

Abstract
Diepoxybutane (DEB) is the most potent active metabolite of the environmental chemical 1,3-butadiene (BD). BD is a known mutagen and human carcinogen and possesses multisystems organ toxicity. We previously reported the elevation of p53 in human TK6 lymphoblasts undergoing DEB-induced apoptosis. In this study, we have characterized the DEB-induced p53 accumulation and investigated the mechanisms by which DEB regulates this p53 accumulation. The elevation of p53 levels in DEB-exposed TK6 lymphoblasts and human embryonic lung (HEL) human fibroblasts was found to be largely due to the stabilization of the p53 protein. DEB increased the acetylation of p53 at lys-382, dramatically reduced complex formation between p53 and its regulator protein mdm2 and induced the phosphorylation of p53 at serines 15, 20, 37, 46, and 392 in human lymphoblasts. A dramatic increase in phosphorylation of p53 at serine 15 in correlation to total p53 levels was observed in DEB-exposed Ataxia Telangiectasia Mutated (ATM) proficient human lymphoblasts as compared to DEB-exposed ATM-deficient human lymphoblasts; this implicates the ATM kinase in the elevation of p53 levels in DEB-exposed cells. Collectively, these findings explain for the first time the mechanism by which p53 accumulates in DEB-exposed cells and contributes to the understanding of the molecular toxicity of DEB and BD.
Keywords: Diepoxybutane, Butadiene, p53, TK6 Human Lymphoblasts, HEL Human Fibroblasts
INTRODUCTION
Diepoxybutane is the most potent active metabolite of 1,3-butadiene (BD). BD is a high production volume environmental chemical that is currently regulated as a hazardous air pollutant. It is a known mutagen, and according to the National Toxicology Program and U.S. Environmental Protection Agency, a human carcinogen [1,2]. Toxic responses resulting from exposure of rodents and humans to BD may include multisite carcinogenesis, cardiovascular disease, bone marrow depletion, ovarian, spleen, and thymic atrophy [3–6]. The toxic effects of BD are known to be mediated through the action of its active metabolites, 1,2-epoxy-3-butene (EB), 1,2:3,4-diepoxybutane (DEB), and 3,4-epoxy-1,2-butanediol (EBD). All three metabolites are genotoxic, carcinogenic, and mutagenic and induce chromosomal aberrations, micronucleus formation, sister chromatid exchanges, and mutations in various animal and human cells [7–10]. While these effects may explain or account for the toxicity and carcinogenicity of BD [8,11,12], detailed biochemical and molecular mechanisms for the toxicity of BD and its metabolites are not completely understood [13,14], and more studies are needed.
DEB, the most potent of the three BD metabolites, is a bifunctional DNA alkylating agent that generates reactive oxygen species and exhibits cytotoxic properties [15]. Previous reports [16,17] have demonstrated that 4–5 μM DEB caused a loss of cell viability in rat cells and human TK6 cells after a 24 h exposure. Additional reports [16,18] demonstrated the induction of apoptosis in the Big Blue Rat system as well as in human CD34+ bone marrow cells exposed to DEB. A subsequent report [19] demonstrated that the DEB-induced loss of cell viability observed in TK6 lymphoblasts after exposure to very low micromolar (2.5–10 μM) concentrations of DEB was due to the occurrence of p53-regulated apoptosis; thus, raising the possibility that apoptosis may be responsible for some of the toxic effects of BD. Since levels of p53 were upregulated by DEB in these cells, understanding the biochemical and molecular mechanisms by which this upregulation occurs will contribute to our knowledge of the biochemical and molecular toxicity of butadiene.
The tumor suppressor p53 is a universal sensor of genotoxicity and a major component of the stress response pathway (for recent reviews, see [20–23]). In this capacity, p53 is critical for maintaining genomic integrity, preventing the accumulation of mutations, and suppression of tumor development [20,21,24–26]. The p53 protein functions as a tetrameric sequence-specific transcription factor that is tightly controlled at low levels in unstressed cells [20,26,27]. In response to a variety of environmental and cellular stresses, including oxidative stress and DNA damage, p53 is stabilized, accumulates in the nucleus, and becomes activated as a transcription factor to mediate cellular responses, including apoptosis [22,26,28,29]. The stabilization of p53 is made possible by cell-type specific and damage-specific post-translational modifications within p53; these have a profound effect on p53 stability and function and serve as a focal point in the regulation of p53. The nature of the posttranslational modifications must be determined for each toxicant-cell-type system, since they cannot be predicted [30].
Although p53 accumulation appears to be regulated at the protein level primarily through posttranslational modifications and complex formation with mdm2 protein [20], increased synthesis of the p53 protein is increasingly being recognized as an additional mechanism for p53 accumulation. Increased synthesis of the p53 protein was reported to occur through increased p53 gene transcription [31] and increased translational control [32–35,36]. Since mechanisms involved in regulating p53 accumulation are cell-type and stimulus-specific, they must be determined for each toxicant cell system.
An increase in amount of p53 protein within cells is often sufficient for the deadly activities of this protein to be expressed in most cases. Thus understanding the molecular mechanisms regulating the accumulation of p53 serves as a prerequisite to the design of applicable therapeutic strategies based on this pathway [22,37,38]. Despite the importance of this protein, there have been very few and limited studies addressing the status of p53 in cells exposed to butadiene and its metabolites. Our laboratory previously reported the induction of elevated p53 levels in TK6 cells undergoing DEB-induced apoptosis [19]. Schmiederer et al. [39] reported an increase in the percentage of p53-positive nuclei in human lung cells after treatment with DEB. Sjoblom et al. [40], however, found no evidence of p53 elevation in rat testis exposed to DEB. Nevertheless, there have been no reports concerning the mechanism(s) of p53-regulation following exposure of cells to BD or its metabolites. In this study, we have characterized the accumulation of p53 in response to increasing DEB concentrations and exposure time in a number of human cell types and determined for the first time the mechanisms by which DEB regulates this p53 accumulation.
MATERIALS AND METHODS
Chemicals
DEB (11.267 M) was purchased from the Sigma Aldrich Chemical Company (St. Louis, MO) and subsequent dilutions were made in Roswell Park Memorial Institute medium (RPMI 1640; Cambrex, Charles City, IA). Trizol reagent was obtained from Invitrogen Life Technologies (Carlsbad, CA). Cycloheximide (CHX) was purchased from Sigma-Aldrich Chemical Company. EasyTag™ EXPRE 35S Protein Labeling mix was purchased from the Perkin–Elmer Corporation (Waltham, MA).
Antibodies
Detection of p53 and mdm2 was performed by utilizing the mouse anti-p53 and anti-mdm2 antibodies (EMD Biosciences, Gibbstown, NJ) at 1:100 dilution. Anti-phospho-p53-(ser15), anti-acetyl-p53-(lys382), and rabbit polyclonal anti-p53 were obtained from Cell Signaling Technology, Inc (Beverly, MA). Anti-glyceraldehyde 3-phosphate dehydrogenase antibody (MAB374) was purchased from Chemicon (Billerica, MA). The secondary (horseradish peroxidase-conjugated goat anti-mouse or goat anti-rabbit) antibodies purchased from Kirkegaard & Perry Laboratories (Gaithersburg, MD) were utilized at 1:5000 dilution.
