasia. After euthanasia (by overexposure to CO2 gas), livers were isolated and pieces of tissue were snap frozen in liquid nitrogen and stored at -80° C until used. Tissue sections from each lobe were fixed in formalin for 48 hrs prior to paraffin sectioning. These sections were used for BrdU or TUNEL immunostaining.
Electrophoretic mobility shift assay
Nuclear extracts were prepared from the frozen tissue by the method of Derykere and Gannon (1994), as modified by Tharappel et al. (2001). Five mg of nuclear extract was incubated with 0.5 mg poly (dI-dC) in a binding buffer (50 mM KCl, 10 mM Hepes-KOH, pH 7.9, 6.5 mM dithiothreitol, and 10% glycerol) for 5 min, followed by 20 min incubation at room temperature after adding 20,000 cpm of the 32P endlabeled NF-κB oligonucleotide probe. After incubation, samples were resolved on 7% nondenaturing polyacrylamide gels, which were then dried under vacuum and exposed overnight at -80 C to Kodak XOMAT-AR film.
BrdU immunostaining
Tissue sections were fixed in formalin and embedded in paraffin blocks. Sections (5 mm thick) were prepared and stained with an anti-BrdU antibody using the Vecta Stain ABC kit according to the manufacturer’s protocol, and then counterstained with hematoxylin. Cells containing newly synthesized DNA were visualized by dark brown labeled nuclei. Roughly 3000 total nuclei were randomly counted from each slide, with at least 1000 nuclei counted from each of 3 lobes, and labeling indices were calculated.
Apoptosis assay
The terminal deoxyribonucleotidyl transferase-mediated dUTP-digoxigenin nick end labeling (TUNEL) apoptosis assay kit ApoTag was purchased from Intergen (Serologicals Corporation, Norcross GA). The assay was performed on paraffin sections following the manufacturer’s protocol. Slides were counterstained with methyl green. At least 3000 nuclei representing each of the lobes were randomly counted per slide and the apoptotic index was expressed as the percentage of number of labeled apoptotic bodies of the total number of nuclei counted.
Real time PCR
RNA for the real time PCR templates were prepared using Trizol reagent (Invitrogen) using the manufacturer’s protocol. The cDNA templates for the Real-Time PCR (RT-PCR) reactions were prepared with iScript cDNA synthesis kit (Bio-Rad). Real-Time PCR reactions were prepared with iQ SYBR Green Super mix (Bio-Rad) and were amplified in a Bio-Rad MyiQ single-color real-time PCR detection system following the manufacture’s protocol. Oligonucleotides for the TNF-α and ß-actin primers were obtained from Integrated DNA Technologies (Coralville, IA); sequence information and RT-PCR conditions are available upon request. Primers for the RT-PCR were from different exons so that amplicons from cDNA and contaminating genomic DNA would be of different lengths.
Statistical analyses
Results were first analyzed by two-way analysis of variance (ANOVA). If a significant interaction was observed, results were further analyzed using Tukey’s test. The results are reported as means ± standard error of mean (SEM).
RESULTS
In this study, we administered PB to mice deficient in the p50 subunit of NF-κB as well as wild-type control mice for three different time periods: 3 days, 10 days, and 34 days. The body weights in the p50 -/- were lower at all time points (Table 1). Body weights were higher in the PB-fed mice at 10 days but not at the other two time points. Liver weights as well as the liver/body weight ratios were increased by PB administration at all three time points. The liver/body weight ratios were higher in the p50 -/- mice at all times, but the gross liver weight was not affected.
Table 1.
