

Abstract
Pulmonary tuberculosis (TB) remains a serious health problem worldwide. Effective vaccination strategies are needed. We report the development of a novel TB vaccine using vesicular stomatitis virus (VSV) as a viral vector system to express Ag85A. VSVAg85A was shown to be immunogenic when given to mice by either an intranasal or an intramuscular (IM) route. Although distinct T-cell profiles resulted from both routes of immunization, only intranasal delivery generated a mucosal T-cell response that was protective upon pulmonary Mycobacterium tuberculosis (M.tb) challenge. While this protection manifested at an early time-point after immunization, it was not sustained. The potential of VSVAg85A to be used as a mucosal booster for parenteral priming by an adenoviral TB vaccine expressing Ag85A (AdAg85A) was investigated. VSVAg85A immunization markedly boosted antigen-specific T-cell responses in the airway lumen while also augmenting immune activation in the systemic compartment, after AdAg85A priming. This translated into significantly better protective efficacy against pulmonary challenge with M.tb than either vaccine used alone. Our study therefore suggests that VSV as a vector system is a promising candidate to be used in a heterologous viral prime-boost immunization regimen against intracellular bacterial infection.
Introduction
Tuberculosis (TB) continues to remain a major threat to health globally. Approximately one-third of the world population has been infected with Mycobacterium tuberculosis (M.tb).1 While most of the infected individuals mount a strong cell-mediated immune response restricting the disease to a dormant state,2 5–10% will eventually develop active TB.1 Approximately 2 million individuals succumb to the disease each year, making TB one of the most deadly infectious diseases in the world.1 Because this disease predominantly affects the populations in developing countries, and in view of the fact that the incidence of infection with multidrug-resistant or extensively drug-resistant strains of M.tb is on the rise, the most effective and practical means to combat TB is through vaccination.
Bacille Calmette-Guérin (BCG) is the current TB vaccine that has been in use since 1920, with >3 billion individuals having been immunized to date.3 Despite its extensive use, the efficacy of BCG remains controversial because, although it is effective at protecting against severe childhood forms of TB, its protection against adult pulmonary TB ranges from 0 to 80%,3 , 4 , 5 which explains the current TB epidemic. There is therefore a recognized need to develop new TB vaccines. Among the major novel TB vaccine formulations are the viral vector systems including adenovirus (Ad),6 , 7 modified vaccinia virus Ankara,8 , 9 poxvirus,10 and influenza virus.11 These viral-vectored TB vaccines have been evaluated as stand-alone vaccines or as boosters in heterologous prime-boost regimens.6 , 8 , 10 , 12 , 13
In comparison with other viral systems, the recombinant adenoviral vaccine system possesses the advantages of established safety, well-characterized vectorology, and transgene expression potency.14 Indeed, a recombinant human type 5 Ad expressing Ag85A (AdAg85A) developed in our lab was shown to be immune-protective following a single immunization in murine models.6 However, a potential drawback of viral systems is that the initially immunized host generates a strong neutralizing antibody response against the viral backbone, limiting the efficacy of repeated immunizations with the same viral vaccine for the purpose of immune boosting. With this in mind we have developed a novel vesicular stomatitis virus (VSV)-vectored TB vaccine and evaluated its potential as a booster to amplify a protective mucosal immune response after prime immunization with AdAg85A.
VSV has become a well established viral vector system for immunization.15 , 16 , 17 Given the low pre-existing anti-VSV immunity in humans and the vector's natural tropism for the mucosa,15 a plethora of effective VSV-vectored vaccines have been generated to induce protective humoral responses after mucosal immunization for viral diseases including human immunodeficiency virus,18 herpes simplex virus,19 human papillomavirus,20 severe acute respiratory syndrome,21 and influenza.22 However, while these studies demonstrate a great potential for VSV to be used as a vaccine platform, its potential to elicit anti-intracellular bacterial immunity remains to be established. In particular, VSV has never been used for the purpose of anti-TB vaccination. In this study, we engineered an interferon (IFN)-inducing VSV mutant to express Ag85A and explored its potential as a boost vaccine. We found that, when delivered mucosally, this vaccine was able to boost mucosal T-cell responses induced by parenteral AdAg85A prime immunization effectively, thereby leading to effective protection in a murine model of pulmonary TB. Our study therefore suggests that VSV as a vector system is a promising candidate for heterologous boost immunization against an intracellular bacterial infection.
Results
Construction and characterization of VSVAg85A
An attenuated replicating VSV vector system was utilized for the development of VSVAg85A. This vector system carries a mutation in the viral matrix protein, leading to inability of the virus to block mRNA export from the nucleus of host cells and thereby restoring antiviral cytokine responses that are normally limited during wild-type VSV infection.23 This feature of the vector was exploited as built-in immune adjuvanticity for immunization.14 VSV was engineered to express the immunodominant M.tb antigen, Ag85A, which is a mycolyl transferase enzyme necessary for mycobacterial cell wall synthesis and a member of the Ag85 complex proteins (Ag85A/B/C).24 , 25 Ag85A, when expressed in genetic TB vaccines, has been shown to be one of the most immunogenic M.tb antigens.24 , 25 Ag85A was placed downstream of the human tissue plasminogen activator (tPA) signal peptide, allowing secretion of the protein in order to maximize antigen presentation through both major histocompatibility complex class I and class II pathways, and thereby to enhance the immunogenicity of this antigen.26 The tPA-Ag85A transgene was cloned into the viral genome between the G and L genes (Figure 1a ). After viral rescue and purification, A549 cells were infected with either titrated VSVAg85A or an Ad expressing Ag85A (AdAg85A) as a control. The cell lysates were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and immunoblots were probed using a monoclonal Ag85A antibody and polyclonal VSV antibody. VSVAg85A-infected cells expressed Ag85A (Figure 1b) and also the VSV proteins (Figure 1c). In VSVAg85A-infected cells, a single Ag85A protein band was detected, suggestive of an unprocessed form (tPA-Ag85A), whereas, in AdAg85A-infected cells, there appeared two similar-sized protein bands, suggestive of the presence of both unprocessed (tPA-Ag85A) and processed (Ag85A) forms (Figure 1b). The Ad- or VSV-infected cells produced Ag85A protein that was at a higher molecular weight than the native Ag85A protein, likely due to the presence of attached signal peptide and post-translational modifications in infected mammalian cells. Therefore, VSVAg85A was able to incorporate the foreign M.tb transgene successfully and express it in abundance from infected cells.
Figure 1.
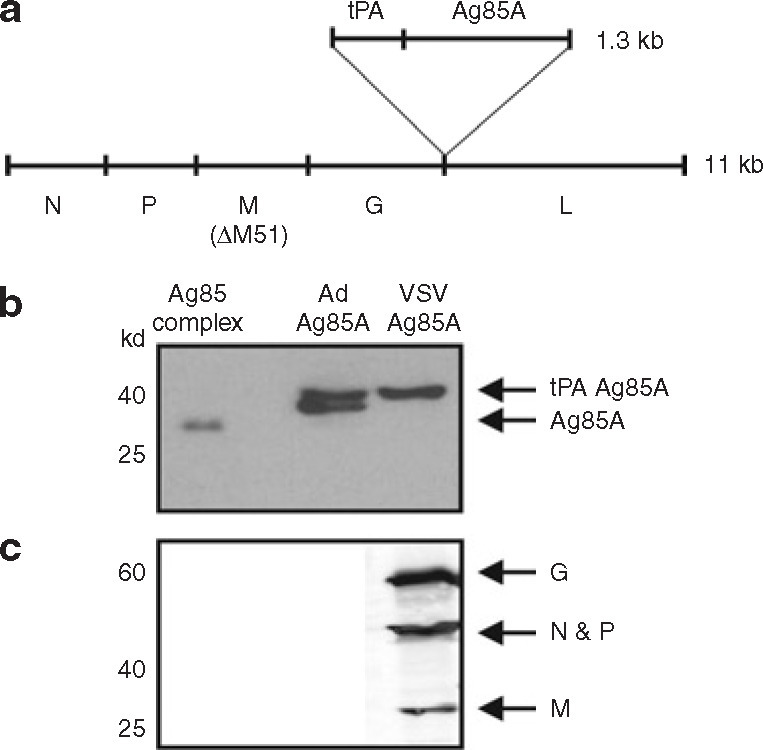
Design and characterization of VSVAg85A. (a) Diagram of the VSV genome and location for insertion of the foreign transgene. A549 cells were infected with VSVAg85A and AdAg85A and cell lysates were harvested 24 and 48 hours after infection, respectively. Equal volumes of the sample were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis, blotted, and probed with (b) a monoclonal Ag85A antibody, or (c) polyclonal VSV antibodies. These data are representative of two independent experiments. G, glycoprotein; kb, kilobase; L, large protein; M, matrix protein; N, nucleoprotein; P, phosphoprotein; tPA, tissue plasminogen activator; VSV, vesicular stomatitis virus.
