

Abstract
The Werner syndrome helicase/3′-exonuclease (WRN) is a major component of the DNA repair and replication machinery. To analyze whether WRN is involved in the repair of topoisomerase-induced DNA damage we utilized U2-OS cells, in which WRN is stably down-regulated (wrn-kd), and the corresponding wild-type cells (wrn-wt). We show that cells not expressing WRN are hypersensitive to the toxic effect of the topoisomerase I inhibitor topotecan, but not to the topoisomerase II inhibitor etoposide. This was shown by mass survival assays, colony formation and induction of apoptosis. Upon topotecan treatment WRN deficient cells showed enhanced DNA replication inhibition and S-phase arrest, whereas after treatment with etoposide they showed the same cell cycle response as the wild-type. A considerable difference between WRN and wild-type cells was also observed for DNA single-and double-strand break formation in response to topotecan. Topotecan induced most DNA single-strand breaks 6 h after treatment. In both wrn-wt and wrn-kd cells these breaks were repaired at similar kinetics. However, in wrn-kd but not wrn-wt cells they were converted into DNA double-strand breaks (DSBs) at high frequency, as shown by neutral comet assay and phosphorylation of H2AX. Our data provide evidence that WRN is involved in the repair of topoisomerase I, but not topoisomerase II-induced DNA damage, most likely via preventing the conversion of DNA single-strand breaks into DSBs during the resolution of stalled replication forks at topo I–DNA complexes. We suggest that the WRN status of tumor cells impacts anticancer therapy with topoisomerase I, but not topoisomerase II inhibitors.
Keywords: Topoisomerase inhibition, Camptothecin, Topotecan, Etoposide, WRN, DNA breaks, Apoptosis
1. Introduction
The anticancer drugs topotecan (TPT) and etoposide (ETO) belong to the family of topoisomerase I (topo I) and topoisomerase II (topo II) inhibitors, respectively [1]. Both drugs are widely used in the therapy of different types of cancer. Topoisomerase I and II have different modes of action, and specific topoisomerase inhibitors induce different biological responses. The mechanisms and molecular determinants of tumor cell response to topo I inhibitors like camptothecin (the parent compound derived from Camptothecia acuminata, from which TPT and irinotecan were derived) have been reviewed in great detail [2]. The catalytic cycle of topo I starts with the formation of a DNA single-strand break (SSB). During this incision, topo I is covalently bound to DNA, forming the so-called topo I–DNA cleavable complex [3]. Topo I inhibitors like TPT stabilize this complex and prevent the religation of topo I mediated SSBs [4,5]. Crystal structure analysis revealed that topotecan and camptothecin mimic a DNA base pair and bind at the site of DNA cleavage by intercalating between the upstream (−1) and downstream (+1) base pairs. This intercalation displaces downstream DNA, thereby preventing religation of the cleaved strand [6,7]. This block to religation of SSBs gives rise to accumulation of topo I–DNA covalent complexes [5]. The cytotoxic mechanism of topo I inhibitors is mainly S-phase dependent [8–11]. After formation of topo I-generated SSBs and in the presence of the inhibitor, a collision between the replication fork and the irreversible cleavable complex occurs, resulting in blockage of replication fork movement and the formation of DNA double-strand breaks (DSBs). In a subsequent reaction, these DSBs induce a checkpoint response, by activation of ATR and DNA-PK, and subsequent phosphorylation of H2AX [12]. Phosphorylated H2AX (γH2AX) recruits different repair proteins like the Mre11/Rad50/Nbs1 complex, thereby activating DSB repair.
The catalytic cycle of topo II starts with the formation of a DSB followed by its religation. Topo II is also covalently bound to the DNA, forming a reversible covalent intermediate. Topo II inhibitors like ETO block topo II mediated religation and lead directly to the accumulation of DSBs (for review see [13]). Therefore, the main repair mechanism thought to be related to topo II inhibitor mediated DNA damage is DSB repair.
The molecular mechanism controlling the repair of topo I–DNA covalent complexes and its impact on cellular sensitivity upon TPT treatment is less well understood. In previous experiments, using non-transformed mouse embryonic fibroblasts (MEFs) and glioblastoma cell lines proficient or deficient for p53, we showed that p53 deficient cells are significantly more sensitive to TPT than their p53 wild-type isogenic counterpart [14]. Therefore, p53 is a factor determining the sensitivity of cells to topo I poisons. A second mechanism obviously involved in the repair of TPT induced DNA damage is DSB repair. The two major pathways of DSB repair are non-homologous end joining (NHEJ) and homologous recombination (HR) (for review see [15,16]). Topo II inhibitor mediated DNA damage is mainly repaired by NHEJ since lig4−/− and ku70−/− cells are extremely sensitive to topo II inhibitors compared to wild-type or rad54−/− cells [17–19]. The data concerning topo I inhibitor mediated DNA damage are less clear. Several studies have shown that cells deficient in either HR or NHEJ are more sensitive to camptothecin (CPT) than their respective controls, and that HR is likely the most prominent pathway employed in CPT-induced DNA damage repair [20,21]. However, these studies used non-isogenic systems that limit the value of the observations.
Since the formation of DSBs following topo I inhibition is dependent on collision of the cleavable complex with the replication fork, the question arises whether DNA helicases, which play an important role during DNA replication and DNA repair, are involved in the repair of topoisomerase inhibitor-induced DNA damage. One of these DNA helicases is the Werner syndrome helicase/3′-exonuclease (WRN), a member of the RecQ DNA helicase family, which posses DNA helicase and exonuclease activity. WRN is a major component of the DNA repair and replication machinery, and has been shown to interact with the DNA repair proteins DNA polymerase delta, p53, RPA, PCNA, FEN1, DNA topoisomerase I, BLM helicase, PARP-1, RAD52 and the DNA-PKcs/Ku70/Ku80 complex (for review see [22,23]). It also impacts cell killing following CPT treatment [24–26]. Whether the WRN helicase is involved in the repair of topo I or topo II inhibitor-induced DNA damage is not known. Although interplay between topo I and WRN has been reported and some early publications are available on WRN and resistance against topo II poisons, there is no systematic analysis of the role of WRN in the sensitivity against topo I and topo II poisons in human cancer cells. Here, we utilized human U2-OS osteosarcoma cells stably transfected with siRNA specific for the wrn gene. Using this isogenic cell system, we studied for the first time comparatively the role of WRN in cellular resistance to topo I and topo II inhibitors and its involvement in the repair or processing of topo I inhibitor induced DNA damage.
