

Abstract
Epidermal growth factor receptor (EGFR) activation is an important event that regulates mitogenic signaling, such as the Raf, MAPK and ERK1/2 cascade. EGFR activation has been implicated in the transition of prostate cancer (PCa) from androgen-dependence to -independence. Therefore, inhibition of EGFR may effectively suppress PCa growth and progression. The goal of this study is to determine whether the natural compound psoralidin alters EGFR-mediated signaling resulting in the inhibition of PCa growth. Results suggest that inhibition of EGFR alone by serum deprivation fails to induce stress-mediated protein kinases (SAPK), namely Jun N-terminal kinase (JNK)/c-Jun signaling in AIPC cells. However, treatment with psoralidin inhibited both constitutive and epidermal growth factor (EGF)-induced EGFR activation and simultaneously triggered SAPK signaling, resulting in the induction of apoptosis in AIPC cells. In addition psoralidin downregulated EGFR-regulated MAPK signaling and inhibited cell proliferation in AIPC cells. Oral administration of psoralidin effectively suppressed PC-3 xenograft tumors in nude mice. Compared to control tumors, inhibition of pEGFR expression, as well as an increase in the phosphorylation, activation and nuclear translocation of c-Jun were observed in psoralidin treated tumor sections. Our studies suggest that psoralidin may be a potent therapeutic agent that modulates EGFR-mediated key epigenetic events in AIPC.
Keywords: Psoralidin, SAPK, MAPK, apoptosis, Prostate cancer
Introduction
Epidermal growth factor receptors (EGFR) play an important role in the pathogenesis of prostate cancer (PCa) and in the transition of PCa from an androgen-dependent to -independent state (1). One of the major alterations in androgen-independent PCa (AIPC) is the overexpression of epidermal growth factor (EGF) receptor (EGFR) and its cognate ligand EGF (2); EGFR is frequently overexpressed in PCa tumors, especially in patients treated with hormone therapy (3). Hence, inhibition of EGFR signaling could be a promising approach for the treatment of PCa.
In response to EGF and/or various growth factors, EGFR regulates many signaling pathways, including the mitogen activated protein kinase (MAPK) and the stress-activated protein kinase (SAPK) cascades (4). The MAPK pathway plays a pivotal role in the molecular network that governs cell growth, proliferation, differentiation, survival and apoptosis. The effects of MAPK signaling are based on the stimuli and cell type (5). In normal physiological conditions, MAPK is tightly regulated. However, activation of the MAP kinase pathway is often associated with an increased PCa Gleason score and tumor stage (6). The MAPK superfamily is divided into three subgroups, namely the extracellular signal-regulated kinase (ERK), c-Jun N-terminal protein kinase (JNK) and p38. EGFR, either through MEKs or directly can phosphorylate and activate ERK (7), which in turn regulates growth, proliferation, the cell cycle and apoptosis in various cell types. The p38 MAPK is well known for its function as a tumor suppressor because of its involvement in the inhibition of cell proliferation and cell cycle progression and induction of mitochondrial apoptotic pathway and regulation of oncogene-induced senescence in many cancer types (8, 9). These studies suggest a dual role for MAPK signaling where, based on the external stimuli, the fate of a cell can be determined.
JNKs belong to the MAPK family of proteins. Based on cell type and stimuli, JNK signaling pathways are important for regulating both cell proliferation and apoptosis (10, 11). Several external stimuli, such as cytokines, stress factors and agents that induce genotoxic and cytotoxic stress have been known to activate JNKs (12). In response to these stimuli, activated JNKs initiate one or both of the following downstream signaling: a nuclear event and/or a mitochondrial event. In the nucleus, activated JNK gains entry into the nucleus where it transactivates c-Jun and other transcription factors to increase expression of pro-apoptotic genes (13). In the mitochondria, activated JNK translocates to the mitochondria and antagonizes the anti-apoptotic function of Bcl-2 and Bcl-xL. In addition, JNK also enhances release of cytochrome C, thereby initiating the caspase cascade (14). Eventually, JNK signaling leads to the induction of apoptosis in many cancer types. Thus, modulating the activity of JNKs may be relevant to cancer therapy.
Treatment of AIPC is often challenging due to the inability to achieve complete remission of the tumor. As a result, the development of new therapeutic strategies is essential. Psoralidin is a natural compound isolated from the leaves of Psoralea corylifolia. Psoralidin is extensively used in traditional medicine for the treatment of various ailments. We previously reported that psoralidin inhibits the phosphatidylinositol-3 kinase (PI3K)/Akt pathway leading to inhibition of cell proliferation and induction of apoptosis in AIPC cells (15). In the present study, we determine whether psoralidin inhibits EGFR signaling which in turn may suppress AIPC growth and progression. Our results suggest that psoralidin inhibits EGFR-mediated signaling events and induces SAPK-mediated apoptotic signaling. Psoralidin treatment thereby leads to the induction of apoptosis in AIPC cells and inhibition of tumor growth in xenograft models.