Determination of an optimal dose of VSVAg85A for immunization
In order to characterize VSVAg85A in vivo, Balb/c mice were immunized intramuscularly (IM) with increasing doses of VSVAg85A. One week after the immunization, the mice were killed and splenocytes were stimulated ex vivo with Ag85A-specific CD4 and CD8 peptides. The frequency of occurrence of IFN-γ+ T cells was measured using enzyme-linked immunosorbent spot. Naive controls exhibited no antigen-specific T cells (data not shown). There was no clear difference in the IFN-γ-secreting CD4 T-cell responses among all the doses tested (Figure 2a ). Although not statistically different, the level of IFN-γ-secreting CD8 T cells triggered by 1 × 107 plaque forming units (PFU) dose was higher than the other two doses tested (Figure 2a). This dose was thus used for all subsequent experiments. These data also indicate that VSVAg85A is immunogenic in vivo.
Figure 2.
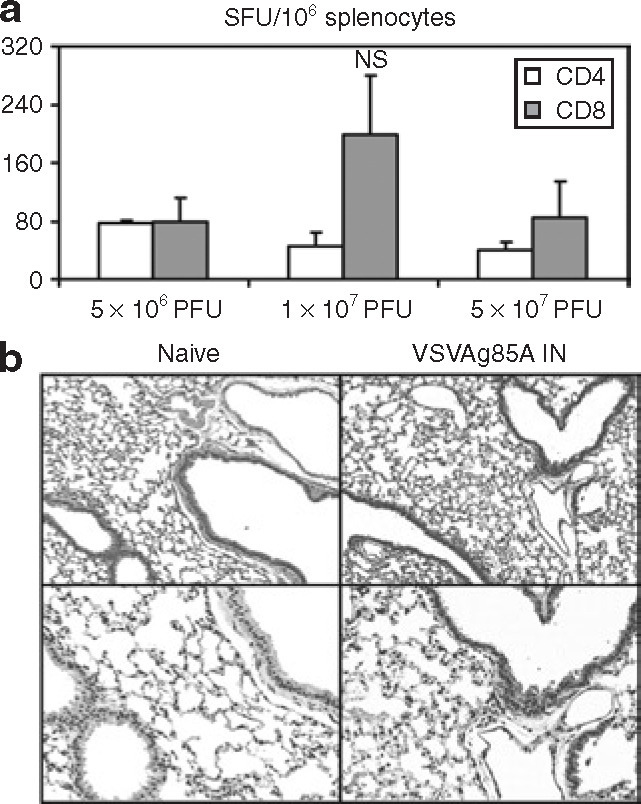
Determination of an appropriate dose for immunization with VSVAg85A, and verification of its safety for mucosal administration. (a) Balb/c mice were immunized intramuscularly with increasing doses of VSVAg85A. One week later, the mice were killed and peptide-stimulated splenocytes were analyzed using enzyme-linked immunosorbent spot. The data are shown as mean values ± SEM of three mice per group. (b) Other mice were immunized intranasally (IN) with 1 × 107 plaque forming units (PFU) of VSVAg85A, and 1 week later the naive mice as well as the immunized mice were examined for lung pathology. For this purpose, the mice were killed, the lungs were sectioned and hematoxylin and eosin stained, and representative images were presented. Original magnification: top images—×10; bottom images—×20. NS, not statistically significant as compared to the other two doses; SFU, spot forming units; VSV, vesicular stomatitis virus.
In view of the fact that VSVAg85A remains replication-efficient, and with the anticipation of its potential usefulness in respiratory mucosal immunization, we used the 1 × 107 PFU dose to examine the extent of lung tissue inflammation caused by intranasal (IN) immunization. In comparison with naive controls (Figure 2b, left panel), we observed only a slight increase in cellularity within the lung parenchyma in mice immunized IN with VSVAg85A (Figure 2b, right panel). Aside from this modest infiltrate, the overall levels of inflammation in the tissue and airway endothelium were minimal. This affirms that this dose of VSVAg85A, when delivered mucosally, is minimally inflammatory.
VSVAg85A immunization by different routes results in distinct kinetic and geographic profiles of antigen-specific T-cell responses
In order to further characterize VSVAg85A immunization, we compared T-cell responses after two different routes of administration of the vector. Balb/c mice were immunized through either the parenteral (IM) or the mucosal (IN) route, and T-cell responses were analyzed in the airway lumen, lung, and spleen over a 3-week time-course. Tetramer staining was used for estimating CD8 T cells specific for an immunodominant CD8 epitope within Ag85A. Following IM immunization, antigen-specific CD8 T cells were found mainly in the lung and spleen (Figure 3b and c, left panel), while minimal responses were detected in the airway lumen (Figure 3a, left panel). The peak responses appeared at 2 weeks. In contrast, IN immunization resulted in a peak in CD8 T-cell responses at 1 week, primarily in the airway lumen (Figure 3a, right panel), whereas such responses were lower in the lung/spleen (Figure 3b and c, right panel).
Figure 3.
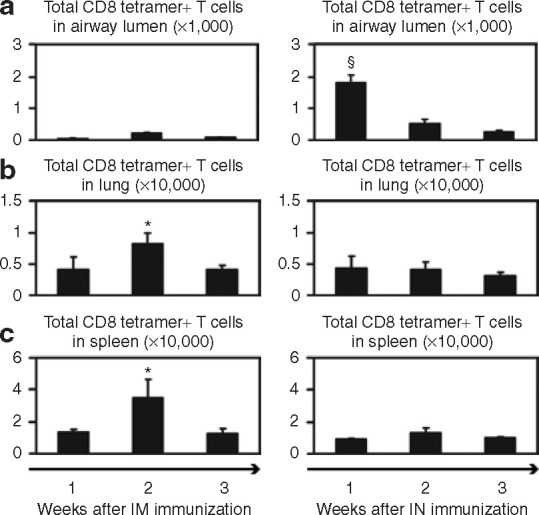
Kinetics of tetramer+ CD8 T-cell responses following intramuscular (IM) and intranasal (IN) immunization with VSVAg85A. Balb/c mice were immunized with 1 × 107 plaque forming units of VSVAg85A, either IM or IN. At 1, 2, and 3 weeks after immunization, the mice were killed and the total tetramer+ CD8 T cells in (a) the airway lumen, (b) lung, and (c) spleen were determined using immunostaining and fluorescence-activated cell sorting analysis. The data are shown as mean values ± SEM of three mice per group. §P ≤ 0.01 as compared to time-points of week 2 and week 3; *P ≤ 0.05 as compared to week 3 in b, and as compared to week 1 or week 3 in c. VSV, vesicular stomatitis virus.
In order to examine the functionality of T cells, the cells harvested from these three tissue compartments were stimulated with Ag85A CD4 and CD8 T-cell peptides and intracellular cytokine staining (ICCS) was carried out to measure antigen-specific, IFN-γ-producing T cells. As with the distribution of tetramer+ CD8 T cells, CD4 or CD8 IFN-γ+ T cells predominantly populated the systemic tissues after IM immunization (Figure 4b and c, left panel), whereas undetectable levels were found at the mucosal site (Figure 4a, left panel). Such responses appeared to be dominated by CD8 T cells. In contrast, IN immunized mice generated IFN-γ+CD4 and CD8 T cells almost exclusively in the airway lumen (Figure 4a, right panel). While some cells were detected in the lung interstitium, very little systemic activation was evident in the spleen following this route of immunization (Figure 4b and c, right panels). It is interesting to note that, in contrast to the effect after IM immunization, a greater CD4 T-cell response was triggered by IN immunization. Further, the kinetics of T-cell activation resulting from these alternative routes of immunization also seemed to differ, with 1 week and 2 weeks being the time-points for peak activation after IN and IM administration, respectively.
Figure 4.
