

Abstract
Stability of the myelin-axon unit is achieved, at least in part, by specialized paranodal junctions comprised of the neuronal heterocomplex of contactin and contactin-associated protein and the myelin protein neurofascin 155. In multiple sclerosis, normal distribution of these proteins is altered, resulting in the loss of the insulating myelin and consequently causing axonal dysfunction. Previously, this laboratory reported that mice lacking the myelin-enriched lipid sulphatide are characterized by a progressive deterioration of the paranodal structure. Here, it is shown that this deterioration is preceded by significant loss of neurofascin 155 clustering at the myelin paranode. Interestingly, prolonged electrophoretic separation revealed the existence of two neurofascin 155 bands, neurofascin 155 high and neurofascin 155 low, which are readily observed following N-linked deglycosylation. Neurofascin 155 high is observed at 7 days of age and reaches peak expression at one month of age, while neurofascin 155 low is first observed at 14 days of age and constantly increases until 5 months of age. Studies using conditional neurofascin knockout mice indicated that neurofascin 155 high and neurofascin 155 low are products of the neurofascin gene and are exclusively expressed by oligodendrocytes within the central nervous system. Neurofascin 155 high is a myelin paranodal protein while the distribution of neurofascin 155 low remains to be determined. While neurofascin 155 high levels are significantly reduced in the sulphatide null mice at 15 days, 30 days and 4 months of age, neurofascin 155 low levels remain unaltered. Although maintained at normal levels, neurofascin 155 low is incapable of preserving paranodal structure, thus indicating that neurofascin 155 high is required for paranodal stability. Additionally, comparisons between neurofascin 155 high and neurofascin 155 low in human samples revealed a significant alteration, specifically in multiple sclerosis plaques.
Keywords: sulphatide, cerebroside sulphotransferase, myelin, paranode, Caspr
Introduction
Axonal saltatory conduction is facilitated by the myelin sheath, a lipid rich multi-lamellar wrap generated by oligodendrocytes in the CNS. Throughout much of its length the mature sheath lacks cytoplasm resulting in compaction of the myelin membrane. At the edges of the sheath, cytoplasm is retained in sacs known as paranodal loops (Peters et al., 1976). These loops ‘bend’ toward the axonal membrane and establish septate-like junctions that maintain clusters of voltage-gated sodium and potassium channels at the node of Ranvier and the juxtaparanode, respectively (Chiu, 1980; Waxman and Richie, 1985; Chiu and Schwarz, 1987; Poliak and Peles, 2003; Rasband, 2004). These specialized junctions are also critical in tethering the myelin sheath to the axon through trans interactions between the myelin protein neurofascin 155 and the axonal heterocomplex of contactin and contactin-associated protein (Caspr) (Rios, 2000; Tait et al., 2000; Bhat et al., 2001; Boyle et al., 2001; Charles et al., 2002). Although the specifics of the interactions between neurofascin 155 and contactin/Caspr remain unresolved (Gollan et al., 2003; Bonnon et al., 2007), it is clear that mice lacking either contactin, Caspr or neurofascin 155 fail to form the paranodal septate-like junctions, do not properly establish paranodal and juxtaparanodal protein domains (Bhat et al., 2001; Boyle et al., 2001; Sherman et al., 2005; Zonta et al., 2008, Pillai et al., 2009), and exhibit compromised paranodal loop orientation (Bhat et al., 2001). Whereas compromised paranode formation and stability are consistent with the absence of either contactin, Caspr or neurofascin 155, it is more difficult to reconcile the loss of paranode structure in mice that lack netrin-1 or deleted in colorectal cancer (DCC) (Jarjour et al., 2008), myelin and lymphocyte (MAL) protein (Schaeren-Wiemers et al., 2004), galactocerebroside and sulphatide (Dupree et al., 1998) or sulphatide alone (Honke et al., 2002). Paranodal deterioration, reminiscent of these mutant mice, also occurs in multiple sclerosis, prior to demyelination, as paranodal loops face away from the axon (Suzuki et al., 1969), paranodal clustering of Caspr and neurofascin 155 is reduced (Wolswijk and Balesar, 2003; Howell et al., 2006), and juxtaparanodal proteins invade the paranode (Howell et al., 2006). Interestingly, in addition to reporting a possible reduction of sulphatide in normal-appearing white matter in patients with multiple sclerosis, Marbois et al. (2000) showed a significant alteration in the sulphatide species present in multiple sclerosis. These findings emphasize the need to elucidate further the role of sulphatide in the regulation of paranode stability.
The sulphatide null mice, which contain a disruption in the gene that encodes the enzyme cerebroside sulphotransferase (CST) (Honke et al., 1996, 1997), initially form abundant, compacted myelin sheaths and structurally normal nodes and paranodes; however, with age the CST knockout mice reveal a loss of myelin sheath compaction, altered paranodal loop orientation, increased nodal length (Marcus et al., 2006), and a reduction of nodal protein domains (Ishibashi et al., 2002). In the present study, quantitative analysis advances the previous observation of neurofascin 155 cluster loss (Marcus et al., 2006), while western blot analysis indicates that the loss of neurofascin 155 clusters does not result strictly from decreased neurofascin 155 levels. Equally important, the current study demonstrates that neurofascin 155 is comprised of two forms, termed neurofascin 155 high (155H) and neurofascin 155 low (155L). It is shown here that neurofascin 155H and neurofascin 155L are oligodendrocyte-specific products of the neurofascin (Nfasc) gene that differ with regard to N-linked glycosylation and temporal expression. Neurofascin 155H is a paranodal protein whose levels are reduced in CST knockout mice and is nearly absent by 4 months of age. In contrast, levels of neurofascin 155L are not altered in the CST knockout mice as compared to littermate wild-type animals. With age in both genotypes, neurofascin 155L becomes more abundant and less glycosylated. However, its function remains unknown. Evaluation of human brain samples showed a multiple sclerosis plaque-specific alteration between neurofascin 155H and neurofascin 155L following deglycosylation. These results indicate that, in both mouse and human samples, the expression pattern of neurofascin 155H and neurofascin 155L is altered coincident with paranodal decay.
Materials and methods
Animals
Wild-type and CST knockout (Honke et al., 2002), also known as sulphatide null, mice were genotyped as described (Shroff et al., 2009). Other animals used in this study include Caspr knockout (Bhat et al., 2001), neurofascin conditional knockout, referred to as CNP-Cre;Nfascflox/flox, (Pillai et al., 2009), and littermate wild-type mice. Sprague-Dawley rats were purchased from Charles River Laboratories (Wilmington, MA, USA). Animal use was conducted in accordance with the guidelines from the National Institutes of Health and approved by the Virginia Commonwealth University’s Animal Care and Use Committee.
Antibodies
The neurofascin antibodies used in this study have been previously characterized and are known as FIGQY (Ogawa et al., 2006) and NF-C1 (Tait et al., 2000). For immunocytochemical labelling of frozen tissue sections, FIGQY and NF-C1 were used at 1:200 and 1:1000 dilutions, respectively. For western blot analysis, both antibodies were diluted 1:2000. These antibodies are directed against distinct epitopes of the cytoplasmic tail common to neuronal neurofascin 186, oligodendrocyte neurofascin 155, and the uncharacterized protein neurofascin 140. Other antibodies used in this study include: anti-Caspr (1:1000; Bhat et al., 2001); anti-pan voltage gated sodium channel (1:200; Sigma-Aldrich, St Louis, MO, USA); anti-extracellular signal-regulated kinase 2 (1:10 000; Santa Cruz Biotechnology, Santa Cruz, CA,USA); anti-myelin basic protein (1:100; Millipore, Billerica, MA); anti-2′, 3′-cyclic nucleotide 3′-phosphodiesterase (CNP; 1:1000, Covance, Emeryville, CA); horseradish peroxidase conjugated secondary antibodies (1:10 000; Santa Cruz); and fluorescently conjugated secondary antibodies (1:200; Vector Laboratories, Burlingame, CA, USA).
Immunocytochemistry
Cervical spinal cord samples from three wild-type and four littermate CST knockout mice at 15 and 30 days of age and a rat at 7 days of age were single, double or triple labelled as previously described (Dupree et al., 1999; Marcus et al., 2006).
Quantitation of Nfasc 155 localization
Immunolabelled sections were analysed using a Leica TCS2 AOBS laser scanning confocal microscope. Maximum projection images were compiled using eight optical sections spanning 3.6 µm and collected using a pin hole of one Airy unit with a resolution of 1024 × 1024 pixels. All images were collected using a PL APO 63 × oil immersion objective lens (numerical aperture of 1.4–0.7) with a digital zoom of two resulting in a microscopic field of 119 µm × 119 µm. To ensure that similar regions of the cervical cord were sampled, the cerebellum was used to discern ventral and dorsal columns. All images were collected from the cervical ventral columns. The posterior portion of the cerebellum was also used as the most anterior extent of the spinal cord and thus served as the starting point for image collection. For quantitation, six maximum projection images per section were collected; neurofascin 155 paranodal clusters were counted, and the average number of clusters per image was determined for each mouse. Two sets of criteria were used to identify paired neurofascin 155-containing paranodes. First, solid rectangular clusters ranging between 5 and 15 µm with well demarcated ends were identified as a single paranodal pair (see Supplementary Fig. 2A’ and B’). Second, paired rectangular clusters (corresponding to two adjacent paranodes) separated by a small region void of labelling (corresponding to the node of Ranvier) were also identified as a single paranodal pair (Supplementary Fig. 2C’). Unpaired clusters were not counted (Supplementary Fig. 2D’). All counts were statistically compared using two-tailed t-tests and significance was defined as P < 0.05.
Western blot analysis
Animals were deeply anaesthetized with 2, 2, 2-tribromoethanol (Sigma-Aldrich); brain and spinal cord were dissected, rapidly frozen separately in liquid nitrogen, and stored at −80°C. Homogenization utilized a Glas-Col (Terre Haute, IN, USA) motor-driven homogenizer, a Potter Elvehjem mortar, and a Teflon pestle (Wheaton Industries, Inc., Millville, NJ, USA). Tissues were homogenized (5 min per brain and 3 min per spinal cord) on ice in modified radioimmunoprecipitation assay buffer [137 mM NaCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, 2.7 mM KCl, pH 7.4 with 0.1% sodium dodecyl sulphate (SDS), 0.5% sodium deoxycholate, 0.1% nonidet P-40] with protease inhibitor cocktail [Sigma-Aldrich P8340, comprised of 4-(2-aminoethyl)benzenesulphonyl fluoride, pepstatinA, E-64, bestatin, leupeptin and aprotinin] followed by centrifugation for 1 min at 16 100g in a table top microcentrifuge. Supernatant concentrations were determined using the Micro BCA Protein Assay Kit (Pierce, Rockford, IL, USA). Twenty–thirty micrograms of spinal cord or 40–50 µg of brain proteins in Laemmli sample buffer (Bio-Rad, Hercules, CA, USA) containing 5% β-mercaptoethanol were subjected to SDS–polyacrylamide gel electrophoresis for 30 min at 70 V followed by either 40 or 150 min (see below) at 180 V using 10% Ready Gels (Bio-Rad). Precision Plus Protein Kaleidoscope Standards (Bio-Rad) allowed monitoring of the gel run and transfer. For the extended electrophoresis protocol, the electrophoresis was allowed to proceed until the 75 kDa standard band reached the bottom of the gel (∼150 min) and transferred to nitrocellulose for 2 h at 100 V. The membranes were then blocked with 5% non-fat dry milk (Bio-Rad) in phosphate buffered saline containing 0.05% Tween 20 (Sigma-Aldrich) prior to incubation with primary and secondary antibodies, and the same solution was utilized for antibody incubations. Immunoreactive bands were visualized with horseradish peroxidase chemiluminescent substrate (Pierce or Millipore, Billerica, MA, USA) and either an Alpha Innotech FluorChem SP imager (San Leandro, CA, USA) or radiograph film.