2. Materials and methods
2.1. Cell lines
The human osteosarcoma cell lines used (wrn-wt and wrn-kd) were described previously [27,28]. The cells were grown in Dulbecco’s minimal essential medium (DMEM) containing 10% fetal bovine serum (FBS), in 7% CO2 at 37 °C.
2.2. Reagents
Topotecan (Hycamtin) and etoposide (VP-16) were obtained from and prepared to stock solution (1 mg/ml) by the pharmacy of the Clinical Center of the Johannes Gutenberg University of Mainz, Germany.
2.3. WST-1 viability assay
Cells (up to 104) were seeded in 96-well plates and 36 h later, being in exponentially growing phase, treated with different concentrations of TPT for 72 h. WST-1 reagent was added for colorimetric reaction (measurement of metabolizing activity) up to a final concentration of 10% (v/v). Plates were incubated up to 2 h at 37 °C. The extinction was measured on an ELISA reader at 450 nm. The data are the mean of three independent experiments performed in triplicates.
2.4. Colony forming assay
Cells from exponentially growing cultures were seeded on 6-cm dishes and treated 12 h later with the anticancer drug. Colonies were fixed with methanol 7 days after seeding and stained with crystal violet. Relative cell survival was determined from the number of colonies after treatment and the number of colonies on untreated control plates, and expressed as percentage of survivors of the non-treated control.
2.5. Determination of apoptosis and cell cycle analysis
For monitoring drug-induced apoptosis and progression of cells through the cell cycle, ethanol-fixed cells were stained with propidium iodide (PI). The G1, S, G2 and the sub-G1 fraction were determined by flow cytometry. The protocol was performed as described [29,30]. In brief, cells were continuously treated with TPT and after different time points fixed with 70% ethanol, incubated with 0.1 mg/ml RNase in PBS for 1 h and stained with PI (25 μg/ml) prior to flow cytometry (CellQuestPro, BD, Heidelberg, Germany).
2.6. BrdU incorporation
Cells were cultured in DMEM (10% FBS) and, after exposure to topotecan or etoposide, the thymidine analogue BrdU (10 μM) was added to the medium. After 1 h-incubation, the incorporation was analyzed using a BrdU Incorporation Kit (Roche) in a microplate reader.
2.7. Preparation of cell extracts and western analysis
Whole-cell extracts for the detection of H2AX phosphorylation were prepared by direct lysis of the cells in to 95 °C pre-heated 1× loading buffer (Roti®-Load 1, Roth, Karlsruhe, Germany). Samples of 10 μl protein extract were separated in 10% SDS-PAGE and electro-blotted onto nitrocellulose membranes, which were then incubated with antibodies as described [31]. Monoclonal mouse anti-γH2AX antibodies (Upstate, Lake Placid, USA Cat. 05-636) were diluted 1:500 in 5% non-fat dry milk, 0.2% Tween/TBS and incubated overnight at 4 °C. Polyclonal rabbit anti-ERK2 antibodies (Santa Cruz Biotechnology) were diluted 1:3000 and incubated for 2 h in 5% non-fat dry milk, 0.1% Tween/TBS at RT. The protein-antibody complexes were visualized by ECL (Amersham). For immunoblotting with anti-topo I antibody, untreated and TPT-treated cells were either directly lysed in 1× loading buffer (band depletion assay) or rinsed up to 5× with culture medium to get rid of the TPT, and incubated in fresh TPT-free medium for 30 min (for reversal of the cleavable complex and monitoring total cellular levels of topo I) prior to direct lysis in 1× loading buffer and sonification. Proteins were transferred onto nitrocellulose membrane and polyclonal mouse anti-topo1 antibodies (Santa Cruz Biotechnology, H300) were diluted 1:1000 in 5% non-fat dry milk, 0.2% Tween/TBS and incubated overnight at 4 °C.
2.8. Preparation of RNA and RT-PCR
Total RNA was isolated using the RNA II Isolation Kit from Machery and Nagel. One microgram RNA was transcribed into cDNA using the Reverse-iT 1st Strand Synthesis Kit (Abgene) in a final volume of 50 μl. 3 μl were subjected to RT-PCR which was performed by the use of specific primers (MWG Biotechnology) and Ampliqon III Taq DNA Polymerase Master Mix Red (Bie & Bernsen. A-S, Roedovre, Denmark). The PCR program used was: 1.5 min 94 °C, [(denaturation: 45 sec, 94 °C; annealing: 1 min 55 °C; elongation: 1 min, 72 °C) 25 cycles], 10 min 72°C.
2.9. Single cell gel electrophoresis (SCGE, comet assay)
Exponentially growing cells were exposed to TPT or ETO and, after the indicated time periods, trypsinized and washed with ice-cold PBS. Alkaline and neutral cell lysis and electrophoresis was essentially performed as described previously [32]. Propidium iodide stained slides were evaluated using a fluorescence microscope and the Komet 4.0.2 software from Kinetic Imaging Ltd. (Liverpool, UK). Data were expressed as Olive Tail Moment (OTM), which represents the percentage of DNA in the tail multiplied by the length between the center of the head and tail [33].
2.10. H2AX foci formation
Wrn-wt and wrn-kd cells were seeded on cover slips. Following treatment with 1 μg/ml TPT, cells were fixed with 4% formaldehyde at different time points. A second fixation step was performed using 100% methanol (−20 °C, 20 min). Cells were then blocked in 5% BSA PBS (0.3% Triton-X100). The antibodies used were monoclonal anti-γH2AX (Upstate) and Alexa Fluor 546 (Molecular probes). Before mounting, DNA was stained with 100 nM DAPI for 15 min. Between all staining steps cells were washed three times in PBS (0.3% Triton-X100) for 5 min. Slides were mounted in anti-fade medium (Glycerol:PBS 1:1, 2.5% DABCO, pH 8.6 with HCl) and scored using a fluorescence microscope and the CellA Software from Olympus Soft Imaging Solution GmbH.