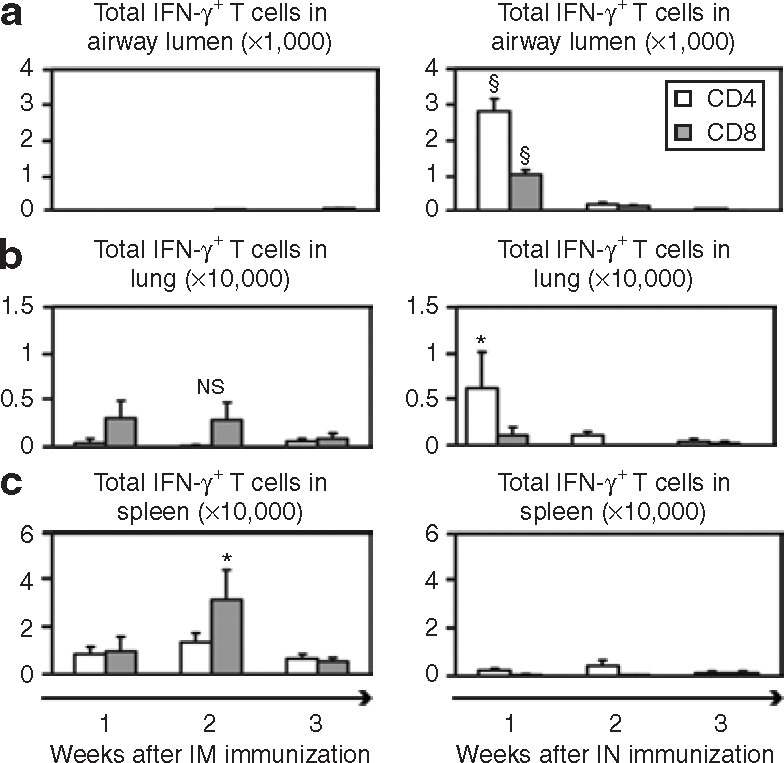
Kinetics of interferon-γ+ (IFN-γ+) CD4 and CD8 T-cell responses following intramuscular (IM) and intranasal (IN) immunization with VSVAg85A. Balb/c mice were immunized with 1 × 107 plaque forming units (PFU) of VSVAg85A, either IM or IN. At 1, 2, and 3 weeks after immunization the mice were killed, and peptide-stimulated cells were analyzed using intracellular cytokine staining in order to determine the number of IFN-γ+ CD4 and CD8 T cells in (a) the airway lumen, (b) lung, and (c) spleen. The data are shown as mean values ± SEM of three mice per group. §P ≤ 0.01 as compared to week 2 CD4 or CD8 T-cell data; *P ≤ 0.05 as compared to week 3 CD4 T-cell data in b, and as compared to week 3 CD8 T-cell data in c; NS, not statistically significant as compared to week 3 data. VSV, vesicular stomatitis virus.
Single respiratory mucosal, but not parenteral, immunization with VSVAg85A confers transient protection from pulmonary M.tb challenge
Having established the immune activation profiles after IM or IN immunization with VSVAg85A, we examined the efficacy of immune protection against M.tb challenge in the lung. To this end, mice were immunized with either IM VSVAg85A or IN VSVAg85A, with IM AdAg85A or subcutaneously administered BCG as controls. Two weeks after immunization, the mice were challenged through the airway with virulent M.tb. Four weeks after the challenge, the lungs were analyzed for bacterial burden. As observed earlier,6 , 27 BCG was able to provide ∼1 log protection and IM AdAg85A provided little protection as compared to naive mice (P ≤ 0.001) (Figure 5a ). As with IM-administered AdAg85A, IM immunization with VSVAg85A also provided little protection (Figure 5a). In contrast, IN VSVAg85A conferred a significant level of protection (∼0.5 log) when compared with naive and IM-immunized mice (P ≤ 0.05) (Figure 5a). In order to understand whether the protective T-cell responses to VSVAg85A administered IN would be sustained, mice were immunized as described earlier and challenged with M.tb at 4 weeks after immunization. Again, as observed earlier, BCG maintained ∼1 log protection (P ≤ 0.001) (Figure 5b). However, the initial protection provided by IN-administered VSVAg85A that was observed at 2 weeks had vanished in the mice when challenged at 4 weeks after immunization (Figure 5b). These data suggest that, although IN immunization with VSV vaccine provides better protection than the IM route, such protection is relatively transient.
Figure 5.
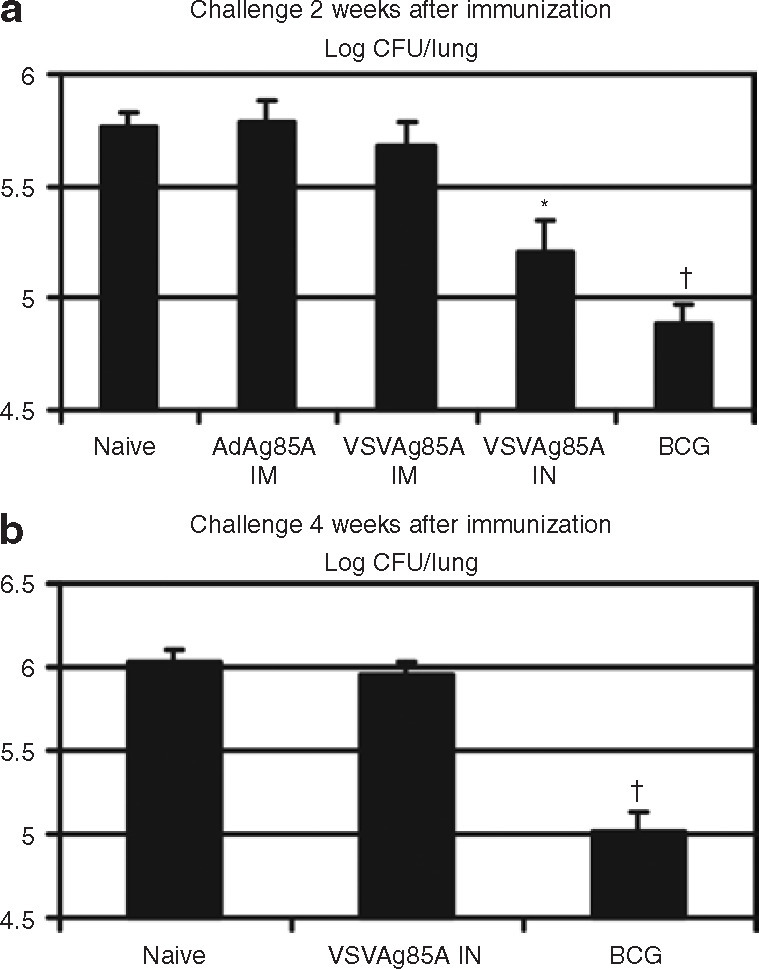
Immune protection against pulmonary Mycobacterium tuberculosis (M.tb) challenge. A group of Balb/c mice were immunized subcutaneously with 1 × 105 colony forming units (CFU) of bacille Calmette-Guérin (BCG). Three weeks later, other groups of mice were immunized with intramuscular (IM) administration of AdAg85A, or IM administration of VSVAg85A, or intranasal (IN) delivery of VSVAg85A, each of them alone, and all at a dose of 1 × 107 plaque forming units. At (a) 2 weeks or (b) 4 weeks after immunization, the mice were challenged through the airway with 15,000 CFU of the M.tb H37Rv strain. At 4 weeks after the challenge, the mice were killed and the lungs were analyzed for CFU counts. The data are shown as mean values ± SEM of five to seven mice per group. *P ≤ 0.05 as compared to naive controls, IM administered adenovirus (Ad), or IM administered VSV; †P ≤ 0.001 as compared to naive controls, IM administered Ad or IM administered VSV in a, and as compared to naive controls or IN administered VSV in b.VSV, vesicular stomatitis virus.
