

Abstract
Resistance to therapeutics such as trimethoprim-sulfamethoxazole has become an increasing problem in strains of methicillin-resistant S. aureus (MRSA). Clinically isolated trimethoprim-resistant strains reveal a double mutation, H30N/F98Y, in dihydrofolate reductase (DHFR). In order to develop novel and effective therapeutics against these resistant strains, we evaluated a series of propargyl-linked antifolate lead compounds for inhibition of the mutant enzyme. For the propargyl-linked antifolates, the F98Y mutation generates minimal (between 1.2- and 6-fold) losses of affinity and the H30N mutation generates greater losses (between 2.4- and 48-fold). Conversely, trimethoprim affinity is largely diminished by the F98Y mutation (36-fold) and is not affected by the H30N mutation. In order to elucidate a mechanism of resistance, we determined a crystal structure of a complex of this double mutant with a lead propargyl-linked antifolate. This structure suggests a resistance mechanism consistent both for the propargyl-linked class of antifolates and for trimethoprim that is based on the loss of a conserved water-mediated hydrogen bond.
Keywords: methicillin-resistant Staphylococcus aureus, dihydrofolate reductase, antifolates, trimethoprim
INTRODUCTION
Staphylococcus aureus is one of the leading contributors of bacterial hospital-acquired infections and in recent years, the frequency of community-acquired infections has also increased significantly (Kluytmans-Vandenbergh and Kluytmans, 2006; Kollef and Micek, 2006). Stemming from the notorious ability of S. aureus to rapidly develop resistance to many potent and diverse antibiotics, the increasing frequency of methicillin-resistant S. aureus (MRSA) infections is of particular concern. In addition to beta-lactam resistance, some MRSA strains are resistant to therapeutics including vancomycin and trimethoprim-sulfamethoxazole (Drew, 2007; Proctor, 2008). Trimethoprim (TMP) and sulfamethoxazole are potent and selective inhibitors of dihydrofolate reductase (DHFR) and dihydropteroate synthase, respectively, two essential enzymes in the folate biosynthetic pathway. This alarming capacity for resistance necessitates the elucidation of antibiotic resistance mechanisms in S. aureus and the development of new therapeutic strategies to combat wild-type and resistant MRSA infections.
Trimethoprim resistance arises in approximately 28 % of MRSA strains (Dale et al., 1997). The majority of TMP-resistant strains exhibit one of two alternative sets of chromosomal mutations: H30N/F98Y and H149R/F98Y (Dale et al., 1997). The shared F98Y mutation is believed to be almost completely responsible for the loss of affinity in the double mutant (Dale et al., 1997; Frey et al., 2009); it alone confers a 36-fold loss of activity against S. aureus DHFR (Sa(wt) DHFR). In addition to chromosomal mutations, evidence suggests that the transfer of exogenous S1 DHFR may also be responsible for trimethoprim resistance in some highly resistant strains (Archer et al., 1986; Dale et al., 1995; Dale et al., 1993; Heaslet et al., 2009).
In previous studies, we investigated the effects of the F98Y mutation in S. aureus DHFR for a series of novel propargyl-linked inhibitors based on the trimethoprim scaffold. From high-resolution crystal structures, we identified an alternate conformation of the cofactor NADPH that reduced the hydrophobic contacts between the residues in the active site of DHFR and the inhibitors, thus contributing to the resistance observed for these ligands in the background of the F98Y mutation (Frey et al., 2009). This discovery, coupled with the desire to design ligands effective against the clinically observed resistant mutants, prompted the structural investigation of additional mutant DHFR enzymes found in S. aureus, specifically Sa (H30N, F98Y) DHFR, one of the sets of mutations isolated from patients (Dale et al., 1997). Herein, we present for the first time a 2.4 Å crystal structure of this clinical mutant bound to a propargyl-linked inhibitor and a mechanism of resistance for Sa (H30N, F98Y) DHFR that involves the loss of a water-mediated hydrogen bond between the diaminopyrimidine moiety and residue Thr 111.