3. Results
3.1. Characterization of wrn-kd cells and sensitivity to topo inhibitors
To determine the role of the WRN DNA-helicase in the repair of topo I and II inhibitor-induced DNA damage, we compared a human U2-OS tumor cell line stably transfected with siRNA specific for the wrn gene and the corresponding parental cell line. To confirm the WRN knock-down phenotype, both cell lines were exposed to 1 μg/ml TPT, and the expression of the wrn mRNA was determined by RT-PCR. As shown in Fig. 1A, there was no expression of wrn mRNA in the wrn-kd cell line and neither in the wrn-kd nor in the wrn-wt the expression of wrn mRNA was enhanced by TPT exposure.
Fig. 1.

Characterization of wrn-kd cells and sensitivity against topo inhibitors. (A) To analyze the WRN status, wrn-wt and wrn-kd cells were exposed to 1 μg/ml TPT. At different times after exposure, cells were harvested and total RNA was isolated. 1 μg was subjected to cDNA synthesis, followed by RT-PCR with specific primers (c, non-exposed control). As internal control, gapdh was amplified. (B and C) To elucidate the role of WRN helicase in the sensitivity against TPT and ETO, wrn-wt and wrn-kd cells were exposed to different doses of TPT. Cellular viability was determined 72 h later by the metabolic WST-1 assay (B) and reproductive cell death was measured 7 days later by colony forming assay (C).
To elucidate the role of the WRN helicase in the sensitivity of cells to TPT and ETO, wrn-wt and wrn-kd cells were exposed for 72 h to the drugs. Cellular viability was determined by the metabolic WST-1 assay. As shown in Fig. 1B, wrn-kd cells were significantly more sensitive than wrn-wt cells to TPT but not ETO. Reproductive cell death (measured in colony forming assays that determine both cell death and irreversible cell cycle blockade) was clearly higher in wrn-kd cells than in the wt upon TPT, but not ETO treatment (Fig. 1C). Whereas nearly 50% of the wrn-wt cells survived a treatment with 7.5 ng/ml TPT, only 1% of the wrn-kd cells were able to form colonies under these conditions. Since the colony-forming assay is more sensitive than the WST-1 assay, lower drug concentrations were used in these experiments.
3.2. Induction of apoptosis and cell cycle blockage by TPT and ETO
To analyze the mode of cell kill in more detail, the frequency of apoptosis was determined. In accordance with data obtained with the WST assay and colony formation, wrn-kd cells were more sensitive than wrn-wt cells. They showed a higher frequency of apoptosis throughout the dose range used (Fig. 2A) and for all time points after exposure up to 96 h (Fig. 2B, left panel). In contrast to TPT, no enhanced sensitivity of wrn-kd cells was observed upon ETO treatment (Fig. 2B, right panel).
Fig. 2.

Induction of apoptosis by TPT and ETO. (A) To analyze the induction of apoptosis wrn-wt and wrn-kd cells were exposed to different concentrations of TPT. 72 h later, cells were harvested and analyzed by FACS. Apoptosis was determined as sub-G1 fraction. (B) To analyze the time dependency of apoptosis, wrn-wt and wrn-kd cells were exposed to 1 μg/ml TPT or ETO. After the indicated time points, cells were harvested and analyzed by FACS. Apoptosis was determined as sub-G1 fraction.
To elucidate the impact of TPT and ETO on cell cycle progression and replication, the cells were exposed to the drug and cell cycle progression was analyzed by flow cytometry (FACS) and DNA replication by BrdU incorporation. As shown in Fig. 3A, TPT treatment did not arrested the cells significantly in G2 while ETO led to a clear G2-arrest. While the G2-arrest observed upon ETO treatment was comparable in wrn-kd and wrn-wt cells, a differential recovery from the S-phase arrest upon TPT was observed. Thus, in wrn-wt cells the amount of cells in the S-phase reached its maximum 24 h after TPT treatment and declined thereafter, whereas wrn-kd cells did not recover from the S-phase arrest (Fig. 3B, left panel). Upon ETO exposure cell did not clearly accumulate in the S-phase, and no difference in the response of wrn-wt and wrn-kd was observed (Fig. 3B, right panel). The extent of DNA replication after TPT and ETO treatment was determined by BrdU incorporation. Corresponding to the FACS analysis, TPT induced a strong block of DNA replication (BrdU incorporation was reduced to about 25% of the control level) in both wrn-kd and wrn-wt cells. However, wrn-wt cells were able to recover from the block of replication while wrn-kd cells did not (Fig. 3C, left panel). In contrast, ETO was not as potent in blocking replication, reducing the BrdU incorporation level to about 50% of the control 24 h after treatment. Both wrn-wt and wrn-kd cells did not show significant recovery from reduced BrdU incorporation (Fig. 3C, right panel).
Fig. 3.

Cell cycle blockage induced by TPT and ETO. To analyze cell cycle progression, wrn-wt and wrn-kd cells were exposed to 1 μg/ml TPT or ETO. Upon the indicated times, cells were harvested and the distribution in G1, S and G2-phase was analyzed by FACS. (A) Percentage of cells in G2 after the addition of TPT or ETO to the culture medium. (B) Percentage of cells in S-phase after treatment with TPT or ETO. The data are the mean of at least three independent experiments. (C) DNA replication in wrn-wt and wrn-kd cells. Exponentially growing cells were treated with 1 μg/ml TPT (left panel) or ETO (right panel) and replication was analyzed by BrdU incorporation at different times after addition of the drugs to the medium. The data are the mean of at least three independent experiments.
