

Abstract
The systemic vasculitides are a complex and often serious group of disorders which, while uncommon, require careful management in order to ensure optimal outcome. In most cases there is no known cause. Multi-system disease is likely to be fatal without judicious use of immunosuppression. A prompt diagnosis is necessary to preserve organ function. Comprehensive and repeated disease assessment is a necessary basis for planning therapy and modification of treatment protocols according to response. Therapies typically include glucocorticoids and, especially for small and medium vessel vasculitis, an effective immunosuppressive agent. Cyclophosphamide is currently the standard therapy for small vessel multi-system vasculitis, but other agents are now being evaluated in large randomized trials. Comorbidity is common in patients with vasculitis, including the cumulative effects of potentially toxic therapy. Long-term evaluation of patients is important in order to detect and manage relapses.
Keywords: ANCA, giant cell arteritis, microscopic polyangiitis, vasculitis, Wegener's granulomatosis
Introduction
Primary systemic vasculitis has an incidence of more than 100 new cases per million [1]. Pathogenic mechanisms remain uncertain, although understanding the viral aetiology of some forms of polyarteritis nodosa (linked to hepatitis B) and cryoglobulinaemic vasculitis (linked to hepatitis C) has allowed a more tailored management approach [2,3]. Despite a significant reduction in mortality as a result of standard immunosuppression, most patients experience poor quality of life, characterized by relapse, persisting low-grade disease activity and increasing burden of drug toxicity [4–6]. Factors influencing remission, relapse and survival include type of immunosuppressive therapy, type of organ involvement, presence of anti-neutrophil cytoplasm antibodies (ANCA), older age and male gender [7]. A structured approach, based on careful disease staging and evaluation, is the cornerstone of good disease management [8]. The relationship between ANCA and Wegener's granulomatosis and microscopic polyangiitis suggests a pathogenic role [9]. Targeting ANCA or monitoring levels to assess disease activity have both been attempted as treatment strategies, but with limited success [10–12].
Initial evaluation includes a comprehensive clinical assessment, serological tests, histology and radiology. For subsequent evaluations, it is effective and practical to measure clinical disease status for most patients with small and medium vessel vasculitis [8]. For large vessel disease such as Takayasu's arteritis, while radiological assessment of vascular anatomy is possible, the correlation of imaging findings may be poor [13].
Therapy is based on the pattern of vasculitis and on careful evaluation of the extent and activity of disease. We will review the evidence for treatment including glucocorticoids and immunosuppressive agents in different forms of vasculitis. There is increasing experience in the use of more specific biological therapies in patients with vasculitis which will also be discussed.
Consequences of missed or delayed diagnosis
The subtlety and diversity of symptoms in the initial phase of vasculitis can be a real diagnostic problem, and thus early recognition of a vasculitic condition relies on the experience of a team of dedicated professionals from several different subspecialties, including laboratory medicine. The fact that systemic vasculitides are uncommon and may present in different guises [14] makes centralization of diagnostics, follow-up and therapy a strategic goal to avoid irreversible damage [15,16].
The course of systemic vasculitis differs considerably from one patient to another. For example, a patient with early Wegener's granulomatosis in the nose, ear or sinuses may not have detectable lung or renal involvement. Early diagnosis and treatment would aim to reduce upper airway damage and hearing loss. If involvement of the lungs or glomeruli were to occur later the clinical situation would alter significantly, as more potent and potentially toxic immunosuppressive therapy would be necessary to rescue vital organ functions. If the clinical onset is manifested mainly by renal disease, the underlying systemic vasculitic condition may take longer to diagnose. The consequences can be detrimental because kidney function is often lost very quickly, and irreversible changes in the glomeruli may have occurred by the time diagnosis is made [5].
Missed or delayed diagnosis influences prognosis strongly if critical organs are involved, and less so when structurally and functionally less critical organs are affected. Careful management, with long-term follow-up, attempts to preserve health. Economic consequences will depend on the health cost for the patient and society as a result of damage.
Clinical history and examination
A systematic approach to diagnosis and follow-up will take into account the relapsing remitting nature of the disease, damage caused by low-grade grumbling disease and side effects of medication. Active inflammation requires an aggressive approach, which is entirely inappropriate in quiescent disease with extensive scarring, although the features of the clinical presentation may overlap.
The initial assessment will be to make a diagnosis, categorize disease severity and formulate a management plan. Subsequent assessments review the success of treatment and detect new organ involvement. The Birmingham Vasculitis Activity Score (BVAS) may be used to summarize this information systematically. Assessment of damage provides clinical and prognostic information on organ scarring caused by the disease and its treatment but does not represent ongoing active inflammation. Suitable tools for this include the Vasculitis Damage Index (VDI) and Disease Extent Index (DEI). Finally, assessment of function considers the overall impact of the disease on the physical, social and psychological function, including quality of life and employment. Tools include the Short Form 36 (SF36) and Health Assessment Questionnaire (HAQ), which are questionnaire-based.
Assessment of large vessel vasculitis
Clinical assessment of patients with giant cell arteritis and Takayasu's arteritis includes palpation of peripheral pulses for asymmetry, bilateral blood pressure assessment, auscultation for bruits and laboratory tests for evidence of systemic inflammation. Further diagnostic information is provided by temporal artery biopsy (TAB) in giant cell arteritis and imaging of the arterial tree by conventional angiography, magnetic resonance imaging (MRI) or positron emission tomography (PET) [17].
Temporal artery biopsy is the gold standard investigation for giant cell arteritis [17]. However, it is not 100% specific or sensitive due to the presence of skip lesions. A positive biopsy is associated with a history of jaw claudication and diplopia, and temporal artery beading, prominence and tenderness on examination [18].
Medium and small vessel vasculitis
The European Vasculitis Study Group recommends the use of structured clinical assessment and that patients with ANCA-associated systemic vasculitis (AASV) are categorized according to disease severity to guide treatment decisions [19].
A number of clinical tools are available to provide a detailed description of the patient's clinical status to aid diagnosis, treatment decisions and assist in measuring response to therapy including the BVAS, VDI DEI and the Five Factor Score (FFS).
The BVAS is the current standard assessment tool to score disease activity in systemic vasculitis [20–23]. It includes 66 clinical features divided into nine organ systems. Each item has a numerical value according to its clinical relevance. Items are scored only if attributable to active vasculitis. This is based on clinical judgement and difficulties arise when distinguishing between ongoing active vasculitis and symptoms due to scars without active disease. Training in scoring is recommended to reduce interobserver variation by overscoring for infection or established disease features due to scars [24]. A simplified checklist of BVAS items is shown in Table 1. While most patients are unlikely to have all the abnormalities listed, the spectrum covered by BVAS accounts for most of the features present in individual patients with different forms of vasculitis.
Table 1.
Disease activity items recorded routinely in assessment of systemic vasculitis (adapted from Flossmann et al. [8]).
| 1.General | 6.Cardiovascular |
| Myalgia | Loss of pulses |
| Arthralgia or arthritis | Valvular heart disease |
| Fever > 38·0°C | Pericarditis |
| Weight loss > 2 kg | Ischaemic cardiac pain |
| 2.Cutaneous | Cardiomyopathy |
| Infarct | Congestive cardiac failure |
| Purpura | 7.Abdominal |
| Ulcer | Peritonitis |
| Gangrene | Bloody diarrhoea |
| Other skin vasculitis | Ischaemic abdominal pain |
| 3.Mucous membranes/eyes | 8.Renal |
| Mouth ulcers/granulomata | Hypertension |
| Genital ulcers | Proteinuria > 1+ |
| Adnexal inflammation | Haematuria ≥ 10 rbc/hpf |
| Significant proptosis | Raised creatinine (> 125 µmol/l) |
| Red eye (epi)scleritis | Rise in creatinine > 30% or fall in GFR > 25% |
| Red eye conjunctivitis/blepharitis/keratitis | 9.Nervous system |
| Blurred vision | Headache |
| Sudden visual loss | Meningitis |
| Uveitis | Organic confusion |
| Retinal change (vasculitis/thrombosis/retinal exudates/haemorrhage) | Seizures (not hypertensive) |
| 4.ENT | Stroke |
| Bloody nasal discharge/nasal discharge/Crusts/ulcers and/or granulomata | Cord lesion |
| Paranasal sinus involvement | Cranial nerve palsy |
| Subglottic stenosis | Sensory peripheral neuropathy |
| Conductive hearing loss | Motor mononeuritis multiplex |
| Sensorineural hearing loss | |
| 5.Chest | |
| Wheeze | |
| Nodules or cavities | |
| Pleural effusion/pleurisy | |
| Infiltrate | |
| Endobronchial involvement | |
| Massive haemoptysis/Alveolar haemorrhage | |
| Respiratory failure |
ENT, ear/nose/throat; GFR, glomerular filtration rate.
The DEI is validated against the BVAS in Wegener's granulomatosis [25] and scores the number of organ systems affected by medium vessel vasculitis. It can be calculated as a subset of BVAS items, and complements the BVAS score.
The FFS evaluates disease activity at the time of diagnosis and was developed to evaluate the initial severity of vasculitis [26]. It provides a prognostic indication and guide to the intensity of treatment for patients with polyarteritis nodosa and Churg–Strauss syndrome [26,27]. It has also been applied to microscopic polyangiitis [28]. It scores the presence of serum creatinine above 1·58 mg/dl, proteinuria above 1 g/day, severe gastrointestinal tract involvement, cardiomyopathy and central nervous system involvement. It is not appropriate for follow-up, and is complementary to the BVAS. It is not entirely satisfactory, as the 5-year mortality is 12% with none of the risk factors. It is up to 46% with two or more risk factors and 45·95% when three or more of the five factors are present [26].
The VDI is a cumulative score describing long-term outcomes for vasculitis patients [29]. It contains 64 items in 11 organ-based systems and defines damage as an irreversible scar present longer than 3 months. Table 2 lists the items routinely recorded in the VDI, and serves as a checklist of possible outcomes in patients with longstanding disease.
Table 2.
Disease damage items routinely recorded using Vasculitis Damage Index (adapted from Flossmann et al. 2007 [8]).
| 1.General | 7.Peripheral vascular disease |
| Significant muscle atrophy or weakness | Absent limb pulses |
| Deforming/erosive arthritis | Major vessel stenosis |
| Osteoporosis/vertebral collapse | Claudication > 3 months |
| Avascular necrosis | Minor tissue loss |
| Osteomyelitis | Major tissue loss |
| 2.Skin/mucous membranes | Complicated venous thrombosis |
| Alopecia | 8.Gastrointestinal |
| Cutaneous ulcers | Gut infarction/resection |
| Mouth ulcers | Mesenteric insufficiency/pancreatitis |
| 3.Ocular | Chronic peritonitis |
| Cataract | Oesophageal stricture/surgery |
| Retinal change | 9.Renal |
| Optic atrophy | Estimated/measured GFR ≤ 50% |
| Visual impairment/diplopia | Proteinuria ≥ 0·5 g/24 h |
| Blindness | End stage renal disease |
| Orbital wall destruction | 10.Neuropsychiatric |
| 4.ENT | Cognitive impairment |
| Hearing loss | Major psychosis |
| Nasal blockage/chronic discharge/crusting | Seizures |
| Nasal bridge collapse/septal perforation | Cerebrovascular accident |
| Chronic sinusitis/radiological damage | Cranial nerve lesion |
| Subglottic stenosis | Peripheral neuropathy |
| 5.Pulmonary | Transverse myelitis |
| Pulmonary hypertension | 11.Other |
| Pulmonary fibrosis | Gonadal failure |
| Pulmonary infarction | Marrow failure |
| Pleural fibrosis | Diabetes |
| Chronic asthma | Chemical cystitis |
| Chronic breathlessness | Malignancy |
| Impaired lung function | |
| 6.Cardiovascular | |
| Angina/angioplasty | |
| Myocardial infarction | |
| Cardiomyopathy | |
| Valvular disease | |
| Pericarditis ≥ 3 months or pericardectomy | |
| Diastolic BP ≥ 95 or requiring anti-hypertensives |
ENT, ear/nose/throat; GFR, glomerular filtration rate.