MATERIAL AND METHODS
Cloning, Expression and Enzyme Purification
We have previously reported the details of cloning experiments for the Sa (F98Y) construct in vector pET41 (Frey et al., 2009). Site-directed mutagenesis was used to change His 30 in Sa (F98Y) DHFR to Asn 30. Final clones for Sa (H30N, F98Y) DHFR were validated by sequencing. The recombinant Sa (H30N, F98Y) enzyme was over-expressed in E. coli BL21(DE3) cells and purified using nickel affinity chromatography. Size exclusion chromatography was used to desalt the protein into 20 mM Tris, 20 % glycerol, 0.5 mM EDTA and 2 mM DTT.
Enzyme Assays
Enzyme activity and inhibition assays were performed as previously described by monitoring the rate of NADPH oxidation by the DHFR enzyme at an absorbance of 340 nm (Frey et al., 2009; Beierlein et al., 2008; Bolstad et al., 2008; Liu et al., 2008). Inhibition was measured at least three times with inhibitor concentrations near the IC50 value. KM values were measured by performing enzyme activity assays at varying substrate concentrations of dihydrofolate. A Lineweaver-Burk plot was used to calculate the KM and Vmax for each enzyme (plots are shown in Supplementary Material Figure S1). Activity assays were measured at least three times in order to calculate an average KM value with standard deviations. KM and IC50 values were used to calculate Ki values for each enzyme and inhibitor, according to the methods of Cheng and Prusoff (Cheng and Prusoff, 1973).
Crystallization
Sa (H30N, F98Y) DHFR was crystallized using hanging-drop vapor diffusion. The purified enzyme (12 mg/mL) was incubated with ligand 2 (1 mM) and NADPH (2 mM) for 2 h on ice. Crystals of the protein:ligand:NADPH complex were optimized in a crystallization solution containing 15% PEG MW 10,000, 150 mM sodium acetate, and 100 mM MES pH 6.5, with a growth period of 5–7 days. High quality crystals were flash-cooled in mother liquor containing 10 % glycerol as the cryoprotectant.
Data Collection and Refinement
Diffraction data with amplitudes extending to 2.4 Ǻ were collected at Brookhaven National Synchrotron Light Source using beamline X29A. Data were indexed and scaled using HKL 2000 (Otwinowski and Minor, 1997). Programs Coot (Emsley and Cowtan, 2004) and Refmac (Murshudov et al., 1997) were used to build and refine the structure until an acceptable Rcryst and Rfree were achieved. The geometry of the structure was validated using Procheck (Laskowski et al., 1993) and Ramachandran plots. Data collection and refinement statistics are reported in Table 3. An omit 2 FO-FC map was generated using the Map and Mask Utilities function within the CCP4 program suite (Bhat and Cohen, 1984).
Table 3.
Statistics of Data Collection and Refinement for Sa(H30N/F98Y):NADPH:2
| Ligand | 2 |
| PDB ID | 3I8A |
| Space Group | P6122 |
| No. Molecules in Asymmetric Unit | 1 |
| Unit Cell (a,b,c in Å) | a=b=79.164, c=108.960 |
| Resolution, (last shell, Å) | 37.22 – 2.41 (2.47 – 2.41) |
| Completeness, % (last shell,%) | 95 (100) |
| Unique Reflections | 7504 |
| Redundancy (last shell) | 10.4 (10.1) |
| Rsym, % (last shell, %) | 0.040 (0.074) |
| <I/σ> (last shell) | 28.4 (23.4) |
| Refinement Statistics | |
| Rcryst/Rfree | 0.208, 0.261 |
| No. of atoms (protein, ligands, solvent) | 1272, 71, 47 |
| Rms deviation bond lengths (Å), angles (°) | 0.014, 1.640 |
| Average B factor (Å2) | 15.7 |
| Average B factor for ligands (Å2) | 12.4, 10.5 |
| Average B factor for solvent molecules (Å2) | 16.3 |
| Ramachandran Plot Statistics | |
| Residues in most favored regions, allowed regions (%) | 90.4, 9.6 |
RESULTS AND DISCUSSION
In previous work, we described a class of novel DHFR inhibitors that show potency for both Sa DHFR and Sa (F98Y) DHFR (Frey et al., 2009; Beierlein et al., 2008; Bolstad et al., 2008; Liu et al., 2008). Here we investigate whether those inhibitors are active against the clinically observed mutant Sa (H30N, F98Y) DHFR. In order to fairly assess potency differences, we first determined KM values for the Sa (wt), Sa (F98Y), and Sa (H30N, F98Y) DHFR enzymes (Table 1) and found that these values are comparable. We then measured IC50 values for each inhibitor using a spectrophotometric DHFR assay and calculated Ki values using the methods established by Cheng and Prusoff (Cheng and Prusoff, 1973). Comparing the calculated Ki values for each enzyme and inhibitor, we found that the H30N mutation impacts the propargyl-linked antifolates much differently than it impacts TMP (Table 2).