Cells
Human B lymphoblastic TK6 cell line (American Type Culture Collection, ATCC) expressing wild-type p53 was propagated at 2.5 × 105 cells/mL in RPMI 1640 supplemented with 10% fetal bovine serum (FBS; Invitrogen Life Technologies), 2 mM l-glutamine and penicillin–streptomycin. Human embryonic lung (HEL) fibroblasts (ATCC) were cultured in Eagle’s Minimum Essential Medium (EMEM; Cambrex) containing 10% FBS, 2 mM l-glutamine, and penicillin–streptomycin. GM02254 and GM01526 human B-lymphoblastic cells were purchased from National Institute of General Medical Sciences (NIGMS) Human Genetic Cell Repository (Bethesda, MD). The GM02254 cell line expresses wild-type ATM kinase. GM01526 is a matched mutant cell line that is ATM-defective due to inactivation of ATM kinase gene by a homozygous 2T > C substitution [41,42]. Both cell lines were propagated at 2.5 × 105 cells/mL in RPMI supplemented with 15% heat inactivated FBS, 2 mM l-glutamine, and penicillin–streptomycin.
Exposure of Cells to Diepoxybutane
Cells passaged into fresh media at 24 h prior to each experiment were washed and seeded in complete media at a density of 2.5 × 105 cells/mL. For experiments utilizing HEL fibroblasts, cells seeded 24 h prior to DEB exposure and at approximately 33%–40% confluence were utilized. Cells were exposed to either media or DEB stock solutions prepared in media; the final vehicle concentration was 0.1%. All manipulations were performed under class 2 type A2 conditions.
Western Blot Analysis
Western blot analysis was performed as previously described [19]. Briefly, cells were harvested and lysed in TBSTDS (10 mM Tris, (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.5% sodium deoxycholate, 0.5% SDS, 0.02% NaN3, 0.0004% NaF) supplemented with protease inhibitors (1 mM PMSF, 2 μg/mL aprotinin, and 0.1 mM leupeptin) by gentle vortexing at 4°C. For the analysis of phosphorylated p53 proteins, cell lysates were prepared in TBSTDS supplemented with phosphatase inhibitor cocktail (2.5 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 25 mM sodium fluoride, and 2 mM β-glycerophosphate). Cell lysates for the analysis of acetylated p53 were prepared in TBSTDS containing 5 μM Trichostatin A. Lysates were cleared by spinning (16,000 × g for 30 min), and the protein concentration in the supernatants was quantified by utilizing the Bio-Rad protein assay. Equal amounts of protein in 1× SDS Laemmli sample buffer (2% SDS, 10% glycerol, 5% β-mercaptoethanol, 0.0625 M Tris, pH 6.8, 0.001% bromophenol blue) were subjected to sodium-dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred on to polyvinylidine difluoride (PVDF) membranes. The membranes were blocked for non-specific binding in 10 mM Tris, pH 7.5, 150 mM NaCl containing 5% Carnation non-fat dry milk for 1 h and incubated in appropriate primary antibody for an additional hour at room temperature. The membranes were then washed in TBST (10 mM Tris, (pH 7.5), 150 mM NaCl, 0.05% Tween 20) and incubated with an appropriate horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h at room temperature. After the blots were washed with TBST, antigen levels were detected by utilizing a chemiluminescent substrate (Kirkegaard & Perry Laboratories) and a Fluorichem 8000 Chemifluorimager (Alpha Innotech, San Leandro, CA). Quantitation of the bands was performed by densitometry tracing using the AlphaEase™ software.
35S-Methionine Metabolic Labeling and Pulse Chase Analysis
TK6 and HEL cells were exposed to vehicle alone or 10 μM DEB for 24 h. Cells were washed and incubated at 37°C in methionine free media supplemented with 5% FBS for 30 min. 35S methionine (EasyTag™ EXPRE35S35S protein labeling mix) in methionine free media was then added to the cells at a final concentration of 100 μCi/mL. To analyze the synthesis of p53 protein, cells were labeled for 45 min at 37°C. For determining the stability of the p53 protein, cells were labeled for 45 min, washed and kept at 37°C in complete media for different incubation times, up to 4 h. Cells were harvested and lysed in ice-cold RIPA buffer (50 mM Tris (pH 7.5), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, 0.004% NaF, 1 mM Phenylmethylsulphonyl Fluoride (PMSF), 2 μg/mL aprotinin, and 0.1 mM leupeptin). Lysates were clarified by spinning (16,000 × g for 30 min), and the amount of incorporated radioactivity was determined by trichloro-acetic acid precipitation, followed by scintillation counting. Equal amounts of incorporated radioactivity (~10 × 106 counts per minute (cpm)) from each sample were precleared overnight at 4°C with 1 μg of purified mouse IgG and 100 μL of protein A-G plus agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA). Mouse anti-p53 antibody (1 μg) was then added to pre-cleared supernatants at 4°C and incubated for 2 h. As a control for non-specific binding, immunoprecipitation reactions utilizing purified mouse IgG antibody were also performed. Protein A-G Plus agarose beads (100 μL) were then added, and incubation was continued for 2 additional hours. The beads were collected by centrifugation and washed six times with ice-cold RIPA buffer. Immunocomplexes were then eluted by boiling the beads in Laemmli sample buffer, and samples were electrophoresed on a 10% SDS-polyacrylamide gel. Labeled protein bands were detected by autoradiography, and quantitation was performed by densitometry analysis.
Northern Blot Hybridization
Total RNA was isolated from vehicle or DEB-treated cells using the Trizol reagent, according to the manufacturer’s instructions. After denaturing at 65°C for 10 min, 20 μg of RNA from each sample was fractionated on a 1% agarose/formaldehyde gel and transferred on to Hybond-N+ nylon membrane by capillary action. The membrane was subjected to UV crosslinking and hybridized with a 32P-labeled p53 gene-specific probe. Levels of p53 mRNA were detected by using a PhosphorImager. The filters were stripped and reprobed with a 32P-labeled 18S rRNA-specific probe to normalize for the quantity of RNA retained on the blot.
Measuring the Half-Life of p53 Protein by Cycloheximide Chase
Cycloheximide (CHX, at 75 μg/mL final concentration) was added to TK6 and HEL cells exposed to vehicle or 10 μM DEB for 24 h and incubated at 37°C for various times. Whole cell extracts were then prepared in TBSTDS (containing 1 mM PMSF, 2 μg/mL aprotinin, and 0.1 mM leupeptin), and equal quantities of protein (~25 μg) were analyzed for p53 levels by western blot analysis. To normalize for the quantity of the protein retained on the blot, the membranes were stripped and reprobed with an anti-glyceraldehyde (GAPDH) antibody. Protein levels were then quantitated by densitometric analysis, using the AlphaEase™ software. The fraction of p53 remaining at each time point after the CHX chase was determined by dividing the p53/GAPDH ratio of each sample over the corresponding p53/GAPDH ratio of the 0 time control sample, which was set at 100%. The half-life of p53 was determined as the time (after CHX chase) taken for p53 to decrease by 50%.