Glycosidase reaction
Peptide N-glycosidase F (PNGase F) and related reagents were obtained from New England Biolabs (Ipswich, MA, USA). Following the manufacturer’s suggested protocol, a typical 25 μl reaction included 50 μg of protein, 2 μl of PNGase F (1000 U, as defined by the manufacturer), and 1 μl of protease inhibitor cocktail (Sigma-Aldrich, P8340). Following 3 h at 37°C, samples were mixed with one volume of Laemmli Sample Buffer containing 5% β-mercaptoethanol and prepared for SDS–polyacrylamide gel electrophoresis as above.
Densitometry, molecular mass and glycosylation analyses
Except where stated otherwise, protein band densities were measured with the AlphaEase software version 4.1.0 by Alpha Innotech Corporation. All western blot densities were normalized to extracellular signal-regulated kinase 2, data were analysed by a two-tailed t-test, and significance was defined as P < 0.05. The same software was paired with Magic Mark XP Western Protein Standard (Invitrogen, Carlsbad, CA, USA) to determine molecular masses and shifts due to deglycosylation utilizing the 220, 120, 100 and 80 kDa bands of the PNGase F treated and untreated samples.
For the quantitative analysis shown in Fig. 8, ImageJ 1.14o was utilized to determine the density of a rectangular selection encompassing neurofascin 155H’ and neurofascin 155L’ (neurofascin' refers to deglycosylated neurofascin). The resulting graphical plot was observed to contain either one or two peaks and the area under the curve was determined with the wand tool. In the case of one peak, the straight line selection function was utilized to divide the plot into the main region corresponding to the neurofascin 155H’ band and the lesser region corresponding to the shadow-like neurofascin 155L’ band. The area under each curve was then determined as above. In the case of two peaks, the wand tool was utilized to divide the plot into two regions, corresponding to neurofascin 155H’ and neurofascin 155L’. For examples of this procedure and representative plots of non-multiple sclerosis and multiple sclerosis plaque samples, see Supplementary Fig. 3. The density values obtained from this analysis were first utilized to create a ratio of neurofascin 155H’/neurofascin 155L’ for each sample. These ratios were then grouped as non-multiple sclerosis and normal-appearing white matter versus multiple sclerosis plaque, and compared using a two-tailed Student’s t-test with unequal variance. Multiple sclerosis sample 119 was removed from quantitative analysis due to a Cook’s distance value >1, calculated with the Predictive Analytics SoftWare Statistics 17.0 software, indicating that a data point is an outlier and therefore a candidate for removal from the data set (Cook, 1977); it is noteworthy that this patient with confirmed multiple sclerosis died of complications due to leukaemia and suffered multiple brain haemorrhages.
Figure 8.

The pattern of neurofascin 155H’ and neurofascin 155L’ is altered in multiple sclerosis plaques. (A) Following the extended electrophoresis protocol, clearly delineated neurofascin 155L was not observed in non-multiple sclerosis (A1 and A2) or normal-appearing white matter-multiple sclerosis (A1 and A3) samples but was observed in multiple sclerosis plaque samples (black stars). (B) Following PNGase F treatment, no clearly delineated neurofascin 155L’ band was observed in either the non-multiple sclerosis or normal-appearing white matter-multiple sclerosis samples. In contrast, neurofascin 155L’ was clearly observed in the majority of multiple sclerosis brains analysed (white stars). (C) The ratio of neurofascin 155H’/neurofascin 155L’ was significantly reduced in multiple sclerosis plaque samples (n = 8; P < 0.006) compared to non-multiple sclerosis and normal-appearing white matter-multiple sclerosis samples (n = 7). (D) Side-by-side analysis of PNGase F treated samples of normal-appearing white matter-multiple sclerosis and multiple sclerosis plaque samples from the same donors shows neurofascin 155L’ only in the plaque samples. It is noteworthy that the pattern of neurofascin 155H’ and neurofascin 155L’ is similar between inactive (#243) and active (#137) plaque samples. Additionally, neurofascin 186’ was frequently not observed in multiple sclerosis plaque samples. The commonly used loading control proteins β-actin, glyceraldehyde 3-phosphate dehydrogenase and extracellular signal-regulated kinase 2 are shown and demonstrate a lack of autolysis within the samples. − = untreated; P = PNGase F treated; NM or NAWM = normal appearing white matter from multiple sclerosis donor; M = Magic Mark XP Protein Standard; MS = multiple sclerosis; Nfasc = neurofascin; ERK2 = extracellular signal-regulated kinase 2; GAPDH = glyceraldehyde 3-phosphate dehydrogenase. *P<0.05.
The NetNGlyc Server 1.0 (http://www.cbs.dtu.dk/services/NetNGlyc/) was used to predict locations of N-linked glycosylation (PubMed accession AAL27854, rat neurofascin 155 kDa isoform) as well as the likelihood that a given site is glycosylated in vivo. ‘Highly likely’ is defined as above 0.5 threshold, 9/9 neural networks agreeing on the outcome, and a ++ result. ‘Somewhat likely’ is defined as above 0.5 threshold, 6 or more neural networks agreeing on the outcome, and a + result. Both likelihoods were considered to be potential sites of glycosylation. Only NXS/T sequons free of proline and located in the extracellular region were considered. Molecular mass was calculated with the ExPASy ‘Computer pI/MW’ tool available at www.expasy.ch/tools/pi_tool.html.
Human samples
Human brain samples from non-multiple sclerosis and multiple sclerosis donors (both plaque and normal-appearing white matter) were provided by the Rocky Mountain Multiple Sclerosis Centre (Englewood, CO, USA). All samples were collected at time of autopsy, which was between one and 9 h post-mortem. At the time of death, donors ranged between 30 and 87 years of age. All samples were collected from sub-cortical white matter regions and immediately snap frozen in liquid nitrogen. Based on medical history and autopsy evaluation, none of the multiple sclerosis individuals included in this study presented any neurodegenerative disorders other than multiple sclerosis. Brains were classified as ‘multiple sclerosis’ or ‘non-multiple sclerosis’ based on medical history and post-mortem pathological analyses at both the macroscopic and light microscopic levels. Macroscopic analysis revealed well circumscribed, grey, glistening depressed plaques only in multiple sclerosis samples. Microscopic analysis was used to assess myelin pallor and cellularity. Using this combination of evaluations, all disease tissue samples used in this study were collected from confirmed multiple sclerosis cases.
Extent of cellularity was used to classify multiple sclerosis plaque samples further as ‘active’ versus ‘inactive’. Active multiple sclerosis samples exhibited hypercellularity demonstrated by lymphocytes, macrophages and/or reactive astrocytes. In contrast, inactive plaques were identified by the absence of hypercellularity. All multiple sclerosis samples were collected within the lesion site whereas normal appearing white matter samples were collected from brains with confirmed multiple sclerosis but from regions that appeared normal based on macroscopic and microscopic evaluation. For specific age, gender, post-mortem harvest times and pathologic summary of each donor used in this study, see Table 1.
Table 1.
Donor information for the Rocky Mountain Multiple Sclerosis Centre brain samples used in this study
| Disease summary | No. | Age | Gender | HTTC | Pathological evaluation of the brain |
|---|---|---|---|---|---|
| Non-MS | 2 | 63 | – | 9 | Myelin stain revealed no evidence of MS |
| Non-MS | 5 | 55 | F | 2 | No plaques observed in coronal sections |
| Non-MS | 27 | 65 | M | 6.5 | History of myocardial infarction |
| Non-MS | 32 | 70 | F | 8 | No plaques observed in multiple brain regions |
| NAWM-MS | 243a | 58 | F | 5 | Numerous shadow plaques in PVWM |
| NAWM-MS | 216a | 71 | F | 4.5 | Numerous plaques with near total myelin loss |
| NAWM-MS | 235 | 41 | F | 3 | Neurons preserved, no anoxic injury; cavitation |
| NAWM-MS | 237 | 50 | M | 4 | PVWM extensively demyelinated, no anoxic injury |
| NAWM-MS | 137a | 30 | M | 5 | Grossly visible plaques in multiple regions |
| Active MS | 137a | 30 | M | 5 | Grossly visible plaques in multiple regions |
| Active MS | 73 | 44 | M | 4 | Myelin pallor and shadow plaques throughout |
| Active MS | 136 | 43 | F | 3.5 | PVWM extensively demyelinated, hypercellularity |
| Active MS | 168 | 37 | F | 3.5 | PVWM extensively demyelinated |
| Inactive MS | 119 | 38 | F | 1 | 12 year history of multiple sclerosis |
| Inactive MS | 164 | – | – | 3 | Axons present in demyelinated regions |
| Inactive MS | 224 | 87 | F | 4 | Myelin pallor in frontal and parietal lobes |
| Inactive MS | 243a | 58 | F | 5 | Numerous shadow plaques in PVWM |
| Inactive MS | 216a | 71 | F | 4.5 | Numerous plaques with near total myelin loss |
| Averages and totals | 52.1 | 9 F, 4 M | 4.5 | ||
| 18 samples | 2 NA |
MS = multiple sclerosis; HTTC = hours to tissue collection; PVWM = periventricular white matter; NAWM = normal appearing white matter; − = data not available (NA).
aTwo samples (normal appearing white matter and plaque) from the same donor.
For analysis within this study, non-multiple sclerosis (n = 4), normal appearing white matter from multiple sclerosis donors (n = 5) and multiple sclerosis plaque samples from active (n = 4) and inactive (n = 5) lesions were evaluated. An ∼2 × 1 cm sample was collected from the received sub-cortical white matter samples and processed for western blot and deglycosylation analyses as described above with 20–25 µg of protein per lane.