Table 1.
KM Data for Substrate Dihydrofolate
| Enzyme | KM (μM) |
|---|---|
| Sa (wt) | 14.5 ± 0.35 |
| Sa (F98Y) | 7.3 ± 0.40 |
| Sa (H30N, F98Y) | 8.2 ± 0.46 |
Table 2.
Ki Data for MRSA DHFR Inhibitors
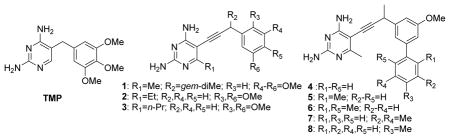 | |||||||
|---|---|---|---|---|---|---|---|
| No. | Saa (wt) Ki (μM) | Sa (F98Y) Ki (μM) | Loss Sa (F98Y)/wt | Sa (H30N, F98Y) Ki (μM) | Loss Sa (H30N, F98Y)/(F98Y) | Tot. Loss Sa (H30N, F98Y)/wt | NADPH occupancy Sa (F98Y) |
| TMP | 0.0033 | 0.12 | 36.4 | 0.11 | ~ 1.0 | 33.3 | 100% NAP1b |
| 1 | 0.0093 | 0.055 | 5.9 | 0.89 | 16.2 | 95.7 | 100% NAP2 |
| 2 | 0.0099 | 0.049 | 4.9 | 0.12 | 2.4 | 12.1 | 70% NAP1 30% NAP2 |
| 3 | 0.0070 | 0.040 | 5.7 | 0.27 | 6.8 | 38.6 | 100% NAP1 |
| 4 | 0.0088 | 0.014 | 1.6 | 0.16 | 11.4 | 18.2 | 100% NAP1 |
| 5 | 0.011 | 0.014 | 1.3 | 0.16 | 11.4 | 14.5 | N/Ac |
| 6 | 0.011 | 0.013 | 1.2 | 0.29 | 22.3 | 26.4 | 100% NAP1 |
| 7d | 0.0061 | 0.012 | 2.0 | 0.58 | 48.3 | 95.1 | 30% NAP1 70% NAP2 |
| 8 | 0.059 | 0.070 | 1.2 | 0.28 | 4.0 | 4.7 | N/A |
Standard deviations are shown in Supplementary Material Table S1.
NAP1 = standard conformation of NADPH, NAP2 = alternate conformation of NADPH
N/A: not applicable because structure with F98Y has not been determined
The structure of Sa (F98Y) DHFR with compound 7 is available in the Protein Data Bank (PDB ID: 3F0X)
The results from the assay data show that trimethoprim exhibits the greatest loss in potency between Sa (wt) and Sa (F98Y) DHFR, while the meta-biphenyl series of compounds (4–8) retain potency against the F98Y mutant with only a minimal 2-fold loss. Described in previous work, Sa (wt) and Sa (F98Y) DHFR structures bound to compound 4 reveal several hydrophobic interactions between the enzyme and the meta-biphenyl (Frey et al., 2009). The propargyl linker extends the meta-biphenyl into the hydrophobic pocket, making interactions that assist in retaining potency against the Sa (F98Y) DHFR enzyme. Interestingly, trimethoprim exhibits no loss in potency between Sa (F98Y) and Sa (H30N, F98Y) DHFR despite the fact that the clinical isolates most frequently contain this double mutation instead of the single F98Ymutation. However, the propargyl-based compounds lose potency with the addition of the H30N mutation. The H30N mutation has the greatest effect on compound 7, lowering its affinity for the enzyme by 48-fold. Lacking substitution at the propargyl linker, compound 2 is most similar to trimethoprim and is least affected by the H30N mutation. We conclude from these results that the F98Y mutation confers the greatest resistance to trimethoprim and the H30N mutation confers the greatest resistance to the propargyl-based compounds.