Immunoprecipitation of p53–mdm2 Complex
Whole cell lysates were prepared in modified RIPA buffer (50 mM Tris (pH 7.5), 150 mM NaCl, 1% Triton, 1 mM EDTA, 2.5 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 25 mM sodium fluoride, 2 mM β-glycerophosphate, 1 mM PMSF, 2 μg/mL aprotinin, and 0.1 mM leupeptin). Cell lysates were then cleared by centrifugation (16,000 × g for 30 min), and 200 μg of protein extracts was precleared for 3 h by incubating with 1 μg of purified mouse IgG and 40 μL of protein A-G plus agarose beads. For immunoprecipitation, pre-cleared supernatants were first incubated with 2 μg of mouse anti-p53 antibody for 4 h, followed by incubation with 40 μL of protein A-G plus agarose beads for an additional 3 h. As a control for nonspecific binding, parallel samples were treated with purified mouse IgG antibody, instead of anti-p53 antibody. All incubation steps were performed by gentle rocking of the samples at 4°C. Immunoprecipitates were collected by centrifugation, washed six times with ice-cold modified RIPA buffer, and eluted using Laemmli sample buffer. The immunoprecipitates were then analyzed for p53–mdm2 complex formation by western blot analysis using anti-p53 and anti-mdm2 antibodies.
RESULTS
Diepoxybutane Induces Elevated Cellular p53 Levels in Exposed Cells
The effect of DEB on cellular p53 levels in TK6 lymphoblasts and HEL human fibroblasts was investigated (Figure 1). TK6 cells were exposed to a range of increasing concentrations of DEB (0–20 μM) for 24 h, and cellular p53 and GAPDH levels were determined by western blot analysis (Figure 1A). Under conditions where GAPDH levels remained unchanged, cellular p53 levels in TK6 lymphoblasts increased as the concentration of DEB increased. Elevation of cellular p53 levels commenced at DEB concentrations as low as 1 μM and attained peak levels at 15 μM DEB, representing a 9 fold increase in p53 levels in exposed cells as compared to control-unexposed samples. The upregulatory effects of DEB on p53 levels were also observed in HEL fibroblasts (Figure 1B). DEB induced a dose-dependent increase in p53 protein levels in HEL cells, with significant effects observed at concentrations as low as 0.1 μM DEB. The highest (6 fold, as compared to control-unexposed cells) elevation in p53 levels was observed in HEL cells exposed to 25 μM DEB. Collectively, these results demonstrate that DEB induces a concentration-dependent elevation of cellular p53 levels in various human cell types.
FIGURE 1.
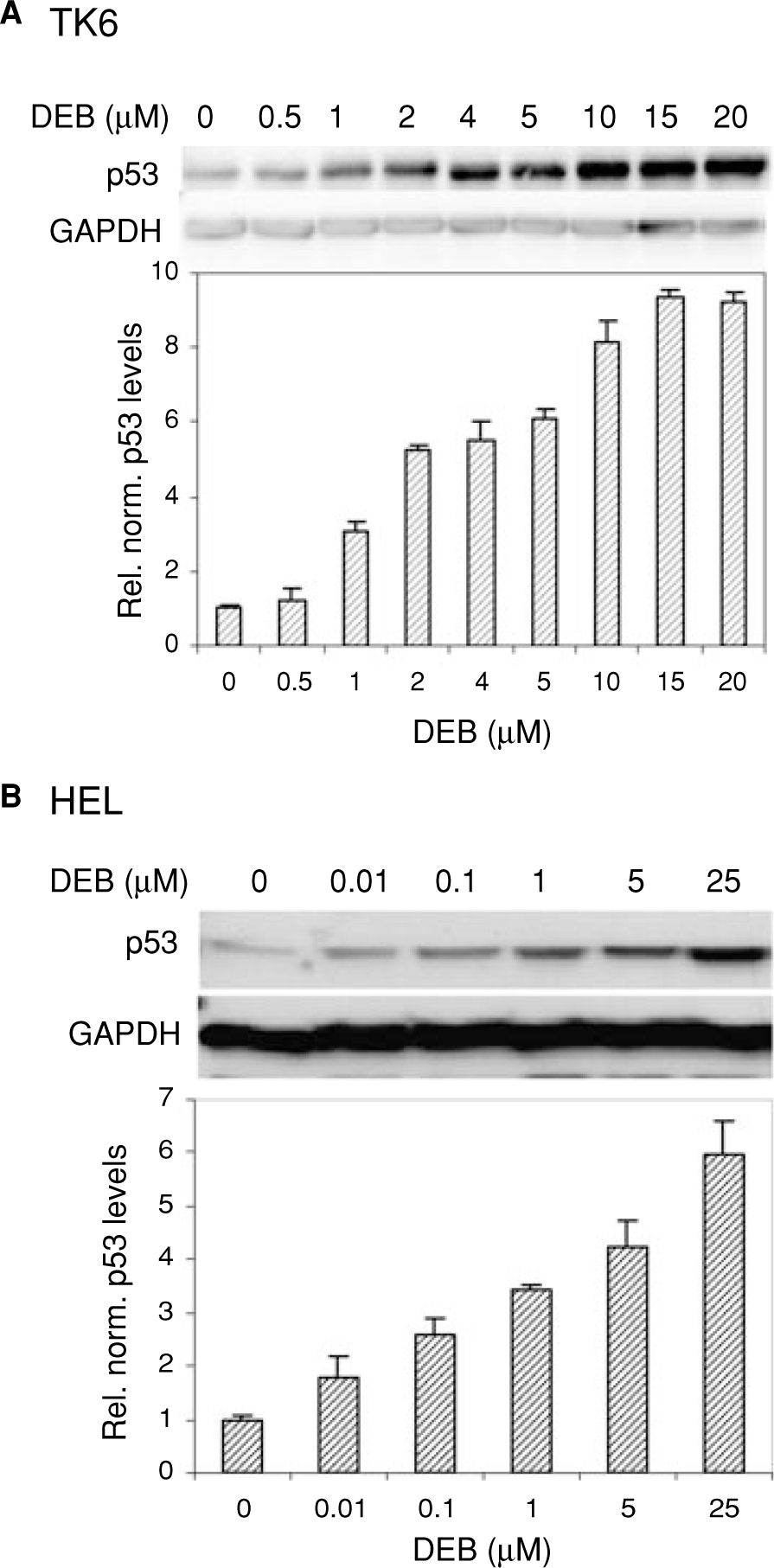
Dose dependent elevation of cellular p53 levels in DEB-exposed human cells. TK6 lymphoblasts (Panel A) and HEL human fibroblasts (Panel B) were exposed to various concentrations of DEB as shown and incubated for 24 h. Cellular p53 levels were analyzed by the western blot technique, using anti-p53 antibody. The quantity of protein retained on the blot was determined by quantifying GAPDH levels for each sample. Relative normalized p53 levels shown in graphical representation were obtained after densitometer tracing, followed by normalization to the corresponding GAPDH levels for each sample. A representative of three experiments is shown.
Diepoxybutane Induces Increased Synthesis of p53 in TK6 Lymphoblasts but Not in HEL Fibroblasts
Cellular p53 levels in toxicant-cell systems are regulated in a cell type and stimulus specific manner [30]. Thus, to begin to investigate the regulatory mechanisms that might be responsible for elevated p53 levels in DEB-exposed cells, the effect of DEB on p53 protein synthesis in TK6 and HEL cells was assessed, using 35S-methionine labeling. Cells exposed to vehicle alone or 10 μM DEB for 24 h were pulse labeled for 45 min using EasyTag™EXPRE35S35S protein labeling mix. Equal quantities of labeled cell extracts were then immunoprecipitated, using anti-p53 antibody or control antibody. The immunoprecipitates were fractionated by SDS-PAGE and analyzed by autoradiography (Figure 2). The relative rate of p53 protein synthesis (shown in a graphical representation) was determined after densitometric tracing of the protein bands corresponding to p53, and normalizing the p53 band intensity to that of the corresponding non-specific band in the same lane. The relative rate of p53 protein synthesis was 3 fold higher in 10 μM DEB-treated TK6 cells, as compared to the corresponding unexposed cells (Figure 2A). The relative rate of p53 protein synthesis in DEB-exposed HEL fibroblasts as compared to the control unexposed cells, however, remained unchanged (Figure 2B). Collectively, these results demonstrate that DEB treatment increases p53 protein synthesis in TK6 cells but not in HEL cells. The results further imply that the elevation of p53 in TK6 cells, but not in HEL cells, is partly due to a higher rate of p53 protein synthesis.