Materials and methods
Cell lines and plasmids
PC-3, DU-145, LNCaP and C4-2B cells were purchased from American Type Culture Collection (Manassas, VA). The cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 1% glutamine and antibiotics. Psoralidin was isolated and purified (99.8%) in Dr. Rohr’s laboratory at the University of Kentucky. Epidermal growth factor (EGF), the JNK inhibitor (iJNK-SP600125) and c-Jun siRNA were purchased from Calbiochem (Gibbstown, NJ) and Dharmacon (Lafayette, CO), respectively. c-Jun promoter-luciferase reporter construct and rennilla control vector (pGL3.4) were obtained from Adgene (Cambridge, MA).
Western blot analysis
PC-3, DU-145, LNCaP and C4-2B cells (at 70–80% confluence) were grown either in complete medium or were serum deprived for 48 h. Whole cell lysates were obtained and Western blot analysis was performed using a pEGFR (Tyr 1173) and EGFR antibodies. PC-3 and DU-145 cells were grown in complete medium and treated with IC50s 60 and 45 μM psoralidin respectively. Cells were also serum deprived for 48 h and stimulated with EGF (100 ng/ml) alone or treated concurrently with EGF and psoralidin for 30 and 60 minutes with the above mentioned concentrations. Whole cell lysates were subjected to Western blot analysis as described previously (16) using pEGFR (Tyr 1173), EGFR, Raf1, MEK-1, MEK4, MEKK-1, pMEK1/2, pMEK3/6, ERK, pERK1/2, pElk (ser 383), Elk, pJNK1/2 (p54, p46), JNK 1/2, pc-Jun (ser 73), c-Jun, Bcl-2, Bax, Caspase-3 and survivin antibodies purchased from either Cell Signaling Technology (Danvers, MA), Santa Cruz Biotechnology (Santa Cruz, CA) or R and D Systems (Minneapolis, MN).
Kinase assay
PC-3 and DU-145 cells (at 70–80% confluence) were treated with vehicle (DMSO) or psoralidin for 24 h. Cell lysates were immunoprecipitated using an ERK antibody or c-Jun fusion beads and ERK or JNK kinase assays, respectively were performed as described previously (17) using ERK and JNK kinase assay kits purchased from Cell Signaling Technology (Danvers, MA).
Transient transfection and promoter assays
PC-3 (at 80–90% confluency) were transiently transfected using Lipofectamine Plus reagents (Life Technologies, Carlsbad, CA) with 4 μg of either c-Jun promoter-luciferase construct or control vector (pGL3.4) containing Renilla luciferase to normalize transfection efficiency and promoter analysis was performed as described previously (18).
Apoptosis assay
PC-3 and DU-145 cells (at 70–80% confluence) were treated with the above mentioned concentrations psoralidin or vehicle (DMSO) in the absence or presence of 5 μM of a JNK inhibitor. An apoptotic assay (Annexin V-FITC) was performed as described earlier (19, 20). Tumor sections from control and psoralidin-treated animals were subjected to TUNEL assay as decribed previously (15).
Xenograft studies
The effect of oral administration of psoralidin on PC-3 xenografts was tested in accordance with the University of Kentucky Animal Care and Use Committee guidelines. In our pilot studies we orally administered various doses (25, 50 or 100mg/kg body weight) of psoralidin to PC-3 xenograft animals and determined that 50mg/kg body weight psoralidin was the optimal dose that suppresses PCa growth with no toxicity to the study animals (data not shown). Tumor injection, measurement of tumor size and growth inhibition studies were performed as described previously (15, 18). Briefly, PC-3 (1×106) cells were subcutaneously injected into 5–6 week old male nude (nu/nu) mice (from Harlan laboratories). The tumors were allowed to reach a 50 mm3 volume and the animals were randomized into two groups with 10 animals each. Psoralidin was dissolved in sunflower oil. One group of animals were orally fed with sunflower oil alone and the other received 50 mg/Kg body weight psoralidin for four weeks. Tumor volume in the animals was monitored and measured daily for the entire study period.
Immunohistochemical analysis
The effect of psoralidin on activated levels of EGFR and c-Jun in PC-3 xenografts were assessed by immunohistochemical analysis using pEGFR (Tyr 1173) and pc-Jun antibodies as described previously (16).
Statistical Analysis
Analysis of variance (ANOVA) was used to calculate statistical significance between the samples. Densitometry analysis was performed using UNSCAN- gel software.
Results
Basal expression of EGFR in androgen-dependent and androgen–independent PCa
EGFR is highly expressed in PCa (2, 3); hence we intended to determine the basal levels of activated (phospho) EGFR in PCa cells lines (PC-3, DU-145, LNCaP and C4-2B). PC-3 and DU-145 cells have higher basal expression of pEGFR compared to LNCaP and C4-2B (Figure 1A). In serum starved conditions, a significant decrease in pEGFR expression was seen (Figure 1A), however, we found that serum starvation alone fails to increase the expression of JNK or c-Jun in both PC-3 and DU-145 cells (Figure 1B). Next, to establish EGF-induced EGFR signaling; serum starved AIPC cells were stimulated with EGF and pEGFR expression was studied. These results collectively suggest that although serum starvation decreases pEGFR expression, it fails to induce the expression of stress-mediated JNK/c-Jun in AIPC cells. In addition, we found that treatment of serum starved AIPC cells with EGF caused a rapid induction of pEGFR expression at 60 minutes following stimulation (Figure 1C) suggesting that EGFR signaling can be modulated in AIPC cells.