Investigations
Laboratory
Initial investigations include full blood count, inflammatory markers [C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR)], renal function such as epidermal growth factor receptor (eGFR) and serology to include anti-glomerular basement membrane antibodies. Inflammatory markers provide a non-specific tool for assessing inflammatory activity and monitoring treatment. Urinalysis detects proteinuria and haematuria which can be assessed further for red cell casts indicating active renal inflammation or a quantification of protein loss with a 24-h urine collection or protein : creatinine ratio. Urine infection should also be excluded. Liver function should be assessed prior to starting disease-modifying agents such as methotrexate. Ovarian function may be assessed prior to cyclophosphamide in women of child-bearing age with measurements of follicle stimulating hormone (FSH), luteinizing hormone (LH) [30] or anti-Müllerian hormone (AMH) levels [31] to provide information prior to fertility counselling.
Anti-neutrophil cytoplasm antibodies (ANCA)
Characteristic autoantibodies are formed towards enzymes and bactericidal proteins within the cytoplasmic granules of neutrophils and monocytes in a substantial proportion of patients with systemic vasculitis manifesting as Wegener's granulomatosis, microscopic polyangiitis and Churg–Strauss syndrome, as well as in patients with limited forms of these conditions. These include renal-limited necrotizing crescentic glomerulonephritis, subglottic stenosis and retrobulbar pseudotumour [15,32]. However, there is a cohort of patients with the same diseases who never manifest ANCA, which may represent an independent disease entity [33].
ANCA are demonstrated by a combination of indirect immunofluorescence (IIF) screening techniques using whole leucocyte smears as substrate to certify the neutrophil-specific reactivity, followed by a form of solid phase assay using isolated autoantigen as target [e.g. enzyme-linked immunosorbent assay (ELISA)][34]. Thus the mere identification of neutrophil-specific autoantibodies (NSA) by IIF does not directly indicate the presence of ANCA [35].
ANCA divide into two main classes: C-ANCA or classical cytoplasmic ANCA (Fig. 1) and P-ANCA or perinuclear-staining ANCA (Fig. 2). The classical granular staining pattern (C-ANCA), seen initially by IIF in rapidly progressive glomerulonephritis patients and Wegener's granulomatosis patients, indicated clearly that the autoantigen was located in granules of neutrophils and monocytes, and the nature of the proteinase 3 (PR3) antigen was revealed [36] as well as its surface expression [37]. As is the case with other IIF screening techniques, the autoantigen may differ even if the staining pattern is the same. International collaborative studies have helped define the diagnostic value of combining ANCA by IIF and antigen-specific ELISA using PR3 and myeloperoxidase (MPO) antigens [38].
Fig. 1.
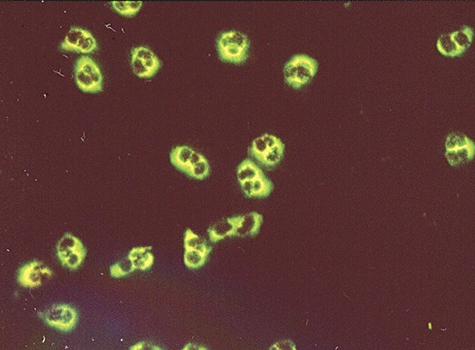
Indirect immunofluorescence pattern of cytoplasmic anti-neutrophil cytoplasm antibody (c-ANCA) from a patient with Wegener's granulomatosis.
Fig. 2.
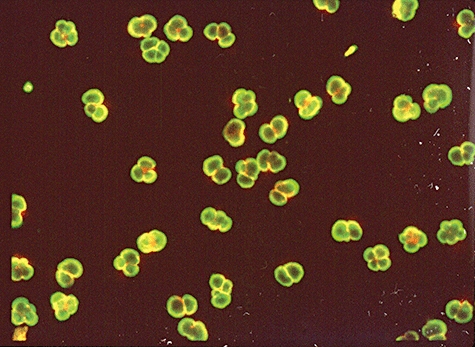
Indirect immunofluorescence pattern of perinuclear anti-neutrophil cytoplasm antibody (p-ANCA) from a patient with microscopic polyangiitis.
Since then, many different solid phase assays to detect PR3-ANCA have been produced and shown to aid in the diagnosis of vasculitis using serum or plasma [39]. Most assays today employ PR3 isolated from human neutrophils [40] by a method that preserves the conformation of the molecule, and attachment of PR3 molecules is accomplished either directly by coating onto some plastic surface (microwells, beads or other particles) or indirectly through attachment via bound specific mouse monoclonal antibody or a linker molecule that does not interfere with important epitopes for human PR3-ANCA reactivity [41]. Less common is the use of recombinant PR3 as antigen. There are data to suggest that ELISAs based on indirect binding of PR3 by a capture technique is superior to direct ELISAs in predicting flares of vasculitis [42], but there is no general agreement about this. Such monitoring would most probably have to involve weekly or biweekly testing to be able to catch an ANCA rise and thus predict imminent flares.
A P-ANCA staining pattern on neutrophils (Fig. 2) and monocytes is found commonly in patients with different chronic inflammatory diseases, e.g. rheumatoid arthritis, ulcerative colitis and chronic hepatitis, and verification that such antibodies are directed specifically to MPO is mandatory to be useful for diagnosing vasculitis [35]. Even then, it is important to emphasize that P-ANCA directed against MPO is not a specific marker for any of the small vessel vasculitides, as anti-MPO positivity occurs in many non-vasculitic disorders. The P-ANCA staining pattern can thus be caused by antibodies to several hydrophilic autoantigens in neutrophils that dislocate from their original site of placement onto neighbouring structures, e.g. the nucleus and its adjacent structures upon fixation of the cells in ethanol or acetone.
A P-ANCA staining pattern can be produced with autoantibodies to MPO, leucocyte elastase, cathepsin G, lactoferrin, azurocidin and lysozyme. If a P-ANCA is not caused by MPO-ANCA, the other specificities may be looked for by separate assays [43], but in practice this is not conducted unless there is a firm suspicion of a drug-induced condition, e.g. lupus-like syndrome or drug-induced vasculitis, where ANCA directed to one or more of these antigens are common [44].
Pathogenicity of ANCA
Although ANCA do not fulfil traditional immunological criteria for pathogenicity of autoantibodies, there is substantial evidence attesting to the biological activity of ANCA in terms of stimulation of the neutrophil respiratory burst, induction of cytokine release and increased adhesion to cultured endothelium [45]. However, the occurrence of ANCA in a variety of non-vasculitic disorders suggests that ANCA are heterogeneous in their biological activity and, consequently, their pathogenicity.
Animal models offer support for a direct pathogenic role for ANCA IgG in human glomerulonephritis and vasculitis. Splenocytes from MPO knock-out mice immunized with mouse MPO induce glomerulonephritis, granulomatous inflammation and systemic necrotizing vasculitis when injected into mice lacking functioning B and T lymphocytes. Anti-MPO IgG is able to cause pauci-immune glomerular necrosis and crescent formation in mice without functioning T or B lymphocytes, and in the presence of an intact immune system [46]. A model for PR3-ANCA-associated vasculitis is not yet available, and transfer of mouse PR3-ANCA containing immunoglobulin (Ig)G to wild-type mice induced a local increase of inflammation, but not systemic vasculitis [47].
ANCA-negative vasculitides
Most cases of predominantly cutaneous leucocytoclastic vasculitis as defined in the Chapel Hill nomenclature proposal (Table 5) [48] are negative in PR3-ANCA and MPO-ANCA tests if the positive cut-off value has been set at a clinically meaningful differential diagnostic level towards vasculitis-mimicking diseases [38]. Although ANCA-negative cases of Wegener's granulomatosis and microscopic polyangiitis are assumed to exist, we need to remember that ANCA levels can fluctuate between positive and negative, and thus periods of positive ANCA may be missed. Even in typical cases of Wegener's granulomatosis ANCA may be negative before and during a disease exacerbation, and other autoantibodies having the potential to mediate abnormal interaction between endothelial cells and neutrophils are likely to play a role in the pathogenesis and be reflected by findings in serum (reviewed in [49]).
Table 5.
Definitions of small vessel vasculitis.
| Small sized vessel vasculitis involving venules, capillaries and arterioles | |
|---|---|
| Wegener's granulomatosis | •Triad of upper and lower airways plus renal disease Limited forms are recognized but may progress to generalized disease |
| •Granulomatous inflammation involving respiratory tract | |
| •Necrotizing vasculitis affecting small- to medium-sized vessels of glomerulus | |
| Churg–Strauss syndrome | •Necrotizing vasculitis affecting small- to medium-sized vessels |
| •Eosinophil-rich and granulomatous inflammation involving respiratory tract | |
| •Often associated with asthma and eosinophilia | |
| Microscopic polyangiitis | •Necrotizing vasculitis of small- and medium-sized vessels of glomerulus and pulmonary capillaries |
| Henoch–Schonlein purpura | •Vasculitis with immunoglobulin A-dominant immune deposits in small vessels |
| •Involves skin, gut, glomeruli | |
| •Associated with arthralgia or arthritis | |
| Cryoglobulinaemic vasculitis | •Vasculitis with cryoglobulin immune deposits in small vessels |
| •Associated with mixed cryoglobulins in serum | |
| •Involves skin and glomeruli. | |
| •Associated with hepatitis C infection | |
| Cutaneous leucocytoclastic angiitis | •Isolated cutaneous leucocytoclastic angiitis without systemic vasculitis or glomerulonephritis |
Histology
Histological examination of biopsy material is useful in confirming a diagnosis in the context of clinical findings and laboratory data. It is considered the gold standard investigation in certain vasculitides; for example, a temporal artery biopsy in suspected giant cell arteritis. The focal nature of the disease and presence of skip lesions can give sampling problems. A negative biopsy does not necessarily exclude disease, and a positive biopsy does not always indicate the presence of disease [50]. Renal biopsy may be particularly useful in diagnosis of AASV and exclusion of other diseases such as malignancy or infection. Renal histological features provide an indication of prognosis in ANCA-associated glomerulonephritis [51] and can differentiate between diagnostic and serological subgroups [52]. In the presence of scarring with functional damage, histological examination may provide the only means of excluding active inflammation and guiding therapeutic decisions.
Large vessel vasculitis
Histological changes start with a patchy inflammatory infiltrate, including giant cells, which may form granulomata in the vessel wall [53]. Inflammation initially involves the outer portion of the vessel wall. Characteristically, the elastic lamina is destroyed and replaced with fibrous tissue, an observation which helps to differentiate vasculitis from the changes of atherosclerosis [54]. In the longer term the vessel wall is greatly thickened. Histological features are similar for Takayasu's arteritis and giant cell arteritis but affect different vessels (Fig. 3).
Fig. 3.
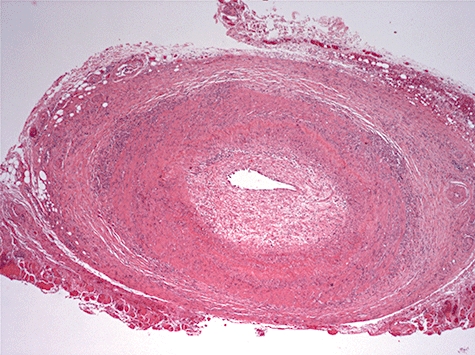
Cross-sectional low-power view of temporal artery biopsy from a patient with new-onset headache due to giant cell arteritis, showing an intense inflammatory infiltrate in the adventitia and media, and proliferation of the intima with narrowing of the lumen.
Medium vessel vasculitis
Classical histological changes include fibrinoid necrosis of the vessel wall accompanied by a chronic inflammatory infiltrate. It is segmental in nature and, characteristically, affected and unaffected vessels may be seen in the same section. As in large vessel vasculitis, there is loss of large portions of the elastic lamina, various numbers of giant cells and granulomata and development of long-term fibrosis and aneurysms.