3.3. Formation and repair of SSBs and DSBs upon TPT
To elucidate whether the hypersensitivity of wrn-kd cells to TPT is based on different induction of DNA lesions, alkaline and neutral single-cell gel electrophoresis (SCGE) experiments were conducted. Cells were exposed to TPT or ETO and harvested at different times after exposure. As shown in Fig. 4A, 2 h after TPT treatment both wrn-wt and wrn-kd cells showed a transient induction of SSBs. Despite the fact that the formation of SSBs was slightly higher in wrn-kd cells, the damage was processed equally in both cell lines during the 24 h post-incubation period. For comparison, the level of DNA strand breaks was assessed upon ETO exposure. Strand breaks increased with time and remained at a plateau up to 24 h after the beginning of treatment, with wrn-kd and wrn-wt cells responding in a similar way (Fig. 4A, right panel). To investigate whether the processing of SSBs leads to error-free repair or conversion into DSBs, neutral SCGE assays were conducted. Cells were exposed to 1 μg/ml TPT and harvested at different post-treatment times. As shown in Fig. 4B, a strong accumulation of DSBs was observed between 15 and 24 h after the beginning of TPT treatment in wrn-kd, but not wrn-wt cells. Thus, 24 h after exposure, wrn-kd cells showed a 3-fold higher relative DSB level than the wt. After ETO treatment, the DSB frequency was low and not different in wrn-wt and wrn-kd cells (not shown).
Fig. 4.

Formation and repair of SSBs and DSBs upon TPT. (A) To analyze the TPT-induced formation and repair of DNA strand breaks, wrn-wt and wrn-kd cells were exposed to 1 μg/ml TPT (left panel) or etoposide (right panel). After the indicated time points, the formation of DNA single-strand breaks was detected via alkaline SCGE. OTM, olive tail moment. Data of the mean ± S.D. of three independent experiments are shown. (B) Determination of DSBs by neutral SCGE in wrn-wt and wrn-kd cells at various times after exposure to 1 μg/ml TPT. Data of at least three independent experiments are shown. (C) To determine the amount of H2AX phosphorylation, wrn-wt and wrn-kd cells were exposed to 1 μg/ml TPT. At different times after exposure, cells were harvested, protein extracts were prepared and 25 μg were subjected to Western blot analysis. The membrane was incubated with γH2AX specific antibodies. For loading control, ERK2 was detected. To quantify the amount H2AX phosphorylation, the intensity of the strongest band was set to 100%. (D) To determine the formation of γH2AX foci, wrn-wt and wrn-kd cells were exposed to 1 μg/ml TPT. After the indicated time points, cells were fixed, and the γH2AX foci were visualized using γH2AX specific antibodies using fluorescence microscopy. For each time point, foci in 40 cells were counted and the results of three independent experiments ± S.D. is shown (left panel). A representative experiment is provided in the right panel.
The high level formation of DSBs in wrn-kd cells was substantiated by determination of H2AX phosphorylation. To this end wrn-kd and wrn-wt cells were exposed to TPT and the phosphorylation of H2AX was measured by the use of a phospho-specific antibody (Lys146 of H2AX). As shown in Fig. 4C, the total phosphorylation level of H2AX is clearly higher in wrn-kd cells than in the control. To quantify the DSBs formed by TPT on the single cell level, we measured the formation of γH2AX foci by immunostaining 3–24 h after treatment with TPT. A representative experiment is shown in Fig. 4D (right panel). While the basal level of γH2AX foci was very low in both cell lines (<5 foci/cell, not shown), TPT-induced a clear formation of γH2AX foci in both cell lines, starting 6 h after exposure. The frequency of TPT-induced γH2AX foci/cell was much higher in wrn-kd than in wrn-wt cells over the whole post-exposure period. In wrn-wt it did not exceed 40 γH2AX foci/cell whereas in wrn-kd it raised up to 140 γH2AX foci/cell (Fig. 4D left panel). The data indicate that WRN is involved either in the prevention of formation or repair of DSBs induced by the topo I inhibitor TPT.
3.4. Stability of topo I upon TPT treatment
Upon treatment with TPT, the repair of the cleavable complex is mediated via degradation of topoisomerase I [34,35], leading to decreased formation of DNA strand breaks and increased survival [14]. As shown by band depletion assay, wrn-wt and wrn-kd cells displayed a similar basal level of free (non-DNA bound) topo I. Immediately upon TPT treatment the majority of the protein is trapped at the DNA in the cleavable complex, as shown by the disappearance of free topo I 2–8 h after TPT exposure. The rate of topo I trapping was similar in wrn-wt and wrn-kd cells (Fig. 5A). However, there was a clear difference in the repair of cleavable complexes, which was inferred from the total cellular amount of topo I. Upon reversal of the cleavable complex by extensive rinsing in vitro, a decline in overall topo I level was observed only in wt cells. This decline was already detectable 4 h after TPT treatment and was most pronounced 4–12 h later. This decline was not observed in wrn-kd cells (Fig. 5B). This decline of topo I in wrn-wt is supposed to be the result of ubiquitination-mediated proteosomal degradation [34,35]. To verify this in our cell system, we utilized the 26S proteosome inhibitor MG132. Pre-treatment of wrn-wt cells with MG132 clearly abrogated the TPT induced topo I decline (Fig. 5C), indicating that it is due to ubiquitin-mediated degradation. The results suggest that topo I bound to DNA is only degraded in wt, but not in cells lacking WRN, supporting a defective repair of cleavable complexes in wrn-kd cells.
Fig. 5.