The affinity loss between the mutant enzymes suggests that the F98Y and H30N mutations have different yet synergistic effects on compound activity. We have previously reported that the F98Y mutation promotes an alternate conformation of NADPH (called NAP2) that significantly lowers the potency of the propargyl-based series, especially in compounds 1 and 2 that lack the meta-biphenyl substitution (Frey et al., 2009). Comparing the assay data with the available Sa (F98Y) crystal structures, it appears that there is a correlation between the occupancy of the alternate NADPH conformation and a decrease in overall compound potency. For example, compounds 1 and 7 have the greatest total loss in affinity and present the alternate NADPH conformation at 100 % and 70 % occupancies, respectively. In contrast, the total loss of affinity toward compounds 2–6 and 8 is lower, corresponding with a higher occupancy of the standard NADPH conformation (NAP1).
In order to determine the mechanism by which the F98Y, H30N mutations generate resistance, we determined a crystal structure of the double mutant bound to a compound from the series. The best crystals formed with Sa (H30N, F98Y) DHFR bound to NADPH and inhibitor 2. These crystals diffract to 2.41 Ǻ and share the same unit cell dimensions and space group P6122 as our previous Sa (wt) and Sa (F98Y) structures (Table 3). Thus, the structure was solved using difference Fourier methods with a model of the wild-type Sa DHFR bound to inhibitor 2 (Frey et al., 2009). The electron density map (Figures 1a, 1b) reveals a structure with the characteristic DHFR fold, containing an eight-stranded β sheet and four α helices connected by flexible loops. Overall similarities are shared with the structures of Sa (wt) and Sa (F98Y) DHFR bound to 2 and NADPH, with root mean square deviations of 0.169 and 0.238 Ǻ, respectively. It should also be noted that the standard, extended conformation of NADPH is present at 100 % occupancy.
Figure 1.

a) 2 Fo–Fc density (1.6 σ, gray) for compound 2 and residues Leu 5, Asp 27, Asn 30, Thr 111, and water molecule (red sphere), b) Omit 2 Fo–Fc density (1.3 σ, blue) for compound 2 and residues Leu 5, Asp 27, Asn 30, Phe 92, Thr 111 and a water molecule (red sphere),c) compound 2 and the residues in the active site.
Similar to the Sa (wt) and Sa (F98Y) structures, the acidic residue Asp 27 forms two hydrogen bonds with the protonated N1 atom and the 2-amino group of the diaminopyrimidine ring. In addition, the 4-amino group can form two additional hydrogen bonds with the backbone carbonyl of residues Leu 5 and Phe 92. Residues Leu 28 and Val 31 form van der Waals interactions with the ethyl substitution at the C6 position of the diaminopyrimidine. Additional hydrophobic interactions are formed between Leu 20 and Phe 92 with the propargyl linker, and Ile 50 with the dimethoxy phenyl ring (Figure 1c).
A comparison of the residues from Sa (wt), Sa (F98Y), and Sa (H30N, F98Y) DHFR involved in hydrogen bonds with the ligand (Fig. 2) reveals the mechanism of resistance resulting from the H30N mutation. In addition to the conserved hydrogen bonds between the ligand and active site residues mentioned above, the Sa (wt) and Sa (F98Y) DHFR structures show water-mediated hydrogen bonds between Thr 111 and the 2-amino group of the diaminopyrimidine of compound 2 (Figure 2a,b), with distances of 2.98 or 3.04 Ǻ, respectively. In fact, this water molecule and its interaction with the 2-amino group appear to be conserved. A survey of eight additional crystal structures of Sa (wt) and Sa (F98Y) DHFR bound to inhibitors in this class demonstrates that the average distance of the hydrogen bond between the water molecule and the 2-amino group is 2.98 ± 0.09 Ǻ (Table S2 in Supplementary Material lists each structure and associated hydrogen bond length). Comparing these structures with Sa (H30N, F98Y) DHFR (Fig. 2c), the distance between the water molecule and the 2-amino group is 3.40 Ǻ, a significantly longer and weaker interaction. Instead, the water molecule in the double mutant structure forms a hydrogen bond with the carboxyamide of the Asn 30 mutation. Absence of the water-mediated hydrogen bond with the diaminopyrimidine moiety may contribute to the overall loss in potency of the propargyl-based compounds with the double mutant as observed in the enzyme inhibition assays.