Figure 1. Basal expression of EGFR in androgen-dependent and -independent PCa cells.

Androgen-dependent (LNCaP) and –independent (PC-3, DU-145 and C4-2B) were grown either in complete medium or serum-free medium. Whole cell lysates were obtained and Western blot analyses were performed using (A) pEGFR (Tyr 1173) and total EGFR antibodies or (B) pJNK1/2, JNK1/2, pc-Jun and c-Jun antibodies. (C) PC-3 and DU-145 cells were serum deprived for 48 h and stimulated with 100 ng/ml of EGF for up to 120 minutes. Whole cell lysates were subjected to Western blot analysis using pEGFR and EGFR antibodies. β-actin was used as the internal loading control.
Psoralidin down-regulates EGF-induced EGFR signaling in AIPC
Next, we determined whether psoralidin inhibits EGFR signaling in AIPC cells. Previously we published that psoralidin, a natural compound inhibits growth of AIPC cells with an IC50 of 60 μM in PC-3 and 45 μM in DU-145 cells. Our results show that psoralidin inhibits EGF-induced pEGFR expression without altering total EGFR levels in both PC-3 and DU-145 cells (Figure 2A). Upon stimulation by EGF, EGFR may activate the downstream Ras-dependent ERK/MAPK pathway (21). So, we analyzed the expression pattern of the MAPK family of proteins. We observed that EGF significantly induced the expression of MEK-4 and pMEK-1/2 in PC-3 and DU-145 cells when compared to Raf, MEK-4 and MEKK-1. This EGF-induced activation of MAPK (MEK-4 and pMEK-1/2) was overcome by psoralidin (Figure 2B). Interestingly in AIPC cells grown in complete medium psoralidin down-regulated the expression of Raf-1, MEK-4, MEKK-1 and pMEK1/2 within 60 min (data not shown).
Figure 2. Psoralidin inhibits EGFR-mediated MAPK signaling in AIPC cells.

Serum-deprived PC-3 and DU-145 cells were stimulated with 100 ng/ml of EGF and treated concurrently with psoralidin for 30 or 60 minutes. Whole cell lysates were subjected to Western blot analysis using (A) pEGFR, and EGFR antibodies, (B) Raf-1, MEK-4, MEKK-1 and pMEK1/2 antibodies or (C) pERK1/2, ERK1/2, pELK-1 and Elk-1 antibodies. β-actin was used as the internal loading control.
Next we found that EGF induced high expression of pERK but no significant change in pElk expression in PC-3 cells. On the other hand, in DU-145 cells EGF did not change expression of pERK but significantly increased expression of pElk (Figure 2C). We also noted that in EGF-induced cells, psoralidin down-regulated pERK and pElk-1 expression at the earlier time points (30 and 60 minutes) in DU-145 but not PC-3 cells (Figure 2C).
Psoralidin induces SAPK (JNK/c-Jun) and inhibits EGF-mediated survival pathways in AIPC
Recent studies suggest that SAPKs induce apoptosis in PCa cells (22, 23). Based on this, we investigated the effect of psoralidin on SAPK-mediated proapoptotic signaling pathway in AIPC cells. EGF treatment reduced the expression of pJNK and pc-Jun in both PC-3 and DU-145 cells. However, psoralidin restores this EGF-meditaed inhibition in both AIPC cells (Figure 3A). Previous studies reported that EGFR induces pro-survival signaling by inducing expression of survivin and Bcl-2 resulting in the suppression of apoptosis (24), hence we determined whether psoralidin overcomes EGF-mediated pro-survival signaling in AIPC cells. Our results suggested that psoralidin inhibited EGF-induced survivin and Bcl-2, upregulated expression of Bax and cleaved caspase-3 in both EGF-induced AIPC cell lines (Figure 3B). These results suggest that psoralidin induces SAPK-mediated apoptotic signaling and simultaneously overcomes EGF-mediated survival signaling in AIPC.
Figure 3. Psoralidin overcomes EGF-mediated inhibition of SAPK signaling and modulates pro-apoptotic machinery in AIPC cells.