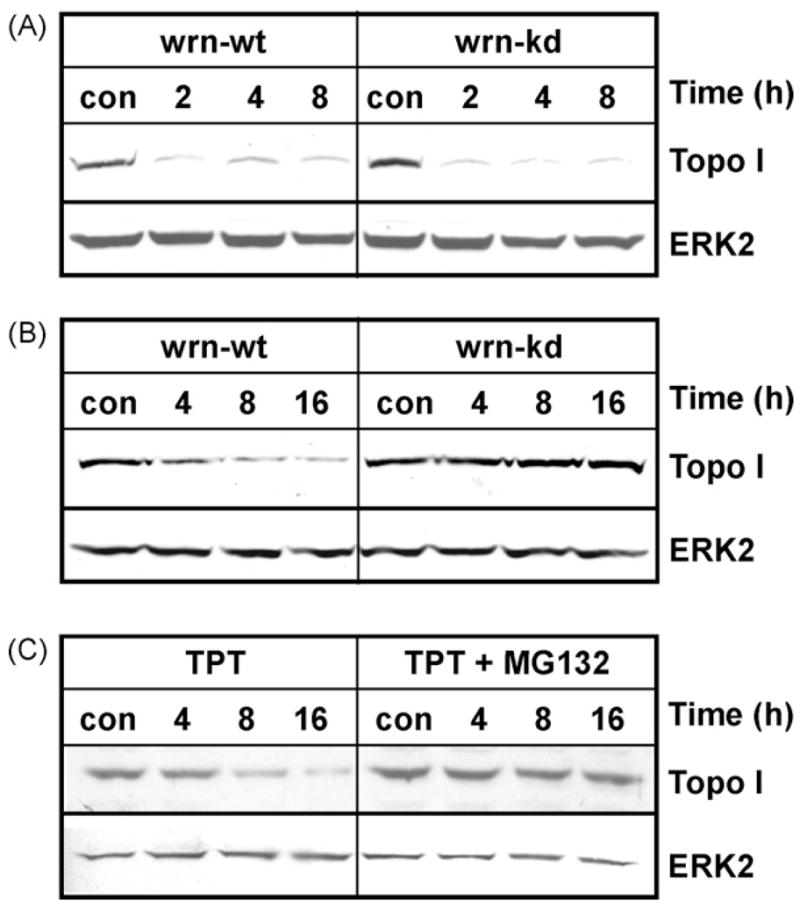
Degradation of topoisomerase I upon TPT treatment. Wrn-wt and wrn-kd cells were exposed to 1 μg/ml TPT for the indicated times. (A) After exposure, the drug treated cells were either directly lysed to detect the formation of the cleavable complex by band depletion or (B) carefully rinsed with culture medium and incubated for another 30 min to reverse the cleavable complex. (C) Cells were exposed to 1 μg/ml TPT for the indicated times in the absence (left blot) or presence of 5 μM MG132. After exposure, the drug-treated cells were carefully rinsed with culture medium and incubated for another 30 min to reverse the cleavable complex. The amount of topo I was detected by Western blot analysis using anti-topo I pAb followed by ECL detection. For loading control, the filter was re-probed with ERK2.
4. Discussion
Even though the anticancer drugs TPT and ETO are widely used in the therapy of different types of cancer, there are only a few molecular markers that can predict the sensitivity of a given tumor. Previously we showed that p53 is one of these prognostic markers [14]. The hypersensitivity of p53 deficient cells to TPT is due to a lack of TPT-triggered topo I degradation, which seems to be a mechanism of DNA damage repair. This went along with the fact that p53 also stimulates topo I activity by enhancing the topo I dissociation from the DNA in vitro [36]. Another protein that has an impact on the sensitivity of topo I inhibitor mediated cell death is the WRN [24–26]. The WRN protein is a major component of the DNA replication machinery and has been shown to localize [37] and resolve several structures at stalled replication forks, thereby facilitating Rad51-dependent HR [38,39]. To elucidate whether the WRN helicase is indeed a potential molecular marker that impacts the tumor cell response to topoisomerase-induced DNA damage, we utilized U2-OS cells stably expressing WRN-specific siRNA.
Using three different methods (WST assay, colony forming assay, and quantification of apoptosis) we showed that the WRN helicase protects against the cytotoxic effects of the topo I inhibitor TPT but not the topo II inhibitor ETO. The hypersensitivity to TPT is in line with results from wrn−/− DT40 chicken cells, which were shown to be hypersensitive to the topo I inhibitor CPT [40]. It is also in line with the finding that murine Wrn-deficient embryonic stem cells, human Wrn-deficient immortalized B-cell lines and EBV-transformed Wrn-deficient lymphoblast cell lines are hypersensitive to camptothecin [24–26]. Three possible mechanisms may explain the role of WRN deficiency in TPT-induced toxicity. Firstly, enhanced formation of CPT-induced chromosomal damage was observed in immortalized WRN cells. This was explained by a potential replication defect in WRN deficient cells, leading to persistence of nicks in the DNA [25,41]. Secondly, WRN helicase could be involved in the repair of DSBs. Accordingly, upon the formation of interstrand DNA crosslinks, WRN was shown to interact with RAD52 and participate in a multiprotein complex including RAD51, RAD54, RAD54B and ATR [38], thereby facilitating DSB repair. This is also supported by data showing that the basal incidence of γH2AX foci is increased in fibroblasts from patients with Werner syndrome [42] suggesting a role for WRN in the protection against spontaneous DSBs. WRN deficient cells show extensive DNA breaks upon treatment with chloroquine (CHL) and trichostatin A, which alter the chromatin structure without producing DNA breaks directly [43]. This is explained by the supposition that upon CHL treatment WRN stimulates topo I leading to prevention of DNA breaks. Thirdly, the effect of WRN on the topo I inhibitor response could be explained by an initial prevention of DNA damage formation. Thus, it has been shown that the WRN helicase can directly interact with topo I, thereby stimulating the relaxation cycle [44]. WRN cell extract displays a reduced DNA relaxation activity, which was explained by lack of stimulation of topo I activity. WRN reduces the duration of the cleavable complex by stimulating topo I relaxation activity and, more specifically, the religation step of the topo I catalytic cycle. By stimulating religation, WRN could prevent the accumulation of cleavable complexes upon TPT treatment. In the light of the fact that an interaction of topo I, but not topo II, and WRN was observed (V. Bohr, unpublished data), the comparable sensitivity of wrn-wt and wrn-kd cells to ETO is comprehensible.