Respiratory mucosal VSVAg85A immunization potently boosts T-cell responses by parenteral AdAg85A priming
Our observation that a single IN immunization with VSVAg85A induces only a transient protective immune response against M.tb challenge suggests that its potential use as a stand-alone vaccine is limited. For this reason, we explored the potential of using VSVAg85A as a booster vaccine. As we have reported earlier, parenteral (IM) immunization with a recombinant adenoviral–vectored vaccine expressing Ag85A (AdAg85A) fails to protect the lung, whereas airway delivery of soluble Ag85 antigens restores protection.6 , 27 , 28 We therefore considered the possibility that IN immunization with VSVAg85A could be employed to boost protective immune responses locally in the lung after IM prime immunization with AdAg85A (heterologous viral prime-boost immunization regimen). A mucosally delivered viral booster will not only deposit Ag85A antigen but will also provide an inflammatory signal in the airway and, cumulatively, this approach can specifically and nonspecifically recruit antigen-specific T cells from systemic pools of T cells. Also, the use of a heterologous viral vector for the purpose of boosting circumvents the problem associated with viral neutralizing antibodies generated after prime immunization. To this end, Balb/c mice were primed with IM AdAg85A, and 3 weeks later a boost was administered with IN VSVAg85A. Two weeks after the boost, the mice were killed and the numbers of Ag85A-specific CD8 T cells in the airway lumen, lung, and spleen were determined using Ag85A tetramer immunostaining. IM administration of AdAg85A resulted in only a few CD8 T cells detectable in the airway lumen and VSVAg85A elicited a small but sizeable number of such T cells (Figure 6a and b). In contrast, the IM AdAg85A and VSVAg85A IN prime-and-boost regimen led to a >200-fold increase in the number of Ag85A-specific CD8 T cells in the airway lumen as compared to the numbers found after either of the stand-alone immunizations (Figure 6a and b). Dramatic increases in CD8 T cells were also evident in the lung interstitium where there was an approximately tenfold increase in the mice that had received prime-and-boost as compared to AdAg85A IM-immunized mice and an ∼100-fold increase as compared to IN VSVAg85A-immunized mice (Figure 6a and c). Upon examination of effector and central memory T-cell surface markers including CD62L, CD127, and CD27, the majority of VSVAg85A-boosted Ag85A-specific CD8 T cells in the lung were found to be of the effector memory phenotype (data not shown). By comparison, the increase of these cells in the spleen on account of the prime-and-boost regimen was much smaller, amplified by approximately twofold and approximately fourfold when compared with AdAg85A IM and VSVAg85A IN, respectively (Figure 6a and d).
Figure 6.
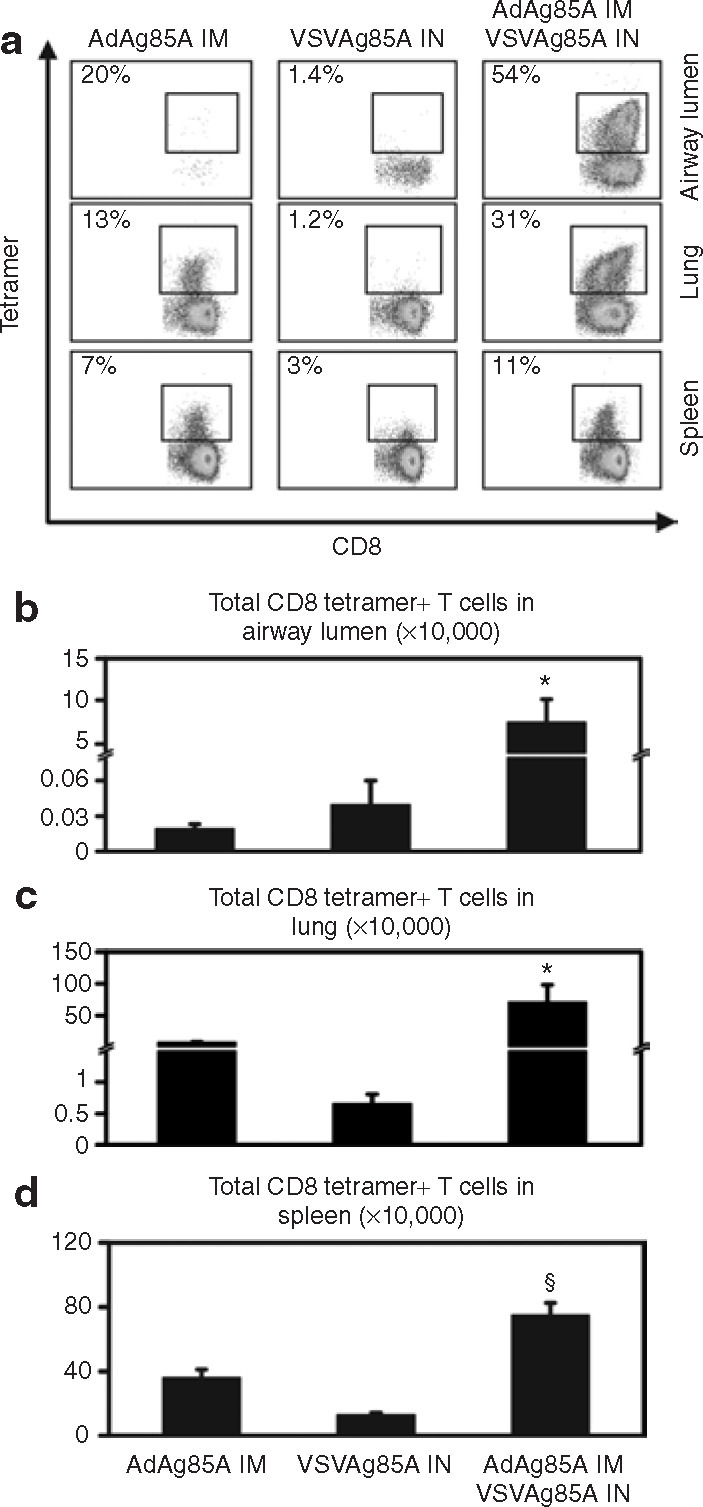
Enhanced tetramer+ T cells by prime-and-boost regimen of intramuscular (IM) AdAg85A (prime) and intranasal (IN) VSVAg85A (boost). Two groups of Balb/c mice were immunized with IM administration of 5 × 107 plaque forming units (PFU) of AdAg85A. Three weeks later, one of these two groups received a boost with IN administration of 1 × 107 PFU of VSVAg85A while the other did not. At the same time-point, a fresh group of mice was immunized with IN administration of 1 × 107 PFU VSVAg85A alone. Two weeks later, all the mice were killed and the total number of tetramer+ CD8 T cells was determined using immunostaining and fluorescence-activated cell sorting analysis. (a) Representative dot plots are presented for the corresponding treatment and tissue. Data from (b) the airway lumen, (c) lung interstitium, and (d) spleen. Values in the left corner of the dot plots represent the percentage of tetramer+ CD8 T cells within the total CD8 T-cell population. The data are shown as mean values ± SEM of three mice per group, representative of two independent experiments. *P ≤ 0.05 as compared to IM AdAg85A or IN VSVAg85A alone; §P ≤ 0.01 as compared to IM AdAg85A or IN VSVAg85A alone. VSV, vesicular stomatitis virus.
We further compared the levels of antigen-specific, IFN-γ-producing CD4 and CD8 T cells in the airway lumen, lung, and spleen using ICCS. There was an ∼1,000-fold difference in IFN-γ+CD4 and IFN-γ+ CD8 T cells in the airway lumen after the prime-and-boost regimen as compared to the numbers of these cells after either immunization administered alone (Figure 7a ). The differences in the lung and spleen cell numbers after the different immunizations were similar to those observed using tetramer staining. In the lung, the prime-and-boost regimen resulted in an approximately tenfold increase in IFN-γ+ CD4 and IFN-γ+ CD8 T-cell numbers relative to those after IM administration of AdAg85A alone, and an ∼100-fold increase relative to IN administration of VSVAg85A alone (Figure 7b). Although to a more modest extent, there were nevertheless threefold and sixfold increases in the cell numbers in the spleen with the prime-and-boost regimen as compared to the cell numbers found after IM AdAg85A and after IN VSVAg85A, respectively (Figure 7c). It is interesting to note that the IN boosting had a dominant effect on CD8 T cells. These results suggest that mucosal VSVAg85A immunization represents a powerful way to boost airway luminal T-cell responses in parenterally genetically immunized hosts.
Figure 7.