Body and liver weights in wild-type (WT) or p50 -/- mice administered phenobarbital (PB)
| Initial body weight (g) | Final body weight (g) | Weight gain (g) | Liver weight (g) | Liver weight/body weight (%) | |
|---|---|---|---|---|---|
| 3 Days | |||||
| WT | 22.4 ± 1.7 | 22.5 ± 1.6 | 0.12 ± 0.31 | 1.07 ± 0.13 | 4.69 ± 0.25 |
| WT+PB | 21.5 ± 1.4 | 22.6 ± 1.3 | 1.12 ± 0.23 | 1.40 ± 0.15* | 6.16 ± 0.44* |
| p50-/- | 17.6 ± 0.3 | 17.8 ± 0.17† | -0.02 ± 0.23 | 1.00 ± 0.02 | 5.63 ± 0.18† |
| p50-/- +PB | 17.1 ± 0.3 | 17.5 ± 0.38† | 0.39 ± 0.09 | 1.23 ± 0.13* | 7.01 ± 0.63*† |
| 10 Days | |||||
| WT | 20.4 ± 0.7 | 21.3 ± 0.79 | 0.90 ± 0.31 | 0.980 ± 0.023 | 4.62 ± 0.13 |
| WT+PB | 20.9 ± 0.2 | 22.0 ± 0.32* | 1.08 ± 0.17 | 1.44 ± 0.05* | 6.57 ± 0.18* |
| p50-/- | 17.1 ± 0.2 | 18.2 ± 0.47† | 1.09 ± 0.38 | 0.982 ± 0.058 | 5.39 ± 0.21† |
| p50-/- +PB | 20.0 ± 0.5 | 20.5 ± 0.81*† | 0.47 ± 0.34 | 1.52 ± 0.15* | 7.39 ± 0.48*† |
| 34 Days | |||||
| WT | 20.5 ± 0.6 | 22.7 ± 0.73 | 2.21 ± 0.30 | 1.03 ± 0.04 | 4.55 ± 0.15 |
| WT+PB | 20.8 ± 0.7 | 23.1 ± 0.65 | 2.25 ± 0.20 | 1.43 ± 0.02* | 6.22 ± 0.15* |
| p50-/- | 20.4 ± 0.4 | 18.8 ± 0.40† | -1.55 ± 0.56 | 1.03 ± 0.04 | 5.50 ± 0.19† |
| p50-/- +PB | 17.0 ± 0.2 | 19.1 ± 0.34† | 2.07 ± 0.25 | 1.57 ± 0.06* | 8.21 ± 0.29*† |
Results are means ± standard errors.
Significant effect of phenobarbital
Significant effect of p50 deletion.
We measured cell proliferation in hepatocytes from these mice using the BrdU immunohistochemical staining method (Figure 1). After 3 days, the labeling index was significantly increased by PB only in p50-/- mice; in wild-type mice, the labeling index was 8-fold higher in mice administered PB, but this increase was not statistically significant. The deletion of the p50 -/- subunit significantly increased cell proliferation only in mice administered PB; in mice not fed PB, p50 -/- had a 2.5 fold increase in the labeling index compared to wild-type mice, but this was not statistically significant. After 10 days of PB administration, the labeling index was significantly increased by PB in both wild-type and p50 -/- mice. The labeling index in p50 -/- mice not fed PB was over 3-fold higher than in wild-type mice, but this was not statistically significant. In the 34 day treatment group, there was a significant increase in cell proliferation in p50 -/- mice in both control and PB-fed mice; PB administration did not have a significant effect.
Figure 1.



Effect of PB administration on hepatocyte proliferation in wild-type and p50 -/- mice. A. 3-day administration. B. 10-day administration. C. 34 day administration. Mice were administered a single dose of BrdU two hours before euthanasia. Histological sections for the liver were immunohistochemically stained for BrdU, and labeling indexes were determined in hepatocytes to determine the rate of DNA synthesis. Data are expressed as means ± standard errors. *Significant effect of phenobarbital; †Significant effect of p50 deletion.
Apoptotic indexes were quantified in hepatocytes using the TUNEL assay. In the 3 day study, PB treatment decreased apoptosis in both wild-type and p50 -/- mice (Figure 2). The p50 -/- mice showed a higher level of apoptosis when compared to wild-type mice, in both the control and PB-fed animals. After 10 days of PB administration, the apoptotic index was higher in p50 -/- mice. The apoptotic index was 34% lower in wild-type mice administered PB and 22% lower in p50 -/- mice administered PB, but there was no statistically-significant difference. In the 34 day study, there were no significant changes in apoptotic indexes.
Figure 2.



Effect of PB administration on hepatocyte apoptosis in wild-type and p50 -/- mice. A. 3-day administration. B. 10-day administration. C. 34 day administration. Histological sections for the liver were used for TUNEL staining, and apoptotic indexes were determined in hepatocytes to determine the rate of apoptosis. Data are expressed as means ± standard errors. *Significant effect of phenobarbital; †Significant effect of p50 deletion.