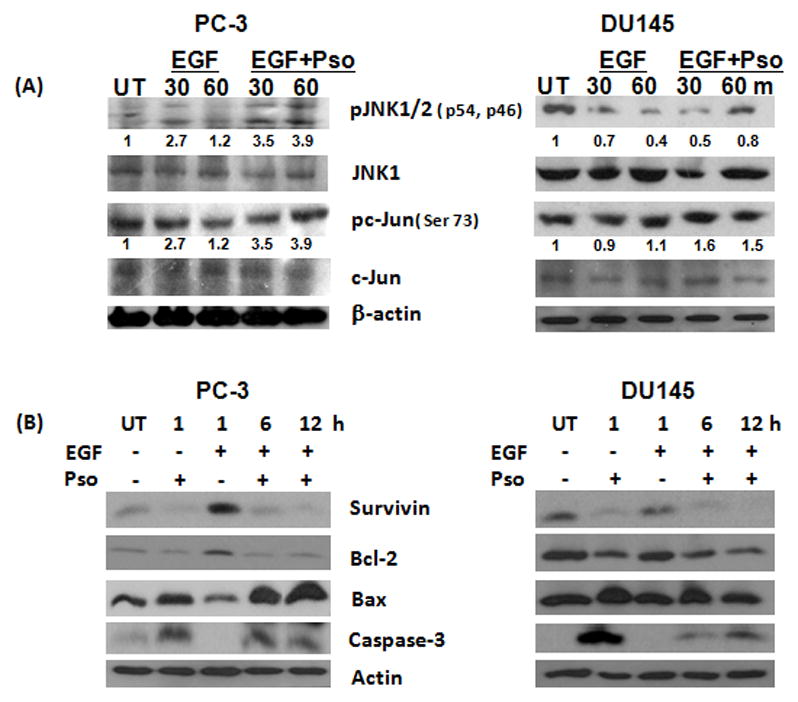
(A) Serum-deprived PC-3 and DU-145 cells were stimulated with 100 ng/ml of EGF and treated concurrently with psoralidin for 30 or 60 minutes. Whole cell lysates were subjected to Western blot analysis using pJNK1/2, JNK1/2, pc-Jun and c-Jun antibodies. (B) PC-3 and DU-145 cells were serum deprived for 48 h, stimulated with 100 ng/ml of EGF and treated concurrently with EGF and psoralidin for up to 12h. Whole cell lysates were subjected to Western blot analysis using survivin, Bcl-2, Bax and cleaved caspase-3 antibodies. β-actin was used as the internal loading control.
Inhibition of JNK/c-Jun partially abrogates psoralidin-mediated apoptosis in AIPC cells
Since, psoralidin induces expression of pJNK and pc-Jun, we studied whether treatment with psoralidin induces JNK kinase activity. We found a significant increase in JNK kinase activity (an approximate 4.5-fold and 3-fold increase in PC-3 and DU-145 cells, respectively) in psoralidin-treated AIPC cells when compared to control (Figure 4A). These results suggest that psoralidin increases JNK kinase activity and thereby activates downstream c-Jun in AIPC cells. Next, considering the effect of psoralidin on JNK/c-Jun, we determined whether inhibition of JNK kinase abrogates psoralidin-mediated effects on AIPC cells. As expected, pharmacological inhibition of JNK significantly decreased pJNK expression (data not shown) and psoralidin-mediated apoptosis in PC-3 cells, however no significant difference in apoptosis was observed in DU-145 cells (Figure 4B). Similarly, inhibition of c-Jun using siRNA significantly abrogated the ability of psoralidin to induce apoptosis in AIPC cells (data not shown). Additionally, we found that psoralidin mediates its function by regulating the c-Jun promoter in AIPC cells (Figure 4C). These results collectively suggest that psoralidin mediates its apoptotic function, at least in part through the JNK/c-Jun pathway. Thus, inhibition of the JNK/c-Jun pathway interferes with psoralidin-mediated apoptosis in AIPC cells.
Figure 4. Psoralidin increases JNK kinase activity and potentiates JNK-mediated apoptosis in AIPC cells.

(A) PC-3 and DU145 cells were treated with 60 and 45 μM (IC50) psoralidin respectively for 24h. Whole cell lysates were subjected to immunoprecipitation using c-Jun fusion beads. JNK kinase activity was assayed by Western blot analysis using a pc-Jun antibody (upper panel). Bars represent a fold increase in JNK kinase activity following the treatment with psoralidin (lower panel). (B) PC-3 and DU-145 cells were treated with SP600125 alone (5 μM), psoralidin alone (60 or 45 μM, respectively) or a combination of SP600125 and psoralidin for 24 h and cells were subjected to apoptosis assay using Annexin V-FITC staining. Bars represent the percentage of apoptosis ± standard deviation (SD). (C) PC-3 cells were transfected with the c-Jun promoter-luciferase reporter construct and rennilla control vector (pGL3.4) was used to normalize transfection efficiency followed by treatment with psoralidin or vehicle. Luciferase reporter assay was performed to determine c-Jun promoter activity. Bars represent fold change in c-Jun promoter activity ± SD.
Effect of psoralidin on EGFR/MAPK/ERK signaling in AIPC cells
The above findings suggest that psoralidin inhibits EGF-induced EGFR/MAPK signaling in AIPC cells. Next we intended to study the effect of psoralidin on PC-3 and DU-145 cells grown in complete medium. Psoralidin inhibits pEGFR expression in both AIPC cells (Figure 5A). Also, we found that the expression of MEK-1, MEK-4 and MEKK-1 was significantly downregulated in both AIPC cells following treatment with psoralidin, (Figure 5B), however no significant change in the expression of pMEK1/2 and -3/6 was observed. There was no significant change in the levels of pERK upon treatment with psoralidin in both PC-3 and DU-145 cells (Figure 5C), however ERK kinase activity assays revealed that psoralidin decreased the kinase activity of ERK as was seen by a decrease in the expression of its downstream effector, pElk (Figure 5C) in PC-3 and DU-145 cells. We also found that psoralidin induces pJNK and pc-Jun expression in both AIPC cell lines (Figure 5D). These findings suggest that psoralidin inhibits EGFR-mediated pro-survival signaling while simultaneously activating SAPK-mediated apoptotic signaling in AIPC cells.