FIGURE 2.

Role of p53 synthesis in DEB-induced cellular p53 elevation in human cells. TK6 lymphoblasts (Panel A) and HEL cells (Panel B) were exposed to vehicle alone or 10 μM DEB. At 24 h post-DEB exposure, cells were pulse labeled with 35S-methionine-35S-cysteine for 45 min. Labeled protein extracts (~10 × 106 counts per minute (cpm)) were immunoprecipitated using an anti-p53 antibody or control antibody, and the immunoprecipitates were fractionated by SDS-PAGE. Labeled p53 protein bands were detected by autoradiography and quantitated by densitometric analysis. A graphical representation of the relative rate of p53 protein synthesis was obtained by comparing the quantity of immunoprecipitated p53 in unexposed and exposed cells after normalization of each value to the density of a corresponding immunoprecipitated representative non-specific band shown as the control.
To determine whether the increased rate of p53 protein synthesis observed in DEB-exposed TK6 cells was due to the occurrence of increased p53 mRNA levels in these cells, the effect of DEB on steady state p53 mRNA levels in TK6 lymphoblasts was investigated. Total cellular RNA extracted from cells exposed to vehicle alone, 10 or 20 μM DEB (in duplicates) for 24 h was analyzed for p53 mRNA levels by northern blot analysis (Figure 3). Under the conditions of equal RNA loading (as determined by18S rRNA levels), 10 and 20 μM DEB-exposed samples contained a 1.3-fold and 1.8-fold higher level of p53-specific mRNA, respectively, as compared to the control unexposed samples. Comparable results were obtained after microarray and real-time PCR analysis of RNA extracted from vehicle alone or 10 μM DEB-exposed TK6 cells (data not shown). These results, thus, demonstrate a very slight increase in p53 steady-state mRNA levels after exposure of TK6 cells to 10 μM DEB. Collectively, the results of Figures 2 and 3 suggest that the DEB-induced 3 fold elevation in p53 protein synthesis observed in DEB-exposed TK6 cells is due to translation events that do not involve a corresponding increase in p53 mRNA steady state levels.
FIGURE 3.

Elevation of p53 mRNA levels in DEB-exposed TK6 lymphoblasts. TK6 cells were exposed to vehicle alone, 10 or 20 μM DEB (in duplicates), and incubated for 24 h. Total RNA was extracted using the Trizol reagent, and steady state p53 mRNA levels were determined by northern blot analysis using a 32P labeled probe against the human p53 gene. The filters were then stripped and re-probed with 32P-labeled 18S rRNA to normalize for the quantity of RNA retained on the blot. The determined relative normalized p53 mRNA levels in control and DEB-exposed TK6 cells are graphically represented.
Stability of the p53 Protein Increases in Diepoxybutane-Treated Human Cells
Since changes in p53 synthesis did not account for the bulk of the DEB-induced elevation of p53 levels, the stability of the p53 protein in DEB-exposed TK6 and HEL cells was investigated (Figure 4). Cells were exposed to vehicle alone or 10 μM DEB for 24 h, and the synthesis of new protein was blocked by the addition of cycloheximide (75 μg/mL final concentration). Cells extracts were then prepared at different times after the addition of CHX, and the remaining cellular p53 and GAPDH levels were determined by western blot analysis. The half-life of the p53 protein in TK6 lymphoblasts increased from 29 min in control-unexposed cells to 160 min in DEB-exposed cells (Figure 4A). In HEL fibroblasts, the half-life of the p53 protein increased from 25 min in unexposed cells to 155 min in exposed cells (Figure 4B). Similar results were obtained for TK6 cells after 35S-methionine pulse chase analysis (not shown). These results, thus, demonstrate that DEB exposure increases the stability of the p53 protein in TK6 lymphoblasts (5.5-fold) and HEL fibroblasts (6.2-fold). These results also imply that the DEB-induced elevation of p53 levels is largely due to the stabilization of the p53 protein in exposed human TK6 cells and HEL fibroblasts.
FIGURE 4.

Stability of the p53 protein increases in DEB-treated human cells. TK6 lymphoblasts (Panel A) and HEL fibroblasts (Panel B) were exposed to vehicle alone or 10 μM DEB. At 24 h postexposure, 75 μg/mL of cycloheximide was added to the cells, and extractions were performed at various times (as shown). Cellular p53 and GAPDH levels in protein extracts were determined by western blot analysis, and quantitation was performed by densitometry tracing of the protein bands. The percent normalized p53 levels were determined by dividing the p53 to GAPDH band density ratio for each sample to the p53 to GAPDH band density ratio at 0 time point, and multiplying by 100. After graphic representation, the half-life of the p53 was determined as the time corresponding to the 50% normalized p53 value.
Effect of Diepoxybutane on p53–mdm2 Complex Formation in TK6 Lymphoblasts
Since stabilization of p53 was observed in DEB-exposed TK6 cells, the effect of DEB on p53–mdm2 complex formation was examined. TK6 cells were exposed to vehicle or 10 μM DEB for 24 h, and ~200 μg of protein from each sample were immunoprecipitated with anti-p53 antibody or control antibody. The resulting p53 immunocomplexes (IP), as well as 100 μg of whole cell lysates (no IP), were analyzed by western blot analysis using anti-p53 and anti-mdm2 antibodies (Figure 5A). To more accurately assess the fraction of mdm2 in complex with p53 in DEB-exposed cells as compared to control unexposed cells, the results of Figure 5A were expressed in terms of relative mdm2 to p53 ratio for both whole cell lysates as well as the p53 immunocomplexes (Figure 5B). Whole cell extracts obtained from DEB-exposed cells contained a 1.5× higher ratio of mdm2 to p53 as compared to control unexposed cells (Figure 5B, left panel). The ratio of mdm2 to p53 in the p53 immunocomplexes obtained from DEB-exposed cells, however, was only 25% of the ratio of mdm2 to p53 in control unexposed cells (Figure 5B, right panel). Collectively, the results of Figure 5 demonstrate that although DEB significantly increases mdm2 levels relative to p53, the complex formation between p53 and mdm2 is dramatically reduced in DEB-exposed cells as compared to the corresponding control unexposed cells. Since reduced p53–mdm2 complex formation has previously been implicated in the stabilization of p53 in response to stress [20,26,43,44], our results suggest that the observed reduced complex formation between p53 and mdm2 is responsible for the increased stability of p53 in DEB-exposed TK6 cells.
FIGURE 5.

Effect of DEB exposure on p53–mdm2 complex formation in human lymphoblasts. TK6 lymphoblasts were exposed to vehicle alone or 10 μM DEB for 24 h. Cell lysates (200 μg of protein) were subjected to immunoprecipitation using mouse control IgG or anti-p53 antibody. (A) The resulting immunoprecipitates (IP) and non-immunoprecipitated extracts (no IP, 100 μg of protein) were analyzed by western blot analysis using antibodies against p53 and mdm2. (B) A graphical representation of the relative mdm2/p53 ratio in whole cell lysates (no IP) and p53 immunocomplexes (IP) of control-unexposed and DEB-exposed cells is shown.