In order to determine how alterations in NF-κB activation may be influencing cell proliferation and apoptosis, we examined the expression of the NF-κB-regulated gene tumor necrosis factor-α (TNF-α). TNF-α influences both cell proliferation and apoptosis in the liver (Schwabe and Brenner, 2006). The expression of TNF-α tended to corrrelate with cell proliferation rather than apoptosis in this study (Figure 3). After 3 days of PB administration, TNF-α expression was significantly increased in p50 mice fed PB compared to either p50 controls or wild-type mice fed PB. At 10 days, there were no significant effects in TNF-α expression, which was due to very high variability in the results for this timepoint. However, the expression of TNF-α was 3-4 fold higher in wild-type mice administered PB and in p50 -/- mice not fed PB. After 34 days of PB administration, TNF-α expression was significantly increased both by PB administration and by deletion of the p50 subunit of NF-κB.
Figure 3.



Effect of PB administration on TNF-α expression in wild-type and p50 -/- mice. A. 3-day administration. B. 10-day administration. C. 34 day administration. Real-time PCR was performed on RNA obtained from liver homogenates for both TNF-α and ß-actin. Results are expressed as a fraction of ß-actin expression. Data are expressed as means ± standard errors. *Significant effect of phenobarbital; †Significant effect of p50 deletion.
The activation of NF-κB was examined using electrophoretic mobility shift assays. In the control and PB-treated groups, the hepatic DNA binding activity of NF-κB in the p50-/- knockout mice was not present for any of the time points (Figures 4-6). In wild-type mice, the DNA binding activity of NF-κB in both treated and control mice livers was clearly seen. However, PB did not increase the DNA binding activity of NF-κB at any of the time points.
Figure 4.

Effect of 3-day PB administration on the DNA binding activity of NF-κB in wild-type and p50 -/- mice. Nuclear extracts were prepared from mouse livers and used in EMSAs with a radiolabeled oligonucleotide containing an NF-κB binding site. After electrophoresis, gels were dried and subjected to autoradiography. Three mice were used for each of the four experimental treatments, and each lane represents a single mouse. Lane abbreviations: WT, wild-type mice; PB, phenobarbital; p50, p50 -/- mice.
Figure 6.
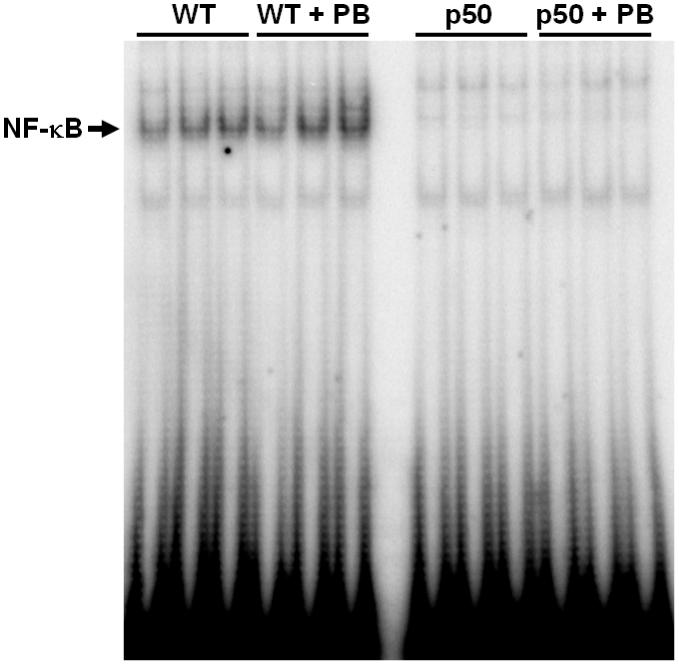
Effect of 34-day PB administration on the DNA binding activity of NF-κB in wild-type and p50 -/- mice. Nuclear extracts were prepared from mouse livers and used in EMSAs with a radiolabeled oligonucleotide containing an NF-κB binding site. After electrophoresis, gels were dried and subjected to autoradiography. Three mice were used for each of the four experimental treatments, and each lane represents a single mouse. Lane abbreviations: WT, wild-type mice; PB, phenobarbital; p50, p50 -/- mice.