Results
Paranodal clusters of neurofascin 155 are significantly reduced in CST knockout mice
The NF-C1 pan-neurofascin antibody was utilized to quantify paranodal clusters of neurofascin 155 in the ventral columns of wild-type and CST knockout mice at 15 and 30 days of age (Fig. 1; see Supplementary Fig. 1 for colour). At both ages, wild-type mice exhibited numerous paranodal clusters of neurofascin 155 (Fig. 1A, C and E; Table 2). While paranodal clusters of neurofascin 155 were also observed in the CST knockout mice, comparison with the wild-type mice indicated significant reductions at each age examined (for an explanation of how the clusters were defined and quantified, see the ‘Materials and methods’ section). In 15-day-old CST knockout mice, the number of paranodal clusters of neurofascin 155 was reduced by ∼50% and by 30 days of age the CST knockout mice contained fewer than 10% of the number of clusters observed in littermate wild-type mice (Fig. 1B, D and E; Table 2). Similar observations were made with the FIGQY pan-neurofascin antibody (data not shown). Additionally, the 30-day-old CST knockout mice showed a prevalence of small, unpaired clusters (Fig. 1D, Supplementary Fig. 2D’). Double labelling with a sodium channel antibody (data not shown) revealed that the unpaired clusters were at the node of Ranvier, consistent with the distribution of neurofascin 186 (Tait et al., 2000).
Figure 1.

Paranodal clustering of neurofascin 155 is significantly reduced in the absence of sulphatide. Cervical spinal cord sections immunolabelled with the NF-C1 pan-neurofascin antibody revealed abundant paired clusters of neurofascin (Nfasc) 155 in the myelin paranode of wild-type (WT) animals at 15 (A) and 30 (C) days of age. Note that the labelling pattern in the 15-day-old animals was frequently rectangular in shape representing immunolabelling of both neurofascin 155 in the myelin paranode and neurofascin 186 in the (axonal) node. In contrast, by 30 days of age, nodal labelling was less apparent while paranodal labelling remained prevalent. In the CST knockout (KO) spinal cord sections, the number of paranodal clusters of neurofascin 155 was reduced by ∼50 and 90% at 15 (B) and 30 (D) days of age, respectively, compared to littermate wild-type mice. Also, note that by 30 days of age, the prevalence of small, single clusters (arrows) was increased in the CST knockout. (E) Quantitative analyses revealed that the reduction of paranodal clustering of neurofascin 155 was significantly reduced at both 15 and 30 days of age. Scale bar = 10 µm; see Table 2. *P<0.05.
Table 2.
Neurofascin paranodal clusters and protein in the CST knockout spinal cord
| Age | Wild-type (n) | SD | Knockout (n) | SD | P | Reduction (%) | |
|---|---|---|---|---|---|---|---|
| Neurofascin 155 positive clusters/field | |||||||
| 15 days | 75.4 (3) | 14.1 | 36.8 (4) | 8 | 0.0001 | 51.2 | |
| 30 days | 118.6 (3) | 51.1 | 9.5 (4) | 9.3 | 0.001 | 92.2 | |
| Neurofascin 155 (protein), % of wild-type | |||||||
| 15 days | 100 (3) | 3.90% | 64.2 (4) | 13.80% | 0.001 | 35.8 | |
| 30 days | 100 (4) | 17.00% | 60.9 (2) | 15.80% | 0.027 | 39.1 | |
Levels of neurofascin 155 are significantly reduced in spinal cords of CST knockout mice
The reduced paranodal clusters of neurofascin 155 observed in the CST knockout mice could result from either decreased protein expression or improper protein localization. To discern between these two possibilities, the levels of neurofascin 155 were determined by western blot analysis. Figure 2A shows that, compared to littermate wild-type mice, neurofascin 155 was reduced by ∼40% in the spinal cords of 15- and 30-day-old CST knockout mice (Fig. 2D; Table 2). However, as shown in Fig. 2B and C, the decrease in neurofascin 155 is not accompanied by a widespread reduction of myelin proteins as neither levels nor developmental expression patterns of the myelin-specific proteins CNP or myelin basic protein was altered in the CST knockout mice.
Figure 2.
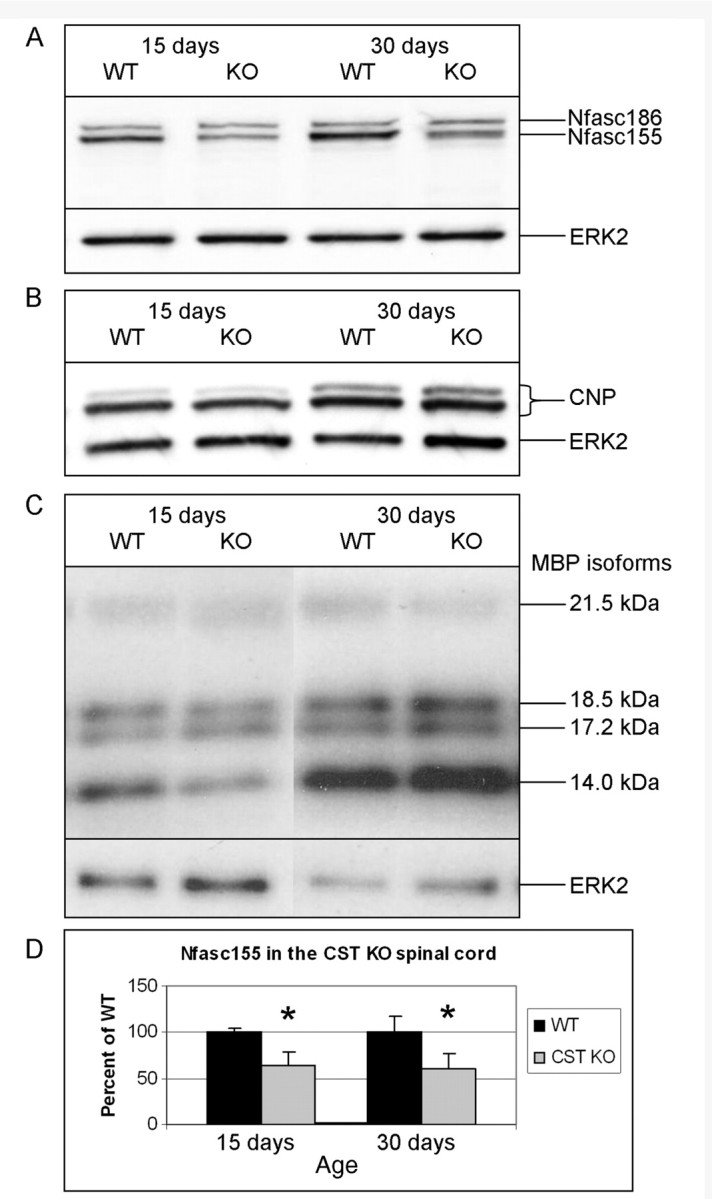
CST knockout spinal cords contain reduced amounts of neurofascin 155 at 15 and 30 days of age. (A) Western blot analysis showed that CST knockout (KO) mice contain ∼40% less (Table 2) neurofascin (Nfasc) 155 at 15 and 30 days of age than wild-type (WT) littermates. However, no change in the levels of either the CNP monomers (B) or myelin basic proteins (MBP) (C) was observed in the absence of sulphatide, demonstrating that the reduction in neurofascin 155 is not due to a global decrease in myelin proteins. (D) Graphical representation of the reduction in neurofascin 155. ERK2 = extracellular signal-regulated kinase 2. *P<0.05.
Extended electrophoresis reveals that neurofascin 155 is comprised of two bands—neurofascin 155 high and neurofascin 155 low
Consistent with previous studies (Schafer et al., 2004; Sherman et al., 2005), standard electrophoretic separation (∼40 min; see ‘Materials and methods’ section) of spinal cord homogenates from wild-type mice up to 30 days of age resulted in the detection of a single band representing neurofascin 155 (Fig. 2A). However, extended electrophoresis of the same samples revealed two bands, now named neurofascin 155 high (155H) and neurofascin 155 low (155L), at 30 days of age in the CST knockout mice (Fig. 3A–C) as well as in the wild-type brain (Fig. 3C) between neurofascin 186 and neurofascin 140. Neurofascin 155L was not detected by extended electrophoresis at 15 days of age in either wild-type or CST knockout animals. Neurofascin 155H and neurofascin 155L were detected by the pan-neurofascin antibodies FIGQY (Fig. 3A and C, and all other figures unless otherwise stated) and NF-C1 (Fig. 3B), and were present in both spinal cord (Fig. 3A and B) and brain (Fig. 3C) homogenates.
Figure 3.

Extended electrophoresis reveals two forms of neurofascin 155. At 15 days of age, the FIGQY (A) and NF-C1 (B) pan-neurofascin antibodies labelled one band representing neurofascin (Nfasc) 155 in both the wild-type (WT) and the CST knockout (KO) spinal cord samples. By 30 days of age, wild-type samples displayed the protein now called neurofascin 155H with a shadow-like band immediately below, while the CST knockout samples showed both neurofascin 155H and neurofascin 155L. (C) Mouse brain samples incubated with FIGQY show neurofascin 155H and neurofascin 155L in both wild-type and CST knockout at 30 days of age and, when compared to 15-day-old samples, suggest a developmental pattern for neurofascin 155L in wild-type as well as CST knockout mice. ERK2 = extracellular signal-regulated kinase 2.
Neurofascin 155H and neurofascin 155L are distinctly regulated through development
To examine whether the observations of Fig. 3 were specific to mouse and to determine the expression profiles of neurofascin 155H and neurofascin 155L in wild-type animals, spinal cord homogenates from rats ranging between 7 days and 5 months of age were separated by the extended electrophoresis protocol (Fig. 4A). Neurofascin 155H was visible at all ages, increasing in intensity from barely detectable at 7 days to maximal levels at 27 days of age, followed by a reduction at 3 and 5 months. An increasingly dark shadow-like band, perhaps representing multiple glycosylation states, was present immediately below neurofascin 155H beginning at 14 days of age. By 3 months of age the shadow-like band was no longer visible but a distinct neurofascin 155L band was observed and remained prominent until at least 5 months of age.
Figure 4.