Figure 5. Psoralidin inhibits EGFR-mediated MAPK and induced SAPK signaling in AIPC cells grown in complete medium.

PC-3 and DU145 cells were grown in complete medium containing 10% fetal bovine serum and treated with 60 and 45 μM (IC50) psoralidin for up to 12 h. Whole cell lysates were subjected to Western blot analysis using (A) pEGFR and EGFR antibodies, (B) MEK-1, MEK-4, pMEK3/6, pMEK1/2 and MEKK-1 antibodies and (C) pERK and ERK antibodies (upper panel) and (C) PC-3 and DU145 cells were treated with 60 and 45 μM psoralidin for 24 h. Whole cell lysates were subjected to immunoprecipitation using ERK antibody. ERK kinase activity was assayed by Western blot analysis using a pElk-1 antibody. Total ELK-1 antibody was used as control (lower panel). (D) PC-3 and DU145 cells were grown in complete medium containing 10% fetal bovine serum and treated with 60 and 45 μM psoralidin for up to 12 h. Whole cell lysates were subjected to Western blot analysis using pJNK1/2, JNK1/2, pc-Jun and c-Jun antibodies. β-actin was used as the internal loading control.
Psoralidin inhibits tumor growth in PC-3 xenografts
We previously reported that intra tumoral injection of psoralidin inhibits PCa growth in nude mouse models (20). In our pilot studies we orally administered various doses (25, 50 or 100mg/kg body weight) of psoralidin to PC-3 xenograft animals and determined that 50mg/kg body weight psoralidin was the optimal dose that suppresses PCa growth with no toxicity to the study animals. As seen in Figure 6A, oral administration of psoralidin (50 mg/kg), five days a week for four weeks, significantly suppressed further PC-3 tumor growth compared to control animals. Also, we observed the animals and monitored tumor sizes up to two weeks after termination of psoralidin treatment. We found that the tumors in psoralidin treated animals failed to regrow. Gross pathology and histopathology studies revealed no significant toxicity in psoralidin treated animals (data not shown). Immunohistochemical analysis of psoralidin-treated tumor tissues showed a significant reduction in pEGFR expression and, induced and also increased nuclear entry of pc-Jun (Figure 6B). Also, we found a significant increase in the number of apoptotic cells in psoralidin-treated tumor sections when compared to control tumor sections (Figure 6B). These results collectively suggest that psoralidin effectively suppresses prostate tumor growth through the inhibition of EGFR-mediated survival signaling with a simultaneous induction of SAPK-mediated apoptotic events.
Figure 6. Psoralidin inhibits tumor growth in xenografts models.

(A) PC-3 cells were implanted subcutaneously in nude mice. When the tumor reached a volume of 50 mm3, the animals were randomized into two groups (control and psoralidin-treated). Control animals were orally fed sunflower oil, while psoralidin-treated animals received oral administration of psoralidin (50 mg/kg) for a four week period. Tumor growth was monitored and tumor volumes were measured and graphed. Each data point represents mean ± SD. (B) Control and psoralidin treated tumors were subjected to immunohistochemical analysis using (i) pEGFR and (ii) pc-Jun antibodies and (iii) number of apoptotic cells using TUNEL assay.
Discussion
Overexpression of EGFR plays an important role in the pathogenesis of PCa (25) and is correlated with Gleason score and androgen independence (26). In this study we demonstrate that psoralidin, a natural compound effectively alters EGFR-mediated signal transduction pathways and inhibits prostate tumor growth in vivo model.
EGFR signaling, being one of the major signaling cascades in PCa, has been the focus of growing interest in therapeutic targeting in both the laboratory and clinical trials (27). Our results suggest that AIPC (PC-3 and DU-145) cells express high levels of phosphorylated EGFR compared to LNCaP and C4-2B cells. Although serum deprivation significantly inhibits pEGFR expression, it fails to trigger SAPK-mediated apoptotic signaling in PCa cells. Similar results were observed by other investigators that inhibition of EGFR by gefinitib fails to significantly inhibit cell viability in cancer models (28). These studies imply that EGFR inhibition alone may be insufficient and that a secondary signal may be required to initiate apoptosis in PCa cells.
In response to EGF and/or various growth factors, EGFR regulates several signaling pathways, including PI3K/Akt and MAPK/ERK1/2/ELK in AIPC (29–31). Here we show that EGF stimulation results in robust pEGFR expression within 60 minutes in both PC-3 and DU-145 cells. We also observed a concomitant increase in the expression of several events downstream of EGFR namely MAPK (MEK-4 and pMEK-1/2). These data suggest that EGFR, through EGF stimulation, governs many pro-survival signaling pathways in PCa and psoralidin overcomes the EGF-induced pro-survival signals as was seen by a complete inhibition in the expression of pro-survival molecules downstream of EGFR. Similar observations were made by other research groups (32).