Small vessel vasculitis
Vasculitic lesions are seen typically in the capillary beds. This may involve skin, lungs and kidney, with necrosis, fibrin deposition and leucocytoclasia, i.e. cell debris, and a mixture of neutrophils and lymphocytes. Henoch–Schonlein purpura, cryoglobulinaemia and vasculitis associated with collagen vascular disease typically demonstrate deposition of immune complexes, whereas ANCA-positive vasculitides do not [53]. The classic Wegener's granulomatosis granulomatous lesion is seen in the lung, but is not always present and vasculitis may be indicated only by the presence of capillaritis with haemorrhage. Granulomatous lesions are not always present and may be a late feature of disease development [55]. Figures 4–7 demonstrate the histological changes of vasculitic neuropathy, skin, kidney and nasal lesions, respectively. Figure 8 shows the rash of Henoch–Schonlein purpura and Fig. 9 demonstrates a skin granulomatous lesion in Wegener's granulomatosis.
Fig. 4.
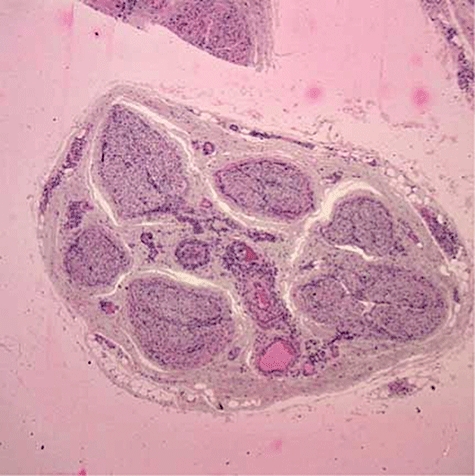
Low-power view of a sural nerve biopsy from a patient with mononeuritis multiplex showing increased cellularity in relation to perineural and epineural vessels (acknowledgements to Dr Colin Smith, Senior Lecturer/Consultant Pathologist, Pathology Department, University of Edinburgh).
Fig. 7.
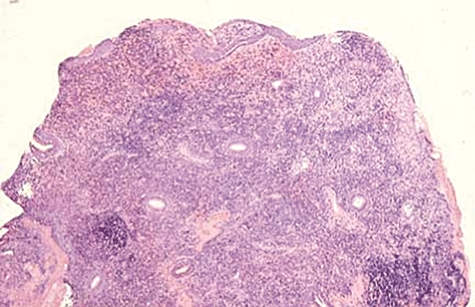
Nasal biopsy showing dense infiltration of lymphocytes and a large granulomatous mass.
Fig. 8.
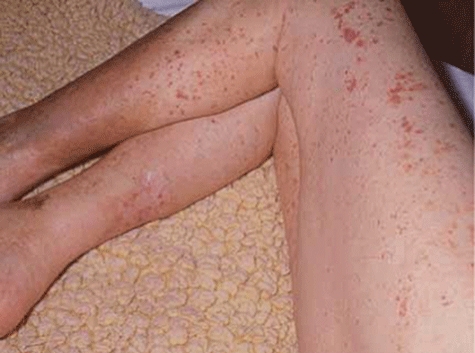
Henoch–Schonlein purpura showing recent-onset purpuric rash on calves with coalescence of lesions on the thighs secondary to pressure. There are haemosiderin deposits from previous episodes of purpura and a scar on the right calf from a healed ulcer.
Fig. 9.
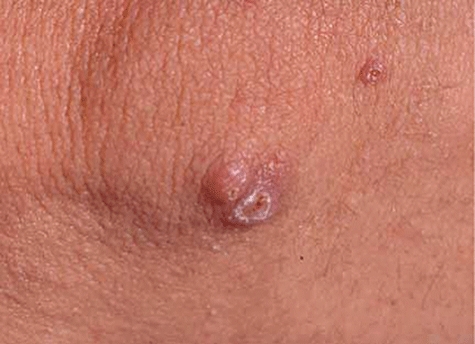
Nodulopapular lesions on the elbow in a patient with Wegener's granulomatosis. These lesions are mobile in the dermis and may ulcerate.
Fig. 5.
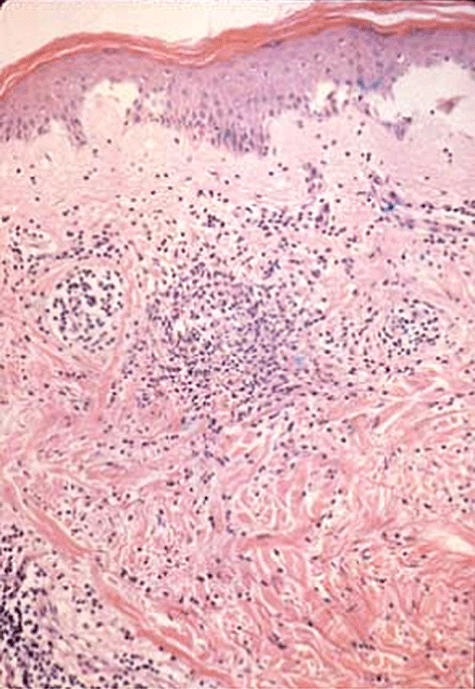
Skin biopsy of a vasculitic rash showing dense infiltration of the subcutaneous tissue.
Fig. 6.
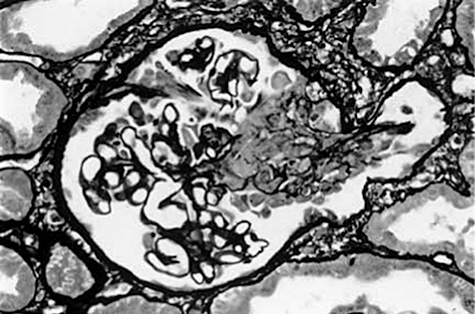
Renal biopsy to show diffuse proliferative pauci-immune glomerulonephritis with basement membrane ruptures and cells in Bowman's space and an associated interstitial reaction (acknowledgements to Dr Lorraine Harper, Educational Director, The Wellcome Trust Clinical Research Facility at the Queen Elizabeth Hospital, Birmingham).
Imaging
Imaging has a dual role in the assessment of vasculitis by providing information on vessel pathology for large and medium vessel vasculitis and by characterizing organ damage in small vessel vasculitis. Figure 10 shows consolidation and a granulomatous lesion in a chest X-ray in Wegener's granulomatosis.
Fig. 10.
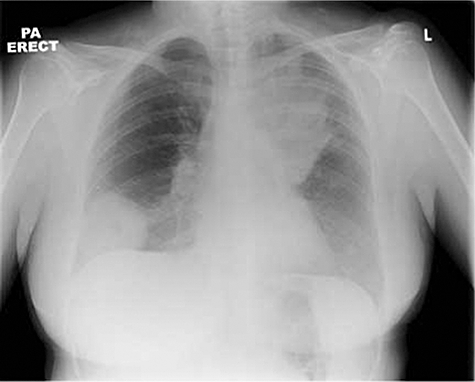
Plain chest radiograph of a patient with Wegener's granulomatosis showing extensive consolidation in the left upper lobe and a large granulomatous lesion in the right lower zone. Both resolved completely with immunosuppression.
Imaging in large vessel vasculitis may demonstrate active inflammation in the vessel wall or structural changes; stenosis, aneurysms and occlusions. If vessel wall inflammation is detected early in the disease course, prompt treatment may prevent irreversible structural changes [56]. Angiography is the current gold standard imaging for Takayasu's arteritis, which demonstrates structural but not arterial wall changes. Newer imaging techniques provide better information about vessel wall inflammation.
MRI demonstrates early vascular inflammation by increased wall thickness, oedema and mural contrast enhancement in Takayasu's arteritis [57] and giant cell arteritis [58]. Colour duplex ultrasonography demonstrates vessel wall oedema with a characteristic halo sign in giant cell arteritis and can also demonstrate stenosis and occlusions [59]. However, it is highly operator-dependent [60]. Both techniques have potential for diagnosis and monitoring large vessel vasculitis and potentially replacing current standard investigations. However, large prospective studies correlating radiological findings with pathological features and clinical changes are lacking. Figure 11 shows a magnetic resonance angiogram in a young woman with subclavian stenosis discovered to have an absent left radial pulse.
Fig. 11.
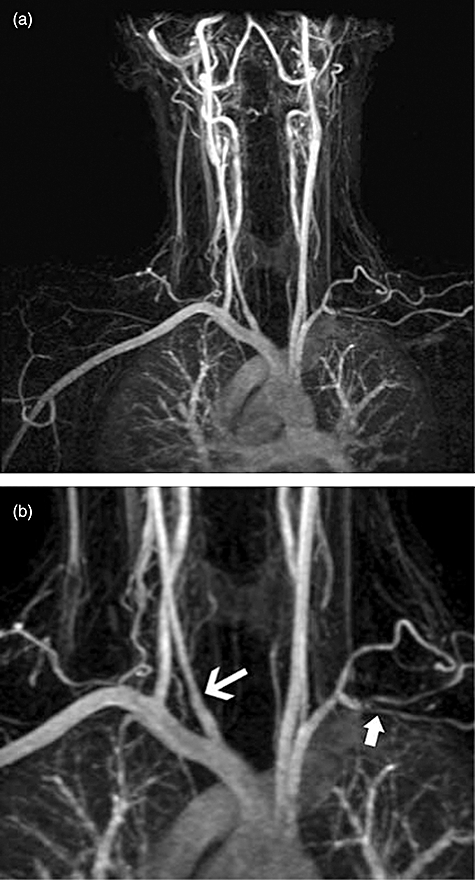
(a,b) Magnetic resonance angiogram of a patient with new onset Takayasu's arteritis. (a) Stenosis of multiple sites; (b) magnification view to show narrowing if the right internal carotid and occlusion of the left subclavian artery (arrows).
PET scans, demonstrating increased cellular glucose uptake, are used primarily to assess tumour metastases. They are also useful in detecting large vessel inflammation (Fig. 12) [61]. Computed tomography (CT) angiography demonstrates vessel involvement in Takayasu's arteritis, but is limited by its use of ionizing radiation [62]. Angiography is the standard investigation to determine the extent of vessel involvement in polyarteritis nodosa, but imaging with magnetic resonance angiography, CT and CT angiography are alternative non-invasive techniques [63,64].
Fig. 12.
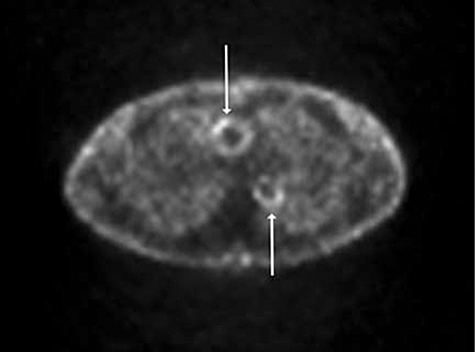
2-fluoro-2-deoxy-D-glucose (FDG) positron emission tomography (PET) scan through the thorax, of a patient with Takayasu's arteritis to show increased glucose uptake throughout the aorta. The ascending and descending aorta show increased uptake in this cross-sectional image (arrows).
Imaging in small vessel vasculitis provides useful information on organ inflammation and damage. CT and MRI scans of the paranasal sinuses demonstrate characteristic features in Wegener's granulomatosis (Fig. 13) [65,66]. A high resolution CT (HRCT) scan of the lungs will provide diagnostic and prognostic information in AASV (Fig. 14) [67].
Fig. 13.
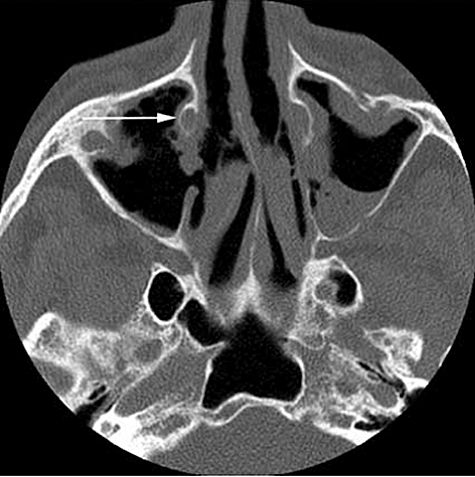
Nasal computed tomography (CT) scan of a patient with Wegener's granulomatosis to show extensive soft tissue infiltration in the sinuses with bony destruction of the medial wall of the maxillary sinuses (arrow).