Neurofascin 155H and neurofascin 155L are developmentally regulated glycoproteins in rat spinal cord. (A) Rat spinal cord homogenates revealed low levels of neurofascin 155H at 7 days of age. Neurofascin 155H steadily increased with age, reaching a peak at 27 days of age followed by a decrease which is maintained through 5 months of age. Neurofascin 155L was first seen as a clearly delineated band at 3 months of age and remained prevalent at 5 months. Interestingly, neurofascin 155L appeared to form gradually from the shadow-like band below neurofascin 155H. (B) Following PNGase F treatment neurofascin 155L’ was observed in 3 and 5 month samples (as expected from Fig. 4A) as well as at 14, 21 and 27 days of age, with increasing abundance with increasing age. Neurofascin 155H’ was barely visible at 7 days of age, became increasingly intense through 27 days of age, then decreased in abundance at 3 and 5 months of age consistent with neurofascin 155H in Fig. 4A. (C) At 7 days of age, when neurofascin 155L’ was not present, the pan-neurofascin antibody FIGQY labelled neurofascin 186 at the node of Ranvier and neurofascin 155H at the paranode in the rat spinal cord. Voltage gated sodium channels are indicated in dark blue (a and e), Caspr is indicated in red (b and f), and FIGQY labelling is shown in green (c and g). The panels on the left (a–d) show a fully formed node:paranode region and in the merged image (d) light blue represents voltage gated sodium channels and neurofascin 186 at the node and the flame colour represents Caspr and neurofascin 155H at the paranode. The panels on the right (e–h) show an immature node:paranode region with a pair of voltage gated sodium channel/neurofascin 186-labelled clusters flanking an unlabelled region that is presumed to develop into the node. Notice that the flame colour is observed in the merged image (h) indicating that Caspr and neurofascin 155H are present in the developing paranode. d = days; m = months; P = PNGase F treated; ERK2 = extracellular signal-regulated kinase 2; Nfasc = neurofascin; Nav = voltage gated sodium. Scale bar = 5 µm.
Neurofascin 155H and neurofascin 155L contain N-linked carbohydrates
Previous work (Volkmer et al., 1992; Davis et al., 1993; Maier et al., 2005) has shown that neurofascin proteins are glycosylated and analysis with the NetNGlyc 1.0 Server indicated that neurofascin 155 contains multiple (predicted) sites for N-linked glycosylation. To test the possibility that neurofascin 155H and neurofascin 155L are the same polypeptide that differ according to the extent of N-linked glycosylation, samples were treated with PNGase F, which specifically removes N-linked carbohydrates without compromising peptide bond integrity (Maley et al., 1989). If the molecular mass difference between neurofascin 155H and neurofascin 155L resulted exclusively from N-linked glycosylation, then a single, faster migrating band would be detected following PNGase F treatment. Interestingly, following N-linked deglycosylation, neurofascin 155H and neurofascin 155L remained as distinct bands (Fig. 4B; see Fig. 6A for side-by-side untreated and treated lanes). The bands resulting from PNGase F treatment are referred to as neurofascin 155H’ and neurofascin 155L’ to acknowledge that neurofascin 155H and neurofascin 155L exist in vivo while neurofascin 155H’ and neurofascin 155L’ are generated by in vitro deglycosylation.
Figure 6.

Neurofascin 155H’ is reduced in CST knockout spinal cords. (A) Following PNGase F treatment, spinal cord homogenates showed an ∼50% decrease in neurofascin 155H’ at 15 and 30 days of age. Surprisingly, at 4 months of age no definitive neurofascin 155H’ (or neurofascin 155H) band was observed in the CST knockout samples (arrow). Quantitative analyses revealed that neurofascin 155H’ (B) was significantly reduced in the CST knockout mice at all ages examined while the levels of neurofascin 155L’ (C) were not altered; see Table 3. − = untreated; P = PNGase F treated; KO = knockout; WT = wild-type; Nfasc = neurofascin; ERK2 = extracellular signal-regulated kinase 2. *P<0.05.
Unexpectedly, PNGase F treatment revealed neurofascin 155L’ from samples of 14, 21 and 27-day-old animals, as well as at the expected ages of 3 and 5 months (Fig. 4B). This transition from shadow-like band to distinct band, possibly due to variable, age-related states of glycosylation (see ‘Discussion’ section), suggested that neurofascin 155L was the source of the increasingly dark shadow-like band observed in Fig. 4A. Fig. 4B shows that neurofascin 155H’ increases steadily between 7 and 27 days of age before sharply decreasing between 27 days and 3 months of age. This temporal pattern of expression of neurofascin 155H’ is consistent with neurofascin 155H playing a role in myelination, as has been previously shown for neurofascin 155 (Collinson et al., 1998). In contrast, neurofascin 155L’, which was not detected at 7 days of age, first appeared in the rat spinal cord between 7 and 14 days of age and steadily increased in abundance through 5 months of age. The slowly increasing abundance of neurofascin 155L’ suggested that, unlike neurofascin 155H, neurofascin 155L does not play a role in the initial stages of myelination.
Neurofascin 155H is a component of the myelin paranode
Since neurofascin 155L was not detected in 7-day-old rat spinal cord (Fig. 4A and B), this age provided the opportunity to determine the localization of neurofascin 155H. As shown in Fig. 4C, the localizations of voltage gated sodium channels and Caspr are used to identify the nodal and paranodal domains, respectively, in established [Fig. 4C, (a–d)] and developing [Fig. 4C, (e–h)] node:paranode regions. The unlabelled region between the labelled clusters in Fig. 4C (e–h) is consistent with the developing node:paranode region described by Shrager and colleagues (Dugandzija-Novakovic et al., 1995). In both series of images, the FIGQY pan-neurofascin antibody strongly labels the nodal and paranodal regions. Paranodal reactivity against the FIGQY pan-neurofascin antibody at an age when neurofascin 155L is not detected by western blot analysis indicates that neurofascin 155H is the myelin paranodal protein previously referred to as neurofascin 155 (Tait et al., 2000; Poliak and Peles, 2003).
Neurofascin 155H and neurofascin 155L are products of the Nfasc gene
Bhat and colleagues recently reported the generation of a mouse line with a floxed Nfasc transgene that is removed via CNP promoter-regulated Cre expression (CNP-Cre;Nfascflox) (Pillai et al., 2009). Since CNP promoter activity in the CNS has been shown to be exclusive to oligodendrocytes (Trapp et al., 1988), these mice allow for specific deletion of the Nfasc gene from oligodendrocytes. CNP-Cre;Nfascflox/flox mice die by 17 days of age; accordingly, spinal cords were harvested from 16-day-old wild-type (+/+), CNP-Cre/Nfasc+/flox (heterozygote) and CNP-Cre;Nfascflox/flox (homozygote) mice and are shown in Fig. 5A and B. Both wild-type and CNP-Cre;Nfasc+/flox samples exhibited the normal pattern of neurofascin 155H plus the shadow-like band, together termed neurofascin 155H&L, prior to PNGase F treatment (Fig. 5A) and both neurofascin 155H’ and neurofascin 155L’ after PNGase F treatment (Fig. 5B). In contrast, neither neurofascin 155H/neurofascin 155H’ nor neurofascin 155L/neurofascin 155L’ were observed in the CNP-Cre;Nfascflox/flox sample (arrows). These results demonstrate that both neurofascin 155H and neurofascin 155L are products of the Nfasc gene and suggest that both neurofascin 155H and neurofascin 155L are exclusively expressed by oligodendrocytes since expression is eliminated by CNP promoter-regulated Cre expression.
Figure 5.

Neurofascin 155H and neurofascin 155L are products of the Nfasc gene. Spinal cords from 16-day-old CNP-Cre;Nfascflox/flox mice contained no ‘neurofascin 155H&L’ and no neurofascin 155H’ or neurofascin 155L’ (arrows) when observed without (A) or with (B) PNGase F treatment. ERK2 = extracellular signal-regulated kinase 2.
Neurofascin 155H, but not neurofascin 155L, is reduced in the absence of sulphatide
The combined results of Figs 3 and 4 suggest that the levels of neurofascin 155H’ and neurofascin 155L’ could be differentially altered in the CST knockout mice. Accordingly, PNGase F was employed to quantitatively compare levels of neurofascin 155H’ and neurofascin 155L’ in the wild-type and CST knockout mice. As shown in Fig. 6A, PNGase F treated spinal cord samples from CST knockout mice contain ∼50% less neurofascin 155H’ than wild-type littermates at 15 and 30 days of age (Fig. 6B; Table 3). In contrast, neurofascin 155L’ was not reduced at either 15 or 30 days of age (Fig. 6C; Table 3). At 4 months of age, neurofascin 155H’ was also significantly reduced and no definitive band was observed for neurofascin 155H’ (or neurofascin 155H) in the CST knockout spinal cords, while the amount of neurofascin 155L’ remained unchanged (Fig. 6C; Table 3). Running the untreated and PNGase F treated samples side-by-side facilitates the comparison of the shifts in migration induced by deglycosylation. Note that neurofascin 155L was not observed in the untreated lanes at 15 days of age from either genotype or in the 30 day wild-type sample; however, following PNGase F treatment, neurofascin 155L’ was clearly distinguished in these lanes, consistent with the idea that the band labelled ‘neurofascin 155H&L’ contains both proteins, and that neurofascin 155L is variably glycosylated with age. For this reason, only neurofascin 155H’ and neurofascin 155L’ were quantified. Age-dependent extinction of the neurofascin 155H band is not a normal event, as neurofascin 155H (and neurofascin 155L) have been observed in spinal cord homogenates from wild-type mice as old as 22 months of age (data not shown).
Table 3.
Neurofascin 155H' and neurofascin 155L' in the CST knockout spinal cord
| Age | Wild-type (n) | SD (%) | Knockout (n) | SD (%) | P | Reduction (%) | |
|---|---|---|---|---|---|---|---|
| Neurofascin 155H’, % of wild-type | |||||||
| 15 days | 100 (3) | 14.40 | 50.1 (4) | 32.30 | 0.009 | 49.9 | |
| 30 days | 100 (3) | 11.30 | 55.0 (4) | 23.50 | 0.005 | 45 | |
| 4 months | 100 (2) | 8.40 | 34.4 (2) | 28.10 | 0.019 | 65.6 | |
| Neurofascin 155L’, % of wild-type | |||||||
| 15 days | 100 (3) | 22.90 | 77.5 (4) | 26.90 | 0.249 | 0 | |
| 30 days | 100 (3) | 14.40 | 91.2 (4) | 20.80 | 0.515 | 0 | |
| 4 months | 100 (2) | 11.60 | 87.1 (2) | 9.70 | 0.342 | 0 | |
Reduced levels of neurofascin 155H are not limited to the CST knockout mice
To determine whether there is a relationship between reductions in neurofascin 155H/neurofascin 155H’ and paranodal stability, Caspr knockout mice (Bhat et al., 2001) were analysed via PNGase F treatment and the extended electrophoresis protocol. These mice, reminiscent of the CST knockout mice, exhibit abnormal paranodes and lack septate-like paranodal junctions. The life expectancy of these mice is ≤21 days (Bhat et al., 2001), so analysis was restricted to younger ages. Similar to 15-day-old CST knockout mice, 19-day-old Caspr knockout mice revealed noticeably reduced levels of neurofascin 155H and neurofascin 155H’, while levels of neurofascin 155L and neurofascin 155L’ were only modestly altered (Fig. 7). This reduction in neurofascin 155H and neurofascin 155H’, taken together with the findings from the CST knockout mice, suggests that the loss of neurofascin 155H may be a general defect associated with paranodal instability.