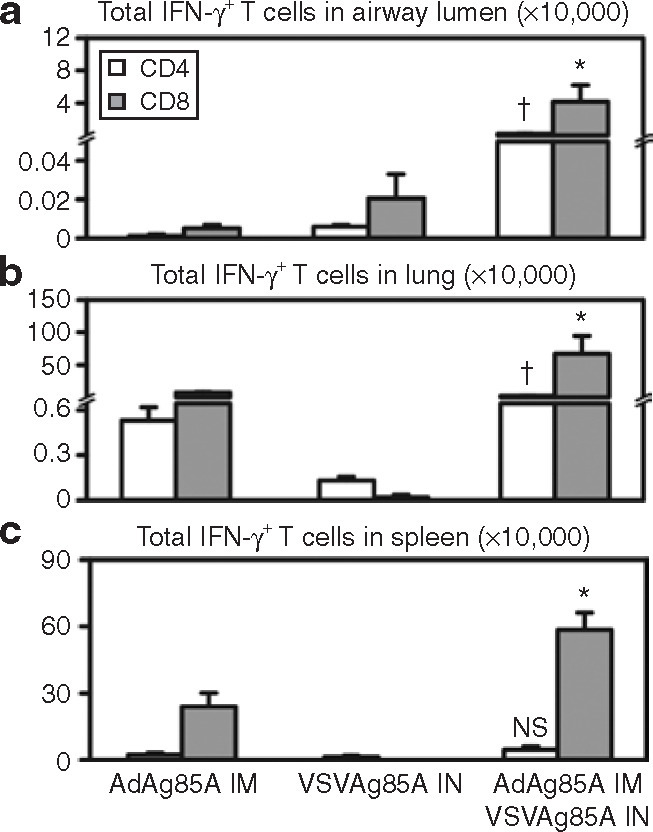
Enhanced T-cell activation by intramuscular (IM) AdAg85A–intranasal (IN) VSVAg85A prime-and-boost regimen. The mice were prepared and treated as explained in the caption for Figure 6. The total numbers of interferon-γ+ (IFN-γ+) CD4 and IFN-γ+ CD8 T cells were determined using intracellular cytokine staining and fluorescence-activated cell sorting analysis after in vitro peptide stimulation of the cells isolated from (a) the airway lumen, (b) lung, and (c) spleen. The data are shown as mean values ± SEM of three mice per group, representative of two independent experiments. *P ≤ 0.05 as compared to CD8 T-cell count data after IM AdAg85A or IN VSVAg85A alone; †P ≤ 0.001 as compared to CD4 T-cell count data after IM AdAg85A or IN VSVAg85A alone; NS, not statistically significant as compared to IM AdAg85A or IN VSVAg85A alone. VSV, vesicular stomatitis virus.
Respiratory mucosal VSVAg85A immunization potently boosts immune protection by parenteral AdAg85A priming
Having observed a robust boosting effect by VSVAg85A on T-cell responses, we examined the protective capacity of this immunization protocol. When challenged at 2 weeks after immunization, mice immunized with the prime-and-boost regimen exhibited a >0.5 log protection when compared with naive mice or with those that had received IM AdA85A alone (P ≤ 0.01) (Figure 8a ). However, at that time-point, while the prime-and-boost regimen provided only a comparable level of protection in the lung to that provided by IN VSVAg85A alone (Figure 8a), it did provide a better protection systemically in the spleen (P ≤ 0.05) (Figure 8b). Further, when challenged at 4 weeks after immunization, mice immunized with IN VSVAg85A alone were no longer protected, whereas the prime-and-boost regimen was able to maintain a ∼0.5 log protection in the lung (P ≤ 0.05) (Figure 8c). It is interesting to note that the prime-and-boost regimen afforded mice a >0.5 log protection (P ≤ 0.01) in the spleen, even better than that provided by BCG (Figure 8d). Therefore, the IM AdAg85A prime-and-IN VSVAg85A boost regimen provided better and more sustained protection from pulmonary M.tb challenge than IM AdAg85A or IN VSVAg85A alone.
Figure 8.
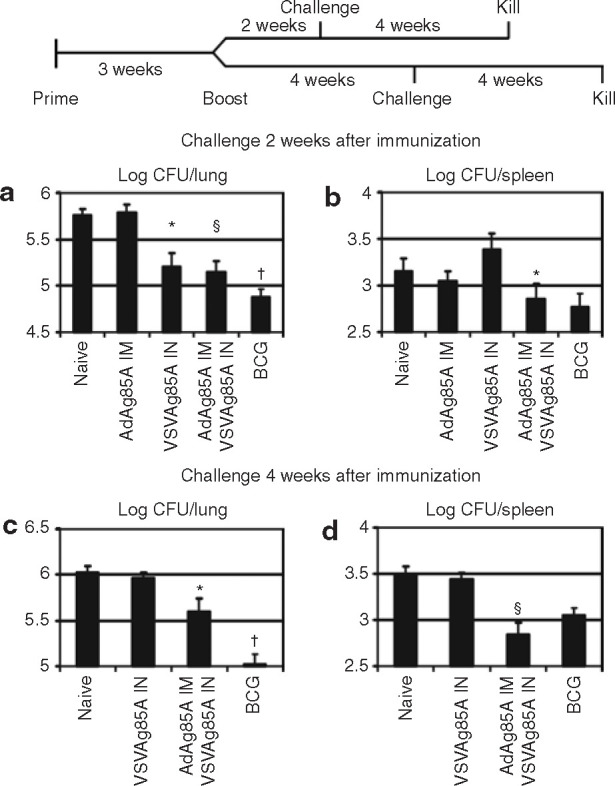
Enhanced immune protection conferred by intramuscular (IM) AdAg85A–intranasal (IN) VSVAg85A prime-and-boost regimen. Female Balb/c mice were immunized either subcutaneously with 1 × 105 colony forming units (CFU) bacille Calmette-Guérin (BCG) or IM with 1 × 107 plaque forming units (PFU) AdAg85A. Three weeks later, the latter group was boosted IN with 1 × 107 PFU VSVAg85A. At the same time-point, a fresh group was immunized IN with 1 × 107 PFU VSVAg85A alone. At 2 weeks [(a)/(b)] or at 4 weeks [(c)/(d)] after immunization, the mice were challenged with 15,000 CFU of the Mycobacterium tuberculosis H37Rv strain. At 4 weeks after the challenge, the mice were killed, and (a)/(b) lungs and (c)/(d) spleens were analyzed for CFU counts. The data are shown as mean values ± SEM of five to seven mice per group. *P ≤ 0.05; §P ≤ 0.01; †P ≤ 0.001. VSV, vesicular stomatitis virus.
Discussion
In comparison with several other viral vector systems utilized for TB vaccination, VSV represents a more attractive candidate because of the low incidence of antibodies to VSV in the general population and the rare occurrence of adverse outcomes following infection in humans.14 On the basis of these considerations, VSV has been used for generating a number of effective antiviral vaccines that mediate immunity by inducing neutralizing antibodies.18 , 19 , 20 , 21 , 22 More recently, VSV-vectored vaccines have been described for immunization against malaria29 and Yersinia pestis infection.30 However, VSV has not been used for vaccination against M.tb and therefore this study represents the first attempt to develop a recombinant VSV TB vaccine. Additionally, an understanding of T-cell activation after VSV-vectored vaccination is yet to be fully established. Our results suggest that VSV can be used as an effective TB booster vaccine in a heterologous prime-and-boost regimen, although its protective efficacy is limited when used as a stand-alone vaccine.
Although VSV causes a self-limiting infection with very mild tissue inflammatory responses in many species including humans, VSV infection can be pathogenic in mice, particularly in Balb/c mice.22 , 23 , 31 This poses a limitation to vaccine development using VSV in murine models and calls for attenuation of the virus. While a variety of methods have been reported for attenuating VSV,16 , 17 , 22 , 32 we employed a VSV harboring a mutant viral matrix protein that substantially attenuated the virus. In addition, this mutant VSV had an enhanced capability to induce an antiviral IFN response in host cells which would otherwise be subverted during wild-type VSV infection.33 This feature is believed to promote the downstream adaptive immune response through cytokine production and major histocompatibility complex class I upregulation,34 and was thus exploited as a built-in immune adjuvant in our current study. On the other hand, such strong immune adjuvant activity will enhance not only the immunogenicity of the heterologous antigen but also the immune response to VSV antigens. This may limit repeated applications of VSV vector in the same host, as is the case for adenoviral vector systems. We therefore believe that the VSV vector holds great potential to be used as a heterologous vector for boosting immune responses triggered by prime immunization with virus vectors such as Ad. Admittedly, the VSV used in our study is a replicating virus that is unlikely to be amenable to clinical application. We are developing replication-deficient VSV TB vaccines.