Fig. 14.
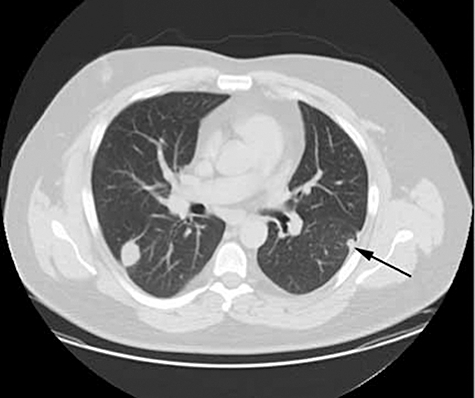
High-resolution chest computed tomography (CT) scan of a patient with Wegener's granulomatosis showing multiple lung nodules bilaterally of varying size including subpleural nodules (arrow).
Differential diagnosis of vasculitis
Various diseases mimic vasculitis, for example infective endocarditis, embolism from atrial myxoma or atheroma, thrombotic disorders such as anti-phospholipid syndrome and drug-induced vasospasm [68]. The potential for confusion is compounded by the occurrence of ANCA positivity in some patients with infective endocarditis and cholesterol emboli. If suspected, these should be investigated with echocardiography, clotting studies, anti-phospholipid antibodies and a history of recent medication.
Other diseases may cause a secondary vasculitis; these include connective tissue diseases, rheumatoid arthritis, viral infections, malignancies or drugs. Serological tests include anti-nuclear antibody (ANA), anti-double-stranded DNA (dsDNA), complement, rheumatoid factor (RF) and anti-citrullinated peptide antibody (ACPA). Infection screens include hepatitis B and C, human immunodeficiency virus (HIV) and cryoprecipitates, particularly in cutaneous vasculitis.
Diagnosis
Vessel size is the key discriminator in the definition of primary systemic vasculitis. While not ideal, this allows the grouping of diseases which can cause significant renal disease and are associated with the highest mortality if untreated. These are the ANCA-associated vasculitides (AASV).
The AASV are a group of overlapping syndromes, associated with, but not exclusively having, a positive test for P or C-ANCA and have similar clinical and histological features. They are characterized by necrotizing small to medium vessel inflammation without immune deposits. Tables 3–5 summarize the main features of these conditions and are adapted from the Chapel Hill Consensus definitions [48].
Table 3.
Definition of large vessel vasculitis.
| Large vessel vasculitis affecting the aorta and/or branches directed towards the major extremities and to the head and neck | |
|---|---|
| Giant cell arteritis | •Granulomatous arteritis of the aorta and its major branches, with predilection for the extra cranial branches of the carotid artery. Often involves the temporal artery |
| •Usually occurs in patients older than 50 years | |
| •Often associated with polymyalgia rheumatica. | |
| Takayasu's arteritis | •Granulomatous inflammation of the aorta and its major branches |
| •Usually occurs in patients younger than 40 years | |
Table 4.
Definition of medium vessel vasculitis.
| Medium-sized vessel vasculitis involving the main visceral arteries (renal, hepatic, coronary, mesenteric) | |
|---|---|
| Polyarteritis nodosa (classic PAN) | •Necrotizing inflammation of medium-sized or small arteries without glomerulonephritis |
| •Vasculitis in arterioles, capillaries or venules | |
| •May be associated with the presence of hepatitis B | |
| Kawasaki disease | •Arteritis involving large- and medium-sized arteries and associated with mucocutaneous lymph node syndrome in children |
| •Coronary arteries often involved | |
| •Aorta and veins may be involved | |
Granulomatous inflammation is similar in Wegener's granulomatosis and Churg–Strauss syndrome. The nasal crusting and destruction of Wegener's granulomatosis can be contrasted with the upper airway involvement in Churg–Strauss syndrome, which begins typically with allergic rhinitis, nasal polyposis and sinusitis. Renal involvement is a common and usually severe feature of ANCA-associated vasculitis, which is characterized histopathologically by a pauci-immune crescentic necrotizing glomerulonephritis, and is identical in Wegener's granulomatosis, microscopic polyangiitis, renal limited vasculitis (which is considered part of microscopic polyangiitis) and, more rarely, Churg–Strauss syndrome.
Diagnostic difficulties may arise because of the overlapping nature of the diseases. Churg–Strauss syndrome is characterized by asthma and peripheral blood eosinophilia. Pulmonary inflammation my be granulomatous and similar to Wegener's granulomatosis or eosinophilic, overlapping with other eosinophilic lung disorders. ANCA-negative Churg–Strauss syndrome may closely resemble idiopathic hypereosinophilic syndrome, which can also involve extra pulmonary organs. It may also overlap non-AASV such as polyarteritis nodosa. Severe renal disease is uncommon, unlike Wegener's granulomatosis and microscopic polyangiitis.
Therapy
The treatment of vasculitis comprises induction of remission followed by maintenance. Remission should be induced rapidly, balancing potential target organ damage against drug toxicity. Maintenance with immunosuppression should limit the amount of corticosteroid use and prevent relapse. Concomitant medication is used to treat or prevent adverse events from immunosuppressive treatment.
Well co-ordinated multi-centre trials are important in standardizing effective treatment for these relatively unusual conditions. The European Vasculitis Study Group (EUVAS) is an international collaboration of physicians and surgeons with an interest in vasculitis and has an important role in informing on management. It conducts a number of clinical trials and studies in the assessment of vasculitis. Completed trials include CYCAZAREM (cyclophosphamide versus azathioprine for remission in generalized vasculitis) [69], SOLUTION (anti-thymocyte globulin for refractory vasculitis) [70], NORAM (methotrexate versus cyclophosphamide for early systemic disease) [71], CHUSPAN (treatment protocols in Churg–Strauss and polyarteritis nodosa plus microscopic polyangiitis) [28], MEPEX (methyl prednisolone or plasma exchange for severe renal vasculitis) [72] and CYCLOPS (daily oral versus pulse cyclophosphamide for renal vasculitis) [73]. Ongoing trials include MYCYC (randomized clinical trial of mycophenolate mofetil versus cyclophosphamide for remission induction in ANCA-associated vasculitis), REMAIN (long-term low-dose immunosuppression versus treatment withdrawal for renal vasculitis), IMPROVE (International Mycophenolate mofetil to Reduce Outbreaks of Vasculitides) and RITUXVAS (comparing a rituximab-based regimen with a standard cyclophosphamide/azathioprine regimen in active generalized ANCA-associated vasculitis. EUVAS guidelines include recommendations on the management of vasculitis and on conducting clinical trials [7,17,19,74].
Large vessel vasculitis
Giant cell arteritis
Induction
If a diagnosis of giant cell arteritis is suspected glucocorticoids should be started without delay, because of the significant risk of visual symptoms, reported in up to 30% of cases, and visual loss in up to 20% [75]. A typical starting dose of prednisolone is 40–60 mg/day for 4 weeks [76], but there are no prospective placebo-controlled trials to prove the effectiveness of steroids, chiefly because of the fear of irreversible ischaemic complications in untreated cases. A retrospective study comparing patients who received glucocorticoid with a retrospective pre-corticosteroid group showed that corticosteroids had a significant effect in preventing visual loss with a rapid onset of symptom control [median time to initial response was 8 days (range 1–44)][77].
Intravenous high-dose methylprednisolone is used commonly in ophthalmology units for patients with impending or recent visual loss, based on a retrospective review of 73 cases presenting with visual loss. Of the 21 cases in which improvement in sight occurred, 40% had received additional intravenous methylprednisolone compared to 13% in those treated with oral glucocorticoids alone [78].
Maintenance
After 4 weeks prednisolone doses should be tapered, reducing every 2–4 weeks down to 10–15 mg/day. Thereafter, tapering by 1 mg per month is typical, depending on recurrence of symptoms. The median time to relapse is 7 months, by which time the median dose of prednisolone is usually 5 mg/day. Treatment may be required for up to 9 years [79]. Adverse effects reported on long-term steroid use include cataract, osteoporosis, infection, hypertension, type II diabetes mellitus and gastrointestinal bleeding [80]. Aspirin is effective in preventing cerebrovascular and cardiovascular ischaemic events [81,82] and is recommended for all patients who have no contraindications to its use [17].
A meta-analysis of three randomized placebo-controlled trials including 161 patients, 84 of whom received methotrexate up to 15 mg per week with steroids, and the rest of whom were treated with glucocorticoid alone, showed that methotrexate reduced the cumulative glucocorticoid dose significantly over 48 weeks and reduced the risk of first and second relapse. However, the adverse event risk was not influenced by the addition of methotrexate [83]. Outcome measures such as visual loss were not reported.
Azathioprine (150 mg/day) has been used as an adjunct to glucocorticoids in a placebo-controlled trial in patients with polymyalgia rheumatica and giant cell arteritis. A significant reduction in the total glucocorticoid dose was achieved after 52 weeks (1·9 ± 0·84 mg versus 4·2 ± 0·58 mg), but clinical benefit was limited and of late onset [84].
Infliximab has been used as maintenance therapy in a randomized controlled trial of 44 patients, but failed to improve disease control above the effect of steroid, or to allow a reduction in the dose of steroid required to prevent relapse [85].
Takayasu's arteritis
Induction
Immunosuppressive therapy is indicated if there is clear evidence of new clinical features indicating increasing ischaemia, or if there is evidence of worsening on radiographic imaging, especially the presence of arterial wall thickening. Typical clinical features indicating active disease include new loss of pulses, painful vessels (typically carotidynia) and new bruits.
Initial therapy is with high-dose glucocorticoids usually in combination with a steroid sparing agent. An open-label study of patients, who were refractory to glucocorticoid therapy, showed that weekly low-dose methotrexate was effective in inducing remission in 13 of 16 cases [86]. In a prospective study of 65 newly diagnosed Takayasu's arteritis patients treated with azathioprine and prednisolone and followed-up for 1 year, therapy was safe, well tolerated and effective in ameliorating systemic symptoms and laboratory measures of disease activity within 3 months. Although it did not reverse angiographic lesions, it did halt disease progression [87].
Maintenance
Despite glucocorticoid therapy, subclinical disease can persist, as demonstrated on magnetic resonance imaging. Approximately half of all Takayasu's arteritis patients have chronic active disease for which glucocorticoid therapy alone does not provide sustained remission [88]. Therefore, the use of adjunctive therapy in addition to glucocorticoids is common, both to improve disease control and to reduce overall steroid use [17].
Methotrexate has been used in refractory cases of Takayasu's arteritis. In one study, eight of the 16 patients who achieved remission on initial methotrexate and glucocorticoid therapy sustained remissions lasting 4–34 months (mean 18 months), and four patients did not require further glucocorticoid or methotrexate therapy. However, three patients experienced disease progression despite treatment. Patients were followed-up for a mean period of 2·8 years. Further long-term studies are required to assess the durability of remission and the need for long-term maintenance therapy in this subset of patients [88].
Surgical intervention
Takayasu's arteritis may result in permanent stenosis, despite remission of the disease. It is important to differentiate the features of disease for which further immunosuppressive agents are required, from abnormalities due to damage to vascular anatomy in which surgical intervention is more appropriate [88]. Reconstructive surgery should be undertaken at expert centres and preferably during the quiescent phase of the disease [17].
Medium vessel vasculitis
Polyarteritis nodosa and Kawasaki disease are the two major categories of medium-sized vessel vasculitis. Both have acute necrotizing arteritis with inflammatory aneurysm formation.
Polyarteritis nodosa
Patients with polyarteritis nodosa present with a multi-system illness with constitutional features such as weight loss, fever, myalgia, development of a rash, neuropathy or abdominal ischaemia. Polyarteritis nodosa is associated commonly with hepatitis B infection.
Induction
Hepatitis B-associated polyarteritis nodosa should be managed in conjunction with a hepatologist [19]. High-dose glucocorticoids are given for 2 weeks followed by anti-viral agents (such as vidarabine, interferon-alpha and lamivudine) and then plasmapheresis. This protocol facilitates seroconversion to hepatitis B immune status, which would prevent relapse [2].