Figure 7.

Spinal cords from Caspr knockout mice, similar to CST knockout animals, contain reduced amounts of neurofascin 155H and neurofascin 155H’. Comparable to 15-day-old CST knockout mice, spinal cords samples from 19-day-old Caspr knockout mice treated with PNGase F showed dramatically reduced neurofascin 155H and no band representing neurofascin 155H’ was observed following PNGase F treatment (arrow). Neurofascin 155L’ appears modestly reduced. The Magic Mark Western Protein standard (M) revealed the shift in molecular mass following treatment with PNGase F. CP = Caspr; − = untreated; P = PNGase F treated; KO = knockout; WT = wild-type; Nfasc = neurofascin; ERK2 = extracellular signal-regulated kinase 2.
The pattern of neurofascin 155H and neurofascin 155L is altered in multiple sclerosis plaques
Analysis of human brain samples by the extended electrophoresis protocol (Figure 8A1) indicated that neurofascin 155H is abundant in non-multiple sclerosis brain samples while no clearly delineated neurofascin 155L was observed. (‘Clearly delineated’ refers to a band that is distinct and separate from adjacent bands.) A similar banding pattern was observed in brain samples taken from regions of white matter that appear normal in multiple sclerosis brains (normal-appearing white matter). Normal-appearing is defined as revealing no visible signs of myelin deterioration as determined by both macroscopic and light microscopic examination. All multiple sclerosis plaque samples also revealed neurofascin 155H. However, samples from two (#164 and 224) of the eight donors analysed revealed a band with an apparent molecular mass consistent with neurofascin 155L as identified in rodent. To assess this possible alteration better, multiple sclerosis plaque samples were run side-by-side with either non-multiple sclerosis or normal-appearing white matter-multiple sclerosis samples (representative examples are presented in Figs 8A2 and Figure 8A3, respectively). This additional analysis further indicated that the banding pattern for neurofascin 155H and neurofascin 155L was altered in certain samples, with clearly delineated neurofascin 155L being observed only in multiple sclerosis plaque samples.
Interestingly, samples from donors #164 and #224 revealed an apparent reduction of neurofascin 155H, which may have increased the visibility of neurofascin 155L. Thus it was postulated that the apparent absence of neurofascin 155L in some multiple sclerosis plaque (donors #168 and 73), normal-appearing white matter-multiple sclerosis (donor #235) and non-multiple sclerosis (donor #5) samples resulted from an intense, bulky neurofascin 155H band which was obscuring a less intense neurofascin 155L band, as is the case with rodent (compare 30-day-old wild-type to 30-day-old knockout in Figs 3A, B). To test this hypothesis, non-multiple sclerosis, normal-appearing white matter-multiple sclerosis and multiple sclerosis plaque samples were treated with PNGase F. Non-multiple sclerosis and normal-appearing white matter-multiple sclerosis samples (donors #5 and 235; Fig. 8B) showed a prominent neurofascin 155H’ band with a faint shadow-like neurofascin 155L’ band that was continuous with the neurofascin 155H’ band. Additionally, the remaining non-multiple sclerosis (donor #2) and normal-appearing white matter-multiple sclerosis (donor #237) samples showed a similar pattern. All multiple sclerosis plaque samples treated with PNGase F yielded a neurofascin 155H’ band while five of the eight samples (Fig. 8B, donors #137, 216, 224; see #243 in Fig. 8D, and #164 not shown) revealed a clearly delineated neurofascin 155L’ band. To compare quantitative changes in the neurofascin 155H’ and neurofascin 155L’ pattern, a densitometric ratio of neurofascin 155H’ to neurofascin 155L’ was determined for each sample. This ratio was utilized instead of direct comparisons to account for the variability that is inherent in human tissue sampling. As shown in Fig. 8C, the ratio of neurofascin 155H’ to neurofascin 155L’ was significantly reduced in the multiple sclerosis plaques (n = 8; 4.2 ± 3.5, P < 0.006) compared to the non-multiple sclerosis and normal-appearing white matter-multiple sclerosis samples (n = 7; 14.2 ± 6.6).
In addition to the decrease in the neurofascin 155H’/neurofascin 155L’ ratio in multiple sclerosis samples, alterations in the levels of neurofascin 186 were also observed. Although neurofascin 186 was always detected in the non-multiple sclerosis samples, one (donor #216) of five normal-appearing white matter-multiple sclerosis samples failed to reveal neurofascin 186 while three (donors # 137, 216, and 224) of eight multiple sclerosis samples yielded no neurofascin 186 (Figs 8A1, 8A2 and 8A3). Importantly, multiple sclerosis donor #164 presented both neurofascin 155L and neurofascin 186 (Fig. 8A1 and 8A3), demonstrating that the presence of neurofascin 155L is not dependent on the absence of neurofascin 186; and suggesting that neurofascin 155L is a not a breakdown product of neurofascin 186. Also note that neurofascin 186 is present in the normal-appearing white matter-multiple sclerosis samples from donors #243 and #137 (Fig. 8D) but is absent in the plaques samples from these same donors, suggesting that the absence of neurofascin 186 is not the result of post-mortem autolysis and is a multiple sclerosis plaque-specific alteration. Further supporting the absence of significant post-mortem autolysis, no changes in the levels of β-actin, extracellular signal-regulated kinase 2 or glyceraldehyde 3-phosphate dehydrogenase were observed between normal-appearing white matter-multiple sclerosis and multiple sclerosis plaque samples from the same donors (Fig. 8D). Additionally, there was no correlation between the absence of neurofascin 186 and the length of time between death and tissue collection (Table 1), in agreement with a previous study demonstrating preservation of nodal and paranodal proteins up to 24 hours post-mortem (Howell et al., 2006).
Discussion
In the absence of sulphatide, paranodes appear structurally normal during development but deteriorate with age (Marcus et al., 2006). In the current study, quantitative analysis revealed that neurofascin 155 accumulated in the paranode in the absence of sulphatide; however, the number of anti-neurofascin-positive paired clusters in the spinal cord of the CST knockout mice was reduced by ∼50 and 90% at 15 and 30 days of age, respectively. Western blot analyses indicated that total levels of neurofascin 155 in the CST knockout mice are reduced ∼40% at both 15 and 30 days of age. Therefore, the significant reduction of neurofascin 155 clustering observed at 15 days of age may be a consequence of reduced neurofascin 155 expression; however, decreased protein levels alone do not provide adequate explanation for the continued reduction of paired paranodal clusters of neurofascin 155 by 30 days of age. These differences suggest a relationship between sulphatide and maintenance and localization of neurofascin 155 at the paranode. In contrast, sulphatide is not required for initial paranode formation (Marcus et al., 2006). The mechanistic differences between paranode development and maintenance are yet to be determined.
Extended electrophoresis revealed the presence of a neurofascin 155 doublet. These bands have been termed neurofascin 155H and neurofascin 155L to reflect their relative molecular masses. The ∼40% reduction in neurofascin 155 at both 15 and 30 days of age can be solely attributed to the reduction of neurofascin 155H, as levels of neurofascin 155L remain unchanged in the CST knockout mice. The observation of neurofascin 155H at the paranode along with the specific reduction of neurofascin 155H in mice that exhibit paranodal instability is consistent with neurofascin 155H, but not neurofascin 155L, being a paranodal protein required for preservation of paranodal integrity. Although the loss of neurofascin 155H correlates with paranodal instability in the sulphatide and Caspr null mice, the possibility persists that the loss of neurofascin 155H is the result of paranodal breakdown, rather than its cause.
Together, these findings demonstrate that sulphatide is essential for paranodal integrity due to its role in maintaining established paranodal protein domains. Ishibashi et al. (2002) reported a loss of both nodal sodium and juxtaparanodal potassium channel clusters in the CST knockout mice; however, this loss was not significant until 6 weeks of age. The reduction of neurofascin 155 paranodal clusters is significant as early as 15 days of age, while the paranodal structure is normal (Marcus et al., 2006), suggesting that the reduction and loss of neurofascin 155 clusters from the myelin paranode precedes, and may facilitate, neuronal deterioration.
What is the difference between neurofascin 155H and neurofascin 155L?
Possible sources for neurofascin 155H and neurofascin 155L include products of different genes, splice variants of the same gene, or the same amino acid sequence with differential post translational modifications. Utilizing the CNP-Cre;Nfascflox mice (Pillai et al., 2009) this study demonstrated that neurofascin 155H and neurofascin 155L are products of the same gene (the Nfasc gene); however, it remains to be determined whether neurofascin 155H and neurofascin 155L differ according to amino acid sequence and/or post-translational modifications.
The production of multiple splice variants is consistent with the current understanding of the Nfasc gene as it produces at least three proteins (Davis et al., 1996; Basak et al., 2007) and as many as 50 transcripts (Hassel et al., 1997), lending validity to the idea that neurofascin 155H and neurofascin 155L are distinct isoforms of the Nfasc gene. However, it is also plausible that neurofascin 155H and neurofascin 155L consist of identical amino acid sequences and that their molecular mass difference results from varying post-translational modifications. Maier et al. (2005) reported the observation of a neurofascin 155 doublet and speculated that variable glycosylation may account for this observation. Here, it has been shown that N-linked deglycosylation of neurofascin 155H and neurofascin 155L does not result in the formation of a single anti-neurofascin band. Thus, the difference between neurofascin 155H and neurofascin 155L is not simply differential N-linked glycosylation. Analyses with the AlphaEase software indicated that removal of N-linked glycans with PNGase F resulted in shifts of ∼12 and ∼17 kDa, respectively, for neurofascin 155H to neurofascin 155H’ and neurofascin 155L to neurofascin 155L’ in animals 3 months of age and older. This difference suggests that neurofascin 155L either maintains N-linked glycans at more sites than neurofascin 155H or that the carbohydrates on neurofascin 155L are more extensive than those attached to neurofascin 155H.
Other modifications that have been reported for neurofascin 186, the neuronal isoform of the neurofascin gene, include phosphorylation (Garver et al., 1997; Tuvia et al., 1997), O-linked glycosylation (Volkmer et al., 1992) and palmitoylation (Ren and Bennett, 1998). While Volkmer et al. (1992) have shown that O-linked glycosylation does not occur in neurofascin 155, it remains to be determined whether neurofascin 155H or neurofascin 155L are palmitoylated or phosphorylated. Following N-linked deglycosylation, neither neurofascin 155H nor neurofascin 155L, at any age analysed, migrated at 132 kDa, which is the predicted mass of neurofascin 155 based on amino acid sequence, suggesting that differences other than the published amino acid sequence for neurofascin 155 and N-linked glycosylation contribute to the molecular mass of neurofascin 155H and neurofascin 155L. However, it cannot be ruled out that the deglycosylated proteins may migrate anomalously in the gel, thus accounting for migration above 132 kDa.