DISCUSSION
The effects of PB in relation to hepatic cell proliferation, apoptosis and tumor promotion are well known (Pitot et al., 1987; Jirtle and Meyer, 1991). In this study we examined if effects of PB on cell proliferation and apoptosis are dependent on the activation of the transcription factor NF-κB. In our previous studies we found that PB increases the DNA binding activity of NF-κB in rat liver (Li et al., 1996; Calfee-Mason et al., 2002). We therefore hypothesized that this activation is necessary for some of the actions of PB. However, although both PB administration and the deletion of the p50 subunit independently influenced cell proliferation and apoptosis for at least one time point, the presence of the p50 subunit does not appear to be necessary for the effects of phenobarbital on these endpoints. PB was found to increase cell proliferation after 3 and 10 days of administration and to inhibit apoptosis after 3 and possibly 10 days. If NF-κB were necessary for these changes, one would expect to observe these changes in wild-type mice but not in p50 -/- mice. Since these effects were never observed only in the wild-type mice, however, we have to conclude that NF-κB is not necessary for changes in cell proliferation and apoptosis induced by PB.
The p50 knockout mice model was previously used to evaluate the effects of other hepatic tumor promoting agents: the peroxisome proliferators ciprofibrate and Wy-14,643-14,643, and the PCB 2,2′,4,4′,5,5′-hexachlorobiphenyl (PCB-153). After short-term administration, both ciprofibrate- and PCB-153-induced hepatocyte proliferation was inhibited in p50 -/- mice (Tharappel et al., 2003; Lu et al., 2004). In addition, both ciprofibrate and PCB-153 were also found to inhibit hepatic apoptosis that was elevated in the p50 -/- mice (Tharappel et al., 2003; Lu et al., 2004). In long term studies, the promotion of hepatic tumors by Wy-14,643 and PCB-153 was inhibited in p50 -/- mice (Glauert et al., 2006; Tharappel et al., 2006). In contrast, the induction of cell proliferation by PB was not inhibited in the p50 -/- mice in the present study. The promotion of hepatic tumors by PB in p50 -/- mice has not been studied, but based on the data observed in the present study it appears unlikely that tumorigenesis would be reduced.
Both PB administration and p50 deletion increased the relative liver weight at all 3 time points, although only PB administration increased the absolute liver weight. The increases in cell proliferation and decreases in apoptosis at the 3 and 10 day time points in PB-fed animals correlate with the increases in liver weight observed. The lesser responses in the p50 -/- mice may be related to the overall higher level of hepatocyte apoptosis observed in these animals, although cell proliferation was generally increased.
Studies show that many genes with κB sites are either not regulated by NF-κB or regulated by other NF-κB-Rel complexes in the absence of p50 (Sha et al., 1995). Studies with B cells from p50-/- mice suggest that p65-p65 or p65 - rel dimers may serve many of the functions that were previously assigned to p50-p65 dimers (Sha et al., 1995). In liver regeneration studies using p50-/- mice, overall liver regeneration in knockout mice was normal, indicating that some form of compensation for the p50 subunit is in place (DeAngelis et al., 2001). These authors also found that there was no increased mortality rate in p50-/- mice, and that DNA synthesis and mitosis analyses showed that cell cycle activities were at normal levels. These authors found that there was an increased level of p65 in the nuclei of p50-/- mice livers before and during regeneration. Along with more p65 in p50-/- livers, there was also a reduction in IκBα, and this can cause more p65 dimers to enter the nuclei and activate NF-κB target genes. In addition, since p50 homodimers as well as unphosphorylated p65-p50 heterodimers can suppress transcription (Plaksin et al., 1993; Zhong et al., 1998; Zhong et al., 2002), increased transcription of these genes could affect growth regulation. Other forms of compensation for the lack of the p50 subunit could also be taking place in utero or in the adult mice. Our finding that the expression of the NF-κB regulated gene TNF-α was increased in p50 mice at the 3 and 34 day timepoints supports the idea that the liver is compensating for the lack of the p50 subunit. Our findings as well as the results of other studies may explain the lack of an effect of the absence of p50 subunit of NF-κB on PB-induced changes in cell proliferation and apoptosis.