Protection against TB is associated with a strong T cell–mediated immune response, and therefore examination of the cellular responses after immunization with VSVAg85A is an important part of investigation. After IM delivery of VSVAg85A, a primarily systemic response was detected, with antigen-specific CD8 T cells populating the spleen and the lung. This route of immunization appeared to activate more IFN-γ+ CD8 T cells. In contrast, VSVAg85A delivered IN activated more antigen-specific T cells in the airway. It is interesting to note that IFN-γ+ cells were primarily CD4 T cells. In general, the primary response to VSVAg85A peaked at 2 weeks after immunization in the lung and spleen and peaked at 1 week in the airway lumen. The distinctive distribution profiles of T cells in response to these two routes of immunization are to be expected, because systemic immunization generates a systemic response whereas mucosal immunization leads to a mucosal response. This finding is in agreement with data from our earlier studies using a recombinant adenoviral TB vaccine.27 The difference in the IFN-γ+ T-cell profiles may be because of the fact that VSV is naturally tropic for the respiratory mucosa and can better infect epithelial cells leading to increased secretion of Ag85A which, in turn, may result in increased CD4 T-cell activation.
The majority of VSV-vectored vaccines developed to date have been designed to elicit antiviral antibody responses. After mucosal delivery of VSV-vectored vaccine, the induction of mucosal and systemic neutralizing antibodies correlated with protection against challenge.18 , 20 , 21 , 22 , 35 , 36 , 37 A recent study with a VSV-vectored vaccine against Y. pestis also emphasized the induction of high antibody titers although the mechanisms of protection remain to be elucidated.30 Studies relating to human immunodeficiency virus were the first to explore the activation of systemic CD8 T cells after mucosal immunization with a VSV-vectored vaccine.16 , 32 , 36 , 38 , 39 Another study observed the induction of a systemic CD4 T-cell response after intranasal immunization with a VSV HSV-2 vaccine.19 In all of these studies, the examination of T-cell responses was restricted to systemic compartments. We are the first to examine the mucosal T-cell immune responses after VSV-vectored immunization.
We found that, despite the modest T-cell response induced by single IN immunization with VSVAg85A, when mice were challenged 2 weeks later with virulent M.tb they exhibited ∼0.5 log protection in the lung as compared to the naive control. The difference between the levels of protection afforded by IN administered VSVAg85A and BCG was not significant. In contrast, IM immunization with the vaccine was unable to protect mice from M.tb challenge. We have previously observed that IM delivery of an adenoviral vector expressing Ag85A (AdAg85A) was also unable to provide significant protection.27 The protection observed after mucosal immunization is probably mediated by the increased cellular responses in the airway lumen; we have earlier shown that this is a good correlate of protection.27 , 28 However, when mice were challenged 4 weeks after immunization, the protection conferred by a single IN delivery of VSVAg85A had disappeared. This is probably because of the waning cellular responses in the airway lumen; by 3 weeks, IFN-γ+T cells were at almost undetectable levels, although minimal numbers of antigen-specific tetramer+ CD8 T cells could still be observed.
The relatively low level of T-cell responses and transient protection conferred by a single IN VSVAg85A immunization is reminiscent of another viral vaccine. This viral vaccine is an modified vaccinia virus Ankara vector expressing Ag85A (MVA85A), and this vector too induces very modest T-cell responses when used as a single delivery either mucosally or parenterally, and indeed, even after two successive intravenous administrations.40 , 41 However, MVA85A had a potent boosting effect on T-cell responses induced by heterologous priming vaccination.9 , 40 , 41 This fact prompted us to continue to analyze VSVAg85A as a boost vaccine. We investigated whether an IN boost with VSVAg85A in IM administered AdAg85A-primed mice would boost memory T cells within the airway lumen and enhance protection. Indeed, the antigen-specific T cells were profoundly boosted in the airway lumen and lung, and to a lesser extent in the spleen. Upon challenge with M.tb, these mice displayed ∼0.5 log protection in the lung as compared to that in naive controls. However, the level of protection in the lung conferred by this prime-and-boost regimen is not as good as that conferred by parenteral BCG immunization, although the level of systemic protection in the spleen provided by these two regimens is comparable. This observation supports the continuing use of BCG as a priming vaccine and the use of virus-based vaccine for boosting. Nevertheless, virus-based TB vaccines have potential in immunizing individuals who cannot qualify for BCG immunization, such as human immunodeficiency virus–infected children.42 Further, it may be argued that multiple heterologous booster vaccines may be required to maintain protective immunity against M.tb throughout a human's lifetime. The demonstrated ability of AdAg85A to boost a BCG prime immunization,12 and of VSVAg85A to boost immunity conferred by AdAg85A, lend support for the adoption of an immunization regimen of BCG prime and repeated Ad/VSV boost.
Materials and Methods
Mice. Six- to ten-week-old female Balb/c mice were purchased from Harlan Laboratory. They were housed in a specific pathogen-free level B facility. All experiments were carried out in accordance with the Animal Research Ethics Board at McMaster University.
Construction of recombinant AdAg85A and VSVAg85A vaccines. A recombinant replication-defective Ad expressing the M.tb Ag85A was generated and purified as described earlier.6 In order to construct VSVAg85A, Ag85A linked to the human tPA signal peptide (tPA-Ag85A) was PCR-amplified from the shuttle plasmid pJW23, using the forward primer 5′-ATTGTCGACTAGAACTAGTGAATTTAG-3′ so as to introduce an upstream SalI restriction site, and the reverse primer 5′-ATTGCTAGCGGATCATCGGAGCTA-3′ so as to introduce a downstream NheI restriction site (sites underlined). The PCR product was digested with SalI and NheI, purified, and ligated into the pVSV-XNDG plasmid23 digested with XhoI and NheI so as to create pVSV-XNDG tPA-Ag85A. Following transformation into DH5-α Escherichia coli cells, the recombinant plasmid was recovered, analyzed, and sequenced (MOBIX, McMaster University) in order to verify its correctness. After large-scale purification, the plasmid was co-transfected with helper plasmids so as to rescue VSVAg85A virus. The virus was subsequently amplified, purified, and titered in accordance with standard protocols.43 , 44
Immunoblotting. In order to verify protein expression after VSVAg85A infection, A549 cells were infected with the virus at a multiplicity of infection of 0.1. Additional cells were infected with AdAg85A at a multiplicity of infection of 20. At time-points of 24 and 48 hours after infection, respectively, cell lysates from the two batches were harvested in sodium dodecyl sulfate-polyacrylamide gel electrophoresis loading buffer (50 mmol/l Tris–Cl pH 6.8, 100 mmol/l dithiothreitol, 2% sodium dodecyl sulfate, 0.1% bromophenol blue, and 10% glycerol). Together with Ag85 complex, the samples were run on a 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel, transferred to a nitrocellulose membrane, and probed with a monoclonal Ag85A antibody (clone TD-17) (1:1,000). The membrane was incubated with a horseradish peroxidase–conjugated secondary antibody (1:5,000) and protein was detected using ECL on film (Kodak). The membrane was subsequently incubated with a polyclonal VSV antibody (1:5,000) followed by incubation with an Alexa Fluor 680–conjugated secondary antibody (1:10,000), and protein was detected using the Odyssey machine.
Immunization. Both VSVAg85A and AdAg85A were prepared in phosphate-buffered saline at doses of 5 × 106 PFU, or 1 × 107 PFU, or 5 × 107 PFU as indicated, and were delivered through either IN or IM routes. For IM immunization, the virus was prepared in a 100 μl volume. The mice were lightly anesthetized and 50 μl of the inoculum was administered into each of the two quadriceps. For IN immunization, the virus was prepared in a 25 μl volume. The mice were lightly anesthetized and were allowed to inhale the volume of virus administered slowly with a pipette tip. For challenge experiments, the mice were immunized with BCG (Connaught strain). BCG was prepared (described in a later section) at a dose of 1 × 105 colony forming units in 100 μl of phosphate-buffered saline. The mice were lightly anesthetized and 50 μl of the innoculum was administered into each of the two hind quarters.
Lung histology. After being isolated, the lungs were submerged in 10% formalin to fix the tissue. The tissue was processed, sectioned, and stained with hematoxylin and eosin. Tissue samples were viewed under ×10 and ×20 magnification.