Figure 2.

Residues in the active site that form hydrogen bonds to the diaminopyrimidine ring of compound 2 (lavender) for: (a) Sa (wt) DHFR (green) (PDB ID: 3FQ0); (b) Sa (F98Y) DHFR (cyan) (PDB ID: 3FQO); and (c) Sa (H30N, F98Y) DHFR (pink) (PDB ID: 3I8A). Water molecules are shown as colored spheres to match the enzyme. Interactions that are within hydrogen bonding distance are represented by dashed lines in black. Weakened hydrogen bonds are represented by dashed lined in red.
Interestingly, the structure of wild-type Sa DHFR bound to trimethoprim and NADPH reveals a water molecule at a similar location; the distance between the water molecule and the 2-amino group is 3.5 Ǻ (Heaslet et al., 2009). By extension from our results with the propargyl-linked inhibitors, it is likely that a hydrogen bond is formed between the mutated residue Asn 30 and this water molecule in structures bound to trimethoprim. In doing so, only a weak interaction between the water molecule and 2-amino group is lost, correlating with the minimal change in Ki values between the F98Y and H30N, F98Y enzymes. Given this minimal effect, at this time it is not clear what the role of the H30N mutation is in clinical isolates of TMP-resistant strains.
Our previous work identified a structural mechanism of resistance for Sa (F98Y) DHFR involving the presence of an alternate NADPH conformation that prevented several hydrophobic interactions. We concluded that the F98Y mutation influenced the equilibrium of the alternate conformation in a ligand-dependent manner (Frey et al., 2009). Here we conclude that the Asn 30 mutation prevents the formation of a water-mediated hydrogen bond with the ligand, thereby lowering the potency of compound 2. The extended conformation of NADPH is present at 100 % occupancy in the Sa (H30N, F98Y) DHFR structure with 2. However, we hypothesize that for other compounds such as 1 and 7 that exhibit full or 70 % occupancy of the alternate NADPH conformation in structures with Sa (F98Y) DHFR (Frey et al., 2009), respectively, the loss of the hydrogen bond from the H30N mutation and the loss of hydrophobic interactions from an alternate NADPH conformation may collectively lower their potency, explaining the 95-fold losses.
With the identification of several new and diverse mutations, the elucidation of mutational resistance mechanisms in S. aureus DHFR is essential for next generation inhibitor design. Interestingly, from these studies we have learned that although TMP and compound 2 have similar overall potency against the doubly mutated enzyme, the structural origins for the loss of potency differ. This difference should allow for the design of superior propargyl-based inhibitors that remain relatively insensitive to the F98Y mutation while simultaneously overcoming the effects of the H30N mutation. For example, an extension of the 2-amino group on the diaminopyrimidine ring would decrease the interaction distance with the water molecule and increase the strength of the hydrogen bond. Using this extension on the meta-biphenyl compounds such as 4 would also enable the inhibitor to maintain efficacy against the F98Y mutation with increased hydrophobic interactions. This structure-based strategy for improving the propargyl-based inhibitors for the trimethoprim-resistant strains of MRSA will be explored in future efforts.