In summary, we have observed that PB-induced changes in hepatocyte cell proliferation and apoptosis are not dependent on the presence of the p50 subunit of NF-κB. Therefore the promoting activity of PB is likely related to other molecular effects, such as the activation of the constitutive androstane receptor (CAR).
Figure 5.

Effect of 10-day PB administration on the DNA binding activity of NF-κB in wild-type and p50 -/- mice. Nuclear extracts were prepared from mouse livers and used in EMSAs with a radiolabeled oligonucleotide containing an NF-κB binding site. After electrophoresis, gels were dried and subjected to autoradiography. Three mice were used for each of the four experimental treatments, and each lane represents a single mouse. Lane abbreviations: WT, wild-type mice; PB, phenobarbital; p50, p50 -/- mice.
ACKNOWLEDGMENTS
This work was supported by a grant from the National Institute of Environmental Health Sciences (ES11480) and by the Kentucky Agricultural Experiment Station. The sponsors of the study had no role in the study design; in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication.
CONFLICT OF INTEREST STATEMENT
There were no financial, personal, or relationships with other people or organizations within 3 years of beginning this work that could inappropriately influence the work submitted.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Barbason H, Rassenfosse C, Betz EH. Promotion mechanism of phenobarbital and partial hepatectomy in DENA hepatocarcinogenesis cell kinetics effect. Br J Cancer. 1983;47:517–525. doi: 10.1038/bjc.1983.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkett M, Gilmore TD. Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6910–6924. doi: 10.1038/sj.onc.1203238. [DOI] [PubMed] [Google Scholar]
- Brawn MK, Chiou WJ, Leach KL. Oxidant-induced activation of protein kinase C in UC11MG cells. Free Radic Res. 1995;22:23–37. doi: 10.3109/10715769509147525. [DOI] [PubMed] [Google Scholar]
- Butler WH, Hempsall V. Histochemical observations on nodules induced in the mouse liver by phenobarbitone. J Pathol. 1978;125:155–161. doi: 10.1002/path.1711250306. [DOI] [PubMed] [Google Scholar]
- Butterworth BE, Conolly RB, Morgan KT. A strategy for establishing mode of action of chemical carcinogens as a guide for approaches to risk assessments. Cancer Lett. 1995;93:129–146. doi: 10.1016/0304-3835(95)03794-W. [DOI] [PubMed] [Google Scholar]
- Calfee-Mason KG, Spear BT, Glauert HP. Vitamin E inhibits hepatic NF-kappa B activation in rats administered the hepatic tumor promoter, phenobarbital. J. Nutr. 2002;132:3178–3185. doi: 10.1093/jn/131.10.3178. [DOI] [PubMed] [Google Scholar]
- DeAngelis RA, Kovalovich K, Cressman DE, Taub R. Normal liver regeneration in p50/nuclear factor kappaB1 knockout mice. Hepatology. 2001;33:915–924. doi: 10.1053/jhep.2001.23192. [DOI] [PubMed] [Google Scholar]
- Deryckere F, Gannon F. A one-hour minipreparation technique for extraction of DNA-binding proteins from animal tissues. Biotechniques. 1994;16:405. [PubMed] [Google Scholar]
- Farber E. Possible etiologic mechanisms in chemical carcinogenesis. Environ Health Perspect. 1987;75:64–70. [PMC free article] [PubMed] [Google Scholar]
- Feig DI, Reid TM, Loeb LA. Reactive oxygen species in tumorigenesis. Cancer Res. 1994;54:1890s–1894s. [PubMed] [Google Scholar]
- Feldman D, Swarm RL, Becker J. Ultrastructural study of rat liver and liver neoplasms after long-term treatment with phenobarbital. Cancer Res. 1981;41:2151–2162. [PubMed] [Google Scholar]