Prostate stromal tissue produces EGF (33) that stimulates several survival pathways converging at the level of ERK. ERK is involved in cell survival or death depending on a cell type and stimuli-dependent manner (27). ERK is shown to be activated in premalignant lesions and early stage PCa (34). In our study, we found no significant change in the expression of pERK, however reduced kinase activity of ERK in both AIPC as was seen by a reduction in the expression of pElk in both AIPC cells. These results suggest that psoralidin not only inhibits EGFR activation but also inhibits the entire downstream prosurvival signaling of EGFR in AIPC cells.
JNK/SAPK signaling plays a dual role, both in the oncogenic process and also as a tumor suppressor (11). As we mentioned above, serum starvation fails to induce pJNK/p-c-Jun activation in AIPC cells. These data suggest that inhibition of pEGFR expression alone may be insufficient to induce apoptosis in AIPC cells. Psoralidin activates the JNK/c-Jun pathway in both AIPC cell lines. These results suggest that psoralidin-mediated apoptosis may be through the activation of the JNK pathway in AIPC cells. Kinase assays were performed to determine the effect of psoralidin on JNK kinase activation. We observed a robust induction in JNK kinase activity in both AIPC cell lines. Additionally, we found that inhibition of JNK kinase activity using a pharmacological inhibitor significantly reduced psoralidin-mediated apoptosis in both AIPC cell lines. Similarly, inhibition of c-Jun using siRNA also abrogated psoralidin-mediated apoptosis in AIPC cells (data not shown). These results imply that activation of JNK/c-Jun is essential at least in part for psoralidin-mediated effects on PC-3 and DU-145 cells. Nuclear factor-κB (NF-κB) inhibits JNK-mediated apoptosis (35), which correlate with our findings. We previously reported that psoralidin inhibits Akt-mediated NF-κB activation in AIPC cells (15). Inhibition of the Akt/NF-κB pathway by psoralidin may lead to the induction of the JNK pathway and resultant apoptosis in AIPC cells. Similar studies reported that dietary compounds, like phenylethyl isothiocyanate (PEITC), curcumin, and epifallocatchin gallate (EGCG), induced apoptosis by activating JNK-mediated cytochrome c release and caspase activation in many cancer models (36–38).
Additionally, we previously reported that psoralidin altered the Bax/Bcl-2 ratio, induced caspase-9 and -3 and caused poly (ADP- ribose) polymerase (PARP) cleavage in both AIPC cells (39). Interestingly, we found that EGF treatment induced survivin and Bcl-2 and inhibited expression of Bax and caspase-3 cleavage and psoralidin treatment overcomes these EGF-mediated effects in both AIPC cell lines.
Oral administration of psoralidin (50 mg/kg) effectively suppresses the growth of PC-3 xenograft tumors in nude mouse models. Our results show that psoralidin failed to completely regress the tumors however it inhibits further tumor growth when compared to the control animals. The inability of psoralidin to cause tumor regression may be due to insufficient duration of psoralidin treatment in our animal studies. Similar observations were made using other dietary compounds polyphenol Epigallocatechin-3-gallate (EGCG), and isothiocyanate sulforaphane (40, 41). Our immunohistochemical analysis suggests an inhibition of pEGFR expression and an increase in pc-Jun expression and nuclear entry of pc-Jun in psoralidin-treated tumors compared to control tumors. These findings correlate with results from our cell culture models.
In conclusion, our findings suggest that psoralidin may be a potential therapeutic agent that targets key signaling pathways in AIPC cells. Based on our data, psoralidin may provide a translational tool that may be relevant to the treatment of PCa with a defined mechanism of action.