In Fig. 4A, neurofascin 155L appears to emerge from the shadow-like band below neurofascin 155H with increasing age. This apparent emergence suggests that neurofascin 155L is a derivative of neurofascin 155H; however, treatment with PNGase F indicates that even at 14 days of age, when the two bands apparently migrate together, neurofascin 155H’ and neurofascin 155L’ represent distinct protein populations. Figure 4B shows, particularly following deglycosylation, that levels of neurofascin 155H’ decrease between 27 days and 3 months of age with a corresponding increase in neurofascin 155L, suggesting that neurofascin 155L could be a post-translational product of neurofascin 155H. However, in opposition to neurofascin 155L being derived from neurofascin 155H, normal levels of neurofascin 155L’ are maintained in the absence of neurofascin 155H’ in the aged CST knockout mice, suggesting that levels of neurofascin 155L’ are not dependent on the presence of neurofascin 155H’. Alternatively, neurofascin 155L could be highly stable, such that the loss of neurofascin 155H would have minimal effect on levels of neurofascin 155L. This supposition is not supported by the recent report that disruption of the Nfasc gene in the oligodendrocytes of 33-day-old mice results in the gradual loss of neurofascin 155 and complete loss within 60 days (Pillai et al., 2009). These findings demonstrate that the stability of all oligodendrocyte-derived Nfasc gene products is less than two months. Longer stability would be required for the maintenance of neurofascin 155L in the absence of neurofascin 155H, as is observed in the aged CST knockout mice, since neurofascin 155H’ is lost as early as 3 months of age and neurofascin 155L is maintained through 15 months of age (unpublished observation). The possibility cannot be excluded, however, that in the aged CST knockout mice neurofascin 155H is rapidly converted into neurofascin 155L, precluding detection of neurofascin 155H or neurofascin 155H’ by western blot analysis.
Hassel and colleagues (1997) identified a fifth fibronectin III-like domain in the Nfasc gene of chicken and rat after the original report that only four fibronectin III-like domains were present (Volkmer et al., 1992). This domain was also observed in cDNA clones and, unlike the other fibronectin III-like domains, is expressed as either a half (one exon) or a whole (two exons) domain. The whole fifth fibronectin III-like domain is expressed with the mucin-like domain, which is specific to neurofascin 186. It is unclear whether or not this fifth fibronectin III-like domain is expressed in known neurofascin proteins (Tait et al. 2000; Koticha et al., 2005). The two halves of the fifth fibronectin III-like domain are ∼5 kDa each and analysis with the NetNGlyc 1.0 Server indicates two potential sites of N-linked glycosylation for the first half and one potential site for the second half. It is interesting to consider that either half of this domain may account for the observed differences in size between neurofascin 155H and neurofascin 155L.
Are neurofascin 155H and neurofascin 155L functionally distinct?
The functional significance of differential glycosylation of neurofascin 155H and neurofascin 155L is unknown. Since glycosylation is a prime regulator of protein functions such as protease activity (Van den Steen et al., 2001), intracellular trafficking (Yan et al., 2008), protein binding (Milev et al., 1995; Zhou et al., 2008) and stability (Buck et al., 2007; Goa and Nehta, 2007; Li et al., 2007), neurofascin 155H and neurofascin 155L may have distinct functions related to their different states of glycosylation, including potential interactions with contactin and/or Caspr; however, this remains to be determined.
The only recognized function of neurofascin 155 is the binding of neuronal contactin at the paranode (Tait et al., 2000; Gollan et al., 2003; Bonnon et al., 2007). At an age when neurofascin 155H’, but not neurofascin 155L’, is detected (7 days of age; Fig. 4B), anti-neurofascin reactive paranodes are prevalent (Fig. 4C) indicating that neurofascin 155H is a myelin paranodal protein expressed early in the process of myelination. In contrast, neurofascin 155L’ is not detected until the end of the second week of life and continues to increase through at least 5 months of age, suggesting that neurofascin 155L is not involved in the onset of myelination. It cannot be concluded, however, that neurofascin 155H is the form that binds contactin or that neurofascin 155L is not a paranodal protein involved in mediating myelin-axon interactions. Interestingly, Maier et al. (2005), who detected an anti-neurofascin doublet in the vicinity of 155 kDa, reported that the lower band of the doublet is not detected in pure cultures of oligodendrocytes, suggesting that oligodendrocyte-axon interaction is a prerequisite for expression of the lower component of the doublet, which is likely to be neurofascin 155L.
Neurofascin 155H and neurofascin 155H’ are virtually absent in 4-month-old CST knockout mice indicating that sulphatide is required for long term maintenance of this protein. One possible explanation for the loss of neurofascin 155H lies with the concept of membrane rafts (Harder and Simons, 1997), which are small, dynamic membrane domains enriched in cholesterol and sphingolipids (reviewed by Dupree and Pomicter, 2009). Sulphatide, a sphingolipid, is enriched in myelin fractions containing putative raft proteins (Arvanitis et al., 2005) such as fyn (Krämer et al., 1999), flotillin-1 (Salzer and Prohaska, 2001) and caveolin (Sargiacomo et al., 1993); and neurofascin 155 has been implicated as a raft protein (Maier, 2007; Schafer et al., 2004). Therefore, the absence of sulphatide may result in abnormal raft formation facilitating neurofascin 155H instability at the myelin paranode. This is consistent with recent evidence implicating a role for membrane rafts in regulating protease activity (Abad-Rodriguez et al., 2004; Bae et al., 2008). It is interesting that the Caspr knockout tissue reveals a decrease in neurofascin 155H and neurofascin 155H’. Since receptor–ligand binding, such as occurs in the paranode between paranodal neurofascin and neuronal contactin:Caspr (Tait et al., 2000; Gollan et al., 2003; Bonnon et al., 2007), may induce raft formation (Subczynski and Kusumi, 2003), a disruption of the myelin-axon junction may also result in neurofascin 155H instability as a consequence of impaired raft structure in the Caspr knockout mice.
Neurofascin 155L is variably glycosylated with age
An increasingly intense, age-dependent shadow-like band attached to and just beneath the band labelled ‘neurofascin 155H’ shown in Fig. 4A has been labelled ‘neurofascin 155H&L’ in later figures to denote the supposition that neurofascin 155H’ and neurofascin 155L’ are derived from this band plus the shadow-like band (Figs 5A, 5B, 6A and 7). Presumably, neurofascin 155L migrates with/immediately below neurofascin 155H in younger animals (compare Fig. 4A and B; 14, 21 and 27 days of age, as well as the first four pairs of lanes in Fig. 6A). At all ages, the shift from neurofascin 155H to neurofascin 155H’ is ∼12 kDa while the shift from the shadow-like band to distinct neurofascin 155L’ is ∼22 kDa in younger animals and ∼17 kDa for neurofascin 155L in older animals, suggesting that neurofascin 155L is variably glycosylated with age. Specifically, neurofascin 155L may become less glycosylated with age, accounting for the gradual, age-related transition from the shadow-like band below neurofascin 155H in younger animals to the emergence of the distinct neurofascin 155L band observed in older animals (Fig. 4A). The same conclusion is suggested by Fig. 6A, where neurofascin 155L is not observed in the 15 or 30-day-old wild-type untreated lanes but neurofascin 155L’ is observed following PNGase F treatment. Age-related changes in glycosylation is documented in adhesion proteins of immune cells (Daniels et al., 2002), epithelial cells (Wang et al., 2008) and nervous system cells (Edelman, 1985). Based on these observations, it is reasonable to propose that neurofascin 155H’ is deglycosylated neurofascin 155H, that neurofascin 155L’ is deglycosylated neurofascin 155L, and that results regarding neurofascin 155H’ and neurofascin 155L’ can provide information regarding neurofascin 155H and neurofascin 155L.
Levels of both oligodendrocyte and neuronal neurofascin are altered in multiple sclerosis
In multiple sclerosis plaque samples, the ratio of neurofascin 155H’/neurofascin 155L’ is significantly reduced compared to non-multiple sclerosis and normal-appearing white matter-multiple sclerosis. This finding is intriguing in light of the significant reduction observed for neurofascin 155H’ in the CST knockout model in the presence of unaltered levels of neurofascin 155L’. In both multiple sclerosis plaques and in CST knockout mice, the pattern of neurofascin 155H’ and neurofascin 155L’ is disrupted, concurrent with paranodal deterioration that precedes axonal pathology. It remains to be determined, however, in both human and mouse models, whether disruption of neurofascin 155H and/or neurofascin 155L causes or results from paranodal deterioration.
In this study, the multiple sclerosis samples were categorized either as active or inactive based on the level of cellularity within the harvested plaque. Neurofascin 155L’ was observed in one of three active (donor #137) and four of five inactive (donors #224, 164, 216, 243) plaques suggesting that the pathological event that results in a reduced ratio of neurofascin 155H’ to neurofascin 155L’ is initiated during active myelin deterioration. The presence of clearly delineated neurofascin 155L’ in inactive plaques is also consistent with the idea that neurofascin 155L’ is a product of oligodendrocytes (although axons cannot be ruled out as a source), as few other cell types are present in inactive plaques (Lassmann et al., 1998; Mycko et al., 2003). Neurofascin 155L’ as a product of oligodendrocytes is consistent with the rodent findings since mice that exhibit a disruption in the neurofascin gene in oligodendrocytes are incapable of synthesizing neurofascin 155L.
It is intriguing that the neuronal form of neurofascin, neurofascin 186, is less frequently observed in multiple sclerosis plaque samples compared to non-multiple sclerosis and normal-appearing white matter-multiple sclerosis samples. The reduced ability to detect neurofascin 186 in multiple sclerosis is particularly interesting in light of the findings of Mathey et al. (2007) who reported that neurofascin antibodies, derived from serum of multiple sclerosis patients, selectively target the node of Ranvier, the specific location of neurofascin 186 (Davis et al., 1996; Poliak and Peles, 2003), resulting in the recruitment of complement, axonal degeneration, and exacerbated pathology in a mouse model. Therefore, the inability to detect neurofascin 186 in some normal-appearing white matter-multiple sclerosis and multiple sclerosis plaque samples is consistent with the early axonal degeneration that occurs in multiple sclerosis.
In conclusion, the present study demonstrates an essential role for sulphatide in maintaining neurofascin 155 at the myelin paranode and introduces the existence of two forms of neurofascin 155—neurofascin 155H and neurofascin 155L—that exhibit differential developmental expression and variable stability in the absence of sulphatide. Moreover, neurofascin 155H is concentrated at the myelin paranode and is progressively lost from both the paranode and the CNS concomitantly with structural deterioration of the paranode. Similarly, the loss of paranodal integrity observed in multiple sclerosis (Suzuki et al., 1969; Howell et al., 2006) coincides with a significant alteration in the pattern of neurofascin 155H’ and neurofascin 155L’. Determining the consequence of this altered pattern of expression in multiple sclerosis plaques will be an important step in understanding the pathological mechanisms that underlie CNS degeneration.