- Frueh FW, Zanger UM, Meyer UA. Extent and character of phenobarbital-mediated changes in gene expression in the liver. Mol Pharmacol. 1997;51:363–369. [PubMed] [Google Scholar]
- Glauert HP, Eyigor A, Tharappel JC, Cooper S, Lee EY, Spear BT. Inhibition of hepatocarcinogenesis by the deletion of the p50 subunit of NF-kB in mice administered the peroxisome proliferator Wy-14,643. Toxicol Sci. 2006;90:331–336. doi: 10.1093/toxsci/kfj116. [DOI] [PubMed] [Google Scholar]
- Hinz M, Krappmann D, Eichten A, Heder A, Scheidereit C, Strauss M. NF-kappaB function in growth control: regulation of cyclin D1 expression and G0/G1-to-S-phase transition. Mol Cell Biol. 1999;19:2690–2698. doi: 10.1128/mcb.19.4.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honkakoski P, Negishi M. Regulatory DNA elements of phenobarbital-responsive cytochrome P450 CYP2B genes. J Biochem Mol Toxicol. 1998;12:3–9. doi: 10.1002/(sici)1099-0461(1998)12:1<3::aid-jbt2>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Jirtle RL, Carr BI, Scott CD. Modulation of isuline-like growth factor-II/mannose 6-phosphate receptors and transforming growth factor-b1 during liver regeneration. J. Biol. Chem. 1991;266:22444–22450. [PubMed] [Google Scholar]
- Jirtle RL, Meyer SA. Liver tumor promotion: effect of phenobarbital on EGF and protein kinase C signal transduction and transformation growth factor-á1 expression. Dig. Dis. Sci. 1991;36:659–668. doi: 10.1007/BF01297035. [DOI] [PubMed] [Google Scholar]
- Kaufmann WK, Byrd LL, Palmieri D, Nims RW, Rice JM. TGF-alpha sustains clonal expansion by promoter-dependent, chemically initiated rat hepatocytes. Carcinogenesis. 1997;18:1381–1387. doi: 10.1093/carcin/18.7.1381. [DOI] [PubMed] [Google Scholar]
- Klaunig JE, Xu Y, Isenberg JS, Bachowski S, Kolaja KL, Jiang J, Stevenson DE, Walborg EF., Jr. The role of oxidative stress in chemical carcinogenesis. Environ Health Perspect. 1998;106(Suppl 1):289–295. doi: 10.1289/ehp.98106s1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Karin M. Is NF-kappaB the sensor of oxidative stress? Faseb J. 1999;13:1137–1143. [PubMed] [Google Scholar]
- Li Y, Leung LK, Spear BT, Glauert HP. Activation of hepatic NF-kappaB by phenobarbital in rats. Biochem Biophys Res Commun. 1996;229:982–989. doi: 10.1006/bbrc.1996.1911. [DOI] [PubMed] [Google Scholar]
- Lu Z, Lee EY, Robertson LW, Glauert HP, Spear BT. Effect of 2,2′,4,4′,5,5′-hexachlorobiphenyl (PCB-153) on hepatocyte proliferation and apoptosis in mice deficient in the p50 subunit of the transcription factor NF-kB. Toxicol Sci. 2004;81:35–42. doi: 10.1093/toxsci/kfh193. [DOI] [PubMed] [Google Scholar]
- Manjeshwar S, Rao PM, Rajalakshmi S, Sarma DSR. Inhibition of DNA synthesis by phenobarbital in primary cultures of hepatocytes from normal rat liver and from hepatic nodules. Carcinogenesis. 1992;13:2287–2291. doi: 10.1093/carcin/13.12.2287. [DOI] [PubMed] [Google Scholar]
- Peraino C, Fry RJM, Staffeldt EF. Reduction and enhancement by phenobarbital of hepatocarcinogenesis induced in the rat by 2-acetylaminofluorene. Cancer Research. 1971;31:1506–1512. [PubMed] [Google Scholar]
- Pitot HC, Goldsworthy TL, Moran S, Kennan W, Glauert HP, Maronpot RR, Campbell HA. A method to quantitate the relative initiating and promoting potencies of hepatocarcinogenic agents in their dose-response relationships to altered hepatic foci. Carcinogenesis. 1987;8:1491–1499. doi: 10.1093/carcin/8.10.1491. [DOI] [PubMed] [Google Scholar]