The p53 Protein Is Acetylated at Lys-382 in Diepoxybutane-Exposed Cells
Acetylation of p53 at lys382 has previously been reported to stabilize p53 in response to DNA damage [45,46]. Since DEB exposure is known to induce DNA damage, the effects of DEB on the acetylation of p53 at lys382 in TK6 cells were examined (Figure 6). Extracts from cells exposed to various low concentrations (0–10 μM) of DEB for 24 h were analyzed for acetyl-p53 (lys382), p53, and GAPDH levels by western blot analysis (panel A). As the exposure concentration of DEB increased, acetyl-p53 levels increased in direct correlation to the elevation of p53 levels; under these conditions GAPDH levels remained unchanged. Exposure of TK6 cells to 10 μM DEB for various times up to 48 h (panel B) resulted in the elevation of acetyl-p53 levels in correlation to the increase in p53 levels as the DEB-exposure time increased; GAPDH levels did not change under these conditions. The specificity of anti-acetyl-p53 (lys382) antibody was confirmed by utilizing Human Cytomegalovirus (HCMV)-infected HEL cells, which contained undetectable acetyl-p53 levels, despite the increase in p53 levels observed in the infected cells as compared to control mock-infected cells (panel C). Collectively, the results of Figure 6 demonstrate that DEB induces the acetylation of p53 at lysine 382 in TK6 cells in a concentration-dependent and DEB-exposure time-dependent manner. Since the induction of p53 acetylation at lysine 382 is in correlation to the DEB-induced elevation of cellular p53 levels in exposed lymphoblasts, the results of Figure 6 suggest a possible role for the acetylation of p53 at lys382 in stabilizing the p53 protein in human TK6 lymphoblasts in response to DEB exposure.
FIGURE 6.

Acetylation of p53 protein in response to DEB exposure in TK6 cells. Cells were exposed to (A). various concentrations of DEB for 24 h or (B) 10 μM DEB for various times, as shown. Acetyl-p53-(lys382) levels were determined by western blot analysis, using an antibody specific for acetyl-p53-(lys382) protein. The blots were stripped and reprobed with antibodies against p53 and GAPDH. (C) To determine the specificity of the anti-acetyl-p53-(lys382) antibody, extracts from vehicle alone, 10 μM and 20 μM DEB-treated TK6 lymphoblasts as well as mock and HCMV-infected HEL fibroblasts were analyzed by the western blot analysis.
ATM Kinase Plays a Role in the Stabilization of p53 in DEB-Exposed Cells
Phosphorylation of ser6, ser9, ser15, ser20, ser37, ser46, and ser392 of p53 has been reported in DNA damage-induced p53 stabilization [47–49]. Thus, the phosphorylation of p53 on these specific residues in DEB-exposed TK6 cells was examined by western blot analysis using specific antibodies that detect p53 only when it is phosphorylated at a specific serine residue (data not shown). Under the conditions where cellular GAPDH levels remained unaltered, phospho-p53 (ser15, ser20, ser37, ser46, and ser392) levels were significantly elevated in 10 and 20 μM DEB-exposed cells, as compared to control unexposed cells at 24 and 48 h post-DEB-exposure. Under these experimental conditions, ser6 and ser9 phospho-p53 levels in DEB-exposed and control unexposed cells were not significantly different (not shown).
Phosphorylation of p53 in response to DNA damage has been reported to be mediated by various kinases, including ATM kinase [49–54]. To address the role of ATM kinase in the stabilization of p53 in DEB-exposed cells, the effects of 10 or 20 μM DEB on p53 phosphorylation (ser15) and p53 elevation were assessed in ATM kinase proficient GM02254 lymphoblasts as compared to ATM kinase deficient GM01526 lymphoblasts (Figures 7). Under experimental conditions where GAPDH levels remained unchanged, p-p53 (ser15) and p53 levels were elevated in exposed GM02254 and GM01526 cells as compared to control unexposed cells. However, DEB-induced p-p53 (ser15) elevation at 24 h post-DEB exposure was dramatically higher in the GM02254 cells (13-fold at 10 μM and 19-fold at 20 μM DEB exposure) as compared to the GM01526 cells (fivefold at 10 μM and 7.5-fold at 20 μM DEB exposure; Figures 7A and B, top panels). The extent of DEB-induced p53 elevation within the same samples correlated to the p-p53 (ser15) levels and was significantly higher in the GM02254 cell line as compared to the corresponding GM02254 cell line samples. At 48 h post DEB-exposure (Figures 7A and 7B, bottom panels), exposed GM02254 cells also revealed higher p53 (ser15) and p53 levels as compared to corresponding exposed GM01526 cell levels. Panel C demonstrates the specificity of the p-p53 (ser 15) antibody, since it failed to detect p53 levels in extracts with elevated p53 protein levels, but lacking p-p53(ser15). Collectively, these results demonstrate that ATM kinase plays a role in mediating the DEB-induced p53-(ser15) phosphorylation, as well as the DEB-induced elevation of p53 levels, since both events were induced by DEB more efficiently in the ATM proficient GM02254 cell line than in the ATM-deficient GM01526 cell line.
FIGURE 7.

Role of ATM kinase in mediating the phosphorylation and stability of p53 in response to DEB exposure. (A) ATM-proficient GM02254 and (B) ATM-deficient GM01526 cells were simultaneously exposed to vehicle alone, 10 μM, or 20 μM DEB for 24 h (top panels) and 48 h postexposure (bottom panels). Phospho-p53-(ser15) levels in the protein extracts were determined by the western blot technique, using specific antibody. The blots were then stripped and reprobed with anti-p53 and anti-GAPDH antibodies. Protein levels were normalized by using GAPDH levels.
DISCUSSION
In this report, we have demonstrated for the first time the mechanism by which DEB, butadiene’s most potent active metabolite, regulates p53 levels in exposed human cells. DEB elevated cellular p53 levels in exposed human TK6 lymphoblasts as well as in the human HEL fibroblastic cell line in a DEB-concentration and exposure-time-dependent manner. This elevation of p53 levels was found to be largely due to the stabilization of the p53 protein in the exposed cells. DEB significantly increased acetylation of p53 at lys-382 and dramatically reduced complex formation between p53 and its regulator protein mdm2. DEB also induced the phosphorylation of p53 at serines 15, 20, 37, 46, and 392. Finally, the DEB-induced elevation of p53 levels in human lymphoblasts was found to be regulated by ATM kinase. Collectively, these findings explain the mechanism of p53 regulation in DEB-exposed cells and contribute to the understanding of the molecular toxicity of DEB and butadiene.
Although p53 protein is an important regulator of cell function in normal and stressed cells, few and limited studies have addressed the status of p53 in cells exposed to butadiene and its metabolites. Our laboratory previously reported the induction of elevated p53 levels in TK6 cells exposed to DEB [19]. Schmiederer et al. [39] reported an increase in the percentage of p53-positive nuclei in human lung cells after treatment with DEB. Sjoblom et al. [40] found no evidence of p53 elevation in rat testis exposed to DEB. Since toxicants regulate p53 in a cell type and tissue specific manner [30,55], the difference between this rat system and the systems with elevated p53 levels may be attributed to cell type differences in the p53 response to DEB. Nevertheless, to-date, there have been no reports concerning the mechanism(s) of p53-regulation following exposure of cells to BD or its metabolites. This study extends our previous observations [19], outlines the mechanisms accounting for the DEB-induced elevation of p53 levels, and demonstrates for the first time the dose and exposure time dependency of the DEB-induced elevation of p53 levels in human TK6 lymphoblasts, as well as in the human HEL lung fibroblastic cell line.