Funding
This work was supported by grants from the National Multiple Sclerosis Society (PP 1440 to J.L.D.) and the A. D. Williams Foundation (to J.L.D.) and the National Institutes of Health (NS066186 to J.L.D. and GM063074 to M.A.B.). All microscopy was performed at the VCU Department of Anatomy and Neurobiology Microscopy Facility, supported, in part, with funding from National Institutes of Health-National Institute of Neurological Disease and Stroke Centre core grant (5P30NS047463).
Supplementary material
Supplementary material is available at Brain online.
Acknowledgements
Human tissue samples were provided by the Rocky Mountain Multiple Sclerosis Centre (Englewood, CO). The authors wish to thank Haley J. Steinert, Multiple Sclerosis Research Coordinator and Tissue Bank Research Associate at the Rocky Mountain Multiple Sclerosis Centre for her assistance in acquiring donor information. Additionally, the authors thank Dr Nitya Ghatak, Neuropathology Division Chair in the Department of Pathology at Virginia Commonwealth University for his assistance in interpreting the pathology reports for each donor.
Glossary
Abbreviations
- CST
cerebroside sulphotransferase
- Caspr
contactin-associated protein
- CNP
2′, 3′-cyclic nucleotide 3′-phosphodiesterase
- CNP-Cre;Nfascflox/flox
neurofascin conditional knockout mice
- neurofascin 155H
neurofascin 155 high
- neurofascin155L
neurofascin 155 low
- neurofascin’
deglycosylated neurofascin
- Nfasc
neurofascin gene
- PNGase F
Peptide N-glycosidase F
References
- Abad-Rodriguez J, Ledesma MD, Craessaerts K, Perga S, Medina M, Delacourte A, et al. Neuronal membrane cholesterol loss enhances amyloid peptide generation. J Cell Biol. 2004;167:953–60. doi: 10.1083/jcb.200404149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvanitis DN, Min W, Gong Y, Heng YM, Boggs JM. Two types of detergent-insoluble, glycosphingolipid/cholesterol-rich membrane domains from isolated myelin. J Neurochem. 2005;94:1696–710. doi: 10.1111/j.1471-4159.2005.03331.x. [DOI] [PubMed] [Google Scholar]
- Bae JS, Yang L, Rezaie AR. Lipid raft localization regulates the cleavage specificity of protease activated receptor 1 in endothelial cells. J Thromb Haemost. 2008;6:954–61. doi: 10.1111/j.1538-7836.2008.02924.x. [DOI] [PubMed] [Google Scholar]
- Basak S, Raju K, Babiarz J, Kane-Goldsmith N, Koticha D, Grumet M. Differential expression and functions of neuronal and glial neurofascin isoforms and splice variants during PNS development. Dev Biol. 2007;311:408–22. doi: 10.1016/j.ydbio.2007.08.045. [DOI] [PubMed] [Google Scholar]
- Bhat MA, Rios JC, Lu Y, Garcia-Fresco GP, Ching W, St Martin M, et al. Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron. 2001;30:369–83. doi: 10.1016/s0896-6273(01)00294-x. [DOI] [PubMed] [Google Scholar]
- Bonnon C, Bel C, Goutebroze L, Maigret B, Girault JA, Faivre-Sarrailh C. PGY repeats and N-glycans govern the trafficking of paranodin and its selective association with contactin and neurofascin-155. Mol Biol Cell. 2007;18:229–41. doi: 10.1091/mbc.E06-06-0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle ME, Berglund EO, Murai KK, Weber L, Peles E, Ranscht B. Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron. 2001;30:385–97. doi: 10.1016/s0896-6273(01)00296-3. [DOI] [PubMed] [Google Scholar]
- Buck TM, Eledge J, Skach WR. Evidence for stabilization of aquaporin-2 folding mutants by N-linked glycosylation in endoplasmic reticulum. Am J Physiol Cell Physiol. 2004;287:C1292–9. doi: 10.1152/ajpcell.00561.2003. [DOI] [PubMed] [Google Scholar]
- Charles P, Tait S, Faivre-Sarrailh C, Barbin G, Gunn-Moore F, Denisenko-Nehrbass N, et al. Neurofascin is a glial receptor for the paranodin/Caspr-contactin axonal complex at the axoglial junction. Curr Biol. 2002;12:217–20. doi: 10.1016/s0960-9822(01)00680-7. [DOI] [PubMed] [Google Scholar]
- Chiu SY. Asymmetry currents in the mammalian myelinated nerve. J Physiol. 1980;309:499–519. doi: 10.1113/jphysiol.1980.sp013523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu SY, Schwarz W. Sodium and potassium currents in acutely demyelinated internodes of rabbit sciatic nerves. J Physiol. 1987;391:631–49. doi: 10.1113/jphysiol.1987.sp016760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinson JM, Marshall D, Gillespie CS, Brophy PJ. xTransient expression of neurofascin by oligodendrocytes at the onset of myelinogenesis: Implications for mechanisms of axon-glial interaction. Glia. 1987;3:11–23. doi: 10.1002/(sici)1098-1136(199805)23:1<11::aid-glia2>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Cook RD. Detection of influential observation in linear regression. Technometrics. 1977;19:15–18. [Google Scholar]
- Daniels MA, Hogquist KA, Jameson SC. Sweet 'n' sour: The impact of differential glycosylation on T cell responses. Nat Immunol. 2002;3:903–10. doi: 10.1038/ni1002-903. [DOI] [PubMed] [Google Scholar]
- Davis JQ, Lambert S, Bennett V. Molecular composition of the node of Ranvier: Identification of ankyrin-binding cell adhesion molecules neurofascin (mucin+/third FNIII domain-) and NrCAM at nodal axon segments. J Cell Biol. 1996;135:1355–67. doi: 10.1083/jcb.135.5.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JQ, McLaughlin T, Bennett V. Ankyrin-binding proteins related to nervous system cell adhesion molecules: candidates to provide transmembrane and intercellular connections in adult brain. J Cell Biol. 1993;121:121–33. doi: 10.1083/jcb.121.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugandzija-Novakovic S, Koszowski AG, Levinson SR, Shrager P. Clustering of Na+ channels and node of Ranvier formation in remyelinating axons. J Neurosci. 1995;15(1 Pt 2):492–503. doi: 10.1523/JNEUROSCI.15-01-00492.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupree JL, Pomicter AD, Myelin DIGs. and membrane rafts in the central nervous system. Prostaglandins Other Lipid Mediat [Epub ahead of print, April 18, 2009; doi:10.1016/j.prostaglandins.2009.04.005] [DOI] [PubMed] [Google Scholar]
- Dupree JL, Girault JA, Popko B. Axo-glial interactions regulate the localization of axonal paranodal proteins. J Cell Biol. 1999;147:1145–52. doi: 10.1083/jcb.147.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupree JL, Coetzee T, Blight A, Suzuki K, Popko B. Myelin galactolipids are essential for proper node of Ranvier formation in the CNS. J Neurosci. 1998;18:1642–9. doi: 10.1523/JNEUROSCI.18-05-01642.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelman GM. Cell adhesion and the molecular processes of morphogenesis. Annu Rev Biochem. 1985;54:135–69. doi: 10.1146/annurev.bi.54.070185.001031. [DOI] [PubMed] [Google Scholar]
- Gao Y, Mehta K. N-linked glycosylation of CD38 is required for its structure stabilization but not for membrane localization. Mol Cell Biochem. 2007;295:1–7. doi: 10.1007/s11010-006-9265-9. [DOI] [PubMed] [Google Scholar]
- Garver TD, Ren Q, Tuvia S, Bennett V. Tyrosine phosphorylation at a site highly conserved in the L1 family of cell adhesion molecules abolishes ankyrin binding and increases lateral mobility of neurofascin. J Cell Biol. 1997;137:703–14. doi: 10.1083/jcb.137.3.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollan L, Salomon D, Salzer JL, Peles E. Caspr regulates the processing of contactin and inhibits its binding to neurofascin. J Cell Biol. 2003;163:1213–8. doi: 10.1083/jcb.200309147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder T, Simons K. Caveolae, DIGs, and the dynamics of sphingolipid-cholesterol microdomains. Curr Opin Cell Biol. 1997;9:534–42. doi: 10.1016/s0955-0674(97)80030-0. [DOI] [PubMed] [Google Scholar]
- Hassel B, Rathjen FG, Volkmer H. Organization of the neurofascin gene and analysis of developmentally regulated alternative splicing. J Biol Chem. 1997;272:28742–9. doi: 10.1074/jbc.272.45.28742. [DOI] [PubMed] [Google Scholar]
- Honke K, Yamane M, Ishii A, Kobayashi T, Makita A. Purification and characterization of 3'-phosphoadenosine-5'-phosphosulfate:GalCer sulfotransferase from human renal cancer cells. J Biochem. 1996;119:421–7. doi: 10.1093/oxfordjournals.jbchem.a021258. [DOI] [PubMed] [Google Scholar]
- Honke K, Tsuda M, Hirahara Y, Ishii A, Makita A, Wada Y. Molecular cloning and expression of cDNA encoding human 3'-phosphoadenylylsulfate: galactosyl -ceramide 3'-sulfotransferase. J Biol Chem. 1997;272:4864–8. doi: 10.1074/jbc.272.8.4864. [DOI] [PubMed] [Google Scholar]
- Honke K, Hirahara Y, Dupree J, Suzuki K, Popko B, Fukushima K, et al. Paranodal junction formation and spermatogenesis require sulfoglycolipids. Proc Natl Acad Sci USA. 2002;99:4227–32. doi: 10.1073/pnas.032068299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell OW, Palser A, Polito A, Melrose S, Zonta B, Scheiermann C, et al. Disruption of neurofascin localization reveals early changes preceding demyelination and remyelination in multiple sclerosis. Brain. 2006;129(Pt 12):3173–85. doi: 10.1093/brain/awl290. [DOI] [PubMed] [Google Scholar]