Lymphocyte isolation and Ag stimulation. The mice were killed after immunization in order to examine immunogenicity. The spleen and lung were isolated and processed as previously described.27 After single-cell suspensions were generated, both samples were incubated in red blood cell lysis buffer for 12 minutes (R&D Systems, Minneapolis, MN). Airway luminal cells were isolated from the lung after exhaustive lavage as described earlier.27 Cells from each tissue were enumerated, resuspended in complete Roswell Park Memorial Institute 1640, and seeded into 96-well plates. For ex vivo antigen stimulation, cells were plated with either Ag85A-specific CD4 (LTSELPGWLQANRHVKPTGS) or CD8 (MPVGGQSST) T-cell peptides at a concentration of 1 μg/well.
Enzyme-linked immunosorbent spot assay. Splenocytes were examined in order to determine the frequency of occurrence of IFN-γ-secreting lymphocytes as described earlier.45 0.5 × 106 Splenocytes were plated in duplicate with either no stimulus, or CD4, or CD8 peptide for 24 hours at 37 °C. Spots were enumerated using the ELISPOT counter and ImmunoSpot software.
ICCS, tetramer staining, and fluorescence-activated cell sorting. Immunostaining was carried out in order to determine the presence of antigen-specific cells and cytokine-secreting effectors, as described earlier.27 Two million spleen and lung cells were seeded in a 96-well plate, while airway luminal cells were divided equally into wells for each condition that required examination. For ICCS, the cells were cultured with no stimulus, or with CD4, or with CD8 peptide (as described in the earlier section) for 5 hours. The antibodies used for staining for ICCS were: CD3 CyChrome, CD4 phycoerythrin-Cy7, CD8a phycoerythrin, and IFN-γ allophycocyanin (BD Pharmingen). For tetramer staining, unstimulated cells were stained with the immunodominant CD8 T-cell peptide (MPVGGQSST) of Ag85A bound to the BALB/c major histocompatibility complex class I allele H-2Ld conjugated to phycoerythrin (from Texas A&M University), CD3 CyChrome, and CD8a allophycocyanin (BD Pharmingen, San Diego, CA). All the stained cells were run on the LSRII flow cytometer, and 100,000–300,000 events were collected per sample, depending on the tissue. fluorescence-activated cell sorting data were analyzed using FlowJo software (Tree Star, Ashland, OR).
Mycobacterium culture, challenge, and plaque forming assay. Mycobacterium bovis BCG (Connaught strain) and M.tb (H37Rv strain) were cultured and prepared as described earlier.27 For the challenge experiments, the mice were infected with 10,000 colony forming units of M.tb IN in the level III containment facility at McMaster University. The bacterial burden in the lungs and spleen was assessed, and is presented as log10 colony forming units per organ, as described earlier.27
Statistics. Statistical analysis was carried out using an unpaired Student's t-test, and was verified using analysis of variance and Tukey's test. The difference was considered significant when P ≤ 0.05.
Acknowledgments
The authors are grateful to Anna Zganiacz, Duncan Chong, Xueya Feng, and Steve Hanson for their invaluable technical assistance. We also thank John Rose for providing us with the VSV genomic plasmid DNA, and Jonathan Bramson for scientific suggestions. Funding for this research was provided by Canadian Institutes of Health Research. The authors have no financial conflict of interest.
Footnotes
published online 25 March 2008
References
- 1.World Health Organization . Tuberculosis facts. World Health Organization; Geneva: 2007. [Google Scholar]
- 2.Flynn JL, Chan J. Immunology of tuberculosis. Annu Rev Immunol. 2001;19:93–129. doi: 10.1146/annurev.immunol.19.1.93. [DOI] [PubMed] [Google Scholar]
- 3.Andersen P, Doherty TM. The success and failure of BCG—implications for a novel tuberculosis vaccine. Nat Rev Microbiol. 2005;3:656–662. doi: 10.1038/nrmicro1211. [DOI] [PubMed] [Google Scholar]
- 4.World Health Organization BCG vaccine. WHO position paper. Wkly Epidemiol Rec. 2004;79:27–38. [PubMed] [Google Scholar]
- 5.Brewer TF. Preventing tuberculosis with bacillus Calmette-Guérin vaccine: a meta-analysis of the literature. Clin Infect Dis. 2000;31(suppl. 3):S64–S67. doi: 10.1086/314072. [DOI] [PubMed] [Google Scholar]
- 6.Wang J, Thorson L, Stokes RW, Santosuosso M, Huygen K, Zganiacz A. Single mucosal, but not parenteral, immunization with recombinant adenoviral-based vaccine provides potent protection from pulmonary tuberculosis. J Immunol. 2004;173:6357–6365. doi: 10.4049/jimmunol.173.10.6357. [DOI] [PubMed] [Google Scholar]
- 7.Bennekov T, Dietrich J, Rosenkrands I, Stryhn A, Doherty TM, Andersen P. Alteration of epitope recognition pattern in Ag85B and ESAT-6 has a profound influence on vaccine-induced protection against Mycobacterium tuberculosis. Eur J Immunol. 2006;36:3346–3355. doi: 10.1002/eji.200636128. [DOI] [PubMed] [Google Scholar]
- 8.McShane H, Brookes R, Gilbert SC, Hill AV. Enhanced immunogenicity of CD4+ T-cell responses and protective efficacy of a DNA-modified vaccinia virus Ankara prime-boost vaccination regimen for murine tuberculosis. Infect Immun. 2001;69:681–686. doi: 10.1128/IAI.69.2.681-686.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vordermeier HM, Rhodes SG, Dean G, Goonetilleke N, Huygen K, Hill AV. Cellular immune responses induced in cattle by heterologous prime-boost vaccination using recombinant viruses and bacille Calmette-Guérin. Immunology. 2004;112:461–470. doi: 10.1111/j.1365-2567.2004.01903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams A, Goonetilleke NP, McShane H, Clark SO, Hatch G, Gilbert SC. Boosting with poxviruses enhances Mycobacterium bovis BCG efficacy against tuberculosis in guinea pigs. Infect Immun. 2005;73:3814–3816. doi: 10.1128/IAI.73.6.3814-3816.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stukova MA, Sereinig S, Zabolotnyh NV, Ferko B, Kittel C, Romonova J. Vaccine potential of influenza vectors expressing Mycobacterium tuberculosis ESAT-6 protein. Tuberculosis (Edinb) 2006;86:236–246. doi: 10.1016/j.tube.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 12.Santosuosso M, McCormick S, Zhang X, Zganiacz A, Xing Z. Intranasal boosting with an adenovirus-vectored vaccine markedly enhances protection by parenteral Mycobacterium bovis BCG immunization against pulmonary tuberculosis. Infect Immun. 2006;74:4634–4643. doi: 10.1128/IAI.00517-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McShane H, Pathan AA, Sander CR, Goonetilleke NP, Fletcher HA, Hill AV. Boosting BCG with MVA85A: the first candidate subunit vaccine for tuberculosis in clinical trials. Tuberculosis (Edinb) 2005;85:47–52. doi: 10.1016/j.tube.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 14.Xing Z, Lichty BD. Use of recombinant virus-vectored tuberculosis vaccines for respiratory mucosal immunization. Tuberculosis (Edinb) 2006;86:211–217. doi: 10.1016/j.tube.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 15.Lichty BD, Power AT, Stojdl DF, Bell JC. Vesicular stomatitis virus: re-inventing the bullet. Trends Mol Med. 2004;10:210–216. doi: 10.1016/j.molmed.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 16.Publicover J, Ramsburg E, Rose JK. A single-cycle vaccine vector based on vesicular stomatitis virus can induce immune responses comparable to those generated by a replication-competent vector. J Virol. 2005;79:13231–13238. doi: 10.1128/JVI.79.21.13231-13238.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flanagan EB, Zamparo JM, Ball LA, Rodriguez LL, Wertz GW. Rearrangement of the genes of vesicular stomatitis virus eliminates clinical disease in the natural host: new strategy for vaccine development. J Virol. 2001;75:6107–6114. doi: 10.1128/JVI.75.13.6107-6114.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang P, Liu Y, Yin X, Yuan F, Nie Y, Luo M. Elicitation of neutralizing antibodies by intranasal administration of recombinant vesicular stomatitis virus expressing human immunodeficiency virus type 1 gp120. Biochem Biophys Res Commun. 