A 3 fold increase in the synthesis of the p53 protein was observed in TK6 cells exposed to 10 μM DEB for 24 h; no significant elevation of p53-mRNA levels was detected under the same exposure conditions. Increasing evidence has shown that increased p53 synthesis in the absence of increased p53 mRNA levels can occur due to increased translational efficiency of p53 mRNA [32–35]. Our results on the DEB-induced increased synthesis of p53 in the absence of altered p53 mRNA are thus in line with these published observations. It should be noted that HEL cells did not show any change in the synthesis of p53 under the same exposure conditions. The observed differences in the synthesis of p53 in response to DEB between TK6 and HEL cell types can be explained in terms of the differential regulation of cellular p53 levels by DEB in a cell type specific manner.
Our results demonstrated that complex formation between p53 and mdm2 was dramatically reduced in DEB-exposed TK6 cells as compared to the corresponding control unexposed cells. Previous studies have demonstrated that complex formation between p53 and mdm2 targets p53 for ubiquitin-mediated degradation in unstressed cells [20,26,27,56]. Under stress conditions, however, p53 undergoes posttranslational modifications that reduce the p53–mdm2 complex formation, stabilizing the p53 protein and causing its accumulation [20,57]. Thus, our observations, which are in line with these reports, suggest that the reduced complex formation observed between p53 and mdm2 in DEB-exposed TK6 cells is partly responsible for the increased stability of p53 in DEB-exposed TK6 cells. It should be noted, however, that p53 stabilization can occur without observed changes in p53–mdm2 complex formation [58,59]. Thus, knowledge concerning the status of complex formation in association with the stability of p53 in DEB-exposed cells contributes toward an increased understanding of the molecular toxicity of DEB and BD.
The stability of the p53 protein dramatically increased in both TK6 cells and HEL cells after DEB exposure, and accounted for most of the elevation of p53 levels in these cells. Although exceptions have been noted [32–35], our results are in line with numerous observations that support the concept that stabilization of p53 is the major mechanism involved in elevating p53 levels in response to a variety of stressful stimuli [30,49,60–62]. Stabilization of the p53 protein in stressed cells has been reported to occur as a result of posttranslational modifications of various p53 residues, although the posttranslational modification fingerprint is cell type and toxicant specific [27,30,60, 63,64].
Posttranslational modifications of p53 within its N-terminal domain (residues 15–27) have been reported to cause segregation of the p53–mdm2 complex in stressed cells, thus stabilizing the p53 protein and causing its accumulation [63,64]. Phosphorylation of N-terminal serine residues, specifically at serine 15, was reported to induce acetylation of p53 at the carboxyl terminus [60,65], and this acetylation also stabilizes the p53 protein [63]. Phosphorylation of p53 at serines 37 and 46 results in the stabilization of p53 as well, and its phosphorylation at ser 392 promotes sequence–specific p53 binding [60,63]. Based on these published reports, our observations thus suggest that the DEB-induced acetylation of p53 at lys 382, and phosphorylation of p53 at ser 15, 20, 37, 46, and 392 are responsible for stabilizing p53 in DEB-exposed TK6 cells. To our knowledge, this is the first detailed study on the mechanism of p53 elevation in DEB-exposed cells.
Our results demonstrated for the first time the ATM kinase-dependent induction of p53-(ser15) phosphorylation in proportion to the elevation of p53 levels in DEB-exposed human lymphoblasts. Both correlating events were induced by DEB more efficiently in the ATM-proficient GM02254 cell line than in the ATM-deficient GM01526 cell line, thus implicating ATM kinase in stabilizing of p53 levels through the phosphorylation of p53 at ser-15. Our findings are in agreement with previous observations that implicated ATM kinase-dependent signaling in the accumulation of p53 in response to UV, IR, adriamycin, and other genotoxic stimuli [66–68]. The residual ser-15 p53 phosphorylation observed in ATM-deficient cells can be attributed to the action of other kinases that phosphorylate p53 at this site ([60,63,69], our unpublished observations).
In summary, this study demonstrates for the first time the mechanism by which DEB, butadiene’s most potent active metabolite, regulates p53 accumulation in exposed human cells. The stabilization of the p53 protein in the exposed cells accounted for the majority of the DEB-induced upregulation of p53 levels, although translational regulation of p53 was also observed in TK6 cells. The increased stability of p53 in DEB-exposed TK6 cells could be explained by the observed decrease in the p53–mdm2 complex formation, as well as by the observed increased phosphorylation of p53 at serines 15, 20, 37, 46, and 392 and the acetylation of p53 at lysine 382. Cellular p53 levels were also found to be upregulated by ATM kinase, through the phosphorylation of p53 at serine 15. Phosphorylation and acetylation of p53 at these residues is known to stabilize p53, release p53 from mdm2-mediated inhibition, activate its sequence-specific DNA-binding activity, and activate it as a transcription factor [45,64,70–72]. In particular, phosphorylation of p53 at serines 15, 37, 46, as well as its acetylation at lysine 382 has been linked to increased pro-apoptotic functions of p53 [60,63,69,72]. The determination of the mechanism of DEB-induced p53 accumulation contributes toward the understanding of the molecular toxicity of DEB and butadiene. Since DEB-induced p53-mediated apoptosis in human TK6 lymphoblasts has previously been described [19,39,73], these studies are a necessary prerequisite to the understanding of the role p53 plays in DEB-induced apoptosis in this experimental system as well as other DEB systems.
Acknowledgments
Contract Grant Sponsor: National Institute of General Medical Sciences MBRS SCORE Grant.
Contract Grant Number: GM076530.
Contract Grant Sponsor: National Institute of Environmental Health Sciences ARCH Grant.
Contract Grant Number: ES10018.
Contract Grant Sponsor: National Science Foundation Grant HRD.
Contract Grant Number: 0450375.