- Ishibashi T, Dupree JL, Ikenaka K, Hirahara Y, Honke K, Peles E, et al. A myelin galactolipid, sulfatide, is essential for maintenance of ion channels on myelinated axon but not essential for initial cluster formation. J Neurosci. 2002;22:6507–14. doi: 10.1523/JNEUROSCI.22-15-06507.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarjour AA, Bull SJ, Almasieh M, Rajasekharan S, Baker KA, Mui J, et al. Maintenance of axo-oligodendroglial paranodal junctions requires DCC and netrin-1. J Neurosci. 2008;28:11003–14. doi: 10.1523/JNEUROSCI.3285-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koticha D, Babiarz J, Kane-Goldsmith N, Jacob J, Raju K, Grumet M. Cell adhesion and neurite outgrowth are promoted by neurofascin NF155 and inhibited by NF186. Mol Cell Neurosci. 2005;30:137–48. doi: 10.1016/j.mcn.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Krämer EM, Klein C, Koch T, Boytinck M, Trotter J. Compartmentation of fyn kinase with glycosylphosphatidylinositol-anchored molecules in oligodendrocytes facilitates kinase activation during myelination. J Biol Chem. 1999;274:29042–9. doi: 10.1074/jbc.274.41.29042. [DOI] [PubMed] [Google Scholar]
- Lassmann H, Raine CS, Antel J, Prineas JW. Immunopathology of multiple sclerosis: report on an international meeting held at the Institute of Neurology of the University of Vienna. J Neuroimmunol. 1998;86:213–7. doi: 10.1016/s0165-5728(98)00031-9. [DOI] [PubMed] [Google Scholar]
- Li JG, Chen C, Liu-Chen LY. N-glycosylation of the human kappa opioid receptor enhances its stability but slows its trafficking along the biosynthesis pathway. Biochemistry. 2007;46:10960–70. doi: 10.1021/bi700443j. [DOI] [PubMed] [Google Scholar]
- Maier O, Baron W, Hoekstra D. Reduced raft-association of NF155 in active MS-lesions is accompanied by the disruption of the paranodal junction. Glia. 2007;55:885–95. doi: 10.1002/glia.20510. [DOI] [PubMed] [Google Scholar]
- Maier O, van der Heide T, van Dam AM, Baron W, de Vries H, Hoekstra D. Alteration of the extracellular matrix interferes with raft association of neurofascin in oligodendrocytes. Potential significance for multiple sclerosis? Mol Cell Neurosci. 2005;28:390–401. doi: 10.1016/j.mcn.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Maley F, Trimble RB, Tarentino AL, Plummer TH., Jr Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal Biochem. 1989;180:195–204. doi: 10.1016/0003-2697(89)90115-2. [DOI] [PubMed] [Google Scholar]
- Marbois BN, Faull KF, Fluharty AL, Raval-Fernandes S, Rome LH. Analysis of sulfatide from rat cerebellum and multiple sclerosis white matter by negative ion electrospray mass spectrometry. Biochim Biophys Acta. 2000;1484:59–70. doi: 10.1016/s1388-1981(99)00201-2. [DOI] [PubMed] [Google Scholar]
- Marcus J, Honigbaum S, Shroff S, Honke K, Rosenbluth J, Dupree JL. Sulfatide is essential for the maintenance of CNS myelin and axon structure. Glia. 2006;53:372–81. doi: 10.1002/glia.20292. [DOI] [PubMed] [Google Scholar]
- Milev P, Meyer-Puttlitz B, Margolis RK, Margolis RU. Complex-type asparagine-linked oligosaccharides on phosphacan and protein-tyrosine phosphatase-zeta/beta mediate their binding to neural cell adhesion molecules and tenascin. J Biol Chem. 1995;270:24650–3. doi: 10.1074/jbc.270.42.24650. [DOI] [PubMed] [Google Scholar]
- Mycko MP, Papoian R, Boschert U, Raine CS, Selmaj KW. cDNA microarray analysis in multiple sclerosis lesions: detection of genes associated with disease activity. Brain. 2003;126(Pt 5):1048–57. doi: 10.1093/brain/awg107. [DOI] [PubMed] [Google Scholar]
- Ogawa Y, Schafer DP, Horresh I, Bar V, Hales K, Yang Y, et al. Spectrins and ankyrinB constitute a specialized paranodal cytoskeleton. J Neurosci. 2006;26:5230–9. doi: 10.1523/JNEUROSCI.0425-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Palay S, Webster H, editors. Philadelphia: W. B. Saunders Company; 1976. The fine structure of the nervous system. [Google Scholar]
- Pillai AM, Thaxton C, Pribisko AL, Cheng JG, Dupree JL, Bhat MA. Spatiotemporal ablation of myelinating glia-specific neurofascin (Nfasc NF155) in mice reveals gradual loss of paranodal axoglial junctions and concomitant disorganization of axonal domains. J Neurosci Res. 2009;87:1773–93. doi: 10.1002/jnr.22015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliak S, Peles E. The local differentiation of myelinated axons at nodes of Ranvier. Nat Rev Neurosci. 2003;4:968–80. doi: 10.1038/nrn1253. [DOI] [PubMed] [Google Scholar]
- Rasband MN. It's; “juxta” potassium channel! J Neurosci Res. 2004;76:749–57. doi: 10.1002/jnr.20073. [DOI] [PubMed] [Google Scholar]
- Ren Q, Bennett V. Palmitoylation of neurofascin at a site in the membrane-spanning domain highly conserved among the L1 family of cell adhesion molecules. J Neurochem. 1998;70:1839–49. doi: 10.1046/j.1471-4159.1998.70051839.x. [DOI] [PubMed] [Google Scholar]
- Rios JC, Melendez-Vasquez CV, Einheber S, Lustig M, Grumet M, Hemperly J, et al. Contactin-associated protein (Caspr) and contactin form a complex that is targeted to the paranodal junctions during myelination. J Neurosci. 2000;20:8354–64. doi: 10.1523/JNEUROSCI.20-22-08354.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzer U, Prohaska R. Stomatin, flotillin-1, and flotillin-2 are major integral proteins of erythrocyte lipid rafts. Blood. 2001;97:1141–3. doi: 10.1182/blood.v97.4.1141. [DOI] [PubMed] [Google Scholar]
- Sargiacomo M, Sudol M, Tang Z, Lisanti MP. Signal transducing molecules and glycosyl-phosphatidylinositol-linked proteins form a caveolin-rich insoluble complex in MDCK cells. J Cell Biol. 1993;122:789–807. doi: 10.1083/jcb.122.4.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeren-Wiemers N, Bonnet A, Erb M, Erne B, Bartsch U, Kern F, et al. The raft-associated protein MAL is required for maintenance of proper axon-glia interactions in the central nervous system. J Cell Biol. 2004;166:731–42. doi: 10.1083/jcb.200406092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Bansal R, Hedstrom KL, Pfeiffer SE, Rasband MN. Does paranode formation and maintenance require partitioning of neurofascin 155 into lipid rafts? J Neurosci. 2004;24:3176–85. doi: 10.1523/JNEUROSCI.5427-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman DL, Tait S, Melrose S, Johnson R, Zonta B, Court FA, et al. Neurofascins are required to establish axonal domains for saltatory conduction. Neuron. 2005;48:737–42. doi: 10.1016/j.neuron.2005.10.019. [DOI] [PubMed] [Google Scholar]
- Shroff SM, Pomicter AD, Chow WN, Fox MA, Colello RJ, Henderson SC, et al. Adult CST-null mice maintain an increased number of oligodendrocytes. J Neurosci Res. 2009;87:3403–14. doi: 10.1002/jnr.22003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subczynski WK, Kusumi A. Dynamics of raft molecules in the cell and artificial membranes: Approaches by pulse EPR spin labeling and single molecule optical microscopy. Biochim Biophys Acta. 2003;1610:231–43. doi: 10.1016/s0005-2736(03)00021-x. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Andrews JM, Waltz JM, Terry RD. Ultrastructural studies of multiple sclerosis. Lab Invest. 1969;20:444–54. [PubMed] [Google Scholar]
- Tait S, Gunn-Moore F, Collinson JM, Huang J, Lubetzki C, Pedraza L, et al. An oligodendrocyte cell adhesion molecule at the site of assembly of the paranodal axo-glial junction. J Cell Biol. 2000;150:657–66. doi: 10.1083/jcb.150.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Bernier L, Andrews SB, Colman DR. Cellular and subcellular distribution of 2', 3'-cyclic nucleotide 3'-phosphodiesterase and its mRNA in the rat central nervous system. J Neurochem. 1988;51:859–68. doi: 10.1111/j.1471-4159.1988.tb01822.x. [DOI] [PubMed] [Google Scholar]
- Tuvia S, Garver TD, Bennett V. The phosphorylation state of the FIGQY tyrosine of neurofascin determines ankyrin-binding activity and patterns of cell segregation. Proc Natl Acad Sci USA. 1997;94:12957–62. doi: 10.1073/pnas.94.24.12957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Steen PE, Opdenakker G, Wormald MR, Dwek RA, Rudd PM. Matrix remodelling enzymes, the protease cascade and glycosylation. Biochim Biophys Acta. 2001;1528:61–73. doi: 10.1016/s0304-4165(01)00190-8. [DOI] [PubMed] [Google Scholar]
- Volkmer H, Hassel B, Wolff JM, Frank R, Rathjen FG. Structure of the axonal surface recognition molecule neurofascin and its relationship to a neural subgroup of the immunoglobulin superfamily. J Cell Biol. 1992;118:149–61. doi: 10.1083/jcb.118.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Julian JA, Carson DD. The MUC1 HMFG1 glycoform is a precursor to the 214D4 glycoform in the human uterine epithelial cell line, HES. Biol Reprod. 2008;78:290–8. doi: 10.1095/biolreprod.107.064584. [DOI] [PubMed] [Google Scholar]
- Waxman SG, Ritchie JM. Organization of ion channels in the myelinated nerve fiber. Science. 1985;228:1502–7. doi: 10.1126/science.2409596. [DOI] [PubMed] [Google Scholar]
- Wolswijk G, Balesar R. Changes in the expression and localization of the paranodal protein Caspr on axons in chronic multiple sclerosis. Brain. 2003;126(Pt 7):1638–49. doi: 10.1093/brain/awg151. [DOI] [PubMed] [Google Scholar]
- Yan Y, Scott DJ, Wilkinson TN, Ji J, Tregear GW, Bathgate RA. Identification of the N-linked glycosylation sites of the human relaxin receptor and effect of glycosylation on receptor function. Biochemistry. 2008;47:6953–68. doi: 10.1021/bi800535b. [DOI] [PubMed] [Google Scholar]
- Zhou F, Su J, Fu L, Yang Y, Zhang L, Wang L, et al. Unglycosylation at Asn-633 made extracellular domain of E-cadherin folded incorrectly and arrested in endoplasmic reticulum, then sequentially degraded by ERAD. Glycoconj J. 2008;25:727–40. doi: 10.1007/s10719-008-9133-9. [DOI] [PubMed] [Google Scholar]
- Zonta B, Tait S, Melrose S, Anderson H, Harroch S, Higginson J, et al. Glial and neuronal isoforms of neurofascin have distinct roles in the assembly of nodes of Ranvier in the central nervous system. J Cell Biol. 2008;181:1169–77. doi: 10.1083/jcb.200712154. [DOI] [PMC free article] [PubMed] [Google Scholar]