References
- 1.Lee TL, Yeh J, Van Waes C, Chen Z. Epigenetic modification of SOCS-1 differentially regulates STAT3 activation in response to interleukin-6 receptor and epidermal growth factor receptor signaling through JAK and/or MEK in head and neck squamous cell carcinomas. Mol Cancer Ther. 2006;5:8–19. doi: 10.1158/1535-7163.MCT-05-0069. [DOI] [PubMed] [Google Scholar]
- 2.Tillotson JK, Rose DP. Endogenous secretion of epidermal growth factor peptides stimulates growth of DU145 prostate cancer cells. Cancer Lett. 1991;60:109–12. doi: 10.1016/0304-3835(91)90216-5. [DOI] [PubMed] [Google Scholar]
- 3.Marks RA, Zhang S, Montironi R, et al. Epidermal growth factor receptor (EGFR) expression in prostatic adenocarcinoma after hormonal therapy: a fluorescence in situ hybridization and immunohistochemical analysis. Prostate. 2008;68:919–23. doi: 10.1002/pros.20715. [DOI] [PubMed] [Google Scholar]
- 4.Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem J. 2000;351:289–305. [PMC free article] [PubMed] [Google Scholar]
- 5.Lax I, Bellot F, Honegger AM, et al. Domain deletion in the extracellular portion of the EGF-receptor reduces ligand binding and impairs cell surface expression. Cell Regul. 1990;1:173–88. doi: 10.1091/mbc.1.2.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gioeli D, Mandell JW, Petroni GR, Frierson HF, Jr, Weber MJ. Activation of Mitogen-Activated Protein Kinase Associated with Prostate Cancer Progression. Cancer Res. 1999;59:279–84. [PubMed] [Google Scholar]
- 7.Knebel A, Rahmsdorf HJ, Ullrich A, Herrlich P. Dephosphorylation of receptor tyrosine kinases as target of regulation by radiation, oxidants or alkylating agents. EMBO J. 1996;15:5314–25. [PMC free article] [PubMed] [Google Scholar]
- 8.Dolado I, Swat A, Ajenjo N, De Vita G, Cuadrado A, Nebreda AR. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell. 2007;11:191–205. doi: 10.1016/j.ccr.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 9.Manke IA, Nguyen A, Lim D, Stewart MQ, Elia AE, Yaffe MB. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol Cell. 2005;17:37–48. doi: 10.1016/j.molcel.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 10.Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene. 2008;27:6245–51. doi: 10.1038/onc.2008.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu J, Lin A. Role of JNK activation in apoptosis: a double-edged sword. Cell Res. 2005;15:36–42. doi: 10.1038/sj.cr.7290262. [DOI] [PubMed] [Google Scholar]
- 12.Prasad MV, Dermott JM, Heasley LE, Johnson GL, Dhanasekaran N. Activation of Jun kinase/stress-activated protein kinase by GTPase-deficient mutants of G alpha 12 and G alpha 13. J Biol Chem. 1995;270:18655–9. doi: 10.1074/jbc.270.31.18655. [DOI] [PubMed] [Google Scholar]
- 13.Fan M, Chambers TC. Role of mitogen-activated protein kinases in the response of tumor cells to chemotherapy. Drug Resist Updat. 2001;4:253–67. doi: 10.1054/drup.2001.0214. [DOI] [PubMed] [Google Scholar]
- 14.Tournier C, Hess P, Yang DD, et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288:870–4. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 15.Kumar R, Srinivasan S, Koduru S, et al. Psoralidin, an herbal molecule, inhibits phosphatidylinositol 3-kinase-mediated Akt signaling in androgen-independent prostate cancer cells. Cancer Prev Res (Phila Pa) 2009;2:234–43. doi: 10.1158/1940-6207.CAPR-08-0129. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Chun JY, Nadiminty N, Dutt S, et al. Interleukin-6 regulates androgen synthesis in prostate cancer cells. Clin Cancer Res. 2009;15:4815–22. doi: 10.1158/1078-0432.CCR-09-0640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Srinivasan S, Koduru S, Kumar R, Venguswamy G, Kyprianou N, Damodaran C. Diosgenin targets Akt-mediated prosurvival signaling in human breast cancer cells. Int J Cancer. 2009;125:961–7. doi: 10.1002/ijc.24419. [DOI] [PubMed] [Google Scholar]
- 18.Srinivasan S, Ranga RS, Burikhanov R, Han SS, Chendil D. Par-4-dependent apoptosis by the dietary compound withaferin A in prostate cancer cells. Cancer Res. 2007;67:246–53. doi: 10.1158/0008-5472.CAN-06-2430. [DOI] [PubMed] [Google Scholar]
- 19.Sowmyalakshmi S, Nur EAM, Akbarsha MA, Thirugnanam S, Rohr J, Chendil D. Investigation on Semecarpus Lehyam-a Siddha medicine for breast cancer. Planta. 2005;220:910–8. doi: 10.1007/s00425-004-1405-4. [DOI] [PubMed] [Google Scholar]
- 20.Chendil D, Ranga RS, Meigooni D, Sathishkumar S, Ahmed MM. Curcumin confers radiosensitizing effect in prostate cancer cell line PC-3. Oncogene. 2004;23:1599–607. doi: 10.1038/sj.onc.1207284. [DOI] [PubMed] [Google Scholar]
- 21.Sato K, Kimoto M, Kakumoto M, et al. Adaptor protein Shc undergoes translocation and mediates up-regulation of the tyrosine kinase c-Src in EGF-stimulated A431 cells. Genes Cells. 2000;5:749–64. doi: 10.1046/j.1365-2443.2000.00358.x. [DOI] [PubMed] [Google Scholar]