In those patients who do not have hepatitis B infection, combination therapy of cyclophosphamide and high-dose glucocorticoids (such as prednisolone 1 mg/kg/day) is usually indicated, unless patients have a favourable prognosis as defined using the five-factor score. Oral or pulsed high-dose cyclophosphamide is given for at least 3 months and glucocorticoids are tapered over the next 4 months to a minimum of 15 mg/day [89].
Intravenous methylprednisolone is used for fulminant disease [19,26]. Short duration (6 × monthly pulses) of high-dose cyclophosphamide is associated with higher relapse rates and lower event-free survival than long duration (12 × monthly pulses) treatment in patients with polyarteritis nodosa; however, there is no significant difference in mortality [28]. Pulsed cyclophosphamide has been used with equal efficacy to continuous oral daily cyclophosphamide in polyarteritis nodosa and had a lower incidence of adverse events over a 12-month period [89,90].
Maintenance
Once remission is achieved, steroids can be reduced gradually to 10 mg/day or less [89]. Polyarteritis nodosa has a low relapse rate and maintenance treatment is usually not needed. In cases of relapse, maintenance treatment with azathioprine or methotrexate could be considered.
Kawasaki disease
Kawasaki disease is characterized by fever, bilateral non-exudative conjunctivitis, erythema of the lips and oral mucosa, indurated oedema of the dorsum of hands and feet with erythema of the palms and soles, rash and cervical lymphadenopathy. Coronary artery aneurysms or ectasia develop in 15–25% of untreated children and may lead to ischaemic heart disease or sudden death. It is a more common cause of heart disease in children than rheumatic fever [91], but is still rare. It is likely to have an infectious cause in genetically predisposed individuals involving an antigen-driven immune response in which immunoglobulin A plasma cells play a central role [92]. Early suppression of inflammation and prevention of thrombosis will reduce the risk of potentially fatal coronary artery abnormalities to between 1 and 5%.
Induction
High-dose intravenous immunoglobulin (IVIG) plus aspirin is the standard treatment, and should be started as early as possible to reduce the risk of coronary artery rupture and sudden death [93]. The mechanism of action is unknown, but it appears to have a generalized anti-inflammatory effect involving modulation of cytokine production, neutralization of bacterial super-antigens, augmentation of T cell suppressor activity and suppression of antibody synthesis [94]. Lower peak IgG levels are associated with worse outcomes, supporting the use of single high-dose infusions of intravenous Ig (IVIG), rather than using multiple infusions at lower doses [95]. Therapy should be commenced within 10 days of onset, and preferably within 7 days. Some patients require retreatment with IVIG for relapse [96]. There does not appear to be any additional benefit from using high-dose aspirin (80–120 mg/kg/day) plus IVIG compared with low dose of aspirin plus IVIG in terms of aneurysm formation [93]. Glucocorticoid therapy is generally not used in the primary treatment of Kawasaki disease but it may be of value in resistant cases [97]. In a small study intravenous methylprednisolone was effective, with more rapid initial resolution of fever in 77% (34 of 44) of cases compared to 63% (12 of 19) of controls [98].
Maintenance
Kawasaki disease is a self-limiting and generally non-recurring vasculitis and long-term immunosuppressive therapy is not indicated. Children with coronary artery abnormalities should be treated with low-dose aspirin, anti-coagulants and beta-blockers according to recommended guidelines [94].
Small vessel vasculitis
Generalized ANCA-associated vasculitis
The treatment of the ANCA-associated vasculitides, Wegener's granulomatosis, Churg–Strauss syndrome and microscopic polyangiitis, are considered as one group. The presence of ANCA has been shown to be associated with more severe forms of disease [99,100]. Collaborative trials conducted by EUVAS have demonstrated that patients with different levels of disease severity respond to different treatment protocols [19]. Treatment is based upon disease severity rather than ANCA status.
Induction: cyclophosphamide
Pulsed intravenous high-dose or low-dose oral continuous cyclophosphamide plus glucocorticoids are equally effective for induction of remission in generalized ANCA-positive vasculitis [73]. However, pulsed cyclophosphamide is associated with reduced morbidity related to leucopenia and infection, due to a lower cumulative dose of cyclophosphamide than continuous daily oral therapy.
Intravenous cyclophosphamide is given every 2 weeks for the first three pulses, and thereafter 3-weekly until remission is achieved, following which patients are switched to maintenance therapy after a median of 3 months. The usual dose is 15 mg/kg/pulse, but reductions are made for impaired renal function and increasing age [89]. Continuous low-dose oral cyclophosphamide can be given at 2 mg/kg/day with dose reductions according to age (patients over the age of 60 and 75 years have a 25% and 50% dose reduction, respectively). The maximum daily dosage is 200 mg/day, given for 3 months, when 80% of patients would be expected to have achieved remission. Thereafter, the dose is reduced to 1·5 mg/kg/day. However, if remission has not been achieved, oral dosing can be continued at 2 mg/kg/day for a further 3 months, by which time 90% should have achieved remission. Use of cyclophosphamide should not usually exceed 6 months, and if patients still have active disease they should be considered for alternative immunomodulatory therapy [69].
Induction: Glucocorticoids
Oral prednisolone regimens usually start at 1 mg/kg/day reducing to 0·4 mg/kg/day by 4 weeks and to 15 mg per day after 12 weeks, with progressive subsequent reduction in dose [19,69].
Early studies supported the use of intravenous methylprednisolone as part of an induction regimen [101]. The use of pulsed methylprednisolone in addition to pulsed cyclophosphamide has been compared to standard oral glucocorticoids plus continuous oral cyclophosphamide in a randomized controlled trial [89]. There was no difference in outcome between the two groups, but it was not possible to determine the effect of the different steroid regimen in this study.
Localized and early systemic disease is characterized by the absence of vital organ disease or damage, but localized disease may still be very destructive. Methotrexate (20–25 mg/week) and oral steroids can be as effective in achieving remission as cyclophosphamide and oral steroids [71]. However, there is a higher risk of relapse and progression of disease with methotrexate. If local disease is resistant to standard therapy, more aggressive treatment is indicated.
Patients should be given cyclophosphamide and corticosteroids, as for generalized disease, when in established renal failure (creatinine > 500 µmol/l), or if they have rapidly progressive renal impairment at diagnosis. Additional treatment with plasmapheresis (typically 7 × 4 l over 2 weeks) improves renal survival, but does not affect mortality) [72]. If patients fail to achieve remission other therapies should be considered, including the use of high-dose intravenous immunoglobulin (2 g/kg/month) [102]. The toxicity of cyclophosphamide and steroids is an important contribution to morbidity and there is a need for improved therapy. The current MYCYC trial is comparing mycophenolate mofetil with cyclophosphamide for induction of remission in AAV.
Maintenance
Following induction of remission, patients should be given maintenance therapy for at least 24 months [19]. This includes prednisolone tapered to 10 mg per day, and withdrawn after 6–18 months depending on the patient's response [19]. However, there is uncertainty as to how long steroids should be maintained and they are often continued for longer than 2 years. The REMAIN study is currently investigating whether low-dose prednisolone and azathioprine reduce long-term morbidity in vasculitis.
Further immunosuppression is recommended in addition to prednisolone. Conventionally, this would be cyclophosphamide, but more recently methotrexate [103], azathioprine [69] and leflunomide [104] have been shown to be beneficial. Methotrexate and azathioprine are associated with relapse rates of 10–30%. High-dose leflunomide (30 mg/day) was more effective than methotrexate in preventing relapse, but associated with more adverse events [104]. Induction with cyclophosphamide followed by maintenance with azathioprine had equivalent efficacy but less toxicity than maintenance with cyclophosphamide [69]. Mycophenolate mofetil (2–3 g/day) has minimal side effects, but despite a 100% remission rate at 3 months results in a high rate of relapse (43% after 10 months) [105,106]. The IMPROVE study is currently randomizing patients with AASV to receive either mycophenolate mofetil or azathioprine following induction of remission with cyclophosphamide and prednisolone. Maintenance therapy plus trimethoprim/sulphamethoxazole reduces the risk of relapse in Wegener's granulomatosis [107].
Cryoglobulinaemic vasculitis
Cryoglobulinaemia is a systemic vasculitis characterized by proliferation of B cell clones producing pathogenic immunoglobulins that precipitate in the cold and may present with fulminant disease. Most patients have an underlying infection with hepatitis C, which is linked closely to the pathogenesis of the disease. Treatment of hepatitis C-associated cryoglobulinaemic vasculitis should be in conjunction with a hepatologist [19]. Treatment with interferon (IFN)-α2b or PEGylated IFN-α2b, both in combination with oral ribavirin, resulted in a complete clinical response in 63%, a sustained virological response in 58% and clearance of cryoglobulins in 46% of patients [108].
There are no controlled trials in patients without hepatitis C infection, but therapy is given based on the treatment for ANCA-associated vasculitis, involving corticosteroids, immunosuppressives and plasma exchange depending on severity [19]. A systematic review of 13 papers reporting on 57 cases of cryoglobulinaemia treated with rituximab infusions reported a clinical response in 80–93% patients but a relapse in 39% patients [109]. A relatively small number of side effects were reported. There have been no randomized controlled trials to date, but B cell therapy shows promise as a treatment.
Henoch–Schonlein purpura
Henoch–Schonlein purpura is primarily a disease affecting children, with an incidence of approximately 15 cases per 100 000 children per year [110]. It is rare in adults (annual incidence of one per million) (Table 6) [111]. Clinical presentation is typically with skin purpura. Some patients also develop abdominal pain, gastrointestinal bleeding, arthropathy and renal failure due to IgA nephropathy. Nephritis occurs in 50–80% adults and 20–40% children [112], who might present with an isolated haematuria, proteinuria, acute nephritis or nephrotic syndrome. In adulthood, Henoch–Schonlein purpura is a more severe clinical syndrome with a higher frequency of diarrhoea and renal involvement and with a worse outcome [111]. Although Henoch–Schonlein purpura usually resolves spontaneously, there are concerns about the development of renal failure which is rare. Evidence for treatment is limited but selected patients may benefit from steroids [111,113].
Table 6.
Epidemiology of vasculitis [1].
| Vasculitis | Incidence per million | |
|---|---|---|
| Giant cell arteritis | 5–367 | Population aged over 50 years [1] |
| 220 | Recent UK data [121] | |
| Takayasu arteritis | 0·4–2·6 | |
| Polyarteritis nodosa | 0·4–77 | |
| Kawasaki disease | 10–1080 | < 5 years |
| Wegener's granulomatosis | 0·7–12 | |
| Churg–Strauss syndrome | 1–4 | |
| Microscopic polyangiitis | 0·5–24 | |
| Henoch–Schonlein purpura | 102–204 | < 17 years |
| 3–14 | Adult | |
| Cryoglobulinaemia | No adequate studies | Prevalence essential MC 1 : 100 000 [122] |
| Cutaneous leucocytoclastic angiitis | 15·4 |
Readers are referred to Reference [1] for discussion of how the classification of vasculitis, geographical and other factors affect calculations of incidence.
New therapies
There is a growing trend in inflammatory diseases to use specific biological therapy designed to interfere with individual cytokines or pathways. While this approach has been extremely successful in the management of rheumatoid arthritis, it is less well tested in systemic vasculitis. The use of tumour necrosis factor (TNF) inhibitors has so far been disappointing in giant cell arteritis [114], and remains relatively untested in Takayasu's arteritis. It is unlikely that such therapies will be used in Kawasaki disease or polyarteritis nodosa.
For ANCA-associated vasculitis it is important to consider a biological approach, given the greater understanding of the underlying pathology. Long-term use of etanercept has proven disappointing in Wegener's granulomatosis [115], although short-term use of TNF inhibitor therapy has been effective in acute disease [116]. Infliximab has been used in a study of 28 patients with systemic vasculitis, resulting in 88% achieving remission but severe infections in 20% [117].