REFERENCES
- 1.National Toxicology Program. Ninth Report on Carcinogens. Research Triangle Park, NC: U.S. Department of Health and Human Services; 2000. [Google Scholar]
- 2.U.S. Environmental Protection Agency. Health Assessment of 1,3-Butadiene: Office of Research and Development, National Center for Environmental Assessment; 2002. 66151–66152. [Google Scholar]
- 3.Doerr JK, Hollis EA, Sipes IG. Species difference in the ovarian toxicity of 1,3-butadiene epoxides in B6C3F1 mice and Sprague–Dawley rats. Toxicology 1996;113(1–3):128–136. [DOI] [PubMed] [Google Scholar]
- 4.Melnick RL, Sills RC. Comparative carcinogenicity of 1,3-butadiene, isoprene, and chloroprene in rats and mice. Chem Biol Interact 2001;135–136:27–42. [DOI] [PubMed] [Google Scholar]
- 5.National Toxicology Program. NTP Toxicology and Carcinogenesis Studies of 1,3-Butadiene (CAS No. 106–99-0) in B6C3F1 Mice (Inhalation Studies): National Toxicol. Program Tech. Rep. Ser; 1993 May 1993. 1–389. [PubMed] [Google Scholar]
- 6.Penn A, Snyder CA. 1,3-Butadiene exposure and cardiovascular disease. Mutat Res 2007;621(1–2):42–49. [DOI] [PubMed] [Google Scholar]
- 7.Bond JA, Medinsky MA. Insights into the toxicokinetics and toxicodynamics of 1,3-butadiene. Chem Biol Interact 2001;135–136:599–614. [DOI] [PubMed] [Google Scholar]
- 8.Kligerman AD, Hu Y. Some insights into the mode of action of butadiene by examining the genotoxicity of its metabolites. Chem Biol Interact 2007;166(1–3):132–139. [DOI] [PubMed] [Google Scholar]
- 9.Murg M, Schuler M, Eastmond DA. Evaluation of micronuclei and chromosomal breakage in the 1cen-q12 region by the butadiene metabolites epoxybutene and diepoxybutane in cultured human lymphocytes. Mutagenesis 1999;14(6):541–546. [DOI] [PubMed] [Google Scholar]
- 10.Recio L, Steen AM, Pluta LJ, Meyer KG, Saranko CJ. Mutational spectrum of 1,3-butadiene and metabolites 1,2-epoxybutene and 1,2,3,4-diepoxybutane to assess mutagenic mechanisms. Chem Biol Interact 2001;135–136:325–341. [DOI] [PubMed] [Google Scholar]
- 11.Fred C, Tornqvist M, Granath F. Evaluation of cancer tests of 1,3-butadiene using internal dose, genotoxic potency, and a multiplicative risk model. Cancer Res 2008;68(19):8014–8021. [DOI] [PubMed] [Google Scholar]
- 12.Vodicka P, Stetina R, Smerak P, Vodickova L, Naccarati A, Barta I, Hemminki K. Micronuclei, DNA single-strand breaks and DNA-repair activity in mice exposed to 1,3-butadiene by inhalation. Mutat Res 2006;608(1):49–57. [DOI] [PubMed] [Google Scholar]
- 13.Hurst HE. Toxicology of 1,3-butadiene, chloroprene, and isoprene. Rev Environ Contam Toxicol 2007;189:131–179. [DOI] [PubMed] [Google Scholar]
- 14.Swenberg JA, Boysen G, Georgieva N, Bird MG, Lewis RJ. Future directions in butadiene risk assessment and the role of cross-species internal dosimetry. Chem Biol Interact 2007;166(1–3):78–83. [DOI] [PubMed] [Google Scholar]
- 15.Erexson GL, Tindall KR. Reduction of diepoxybutane-induced sister chromatid exchanges by glutathione peroxidase and erythrocytes in transgenic Big Blue mouse and rat fibroblasts. Mutat Res 2000;447(2):267–274. [DOI] [PubMed] [Google Scholar]
- 16.Erexson GL, Tindall KR. Micronuclei and gene mutations in transgenic big Blue((R)) mouse and rat fibroblasts after exposure to the epoxide metabolites of 1,3-butadiene. Mutat Res 2000;472(1–2):105–117. [DOI] [PubMed] [Google Scholar]
- 17.Steen AM, Meyer KG, Recio L. Analysis of HPRT mutations occurring in human TK6 lymphoblastoid cells following exposure to 1,2,3,4-diepoxybutane. Mutagenesis 1997;12(2):61–67. [DOI] [PubMed] [Google Scholar]
- 18.Irons RD, Pyatt DW, Stillman WS, Som DB, Claffey DJ, Ruth JA. Comparative toxicity of known and putative metabolites of 1, 3-butadiene in human CD34(+) bone marrow cells. Toxicology 2000;150(1–3):99–106. [DOI] [PubMed] [Google Scholar]
- 19.Yadavilli S, Muganda PM. Diepoxybutane induces caspase and p53-mediated apoptosis in human lymphoblasts. Toxicol Appl Pharmacol 2004;195(2):154–165. [DOI] [PubMed] [Google Scholar]
- 20.Lahav G Oscillations by the p53–Mdm2 feedback loop. Adv Exp Med Biol 2008;641:28–38. [DOI] [PubMed] [Google Scholar]
- 21.Oren M Decision making by p53: life, death, cancer. Cell Death Differ. 2003;10(4):431–442. [DOI] [PubMed] [Google Scholar]
- 22.Vazquez A, Bond EE, Levine AJ, Bond GL. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov 2008;7(12):979–987. [DOI] [PubMed] [Google Scholar]
- 23.Viadiu H Molecular architecture of tumor suppressor p53. Curr Top Med Chem 2008;8(15):1327–1334. [DOI] [PubMed] [Google Scholar]
- 24.Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis—the p53 network. J Cell Sci 2003;116(Pt 20):4077–4085. [DOI] [PubMed] [Google Scholar]
- 25.Hofseth LJ, Robles AI, Yang Q, Wang XW, Hussain SP, Harris C. p53: at the crossroads of molecular carcinogenesis and molecular epidemiology. Chest 2004;125(5 Suppl):83S–85S. [DOI] [PubMed] [Google Scholar]
- 26.Meulmeester E, Jochemsen AG. p53: a guide to apoptosis. Curr Cancer Drug Targets 2008;8(2):87–97. [DOI] [PubMed] [Google Scholar]
- 27.Scoumanne A, Chen X. Protein methylation: a new mechanism of p53 tumor suppressor regulation. Histol Histopathol 2008;23(9):1143–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuribayashi K, El-Deiry WS. Regulation of programmed cell death by the p53 pathway. Adv Exp Med Biol 2008;615:201–221. [DOI] [PubMed] [Google Scholar]
- 29.Shen Y, White E. p53-Dependent apoptosis pathways. Adv Cancer Res 2001;82:55–84. [DOI] [PubMed] [Google Scholar]
- 30.Murray-Zmijewski F, Slee EA, Lu X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat Rev Mol Cell Biol 2008;9(9):702–712. [DOI] [PubMed] [Google Scholar]
- 31.Reisman D, Boggs K. Transcriptional regulation of the p53 tumor suppressor gene: a potential target for cancer therapeutics? Recent Pat DNA Gene Seq 2007;1(3):176–185. [DOI] [PubMed] [Google Scholar]
- 32.Fu L, Benchimol S. Participation of the human p53 3’UTR in translational repression and activation following gamma-irradiation. Embo J 1997;16(13):4117–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Halaby MJ, Yang DQ. p53 Translational control: a new facet of p53 regulation and its implication for tumorigenesis and cancer therapeutics. Gene 2007;395(1–2):1–7. [DOI] [PubMed] [Google Scholar]
- 34.Takagi M, Absalon MJ, McLure KG, Kastan MB. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 2005;123(1):49–63. [DOI] [PubMed] [Google Scholar]
- 35.Zhang J, Chen X. Posttranscriptional regulation of p53 and its targets by RNA-binding proteins. Curr Mol Med 2008;8(8):845–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naski N, Gajjar M, Bourougaa K, Malbert-Colas L, Fahraeus R, Candeias MM. The p53 mRNA-Mdm2 interaction. Cell Cycle 2009;8(1):31–34. [DOI] [PubMed] [Google Scholar]