- 22.Hagemann C, Blank JL. The ups and downs of MEK kinase interactions. Cell Signal. 2001;13:863–75. doi: 10.1016/s0898-6568(01)00220-0. [DOI] [PubMed] [Google Scholar]
- 23.Srikanth S, Franklin CC, Duke RC, Kraft RS. Human DU145 prostate cancer cells overexpressing mitogen-activated protein kinase phosphatase-1 are resistant to Fas ligand-induced mitochondrial perturbations and cellular apoptosis. Mol Cell Biochem. 1999;199:169–78. doi: 10.1023/a:1006980326855. [DOI] [PubMed] [Google Scholar]
- 24.Li F, Ambrosini G, Chu EY, et al. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396:580–4. doi: 10.1038/25141. [DOI] [PubMed] [Google Scholar]
- 25.Li LC, Carroll PR, Dahiya R. Epigenetic changes in prostate cancer: implication for diagnosis and treatment. J Natl Cancer Inst. 2005;97:103–15. doi: 10.1093/jnci/dji010. [DOI] [PubMed] [Google Scholar]
- 26.Syed S. Combination chemotherapy for hormone-refractory prostate carcinoma: progress and pitfalls. Cancer. 2003;98:2088–90. doi: 10.1002/cncr.11788. [DOI] [PubMed] [Google Scholar]
- 27.Putz T, Culig Z, Eder IE, et al. Epidermal growth factor (EGF) receptor blockade inhibits the action of EGF, insulin-like growth factor I, and a protein kinase A activator on the mitogen-activated protein kinase pathway in prostate cancer cell lines. Cancer Res. 1999;59:227–33. [PubMed] [Google Scholar]
- 28.Lee SY, Hur GY, Jung KH, et al. PPAR-gamma agonist increase gefitinib’s antitumor activity through PTEN expression. Lung Cancer. 2006;51:297–301. doi: 10.1016/j.lungcan.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 29.Sastry KS, Karpova Y, Kulik G. Epidermal growth factor protects prostate cancer cells from apoptosis by inducing BAD phosphorylation via redundant signaling pathways. J Biol Chem. 2006;281:27367–77. doi: 10.1074/jbc.M511485200. [DOI] [PubMed] [Google Scholar]
- 30.Gioeli D, Mandell JW, Petroni GR, Frierson HF, Jr, Weber MJ. Activation of mitogen-activated protein kinase associated with prostate cancer progression. Cancer Res. 1999;59:279–84. [PubMed] [Google Scholar]
- 31.Levitzki A, Gazit A. Tyrosine kinase inhibition: an approach to drug development. Science. 1995;267:1782–8. doi: 10.1126/science.7892601. [DOI] [PubMed] [Google Scholar]
- 32.Festuccia C, Angelucci A, Gravina GL, et al. Epidermal growth factor modulates prostate cancer cell invasiveness regulating urokinase-type plasminogen activator activity. EGF-receptor inhibition may prevent tumor cell dissemination. Thromb Haemost. 2005;93:964–75. doi: 10.1160/TH04-09-0637. [DOI] [PubMed] [Google Scholar]
- 33.Oh HY, Lee EJ, Yoon S, Chung BH, Cho KS, Hong SJ. Cholesterol level of lipid raft microdomains regulates apoptotic cell death in prostate cancer cells through EGFR-mediated Akt and ERK signal transduction. Prostate. 2007;67:1061–9. doi: 10.1002/pros.20593. [DOI] [PubMed] [Google Scholar]
- 34.Uzgare AR, Kaplan PJ, Greenberg NM. Differential expression and/or activation of P38MAPK, erk1/2, and jnk during the initiation and progression of prostate cancer. Prostate. 2003;55:128–39. doi: 10.1002/pros.10212. [DOI] [PubMed] [Google Scholar]
- 35.Tang G, Minemoto Y, Dibling B, et al. Inhibition of JNK activation through NF-kappaB target genes. Nature. 2001;414:313–7. doi: 10.1038/35104568. [DOI] [PubMed] [Google Scholar]
- 36.Hu R, Kong AN. Activation of MAP kinases, apoptosis and nutrigenomics of gene expression elicited by dietary cancer-prevention compounds. Nutrition. 2004;20:83–8. doi: 10.1016/j.nut.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 37.Xu C, Shen G, Yuan X, et al. ERK and JNK signaling pathways are involved in the regulation of activator protein 1 and cell death elicited by three isothiocyanates in human prostate cancer PC-3 cells. Carcinogenesis. 2006;27:437–45. doi: 10.1093/carcin/bgi251. [DOI] [PubMed] [Google Scholar]
- 38.Ravindran J, Prasad S, Aggarwal BB. Curcumin and cancer cells: how many ways can curry kill tumor cells selectively? AAPS J. 2009;11:495–510. doi: 10.1208/s12248-009-9128-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Srinivasan S, Kumar R, Koduru S, Chandramouli A, Damodaran C. Inhibiting TNF-mediated signaling: a novel therapeutic paradigm for androgen independent prostate cancer. Apoptosis. 15:153–61. doi: 10.1007/s10495-009-0416-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anderson MW, Goodin C, Zhang Y, et al. Effect of dietary green tea extract and aerosolized difluoromethylornithine during lung tumor progression in A/J strain mice. Carcinogenesis. 2008;29:1594–600. doi: 10.1093/carcin/bgn129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pham NA, Jacobberger JW, Schimmer AD, Cao P, Gronda M, Hedley DW. The dietary isothiocyanate sulforaphane targets pathways of apoptosis, cell cycle arrest, and oxidative stress in human pancreatic cancer cells and inhibits tumor growth in severe combined immunodeficient mice. Mol Cancer Ther. 2004;3:1239–48. [PubMed] [Google Scholar]