Rituximab is a chimeric monoclonal IgG1 antibody directed against CD 20 leading to the destruction of B cells via complement-mediated lysis and antibody-dependent cellular cytotoxicity. Because ANCA are involved in the pathogenesis of small vessel vasculitis, it stands to reason that rituximab may be an effective and safe treatment. It might be postulated that ANCA-positive disease would respond better than ANCA-negative vasculitis. There is evidence of benefit in using rituximab in Wegener's granulomatosis to achieve remission in patients who have failed conventional therapy, but given the small numbers of published cases there is a need for large randomized controlled trials, which are currently under way [118]. These include the RAVE study comparing cyclophosphamide with rituximab in inducing remission in patients with severe ANCA-associated vasculitis. A potential problem in AASV is that the full therapeutic effect of rituximab may be delayed for up to 3 months, and so may not have a role as a single agent in patients with rapidly progressive disease. It might be expected that rituximab would work better in antibody-positive disease, but this has not been shown.
Imatinib mesylate is an inhibitor of a class of tyrosine kinases and inhibits T cell activation and proliferation. In vitro it impairs conversion from naive to memory T cells after T cell activation using cells from patients with AASV [119]. It was found to inhibit platelet-derived growth factor (PGDF)-mediated responses strongly in myointimal cells in giant cell arteritis and may have therapeutic potential to limit ischaemic complications in large vessel vasculitis [120].
Summary
The vasculitides remain a challenge in terms of diagnosis and treatment. The recognition of disease remains unsatisfactory in the absence of any gold standard tests. The clinical presentation and correct use of appropriate laboratory tests, imaging and pathology are essential to assist in making an early diagnosis. The patient should be assessed by clinicians familiar with vasculitis to plan treatment. For many patients, disease control can be achieved with the use of current immunosuppressive therapy, but relapse and accumulating co-morbidity are common long-term consequences.
Disclosure
Drs Miller, Chan, Wiik and Misbah have no disclosures. Dr Luqmani has received consultancy fees from Roche and honoraria from Schering Plough and Wyeth.
References
- 1.Watts RA, Scott DGI. Epidemiology of vasculitis. In: Ball GV, Bridges SL Jr, editors. Vasculitis. 2nd. Oxford: Oxford University Press; 2008. pp. 7–21. [Google Scholar]
- 2.Guillevin L, Mahr A, Callard P, et al. French Vasculitis Study Group. Hepatitis B virus-associated polyarteritis nodosa: clinical characteristics, outcome, and impact of treatment in 115 patients. Medicine (Balt) 2005;84:313–22. doi: 10.1097/01.md.0000180792.80212.5e. [DOI] [PubMed] [Google Scholar]
- 3.Ferri C, Greco F, Longombardo G, et al. Antibodies against hepatitis C virus in mixed cryoglobulinemia patients. Infection. 1991;19:417–20. doi: 10.1007/BF01726453. [DOI] [PubMed] [Google Scholar]
- 4.Gordon M, Luqmani RA, Adu D, et al. Relapses in patients with a systemic vasculitis. Q J Med. 1993;86:779–89. [PubMed] [Google Scholar]
- 5.Exley AR, Carruthers DM, Luqmani RA, et al. Damage occurs early in systemic vasculitis and is and index of outcome. Q J Med. 1997;90:391–9. doi: 10.1093/qjmed/90.6.391. [DOI] [PubMed] [Google Scholar]
- 6.Seo P, Luqmani RA, Flossmann O, et al. The future of damage assessment in vasculitis. J Rheumatol. 2007;34:1357–71. [PubMed] [Google Scholar]
- 7.Mukhtyar C, Flossmann O, Hellmich B, et al. Outcomes from studies of antineutrophil cytoplasm antibody associated vasculitis: a systematic review by the European League Against Rheumatism systemic vasculitis task force. Ann Rheum Dis. 2008;67:1004–10. doi: 10.1136/ard.2007.071936. [DOI] [PubMed] [Google Scholar]
- 8.Flossmann O, Bacon P, de Groot K, et al. Development of comprehensive disease assessment in systemic vasculitis. Ann Rheum Dis. 2007;66:283–92. doi: 10.1136/ard.2005.051078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nolan SL, Kalia N, Nash GB, Kamel D, Heering P, Savage CO. Mechanisms of ANCA-mediated leukocyte-endothelial cell interactions in vivo. J Am Soc Nephrol. 2008;19:973–84. doi: 10.1681/ASN.2007111166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tervaert JW, Huitema MG, Hené RJ, et al. Prevention of relapses in Wegener's granulomatosis by treatment based on antineutrophil cytoplasmic antibody titre. Lancet. 1990;336:709–11. doi: 10.1016/0140-6736(90)92205-v. [DOI] [PubMed] [Google Scholar]
- 11.Birck R, Schmitt WH, Kaelsch IA, van der Woude FJ. Serial ANCA determinations for monitoring disease activity in patients with ANCA-associated vasculitis: systematic review. Am J Kidney Dis. 2006;47:15–23. doi: 10.1053/j.ajkd.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 12.Sanders JS, Huitma MG, Kallenberg CG, Stegeman CA. Prediction of relapses in PR3-ANCA-associated vasculitis by assessing responses of ANCA titres to treatment. Rheumatology. 2006;45:724–9. doi: 10.1093/rheumatology/kei272. [DOI] [PubMed] [Google Scholar]
- 13.Tso E, Flamm SD, White RD, et al. Takayasu arteritis. Utility and limitation of magnetic resonance imaging in diagnosis and treatment. Arthritis Rheum. 2002;46:1634–42. doi: 10.1002/art.10251. [DOI] [PubMed] [Google Scholar]
- 14.Deacock SJ. An approach to the patient with urticaria. Clin Exp Immunol. 2008;153:151–61. doi: 10.1111/j.1365-2249.2008.03693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wiik A. Clinical use of diagnostic tests for antineutrophil cytoplasmic antibodies. What do the studies say? Rheum Dis Clin North Am. 2001;27:799–813. doi: 10.1016/s0889-857x(05)70236-2. [DOI] [PubMed] [Google Scholar]
- 16.Savige J, Davies D, Falk RJ, Jennette JC, Wiik A. Antineutrophil cytoplasmic antibodies and associated diseases: a review of the clinical and laboratory features. Kidney Int. 2000;57:846–62. doi: 10.1046/j.1523-1755.2000.057003846.x. [DOI] [PubMed] [Google Scholar]
- 17.Mukhtyar C, Guillevin L, Cid M, et al. EULAR Recommendations for the management of large vessel vasculitis. Ann Rheum Dis. 2009;68:318–23. doi: 10.1136/ard.2008.088351. [DOI] [PubMed] [Google Scholar]
- 18.Smetana GW, Schmerling RH. Does this patient have temporal arteritis? JAMA. 2002;287:92–101. doi: 10.1001/jama.287.1.92. [DOI] [PubMed] [Google Scholar]
- 19.Mukhtyar C, Guillevin L, Cid MC, et al. EULAR recommendations for the management of primary small and medium vessel vasculitis. Ann Rheum Dis. 2009;68:310–17. doi: 10.1136/ard.2008.088096. [DOI] [PubMed] [Google Scholar]
- 20.Luqmani RA, Bacon PA, Moots RJ, et al. Birmingham Vasculitis Activity Score (BVAS) in systemic necrotizing vasculitis. Q J Med. 1994;87:671–8. [PubMed] [Google Scholar]
- 21.Luqmani RA, Exley AR, Kitas GD, Bacon PA. Disease assessment and management of the vasculitides. Baillieres Clin Rheumatol. 1997;11:423–46. doi: 10.1016/s0950-3579(97)80052-0. [DOI] [PubMed] [Google Scholar]
- 22.Luqmani R. Evaluation of vasculitis disease activity in Europe. Eur J Intern Med. 2001;12:401–2. doi: 10.1016/s0953-6205(01)00158-3. [DOI] [PubMed] [Google Scholar]
- 23.Mukhtyar C, Lee R, Brown D, et al. Modification and validation of the Birmingham Vasculitis Activity Score (Version 3) Ann Rheum Dis. 2009;68:1827–32. doi: 10.1136/ard.2008.101279. [DOI] [PubMed] [Google Scholar]
- 24.European Community Study Group on Clinical Trials in Systemic Vasculitis ECSYSVASTRIAL. European therapeutic trials in ANCA-associated systemic vasculitis: disease scoring, consensus regimens and proposed clinical trials. Clin Exp Immunol. 1995;101(Suppl. 1):29–34. doi: 10.1111/j.1365-2249.1995.tb06161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Groot K, Gross WL, Herlyn K, Reinhold-Keller E. Development and validation of a disease extent index for Wegener's granulomatosis. Clin Nephrol. 2001;55:31–8. [PubMed] [Google Scholar]
- 26.Guillevin L, Lhote F, Gayraud M, et al. Prognostic factors in polyarteritis nodosa and Churg–Strauss syndrome: a prospective study in 342 patients. Medicine (Baltimore) 1996;75:17–28. doi: 10.1097/00005792-199601000-00003. [DOI] [PubMed] [Google Scholar]
- 27.Gayraud M, Guillevin L, le Toumelin P, et al. French Vasculitis Study Group. Long-term follow-up of polyarteritis nodosa, microscopic polyangiitis, and Churg–Strauss syndrome: analysis of four prospective trials including 278 patients. Arthritis Rheum. 2001;44:666–75. doi: 10.1002/1529-0131(200103)44:3<666::AID-ANR116>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 28.Guillevin L, Cohen P Mahr A, et al. Treatment of polyarteritis nodosa and microscopic polyangiitis with poor prognosis factors; a prospective trial comparing glucocorticoids and six or twelve cyclophosphamide pulses in sixty five patients. Arthritis Rheum. 2003;15:93–100. doi: 10.1002/art.10922. [DOI] [PubMed] [Google Scholar]
- 29.Exley AR, Bacon PA, Luqmani RA, et al. Development and initial validation of the VDI. Arthritis Rheum. 1997;40:371–80. doi: 10.1002/art.1780400222. [DOI] [PubMed] [Google Scholar]
- 30.Manger K, Wildt L, Kalden JR, Manger B. Prevention of gonadal toxicity and preservation of gonadal function and fertility in young women with systemic lupus erythematosus treated by cyclophosphamide: the PREGO-Study. Autoimmun Rev. 2006;5:269–72. doi: 10.1016/j.autrev.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 31.Bidet M, Bachelot A, Touraine P. Premature ovarian failure: predictability of intermittent ovarian function and response to ovulation induction agents. Curr Opin Obstet Gynecol. 2008;20:416–20. doi: 10.1097/GCO.0b013e328306a06b. [DOI] [PubMed] [Google Scholar]
- 32.Wiik A. Antineutrophil cytoplasm antibodies (ANCAs) and strategy for diagnosing ANCA-associated vasculitides. In: Detrick B, Hamilton RG, Folds JD, editors. Manual of molecular and clinical laboratory immunology. Washington, DC: ASM Press; 2006. pp. 1053–8. [Google Scholar]
- 33.Chen M, Yu F, Wang S-X, Zou WZ, Zhao MH, Wang HY. Antineutrophil cytoplasmic autoantibody negative pauci-immune crescentic glomerulonephritis. Am Soc Nephrol. 2007;18:599–605. doi: 10.1681/ASN.2006091021. [DOI] [PubMed] [Google Scholar]