Supplementary Material
Acknowledgments
The authors thank the NIH for funding (GM067542 and AI073375 to A.C.A. and AI065143 to D.L.W.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Archer GL, Coughter JP, Johnston JL. Plasmid-encoded trimethoprim resistance in staphylococci. Antimicrob Agents Chemother. 1986;29:733–740. doi: 10.1128/aac.29.5.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beierlein JM, Frey KM, Bolstad DB, Pelphrey PM, Joska TM, Smith AE, Priestley ND, Wright DL, Anderson AC. Synthetic and crystallographic studies of a new inhibitor series targeting Bacillus anthracis dihydrofolate reductase. J Med Chem. 2008;51:7532–7540. doi: 10.1021/jm800776a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat TN, Cohen GH. OMITMAP: An electron density map suitable for the examination of errors in a macromolecular model. J Appl Cryst. 1984;17:244–248. [Google Scholar]
- Bolstad DB, Bolstad ES, Frey KM, Wright DL, Anderson AC. Structure-based approach to the development of potent and selective inhibitors of dihydrofolate reductase from Cryptosporidium. J Med Chem. 2008;51:6839–6852. doi: 10.1021/jm8009124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Dale GE, Broger C, Hartman PG, Langen H, Page MG, Then RL, Stuber D. Characterization of the gene for the chromosomal dihydrofolate reductase (DHFR) of Staphylococcus epidermidis ATCC 14990: the origin of the trimethoprim-resistant S1 DHFR from Staphylococcus aureus? J Bacteriol. 1995;177:2965–2970. doi: 10.1128/jb.177.11.2965-2970.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale GE, Broger C, D’Arcy A, Hartman PG, DeHoogt R, Jolidon S, Kompis I, Labhardt AM, Langen H, Locher H, Page MG, Stuber D, Then RL, Wipf B, Oefner C. A single amino acid substitution in Staphylococcus aureus dihydrofolate reductase determines trimethoprim resistance. J Mol Biol. 1997;266:23–30. doi: 10.1006/jmbi.1996.0770. [DOI] [PubMed] [Google Scholar]
- Dale GE, Then RL, Stuber D. Characterization of the gene for chromosomal trimethoprim-sensitive dihydrofolate reductase of Staphylococcus aureus ATCC 25923. Antimicrob Agents Chemother. 1993;37:1400–1405. doi: 10.1128/aac.37.7.1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew RH. Emerging options for treatment of invasive, multidrug-resistant Staphylococcus aureus infections. Pharmacotherapy. 2007;27:227–249. doi: 10.1592/phco.27.2.227. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr Sect D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Frey KM, Liu J, Lombardo MN, Bolstad DB, Wright DL, Anderson AC. Crystal structures of wild-type and mutant methicillin-resistant Staphylococcus aureus dihydrofolate reductase reveal an alternate conformation of NADPH that may be linked to trimethoprim resistance. J Mol Biol. 2009;387:1298–1308. doi: 10.1016/j.jmb.2009.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaslet H, Harris M, Fahnoe K, Sarver R, Putz H, Chang J, Subramanyam C, Barreiro G, Miller JR. Structural comparison of chromosomal and exogenous dihydrofolate reductase from Staphylococcus aureus in complex with the potent inhibitor trimethoprim. Proteins. 2009;76:706–717. doi: 10.1002/prot.22383. [DOI] [PubMed] [Google Scholar]
- Kluytmans-Vandenbergh MF, Kluytmans JA. Community-acquired methicillin-resistant Staphylococcus aureus: current perspectives. Clin Microbiol Infect. 2006;12:9–15. doi: 10.1111/j.1469-0691.2006.01341.x. [DOI] [PubMed] [Google Scholar]
- Kollef MH, Micek ST. Methicillin-resistant Staphylococcus aureus: a new community-acquired pathogen? Curr Opin Infect Dis. 2006;19:161–168. doi: 10.1097/01.qco.0000216627.13445.e2. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283. [Google Scholar]
- Liu J, Bolstad DB, Smith AE, Priestley ND, Wright DL, Anderson AC. Structure-guided development of efficacious antifungal agents targeting Candida glabrata dihydrofolate reductase. Chem Biol. 2008;15:990–996. doi: 10.1016/j.chembiol.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr Sect D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Proctor RA. Role of folate antagonists in the treatment of methicillin-resistant Staphylococcus aureus infection. Clin Infect Dis. 2008;46:584–593. doi: 10.1086/525536. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.