- Plaksin D, Baeuerle PA, Eisenbach L. KBF1 (p50 NF-kappa B homodimer) acts as a repressor of H-2Kb gene expression in metastatic tumor cells. J. Exp. Med. 1993;177:1651–1662. doi: 10.1084/jem.177.6.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandgren EP, Schroeder JA, Qui TH, Palmiter RD, Brinster RL, Lee DC. Inhibition of mammary gland involution is associated with transforming growth factor alpha but not c-myc-induced tumorigenesis in transgenic mice. Cancer Res. 1995;55:3915–3927. [PubMed] [Google Scholar]
- Santoni-Rugiu E, Jensen MR, Thorgeirsson SS. Disruption of the pRb/E2F pathway and inhibition of apoptosis are major oncogenic events in liver constitutively expressing c-myc and transforming growth factor alpha. Cancer Res. 1998;58:123–134. [PubMed] [Google Scholar]
- Schulte-Hermann R, Timmermann-Trosiener I, Barthel G, Bursch W. DNA synthesis, apoptosis, and phenotypic expression as determinants of growth of altered foci in rat liver during phenobarbital promotion. Cancer Res. 1990;50:5127–5135. [PubMed] [Google Scholar]
- Schwabe RF, Brenner DA. Mechanisms of Liver Injury. I. TNF-alpha-induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol. 2006;290:G583–589. doi: 10.1152/ajpgi.00422.2005. [DOI] [PubMed] [Google Scholar]
- Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- Smith GH, Sharp R, Kordon EC, Jhappan C, Merlino G. Transforming growth factor-alpha promotes mammary tumorigenesis through selective survival and growth of secretory epithelial cells. Am J Pathol. 1995;147:1081–1096. [PMC free article] [PubMed] [Google Scholar]
- Storz G, Polla BS. Transcriptional regulators of oxidative stress-inducible genes in prokaryotes and eukaryotes. Exs. 1996;77:239–254. doi: 10.1007/978-3-0348-9088-5_16. [DOI] [PubMed] [Google Scholar]
- Sueyoshi T, Negishi M. Phenobarbital response elements of cytochrome P450 genes and nuclear receptors. Annu Rev Pharmacol Toxicol. 2001;41:123–143. doi: 10.1146/annurev.pharmtox.41.1.123. [DOI] [PubMed] [Google Scholar]
- Tharappel JC, Cunningham ML, Spear BT, Glauert HP. Differential activation of hepatic NF-kappaB in rats and hamsters by the peroxisome proliferators Wy-14,643, gemfibrozil, and dibutyl phthalate. Toxicol Sci. 2001;62:20–27. doi: 10.1093/toxsci/62.1.20. [DOI] [PubMed] [Google Scholar]
- Tharappel JC, Nalca A, Owens AB, Ghabrial L, Konz EC, Glauert HP, Spear BT. Cell proliferation and apoptosis are altered in mice deficient in the NF-kappaB p50 subunit after treatment with the peroxisome proliferator ciprofibrate. Toxicol Sci. 2003;75:300–308. doi: 10.1093/toxsci/kfg201. [DOI] [PubMed] [Google Scholar]
- Tharappel JC, Spear BT, Lehmler HJ, Lee EY, Robertson LW, Glauert HP. Effect of the deletion of the p50 subunit of NF-kB on hepatocarcinogenesis in mice receiving diethylnitrosamine and polychlorinated biphenyls. Toxicol. Sci. (Toxicologist supplement) 2006;90:249. [Google Scholar]
- Ueda A, Hamadeh HK, Webb HK, Yamamoto Y, Sueyoshi T, Afshari CA, Lehmann JM, Negishi M. Diverse roles of the nuclear orphan receptor CAR in regulating hepatic genes in response to phenobarbital. Mol Pharmacol. 2002;61:1–6. doi: 10.1124/mol.61.1.1. [DOI] [PubMed] [Google Scholar]
- Waxman DJ, Azaroff L. Phenobarbital induction of cytochrome P-450 gene expression. Biochem J. 1992;281(Pt 3):577–592. doi: 10.1042/bj2810577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, May MJ, Jimi E, Ghosh S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol. Cell. 2002;9:625–636. doi: 10.1016/s1097-2765(02)00477-x. [DOI] [PubMed] [Google Scholar]
- Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol. Cell. 1998;1:661–671. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]