Here, we observed that upon TPT treatment topo I–DNA cleavable complexes are formed in wrn-wt and wrn-kd cells at similar rate. The formation of SSBs and the kinetics of repair of SSBs induced by TPT were nearly identical in wrn-wt and wrn-kd cells. However, more SSBs were converted into DSBs in wrn-kd cells compared to the wild-type as shown by neutral comet assay and the formation of γH2AX foci. The increased amount of DSBs may lead to persistent replication blockage and finally apoptosis [45], which has been shown to occur in these cells in response to TPT. The important role of DSBs as toxic lesions in TPT treated cells is also supported by the fact that homologous recombination defective mutants are hypersensitive to TPT (Christmann and Kaina, unpublished data). Contrary to the results obtained with TPT, WRN helicase has no impact on the sensitivity to the topo II inhibitor etoposide, which directly induces DSBs. This also supports the conclusion that in case of topoisomerase II inhibition WRN is not directly involved in the repair of DSBs.
How is WRN helicase preventing DSB formation upon TPT treatment? An answer to this question might be provided by the fact that upon TPT treatment topo I was present in lower amounts in wild-type cells compared to wrn-kd, which is presumably due to degradation of the enzyme trapped to the DNA. Degradation of topo I is the main repair mechanism for the TPT induced cleavable complex [14,34,35]. As already mentioned, WRN helicase can directly interact with topo I, thereby stimulating the relaxation cycle in untreated cells [44]. Whether this also happens upon TPT treatment, when topo I is covalently linked to DNA, is unclear. Our data suggest that under these conditions, WRN helicase still interacts with topo I and stimulates the proteosomal degradation of topo I. A similar mechanism was also described for p53, which has a protective effect against TPT by stimulating topo I degradation [14]. U2-OS cells used in this study are p53 wt and it was shown that p53 physically and functionally interacts with WRN [46,47]. Therefore, it is pertinent to conclude that p53 together with WRN is involved in the degradation of the topo I cleavable complex. This degradation would provide free access to additional DNA repair proteins, which can further process SSBs induced by topo I. In this context it is important to note that SSBs arising by topo I activity are processed via the single-strand break repair pathway. During this pathway XRCC1 interacts with the tyrosyl-DNA phosphodiesterase (Tdp1) which is able to remove blocked 3′-termini at DNA strand breaks. Both, XRCC1 deficiency [48] and Tdp1 deficiency is associated with hypersensitivity to camptothecin in yeast [49] hamster [50] and human cells [51–53].
Overall, we show that WRN helicase is a critical factor determining the toxicity of topo I, but not topo II poisons in human cancer cells. Upon inhibition of topo I by TPT, the WRN helicase protects cells by preventing the conversion of SSBs into lethal DSBs, resulting from collision of the cleavable complex with the replication fork. We favor the hypothesis that WRN prevents this conversion by either stimulation of the relaxation cycle of topo I or by increasing topo I degradation, thereby allowing access and repair of SSBs by additional repair proteins. In addition, the data are in line with the view that WRN is not directly involved in the repair of the cleavable complex induced by etoposide and, therefore, does not play a role in the toxicity caused by topo II inhibitors. It will be an interesting issue of future research to determine whether the expression level of WRN in tumors is related to their sensitivity to topo I and topo II inhibitors.
Acknowledgments
The work was supported by Deutsche Krebshilfe 106748, Deutsche Forschungsgemeinschaft CH 665/2-1 and Maifor 0428000.
Footnotes
Conflict of interest statement The authors declare that there are no conflicts of interest.
References
- 1.Slichenmyer WJ, Von Hoff DD. New natural products in cancer chemotherapy. J Clin Pharmacol. 1990;30:770–788. doi: 10.1002/j.1552-4604.1990.tb01873.x. [DOI] [PubMed] [Google Scholar]
- 2.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 3.Nitiss JL, Wang JC. Mechanisms of cell killing by drugs that trap covalent complexes between DNA topoisomerases and DNA. Mol Pharmacol. 1996;50:1095–1102. [PubMed] [Google Scholar]
- 4.Hertzberg RP, Caranfa MJ, Hecht SM. On the mechanism of topoisomerase I inhibition by camptothecin: evidence for binding to an enzyme–DNA complex. Biochemistry. 1989;28:4629–4638. doi: 10.1021/bi00437a018. [DOI] [PubMed] [Google Scholar]
- 5.Hsiang YH, Hertzberg R, Hecht S, Liu LF. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem. 1985;260:14873–14878. [PubMed] [Google Scholar]
- 6.Staker BL, Feese MD, Cushman M, Pommier Y, Zembower D, Stewart L, Burgin AB. Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex. J Med Chem. 2005;48:2336–2345. doi: 10.1021/jm049146p. [DOI] [PubMed] [Google Scholar]
- 7.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB, Jr, Stewart L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc Natl Acad Sci USA. 2002;99:15387–15392. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldwasser F, Shimizu T, Jackman J, Hoki Y, O’Connor PM, Kohn KW, Pommier Y. Correlations between S and G2 arrest and the cytotoxicity of camptothecin in human colon carcinoma cells. Cancer Res. 1996;56:4430–4437. [PubMed] [Google Scholar]