- 37.Bassett EA, Wang W, Rastinejad F, El-Deiry WS. Structural and functional basis for therapeutic modulation of p53 signaling. Clin Cancer Res 2008;14(20):6376–6386. [DOI] [PubMed] [Google Scholar]
- 38.Staples OD, Steele RJ, Lain S. p53 as a therapeutic target. Surgeon 2008;6(4):240–243. [DOI] [PubMed] [Google Scholar]
- 39.Schmiederer M, Knutson E, Muganda P, Albrecht T. Acute exposure of human lung cells to 1,3-butadiene diepoxide results in G1 and G2 cell cycle arrest. Environ Mol Mutagen 2005;45(4):354–364. [DOI] [PubMed] [Google Scholar]
- 40.Sjoblom T, West A, Lahdetie J. Apoptotic response of spermatogenic cells to the germ cell mutagens etoposide, adriamycin, and diepoxybutane. Environ Mol Mutagen 1998;31(2):133–148. [DOI] [PubMed] [Google Scholar]
- 41.Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998;281(5383):1674–1677. [DOI] [PubMed] [Google Scholar]
- 42.Nakagawa K, Taya Y, Tamai K, Yamaizumi M. Requirement of ATM in phosphorylation of the human p53 protein at serine 15 following DNA double-strand breaks. Mol Cell Biol 1999;19(4):2828–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dey A, Verma CS, Lane DP. Updates on p53: modulation of p53 degradation as a therapeutic approach. Br J Cancer 2008;98(1):4–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moll UM, Petrenko O. The mdm2-p53 interaction. Mol Cancer Res 2003;1(14):1001–1008. [PubMed] [Google Scholar]
- 45.Lin T, Mak NK, Yang MS. MAPK regulate p53-dependent cell death induced by benzo[a]pyrene: involvement of p53 phosphorylation and acetylation. Toxicology 2008;247(2–03):145–153. [DOI] [PubMed] [Google Scholar]
- 46.Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, Anderson CW, Appella E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev 1998;12(18):2831–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Appella E, Anderson CW. Signaling to p53: breaking the posttranslational modification code. Pathol Biol (Paris) 2000;48(3):227–245. [PubMed] [Google Scholar]
- 48.Appella E, Anderson CW. Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem 2001;268(10):2764–2772. [DOI] [PubMed] [Google Scholar]
- 49.Lavin MF, Gueven N. The complexity of p53 stabilization and activation. Cell Death Differ 2006;13(6):941–950. [DOI] [PubMed] [Google Scholar]
- 50.Borges HL, Chao C, Xu Y, Linden R, Wang JY. Radiation-induced apoptosis in developing mouse retina exhibits dose-dependent requirement for ATM phosphorylation of p53. Cell Death Differ 2004;11(5):494–502. [DOI] [PubMed] [Google Scholar]
- 51.Komiyama S, Taniguchi S, Matsumoto Y, Tsunoda E, Ohto T, Suzuki Y, Yin HL, Tomita M, Enomoto A, Morita A, Suzuki T, Ohtomo K, Hosoi Y, Suzuki N. Potentiality of DNA-dependent protein kinase to phosphorylate Ser46 of human p53. Biochem Biophys Res Commun 2004;323(3):816–822. [DOI] [PubMed] [Google Scholar]
- 52.Kurz EU, Douglas P, Lees-Miller SP. Doxorubicin activates ATM-dependent phosphorylation of multiple downstream targets in part through the generation of reactive oxygen species. J Biol Chem 2004;279(51):53272–53281. [DOI] [PubMed] [Google Scholar]
- 53.Lavin MF, Delia D, Chessa L. ATM and the DNA damage response. Workshop on ataxia-telangiectasia and related syndromes. EMBO Rep 2006;7(2):154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Siu WY, Lau A, Arooz T, Chow JP, Ho HT, Poon RY. Topoisomerase poisons differentially activate DNA damage checkpoints through ataxia-telangiectasia mutated-dependent and -independent mechanisms. Mol Cancer Ther 2004;3(5):621–632. [PubMed] [Google Scholar]
- 55.Meek DW. The p53 response to DNA damage. DNA Repair (Amsterdam) 2004;3(8–9):1049–1056. [DOI] [PubMed] [Google Scholar]
- 56.Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature 1997;387(6630):299–303. [DOI] [PubMed] [Google Scholar]
- 57.Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by mdm2. Cell 1997;91(3):325–334. [DOI] [PubMed] [Google Scholar]
- 58.Brune B, Schneiderhan N. Nitric oxide evoked p53-accumulation and apoptosis. Toxicol Lett 2003;139(2–3):119–123. [DOI] [PubMed] [Google Scholar]
- 59.Schneiderhan N, Budde A, Zhang Y, Brune B. Nitric oxide induces phosphorylation of p53 and impairs nuclear export. Oncogene 2003;22(19):2857–2868. [DOI] [PubMed] [Google Scholar]
- 60.Blattner C Regulation of p53: the next generation. Cell Cycle 2008;7(20):3149–3153. [DOI] [PubMed] [Google Scholar]
- 61.Boehme KA, Kulikov R, Blattner C. p53 stabilization in response to DNA damage requires Akt/PKB and DNA-PK. Proc Natl Acad Sci USA 2008;105(22):7785–7790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang S, Shi X. Mechanisms of Cr(VI)-induced p53 activation: the role of phosphorylation, mdm2 and ERK. Carcinogenesis 2001;22(5):757–762. [DOI] [PubMed] [Google Scholar]
- 63.Kruse JP, Gu W. SnapShot: p53 posttranslational modifications. Cell 2008;133(5):930–930 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu Y Regulation of p53 responses by post-translational modifications. Cell Death Differ 2003;10(4):400–403. [DOI] [PubMed] [Google Scholar]
- 65.Lambert PF, Kashanchi F, Radonovich MF, Shiekhattar R, Brady JN. Phosphorylation of p53 serine 15 increases interaction with CBP. J Biol Chem 1998;273(49):33048–33053. [DOI] [PubMed] [Google Scholar]
- 66.Bean LJ, Stark GR. Phosphorylation of serines 15 and 37 is necessary for efficient accumulation of p53 following irradiation with UV. Oncogene 2001;20(9):1076–1084. [DOI] [PubMed] [Google Scholar]
- 67.Saito S, Goodarzi AA, Higashimoto Y, Noda Y, Lees-Miller SP, Appella E, Anderson CW. ATM mediates phosphorylation at multiple p53 sites, including Ser(46), in response to ionizing radiation. J Biol Chem 2002;277(15):12491–12494. [DOI] [PubMed] [Google Scholar]
- 68.Saito S, Yamaguchi H, Higashimoto Y, Chao C, Xu Y, Fornace AJ, Jr.,, Appella E, Anderson CW Phosphorylation site interdependence of human p53 post-translational modifications in response to stress. J Biol Chem 2003;278(39):37536–37544. [DOI] [PubMed] [Google Scholar]
- 69.Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer 2004;4(10):793–805. [DOI] [PubMed] [Google Scholar]
- 70.Bean LJ, Stark GR. Regulation of the accumulation and function of p53 by phosphorylation of two residues within the domain that binds to Mdm2. J Biol Chem 2002;277(3):1864–1871. [DOI] [PubMed] [Google Scholar]
- 71.Joubel A, Chalkley RJ, Medzihradszky KF, Hondermarck H, Burlingame A. Identification of new p53 acetylation sites in Cos-1 cells. Mol Cell Proteomics 2009. (Epub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell 2008;133(4):612–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yadavilli S, Martinez-Ceballos E, Snowden-Aikens J, Hurst A, Joseph T, Albrecht T, Muganda PM. Diepoxybutane activates the mitochondrial apoptotic pathway and mediates apoptosis in human lymphoblasts through oxidative stress. Toxicol In Vitro 2007;21(8):1429–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]