2006;339:526–532. doi: 10.1016/j.bbrc.2005.11.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Natuk RJ, Cooper D, Guo M, Calderon P, Wright KJ, Nasar F. Recombinant vesicular stomatitis virus vectors expressing herpes simplex virus type 2 gD elicit robust CD4+ Th1 immune responses and are protective in mouse and guinea pig models of vaginal challenge. J Virol. 2006;80:4447–4457. doi: 10.1128/JVI.80.9.4447-4457.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reuter JD, Vivas-Gonzalez BE, Gomez D, Wilson JH, Brandsma JL, Greenstone HL. Intranasal vaccination with a recombinant vesicular stomatitis virus expressing cottontail rabbit papillomavirus L1 protein provides complete protection against papillomavirus-induced disease. J Virol. 2002;76:8900–8909. doi: 10.1128/JVI.76.17.8900-8909.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kapadia SU, Rose JK, Lamirande E, Vogel L, Subbarao K, Roberts A. Long-term protection from SARS coronavirus infection conferred by a single immunization with an attenuated VSV-based vaccine. Virology. 2005;340:174–182. doi: 10.1016/j.virol.2005.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roberts A, Buonocore L, Price R, Forman J, Rose JK. Attenuated vesicular stomatitis viruses as vaccine vectors. J Virol. 1999;73:3723–3732. doi: 10.1128/jvi.73.5.3723-3732.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stojdl DF, Lichty BD, tenOever BR, Paterson JM, Power AT, Knowles S. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4:263–275. doi: 10.1016/s1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- 24.Antas PR, Cardoso FL, Pereira KC, Franken KL, Cunha KS, Klatser P. T cell immune responses to mycobacterial antigens in Brazilian tuberculosis patients and controls. Trans R Soc Trop Med Hyg. 2005;99:699–707. doi: 10.1016/j.trstmh.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 25.Belisle JT, Vissa VD, Sievert T, Takayama K, Brennan PJ, Besra GS. Role of the major antigen of Mycobacterium tuberculosis in cell wall biogenesis. Science. 1997;276:1420–1422. doi: 10.1126/science.276.5317.1420. [DOI] [PubMed] [Google Scholar]
- 26.Li Z, Howard A, Kelley C, Delogu G, Collins F, Morris S. Immunogenicity of DNA vaccines expressing tuberculosis proteins fused to tissue plasminogen activator signal sequences. Infect Immun. 1999;67:4780–4786. doi: 10.1128/iai.67.9.4780-4786.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Santosuosso M, Zhang X, McCormick S, Wang J, Hitt M, Xing Z. Mechanisms of mucosal and parenteral tuberculosis vaccinations: adenoviral-based mucosal immunization preferentially elicits sustained accumulation of immune protective CD4 and CD8 T cells within the airway lumen. J Immunol. 2005;174:7986–7994. doi: 10.4049/jimmunol.174.12.7986. [DOI] [PubMed] [Google Scholar]
- 28.Santosuosso M, McCormick S, Roediger E, Zhang X, Zganiacz A, Lichty BD. Mucosal luminal manipulation of T cell geography switches on protective efficacy by otherwise ineffective parenteral genetic immunization. J Immunol. 2007;178:2387–2395. doi: 10.4049/jimmunol.178.4.2387. [DOI] [PubMed] [Google Scholar]
- 29.Li S, Locke E, Bruder J, Clarke D, Doolan DL, Havenga MJ. Viral vectors for malaria vaccine development. Vaccine. 2007;25:2567–2574. doi: 10.1016/j.vaccine.2006.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palin A, Chattopadhyay A, Park S, Delmas G, Suresh R, Senina S. An optimized vaccine vector based on recombinant vesicular stomatitis virus gives high-level, long-term protection against Yersinia pestis challenge. Vaccine. 2007;25:741–750. doi: 10.1016/j.vaccine.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 31.Roberts A, Kretzschmar E, Perkins AS, Forman J, Price R, Buonocore L. Vaccination with a recombinant vesicular stomatitis virus expressing an influenza virus hemagglutinin provides complete protection from influenza virus challenge. J Virol. 1998;72:4704–4711. doi: 10.1128/jvi.72.6.4704-4711.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Publicover J, Ramsburg E, Rose JK. Characterization of nonpathogenic, live, viral vaccine vectors inducing potent cellular immune responses. J Virol. 2004;78:9317–9324. doi: 10.1128/JVI.78.17.9317-9324.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hengel H, Koszinowski UH, Conzelmann KK. Viruses know it all: new insights into IFN networks. Trends Immunol. 2005;26:396–401. doi: 10.1016/j.it.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 34.Galligan CL, Murooka TT, Rahbar R, Baig E, Majchrzak-Kita B, Fish EN. Interferons and viruses: signaling for supremacy. Immunol Res. 2006;35:27–40. doi: 10.1385/IR:35:1:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rose NF, Roberts A, Buonocore L, Rose JK. Glycoprotein exchange vectors based on vesicular stomatitis virus allow effective boosting and generation of neutralizing antibodies to a primary isolate of human immunodeficiency virus type 1. J Virol. 2000;74:10903–10910. doi: 10.1128/jvi.74.23.10903-10910.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Egan MA, Chong SY, Rose NF, Megati S, Lopez KJ, Schadeck EB. Immunogenicity of attenuated vesicular stomatitis virus vectors expressing HIV type 1 Env and SIV Gag proteins: comparison of intranasal and intramuscular vaccination routes. AIDS Res Hum Retroviruses. 2004;20:989–1004. doi: 10.1089/aid.2004.20.989. [DOI] [PubMed] [Google Scholar]
- 37.Schlereth B, Buonocore L, Tietz A, Meulen Vt V, Rose JK, Niewiesk S. Successful mucosal immunization of cotton rats in the presence of measles virus-specific antibodies depends on degree of attenuation of vaccine vector and virus dose. J Gen Virol. 2003;84:2145–2151. doi: 10.1099/vir.0.19050-0. [DOI] [PubMed] [Google Scholar]
- 38.Rose NF, Marx PA, Luckay A, Nixon DF, Moretto WJ, Donahoe SM. An effective AIDS vaccine based on live attenuated vesicular stomatitis virus recombinants. Cell. 2001;106:539–549. doi: 10.1016/s0092-8674(01)00482-2. [DOI] [PubMed] [Google Scholar]
- 39.Haglund K, Leiner I, Kerksiek K, Buonocore L, Pamer E, Rose JK. High-level primary CD8+ T-cell response to human immunodeficiency virus type 1 gag and env generated by vaccination with recombinant vesicular stomatitis viruses. J Virol. 2002;76:2730–2738. doi: 10.1128/JVI.76.6.2730-2738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goonetilleke NP, McShane H, Hannan CM, Anderson RJ, Brookes RH, Hill AV. Enhanced immunogenicity and protective efficacy against Mycobacterium tuberculosis of bacille Calmette-Guérin vaccine using mucosal administration and boosting with a recombinant modified vaccinia virus Ankara. J Immunol. 2003;171:1602–1609. doi: 10.4049/jimmunol.171.3.1602. [DOI] [PubMed] [Google Scholar]
- 41.McShane H, Behboudi S, Goonetilleke N, Brookes R, Hill AV. Protective immunity against Mycobacterium tuberculosis induced by dendritic cells pulsed with both CD8+- and CD4+-T-cell epitopes from antigen 85A. Infect Immun. 2002;70:1623–1626. doi: 10.1128/IAI.70.3.1623-1626.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hesseling AC, Marais BJ, Gie RP, Schaaf HS, Fine PE, Godfrey-Faussett P. The risk of disseminated Bacille Calmette-Guerin (BCG) disease in HIV-infected children. Vaccine. 2007;25:14–18. doi: 10.1016/j.vaccine.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 43.Lawson ND, Stillman EA, Whitt MA, Rose JK. Recombinant vesicular stomatitis viruses from DNA. Proc Natl Acad Sci USA. 1995;92:4477–4481. doi: 10.1073/pnas.92.10.4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schnell MJ, Buonocore L, Whitt MA, Rose JK. The minimal conserved transcription stop-start signal promotes stable expression of a foreign gene in vesicular stomatitis virus. J Virol. 1996;70:2318–2323. doi: 10.1128/jvi.70.4.2318-2323.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zganiacz A, Santosuosso M, Wang J, Yang T, Chen L, Anzulovic M. TNF-α is a critical negative regulator of type 1 immune activation during intracellular bacterial infection. J Clin Invest. 2004;113:401–413. doi: 10.1172/JCI18991. [DOI] [PMC free article] [PubMed] [Google Scholar]