- 9.Hsiang YH, Lihou MG, Liu LF. Arrest of replication forks by drug-stabilized topoisomerase I-DNA cleavable complexes as a mechanism of cell killing by camptothecin. Cancer Res. 1989;49:5077–5082. [PubMed] [Google Scholar]
- 10.Shao RG, Cao CX, Zhang H, Kohn KW, Wold MS, Pommier Y. Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA: DNA-PK complexes. EMBO J. 1999;18:1397–1406. doi: 10.1093/emboj/18.5.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D’Arpa P, Beardmore C, Liu LF. Involvement of nucleic acid synthesis in cell killing mechanisms of topoisomerase poisons. Cancer Res. 1990;50:6919–6924. [PubMed] [Google Scholar]
- 12.Furuta T, Takemura H, Liao ZY, Aune GJ, Redon C, Sedelnikova OA, Pilch DR, Rogakou EP, Celeste A, Chen HT, Nussenzweig A, Aladjem MI, Bonner WM, Pommier Y. Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J Biol Chem. 2003;278:20303–20312. doi: 10.1074/jbc.M300198200. [DOI] [PubMed] [Google Scholar]
- 13.D’Arpa P, Liu LF. Topoisomerase-targeting antitumor drugs. Biochim Biophys Acta. 1989;989:163–177. doi: 10.1016/0304-419x(89)90041-3. [DOI] [PubMed] [Google Scholar]
- 14.Tomicic MT, Christmann M, Kaina B. Topotecan-triggered degradation of topoisomerase I is p53-dependent and impacts cell survival. Cancer Res. 2005;65:8920–8926. doi: 10.1158/0008-5472.CAN-05-0266. [DOI] [PubMed] [Google Scholar]
- 15.Christmann M, Tomicic MT, Roos WP, Kaina B. Mechanisms of human DNA repair: an update. Toxicology. 2003;193:3–34. doi: 10.1016/s0300-483x(03)00287-7. [DOI] [PubMed] [Google Scholar]
- 16.Helleday T, Lo J, van Gent DC, Engelward BP. DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair (Amst) 2007;6:923–935. doi: 10.1016/j.dnarep.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 17.Caldecott K, Banks G, Jeggo P. DNA double-strand break repair pathways and cellular tolerance to inhibitors of topoisomerase II. Cancer Res. 1990;50:5778–5783. [PubMed] [Google Scholar]
- 18.Adachi N, Suzuki H, Iiizumi S, Koyama H. Hypersensitivity of nonhomologous DNA end-joining mutants to VP-16 and ICRF-193: implications for the repair of topoisomerase II-mediated DNA damage. J Biol Chem. 2003;278:35897–35902. doi: 10.1074/jbc.M306500200. [DOI] [PubMed] [Google Scholar]
- 19.Adachi N, Iiizumi S, So S, Koyama H. Genetic evidence for involvement of two distinct nonhomologous end-joining pathways in repair of topoisomerase II-mediated DNA damage. Biochem Biophys Res Commun. 2004;318:856–861. doi: 10.1016/j.bbrc.2004.04.099. [DOI] [PubMed] [Google Scholar]
- 20.Arnaudeau C, Lundin C, Helleday T. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol. 2001;307:1235–1245. doi: 10.1006/jmbi.2001.4564. [DOI] [PubMed] [Google Scholar]
- 21.Hinz JM, Helleday T, Meuth M. Reduced apoptotic response to camptothecin in CHO cells deficient in XRCC3. Carcinogenesis. 2003;24:249–253. doi: 10.1093/carcin/24.2.249. [DOI] [PubMed] [Google Scholar]
- 22.Bachrati CZ, Hickson ID. RecQ helicases: suppressors of tumorigenesis and premature aging. Biochem J. 2003;374:577–606. doi: 10.1042/BJ20030491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bohr VA. Deficient DNA repair in the human progeroid disorder, Werner syndrome. Mutat Res. 2005;577:252–259. doi: 10.1016/j.mrfmmm.2005.03.021. [DOI] [PubMed] [Google Scholar]
- 24.Poot M, Gollahon KA, Rabinovitch PS. Werner syndrome lymphoblastoid cells are sensitive to camptothecin-induced apoptosis in S-phase. Hum Genet. 1999;104:10–14. doi: 10.1007/s004390050903. [DOI] [PubMed] [Google Scholar]
- 25.Pichierri P, Franchitto A, Mosesso P, Palitti F. Werner’s syndrome cell lines are hypersensitive to camptothecin-induced chromosomal damage. Mutat Res. 2000;456:45–57. doi: 10.1016/s0027-5107(00)00109-3. [DOI] [PubMed] [Google Scholar]
- 26.Lebel M, Leder P. A deletion within the murine Werner syndrome helicase induces sensitivity to inhibitors of topoisomerase and loss of cellular proliferative capacity. Proc Natl Acad Sci USA. 1998;95:13097–13102. doi: 10.1073/pnas.95.22.13097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng WH, Kusumoto R, Opresko PL, Sui X, Huang S, Nicolette ML, Paull TT, Campisi J, Seidman M, Bohr VA. Collaboration of Werner syndrome protein and BRCA1 in cellular responses to DNA interstrand cross-links. Nucleic Acids Res. 2006;34:2751–2760. doi: 10.1093/nar/gkl362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harrigan JA, Wilson DM, 3rd, Prasad R, Opresko PL, Beck G, May A, Wilson SH, Bohr VA. The Werner syndrome protein operates in base excision repair and cooperates with DNA polymerase beta. Nucleic Acids Res. 2006;34:745–754. doi: 10.1093/nar/gkj475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tomicic MT, Bey E, Wutzler P, Thust R, Kaina B. Comparative analysis of DNA breakage, chromosomal aberrations and apoptosis induced by the anti-herpes purine nucleoside analogues aciclovir, ganciclovir and penciclovir. Mutat Res. 2002;505:1–11. doi: 10.1016/s0027-5107(02)00105-7. [DOI] [PubMed] [Google Scholar]
- 30.Tomicic MT, Friedrichs C, Christmann M, Wutzler P, Thust R, Kaina B. Apoptosis induced by (E)-5-(2-bromovinyl)-2′-deoxyuridine in varicella zoster virus thymidine kinase-expressing cells is driven by activation of c-Jun/activator protein-1 and Fas ligand/caspase-8. Mol Pharmacol. 2003;63:439–449. doi: 10.1124/mol.63.2.439. [DOI] [PubMed] [Google Scholar]