- 34.Savige J, Gillis D, Benson E, et al. International consensus statement on testing and reporting of antineutrophil cytoplasmic antibodies (ANCA) Am J Clin Pathol. 1999;111:507–13. doi: 10.1093/ajcp/111.4.507. [DOI] [PubMed] [Google Scholar]
- 35.Wiik A. Neutrophil-specific autoantibodies in chronic inflammatory diseases. Autoimmun Rev. 2002;1:67–72. doi: 10.1016/s1568-9972(01)00007-6. [DOI] [PubMed] [Google Scholar]
- 36.Niles JL, McCluskey RT, Ahmad MF, et al. Wegener's granulomatosis autoantigen is a novel neutrophil serine protease. Blood. 1989;74:1888–93. [PubMed] [Google Scholar]
- 37.von Vietinghoff S, Eulenberg C, Wellner M, Luft FC, Kettritz R. Neutrophil surface presentation of the anti-neutrophil cytoplasmic antibody–antigen proteinase 3 depends on N-terminal processing. Clin Exp Immunol. 2008;152:508–16. doi: 10.1111/j.1365-2249.2008.03663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hagen EC, Daha M, Hermans J, et al. The diagnostic value of standardized assays for antineutrophil cytoplasmic antibodies (ANCA) in idiopathic systemic vasculitis. Results of an international collaborative study. Kidney Int. 1998;53:743–53. doi: 10.1046/j.1523-1755.1998.00807.x. [DOI] [PubMed] [Google Scholar]
- 39.Lee AS, Finkielman JD, Peikert T, et al. Wegener's Granulomatosis Etanercept Trial Research Group. Agreement of anti-neutrophil cytoplasmic antibody measurements obtained from serum and plasma. Clin Exp Immunol. 2006;146:15–20. doi: 10.1111/j.1365-2249.2006.03179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stummann L, Wiik A. A high yield procedure for purification of proteinase 3, the autoantigen in Wegener's granulomatosis. J Immunol Methods. 1997;211:111–23. doi: 10.1016/s0022-1759(97)00082-3. [DOI] [PubMed] [Google Scholar]
- 41.Westman K, Selga D, Bygren P, et al. Clinical evaluation of a capture ELISA for detection of proteinase 3 antineutrophil cytoplasmic antibody. Kidney Int. 1998;53:1230–6. doi: 10.1046/j.1523-1755.1998.00873.x. [DOI] [PubMed] [Google Scholar]
- 42.Segelmark M, Phillips BD, Hogan SL, et al. Monitoring proteinase 3 antineutrophil cytoplasmic antibodies for detection of relapses in small vessel vasculitis. Clin Diagn Lab Immunol. 2003;10:769–74. doi: 10.1128/CDLI.10.5.769-774.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Talor MV, Stone JH, Stebbing J, Barin J, Rose NR, Burek CL. Antibodies to selected minor target antigens in patients with anti-neutrophil cytoplasmic antibodies (ANCA) Clin Exp Immunol. 2007;150:42–8. doi: 10.1111/j.1365-2249.2007.03453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wiik A. Drug-induced vasculitis. Curr Opin Rheumatol. 2008;20:35–9. doi: 10.1097/BOR.0b013e3282f1331f. [DOI] [PubMed] [Google Scholar]
- 45.Falk RJ, Jennette JC. ANCA are pathogenic – oh yes they are! J Am Soc Nephrol. 2002;13:1977–79. doi: 10.1681/ASN.V1371977. [DOI] [PubMed] [Google Scholar]
- 46.Xiao H, Heeringa P, Hu P, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest. 2002;110:955–63. doi: 10.1172/JCI15918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pfister H, Ollert M, Frohlich LF, et al. Antineutrophil cytoplasmic autoantibodies against the murine homolog of proteinase 3 (Wegener autoantigen) are pathogenic in vivo. Blood. 2004;104:1411–8. doi: 10.1182/blood-2004-01-0267. [DOI] [PubMed] [Google Scholar]
- 48.Jennette JC, Falk RJ, Andrassy K, et al. Nomenclature of systemic vasculitides: proposal of an international consensus conference. Arthritis Rheum. 1994;37:187–92. doi: 10.1002/art.1780370206. [DOI] [PubMed] [Google Scholar]
- 49.Wiik AS. Autoantibodies in vasculitis. In: Ball GV, Bridges SL, editors. Vasculitis. 2nd. Oxford: Oxford University Press; 2008. pp. 53–65. [Google Scholar]
- 50.Genereau T, Lortholary O, Pottier MA, et al. Temporal artery biopsy: a diagnostic tool for systemic necrotizing vasculitis. French Vasculitis Study Group. Arthritis Rheum. 1999;42:2674–81. doi: 10.1002/1529-0131(199912)42:12<2674::AID-ANR25>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 51.Hauer HA, Bajema IM, Van Houwelingen HC, et al. European Vasculitis Study Group (EUVAS) Determinants of outcome in ANCA-associated glomerulonephritis: a prospective clinico-histopathological analysis of 96 patients. Kidney Int. 2002;62:1732–42. doi: 10.1046/j.1523-1755.2002.00605.x. [DOI] [PubMed] [Google Scholar]
- 52.Hauer HA, Bajema IM, Van Houwelingen HC, et al. European Vasculitis Study Group (EUVAS) Renal histology in ANCA-associated vasculitis: differences between diagnostic and serologic subgroups. Kidney Int. 2002;61:80–9. doi: 10.1046/j.1523-1755.2002.00089.x. [DOI] [PubMed] [Google Scholar]
- 53.Churg A. Pathological features of vasculitis. In: Ball GV, Bridges SL Jr, editors. Vasculitis. 2nd. Oxford: Oxford University Press; 2008. pp. 97–114. [Google Scholar]
- 54.Ashton-Key M, Gallagher PJ. Surgical pathology of cranial arteritis and polymyalgia rheumatica. Ballieres Clin Rheumatol. 1991;5:387–404. doi: 10.1016/s0950-3579(05)80061-5. [DOI] [PubMed] [Google Scholar]
- 55.Colby TV, Tazelaar HD, Specks U, DeRemee RA. Nasal biopsy in Wegener's granulomatosis. Hum Pathol. 1991;22:101–4. doi: 10.1016/0046-8177(91)90028-n. [DOI] [PubMed] [Google Scholar]
- 56.Bley TA, Markl M, Schlep M, et al. Mural inflammatory hyperenhancement in MRI of giant cell (temporal) arteritis resolves under corticosteroid treatment. Rheumatology. 2008;47:65–7. doi: 10.1093/rheumatology/kem283. [DOI] [PubMed] [Google Scholar]
- 57.Kissin EY, Merker PA. Diagnostic imaging in Takayasu arteritis. Curr Opin Rheumatol. 2004;16:31–7. doi: 10.1097/00002281-200401000-00007. [DOI] [PubMed] [Google Scholar]
- 58.Bley TA, Wieben O, Uhl M, et al. High-resolution MRI in giant cell arteritis: imaging of the wall of the superficial temporal artery. Am J Roentgenol. 2005;184:283–7. doi: 10.2214/ajr.184.1.01840283. [DOI] [PubMed] [Google Scholar]
- 59.Schmidt WA, Kraft HE, Vorpahl K, Volker L, Gromnica-Ihle EJ. Color duplex ultrasonography in the diagnosis of temporal arteritis. N Engl J Med. 1997;337:1336–42. doi: 10.1056/NEJM199711063371902. [DOI] [PubMed] [Google Scholar]
- 60.Salvarani C, Silingardi M, Ghirarduzzi A, et al. Is duplex ultrasonography useful for the diagnosis of giant-cell arteritis? Ann Intern Med. 2002;137:232–8. doi: 10.7326/0003-4819-137-4-200208200-00006. [DOI] [PubMed] [Google Scholar]
- 61.Walter MA. [(18)F] fluorodeoxyglucose PET in large vessel vasculitis. Radiol Clin North Am. 2007;45:735–44. doi: 10.1016/j.rcl.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 62.Gotway MB, Araoz PA, Macedo TA, et al. Imaging findings in Takayasu's. Arteritis Am J Roent. 2005;184:1945–50. doi: 10.2214/ajr.184.6.01841945. [DOI] [PubMed] [Google Scholar]
- 63.Schmidt WA. Use of imaging studies in the diagnosis of vasculitis. Curr Rheumatolol Rep. 2004;6:203–11. doi: 10.1007/s11926-004-0069-1. [DOI] [PubMed] [Google Scholar]
- 64.Chen LJ, Wang JH. Diagnosis of systemic arterial diseases with whole-body 3D contrast-enhanced magnetic resonance angiography. Chin Med J (Engl) 2006;119:1772–8. [PubMed] [Google Scholar]
- 65.Lloyd G, Lund VJ, Beale T, Howard D. Rhinologic changes in Wegener's granulomatosis. J Laryngol Otol. 2002;116:565–9. doi: 10.1258/002221502760132737. [DOI] [PubMed] [Google Scholar]
- 66.Muhle C, Reinhold-Keller E, Richter C, et al. MRI of the nasal cavity, the paranasal sinuses and orbits in Wegener's granulomatosis. Eur Radiol. 1997;7:566–70. doi: 10.1007/s003300050206. [DOI] [PubMed] [Google Scholar]
- 67.Pesci A, Manganelli P. Respiratory system involvement in antineutrophil cytoplasmic-associated systemic vasculitides: clinical, pathological and therapeutic considerations. Drugs R D. 2007;8:25–42. doi: 10.2165/00126839-200708010-00003. [DOI] [PubMed] [Google Scholar]
- 68.Luqmani RA, Pathare S, Kwok-Fai TL. How to diagnose and treat secondary forms of vasculitis. Best Pract Res Clin Rheum. 2005;19:321–36. doi: 10.1016/j.berh.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 69.Jayne D, Rasmussen N, Andrassy K, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic antibodies. N Engl J Med. 2003;349:36–44. doi: 10.1056/NEJMoa020286. [DOI] [PubMed] [Google Scholar]
- 70.Schmitt WH, Hagen EC, Neumann I European Vasculitis Study Group. Treatment of refractory Wegener's granulomatosis with antithymocyte globulin (ATG): an open study in 15 patients. Kidney Int. 2004;65:1440–8. doi: 10.1111/j.1523-1755.2004.00534.x. [DOI] [PubMed] [Google Scholar]
- 71.De Groot K, Rasmussen N, Bacon PA, et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2005;52:2461–9. doi: 10.1002/art.21142. [DOI] [PubMed] [Google Scholar]
- 72.Jayne DR, Gaskin G, Rasmussen N, et al. European Vasculitis Study Group. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol. 2007;18:2180–8. doi: 10.1681/ASN.2007010090. [DOI] [PubMed] [Google Scholar]
- 73.De Groot K, Harper L, Jayne DR, et al. EUVAS. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med. 2009;19:670–80. doi: 10.7326/0003-4819-150-10-200905190-00004. [DOI] [PubMed] [Google Scholar]
- 74.Hellmich B, Flossmann O, Gross WL, et al. EULAR recommendations for conducting clinical studies and/or clinical trials in systemic vasculitis: focus on anti-neutrophil cytoplasm antibody-associated vasculitis. Ann Rheum Dis. 2007;66:605–17. doi: 10.1136/ard.2006.062711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Salvarani C, Cimino L, Macchioni P, et al. Risk factors for visual loss in an Italian population-based cohort of patients with giant cell arteritis. Arthritis Rheum. 2005;53:293–97. doi: 10.1002/art.21075. [DOI] [PubMed] [Google Scholar]
- 76.Hoffman GS, Cid MC, Hellmann DB, et al. International Network for the Study of Systemic Vasculitides. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum. 2002;46:1309–18. doi: 10.1002/art.10262. [DOI] [PubMed] [Google Scholar]
- 77.Birkhead NC, Wagereor HP, Shick RM. Treatment of temporal arteritis with adrenal corticosteroids. Results in 55 cases in which lesions was proved at biopsy. JAMA. 1957;163:821–7. doi: 10.1001/jama.1957.02970450023007. [DOI] [PubMed] [Google Scholar]
- 78.Chan CC, Paine M, O'Day J. Steroid management in giant cell arteritis. Br J Ophthalmol. 2001;85:1061–4. doi: 10.1136/bjo.85.9.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Warrington KJ, Matteson EL. Management guidelines and outcome measure in giant cell arteritis. Clin Exp Rheumatol. 2007;25(Suppl. 47):S137–41. [PubMed] [Google Scholar]