- 31.Christmann M, Tomicic MT, Kaina B. Phosphorylation of mismatch repair proteins MSH2 and MSH6 affecting MutS{alpha} mismatch-binding activity. Nucleic Acids Res. 2002;30:1959–1966. doi: 10.1093/nar/30.9.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tomicic MT, Thust R, Sobol RW, Kaina B. DNA polymerase beta mediates protection of mammalian cells against ganciclovir-induced cytotoxicity and DNA breakage. Cancer Res. 2001;61:7399–7403. [PubMed] [Google Scholar]
- 33.Olive PL, Banath JP, Durand RE. Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells measured using the “comet” assay. Radiat Res. 1990;122:86–94. [PubMed] [Google Scholar]
- 34.Desai SD, Li TK, Rodriguez-Bauman A, Rubin EH, Liu LF. Ubiquitin/26S proteasome-mediated degradation of topoisomerase I as a resistance mechanism to camptothecin in tumor cells. Cancer Res. 2001;61:5926–5932. [PubMed] [Google Scholar]
- 35.Desai SD, Liu LF, Vazquez-Abad D, D’Arpa P. Ubiquitin-dependent destruction of topoisomerase I is stimulated by the antitumor drug camptothecin. J Biol Chem. 1997;272:24159–24164. doi: 10.1074/jbc.272.39.24159. [DOI] [PubMed] [Google Scholar]
- 36.Soe K, Grosse F. p53 stimulates human topoisomerase I activity by modulating its DNA binding. Nucleic Acids Res. 2003;31:6585–6592. doi: 10.1093/nar/gkg846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sakamoto S, Nishikawa K, Heo SJ, Goto M, Furuichi Y, Shimamoto A. Werner helicase relocates into nuclear foci in response to DNA damaging agents and co-localizes with RPA and Rad51. Genes Cells. 2001;6:421–430. doi: 10.1046/j.1365-2443.2001.00433.x. [DOI] [PubMed] [Google Scholar]
- 38.Otterlei M, Bruheim P, Ahn B, Bussen W, Karmakar P, Baynton K, Bohr VA. Werner syndrome protein participates in a complex with RAD51, RAD54, RAD54B and ATR in response to ICL-induced replication arrest. J Cell Sci. 2006;119:5137–5146. doi: 10.1242/jcs.03291. [DOI] [PubMed] [Google Scholar]
- 39.Saintigny Y, Makienko K, Swanson C, Emond MJ, Monnat RJ., Jr Homologous recombination resolution defect in werner syndrome. Mol Cell Biol. 2002;22:6971–6978. doi: 10.1128/MCB.22.20.6971-6978.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Otsuki M, Seki M, Kawabe Y, Inoue E, Dong YP, Abe T, Kato G, Yoshimura A, Tada S, Enomoto T. WRN counteracts the NHEJ pathway upon camptothecin exposure. Biochem Biophys Res Commun. 2007;355:477–482. doi: 10.1016/j.bbrc.2007.01.175. [DOI] [PubMed] [Google Scholar]
- 41.Pichierri P, Franchitto A, Mosesso P, Palitti F. Werner’s syndrome protein is required for correct recovery after replication arrest and DNA damage induced in S-phase of cell cycle. Mol Biol Cell. 2001;12:2412–2421. doi: 10.1091/mbc.12.8.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sedelnikova OA, Horikawa I, Redon C, Nakamura A, Zimonjic DB, Popescu NC, Bonner WM. Delayed kinetics of DNA double-strand break processing in normal and pathological aging. Aging Cell. 2007 doi: 10.1111/j.1474-9726.2007.00354.x. [DOI] [PubMed] [Google Scholar]
- 43.Turaga RV, Massip L, Chavez A, Johnson FB, Lebel M. Werner syndrome protein prevents DNA breaks upon chromatin structure alteration. Aging Cell. 2007;6:471–481. doi: 10.1111/j.1474-9726.2007.00301.x. [DOI] [PubMed] [Google Scholar]
- 44.Laine JP, Opresko PL, Indig FE, Harrigan JA, von Kobbe C, Bohr VA. Werner protein stimulates topoisomerase I DNA relaxation activity. Cancer Res. 2003;63:7136–7146. [PubMed] [Google Scholar]
- 45.Roos WP, Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol Med. 2006;12:440–450. doi: 10.1016/j.molmed.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 46.Sommers JA, Sharma S, Doherty KM, Karmakar P, Yang Q, Kenny MK, Harris CC, Brosh RM., Jr p53 modulates RPA-dependent and RPA-independent WRN helicase activity. Cancer Res. 2005;65:1223–1233. doi: 10.1158/0008-5472.CAN-03-0231. [DOI] [PubMed] [Google Scholar]
- 47.Brosh RM, Jr, Karmakar P, Sommers JA, Yang Q, Wang XW, Spillare EA, Harris CC, Bohr VA. p53 Modulates the exonuclease activity of Werner syndrome protein. J Biol Chem. 2001;276:35093–35102. doi: 10.1074/jbc.M103332200. [DOI] [PubMed] [Google Scholar]
- 48.Park SY, Lam W, Cheng YC. X-ray repair cross-complementing gene I protein plays an important role in camptothecin resistance. Cancer Res. 2002;62:459–465. [PubMed] [Google Scholar]
- 49.Liu C, Pouliot JJ, Nash HA. Repair of topoisomerase I covalent complexes in the absence of the tyrosyl-DNA phosphodiesterase Tdp1. Proc Natl Acad Sci USA. 2002;99:14970–14975. doi: 10.1073/pnas.182557199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Plo I, Liao ZY, Barcelo JM, Kohlhagen G, Caldecott KW, Weinfeld M, Pommier Y. Association of XRCC1 and tyrosyl DNA phosphodiesterase (Tdp1) for the repair of topoisomerase I-mediated DNA lesions. DNA Repair (Amst) 2003;2:1087–1100. doi: 10.1016/s1568-7864(03)00116-2. [DOI] [PubMed] [Google Scholar]
- 51.Barthelmes HU, Habermeyer M, Christensen MO, Mielke C, Interthal H, Pouliot JJ, Boege F, Marko D. TDP1 overexpression in human cells counteracts DNA damage mediated by topoisomerases I and II. J Biol Chem. 2004;279:55618–55625. doi: 10.1074/jbc.M405042200. [DOI] [PubMed] [Google Scholar]
- 52.Interthal H, Chen HJ, Kehl-Fie TE, Zotzmann J, Leppard JB, Champoux JJ. SCAN1 mutant Tdp1 accumulates the enzyme–DNA intermediate and causes camptothecin hypersensitivity. EMBO J. 2005;24:2224–2233. doi: 10.1038/sj.emboj.7600694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miao ZH, Agama K, Sordet O, Povirk L, Kohn KW, Pommier Y. Hereditary ataxia SCAN1 cells are defective for the repair of transcription-dependent topoisomerase I cleavage complexes. DNA Repair (Amst) 2006;5:1489–1494. doi: 10.1016/j.dnarep.2006.07.004. [DOI] [PubMed] [Google Scholar]