- 80.Hunder GG, Sheps SG, Allen GL, Joyce JW. Daily and alternate day corticosteroid regimens in treatment of GCA: comparison in a prospective study. Ann Intern Med. 1975;82:613–8. doi: 10.7326/0003-4819-82-5-613. [DOI] [PubMed] [Google Scholar]
- 81.Lee MS, Smith SD, Gala A, Hoffman GS. Antiplatelet and anticoagulant therapy I patients with GCA. Arthritis Rheum. 2006;54:3306–9. doi: 10.1002/art.22141. [DOI] [PubMed] [Google Scholar]
- 82.Nester G, Berkum Y, Mates M, et al. Low dose aspirin and prevention of cranial ischaemic complications in GCA. Arthritis Rheum. 2004;50:1332–7. doi: 10.1002/art.20171. [DOI] [PubMed] [Google Scholar]
- 83.Mahr AD, Jover JA, Spiera RF, et al. Adjunctive methotrexate for treatment of giant cell arteritis: an individual patient data meta-analysis. Arthritis Rheum. 2007;56:2789–97. doi: 10.1002/art.22754. [DOI] [PubMed] [Google Scholar]
- 84.De Silva M, Hazleman BL. Azathioprine in GCA – a double blind study. Ann Rheum Dis. 1986;45:136–8. doi: 10.1136/ard.45.2.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hoffman GS, Cid MC, Rendt-Zagar KE, et al. Infliximab–GCA Study Group. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med. 2007;146:621–30. doi: 10.7326/0003-4819-146-9-200705010-00004. [DOI] [PubMed] [Google Scholar]
- 86.Hoffman GS, Leavitt RY, Kerr GS, et al. Treatment of glucocorticoid-resistant or relapsing Takayasu arteritis with methotrexate. Arthritis Rheum. 1994;37:578–82. doi: 10.1002/art.1780370420. [DOI] [PubMed] [Google Scholar]
- 87.Valsakumar AK, Valappil UC, Jorapur V, et al. Role of immunosuppressive therapy on clinical, immunological, and angiographic outcome in active Takayasu's arteritis. Rheumatol. 2003;30:1793–8. [PubMed] [Google Scholar]
- 88.Hoffman GS. Treatment of resistant Takayasu's arteritis. Rheum Dis Clin North Am. 1995;21:73–80. [PubMed] [Google Scholar]
- 89.Adu D, Pall A, Luqmani RA, et al. Controlled trial of pulse versus continuous prednisolone and cyclophosphamide in the treatment of vasculitis. Q J Med. 1997;90:401–9. doi: 10.1093/qjmed/90.6.401. [DOI] [PubMed] [Google Scholar]
- 90.Gayraud M, Guillevin L, Cohen P, et al. Treatment of good-prognosis polyarteritis nodosa and Churg Strauss syndrome: comparison of steroids and oral or pulse cyclophosphamide in 25 patients. French Co-operative Study Group for Vasculitides. Br J Rheumatol. 1997;36:1209–17. doi: 10.1093/rheumatology/36.12.1290. [DOI] [PubMed] [Google Scholar]
- 91.Taubert KA, Rowley AH, Shulman ST. Nationwide survey of Kawasaki disease and acute rheumatic fever. J Pediatr. 1991;119:279–82. doi: 10.1016/s0022-3476(05)80742-5. [DOI] [PubMed] [Google Scholar]
- 92.Rowley AH, Shulman ST, Spike BT, Mask CA, Baker SC. Oligoclonal IgA response in the vascular wall in acute Kawasaki disease. J Immunol. 2001;166:1334–43. doi: 10.4049/jimmunol.166.2.1334. [DOI] [PubMed] [Google Scholar]
- 93.Durongpisitkul K, Gururaj VJ, Park JM, Martin CF. The prevention of coronary artery aneurysm in Kawasaki disease: a meta-analysis on the efficacy of aspirin and immunoglobulin treatment. Pediatrics. 1995;96:1057–61. [PubMed] [Google Scholar]
- 94.Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment and long-term management of Kawasaki disease: a statement for health professionals from the committee on rheumatic fever, endocarditis and Kawasaki disease. Council on Cardiovascular Disease in the Young: American Heart Association. Pediatrics. 2004;114:1708–33. doi: 10.1542/peds.2004-2182. [DOI] [PubMed] [Google Scholar]
- 95.Sawaji Y, Haneda N, Yamaguchi S, et al. Coronary risk factors in acute Kawasaki disease: correlation of serum immunoglobulin levels with coronary complications. Acta Paediatr Jpn. 1998;40:218–25. doi: 10.1111/j.1442-200x.1998.tb01915.x. [DOI] [PubMed] [Google Scholar]
- 96.Muta H, Ishii M, Egami K, et al. Early intravenous gamma-globulin treatment for Kawasaki disease: the nationwide surveys in Japan. J Pediatr. 2004;144:496–9. doi: 10.1016/j.jpeds.2003.12.033. [DOI] [PubMed] [Google Scholar]
- 97.Newburger JW, Sleeper LA, McCrindle BW, et al. Pediatric Heart Network Investigators. Randomized trial of pulsed corticosteroid therapy for primary treatment of Kawasaki disease. N Engl J Med. 2007;356:663–75. doi: 10.1056/NEJMoa061235. [DOI] [PubMed] [Google Scholar]
- 98.Furukawa T, Kishiro M, Akimoto K, et al. Effects of steroid pulse therapy on immunoglobulin-resistant Kawasaki disease. Arch Dis Child. 2008;93:142–6. doi: 10.1136/adc.2007.126144. [DOI] [PubMed] [Google Scholar]
- 99.The Wegener's Granulomatosis Etanercept Trial Research Group. Limited versus severe Wegener's granulomatosis. Arthritis Rheum. 2003;48:2299–309. doi: 10.1002/art.11075. [DOI] [PubMed] [Google Scholar]
- 100.Eisenberger U, Fakhouri F, Vanhille P, et al. ANCA-negative pauci-immune renal vasculitis: histology and outcome. Nephrol Dial Transplant. 2005;20:1392–9. doi: 10.1093/ndt/gfh830. [DOI] [PubMed] [Google Scholar]
- 101.Bolton WK, Sturgill BC. Methylprednisolone therapy for acute crescentic rapidly progressive glomerulonephritis. Am J Nephrol. 1989;9:368–75. doi: 10.1159/000167998. [DOI] [PubMed] [Google Scholar]
- 102.Jayne DR, Chapel H, Adu D, et al. Intravenous immunoglobulin for ANCA-associated systemic vasculitis with persistent disease activity. Q J Med. 2000;93:433–9. doi: 10.1093/qjmed/93.7.433. [DOI] [PubMed] [Google Scholar]
- 103.Langford CA, Talar-Williams C, Barron KS, Sneller MC. Use of a cyclophosphamide-induction methotrexate-maintenance regimen for the treatment of Wegener's granulomatosis: extended follow-up and rate of relapse. Am J Med. 2003;114:463–9. doi: 10.1016/s0002-9343(03)00077-9. [DOI] [PubMed] [Google Scholar]
- 104.Metzler C, Miehle N, Manger K, et al. German Network of Rheumatic Diseases. Elevated relapse rate under oral methotrexate versus leflunomide for maintenance of remission in Wegener's granulomatosis. Rheumatology (Oxf) 2007;46:1087–91. doi: 10.1093/rheumatology/kem029. [DOI] [PubMed] [Google Scholar]
- 105.Langford CA, Talar-Williams C, Sneller MC. Mycophenolate mofetil for remission maintenance in the treatment of Wegener's granulomatosis. Arthritis Rheum. 2004;51:278–83. doi: 10.1002/art.20240. [DOI] [PubMed] [Google Scholar]
- 106.Koulaki M, Jayne DR. Mycofenolate mofetil in anti-neutrophil associated systemic vasculitis. Nephron Clin Pract. 2006;102:100–7. doi: 10.1159/000089667. [DOI] [PubMed] [Google Scholar]
- 107.Stegeman CA, Tervaert JW, Sluiter WJ, et al. Association of chronic nasal carriage of Staphylococcus aureus and higher relapse rates in Wegener granulomatosis. Ann Intern Med. 1994;120:12–7. doi: 10.7326/0003-4819-120-1-199401010-00003. [DOI] [PubMed] [Google Scholar]
- 108.Saadoun D, Resche-Rigon M, Thibault V, Piette JC, Cacoub P. Antiviral therapy for hepatitis C virus-associated mixed cryoglobulinemia vasculitis: a long-term follow up study. Arthritis Rheum. 2006;54:3696–706. doi: 10.1002/art.22168. [DOI] [PubMed] [Google Scholar]
- 109.Cacoub P, Delluc A, Saadoun D, Landau DA, Sene D. Anti-CD20 monoclonal antibody (rituximab) treatment for cryoglobulinemic vasculitis: where do we stand? Ann Rheum Dis. 2008;67:283–7. doi: 10.1136/ard.2006.065565. [DOI] [PubMed] [Google Scholar]
- 110.Nielsen HE. Epidemiology of Schonlein–Henoch purpura. Acta Paediatr Scand. 1988;77:125–31. doi: 10.1111/j.1651-2227.1988.tb10610.x. [DOI] [PubMed] [Google Scholar]
- 111.Pillebout E, Thervet E, Hill G, et al. Henoch–Schönlein purpura in adults: outcome and prognostic. Factors Am Soc Nephrol. 2002;13:1271–8. doi: 10.1097/01.asn.0000013883.99976.22. [DOI] [PubMed] [Google Scholar]
- 112.Saulsbury FT. Epidemiology of Henoch–Schonlein purpura. Cleve Clin J Med. 2002;69(Suppl. 2):S1187–9. doi: 10.3949/ccjm.69.suppl_2.sii87. [DOI] [PubMed] [Google Scholar]
- 113.Chartapisak W, Opastirakul S, Willis N, Craig JC, Hodson EM. Prevention and treatment of renal disease in Henoch–Schonlein Purpura: a systematic review. Arch Dis Child. 2009;94:132–7. doi: 10.1136/adc.2008.141820. [DOI] [PubMed] [Google Scholar]
- 114.Luqmani R. Treatment of polymyalgia rheumatica and giant cell arteritis: are we any further forward? Ann Intern Med. 2007;146:674–6. doi: 10.7326/0003-4819-146-9-200705010-00011. [DOI] [PubMed] [Google Scholar]
- 115.Wegener's Granulomatosis Etanercept Trial (WGET) Research Group. Etanercept plus standard therapy for Wegener's granulomatosis. N Engl J Med. 2005;352:351–61. doi: 10.1056/NEJMoa041884. [DOI] [PubMed] [Google Scholar]
- 116.Chan AT, Flossmann O, Mukhtyar C, Jayne DR, Luqmani RA. The role of biologic therapies in the management of systemic vasculitis. Autoimmun Rev. 2006;5:273–8. doi: 10.1016/j.autrev.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 117.Booth A, Harper L, Hammad T, et al. Prospective study of TNFalpha blockade with infliximab in anti-neutrophil cytoplasmic antibody-associated systemic vasculitis. J Am Soc Nephrol. 2004;15:717–21. doi: 10.1097/01.asn.0000114554.67106.28. [DOI] [PubMed] [Google Scholar]
- 118.Flossmann O, Jones RB, Jayne DR, Luqmani RA. Should rituximab be used to treat antineutrophil cytoplasmic antibody associated vasculitis? Ann Rheum Dis. 2006;65:841–4. doi: 10.1136/ard.2005.048900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kalsch AI, Soboletzki M, Schmitt WH, et al. Imatinib mesylate, a new kid on the block for the treatment of anti-neutrophil cytoplasmic autoantibodies-associated vasculitis? Clin Exp Immunol. 2008;151:391–8. doi: 10.1111/j.1365-2249.2007.03572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lozano E, Segarra M, Garcia-Martinez A, et al. Imatinib mesylate inhibits in vitro and ex vivo biological responses related to vascular occlusion in giant cell arteritis. Ann Rheum Dis. 2008;67:1581–8. doi: 10.1136/ard.2007.070805. [DOI] [PubMed] [Google Scholar]
- 121.Smeeth L, Cook C, Hall AJ. Incidence of diagnosed polymyalgia rheumatica and temporal arteritis in the United Kingdom. Ann Rheum Dis. 2006;65:1093–8. doi: 10.1136/ard.2005.046912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ferri C. Mixed cryoglobulinemia. Orphanet J Rare Dis. 2008;3:25. doi: 10.1186/1750-1172-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]