

Abstract
Retinoblastoma is the most common childhood primary intraocular malignancy, with the majority of cases being diagnosed before 5 years of age. Retinoblastoma in adults is extremely rare. Here, we report the case of a 20-year-old man who presented with a 3 year history of blurred vision in the right eye. Imaging did not reveal the typical presentation of retinoblastoma. After considering Coats' disease, a diagnosis of late-presenting retinoblastoma was made through cytological analysis. Diagnosis of retinoblastoma should be considered in the presence of uncertain mass lesions in the fundus of an adult.
Keywords: adult, retinoblastoma, Coats'disease, cytological analysis
INTRODUCTION
Retinoblastoma is the most common malignant intraocular tumor in children. More than 90% of cases are diagnosed before 5 years of age, and the average age of diagnosis is 1 year and 2 years in bilateral and unilateral cases, respectively[1]. The presentation of retinoblastoma in adults is extremely rare[2]. Here, we report the case of an adult with retinoblastoma who presented with diminished visual acuity masquerading as Coats' disease.
TYPICAL CASE
A twenty-year-old man was referred to our department for loss of vision in the right eye for approximately 3 years. Three years earlier, he had noticed decreased vision in his right eye, without pain, photophobia, or discharge. After a routine eye examination at a local hospital, he was diagnosed with Coats' disease. Laser photocoagulation of the right eye was performed twice. His visual acuity had diminished rapidly within the last year. At this time, visual acuity was limited to light perception in the right eye and was 0.6 in the left eye. He was referred for further evaluation to the Beijing Tongren hospital.
The patient had no history of trauma and surgery. He was born at full term and had met all physical and developmental milestones. His family members were well, and there was no family history of ophthalmologic problems.
The general physical examination was normal. On ocular examination, visual acuity of the patient demonstrated light perception in the right eye and 0.6 in the left eye. Intraocular pressures were 15 mmHg in the right eye and 12 mmHg in the left eye. The slit-lamp examination of the anterior segment of the right eye showed a normal conjunctiva, a clear cornea, a normal appearing open anterior chamber angle containing a significant amount of white floccular exudates, and some neovascularization on the iris. The pupil appeared circular and the direct response to light was sluggish. The lens was opaque. Funduscopy could not be performed through the opaque medium. The left eye was normal. The eye movements were normal.
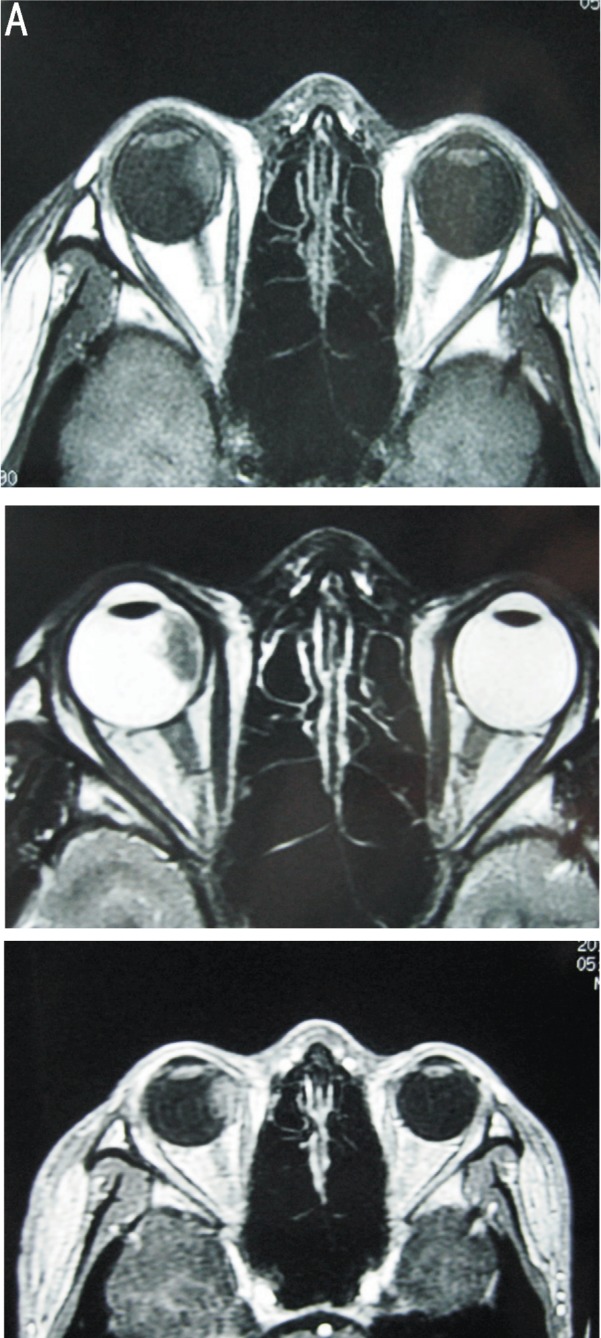
Ultrasonography showed a moderately reflective material occupying the nasal peripheral retina that was suggestive of a mass with a collection of fluid beneath the retina in the right eye, and a highly reflective lesion within the mass (Figure 1). Computed tomography (CT) demonstrated that the mass had a higher density than the vitreous body and did not reveal any calcifications within the mass (Figure 2). Magnetic resonance imaging (MRI) showed an irregular mass involving the ciliary body and the optic nerve head, but the extraocular muscles appeared normal (Figure 3).
Figure 1. Ultrasonographic examination of the right eye showing a moderately reflective material occupying the nasal peripheral retina that was suggestive of a mass with a collection of fluid beneath the retina, a highly reflective lesion can be seen in the mass. The left eye was sound.

Figure 2. Computed tomography (CT) showing the mass involving the nasal retina with a minimal higher density and without calcification in the right eye.

Figure 3. A: Axial T1-weighted MR image showing the mass involving the ciliary body, which had slightly higher signal intensity; B: Axial T2-weighted MR image showing the mass had a relatively low-intensity signal; C: The contrast-enhanced MR image showing there was diffuse enhancement of the mass.
An anterior chamber puncture was performed to perform cytological analysis, which revealed extensive necrosis and small malignant cells with large, darkly staining nuclei and scanty cytoplasm (Figure 4). We performed an enucleation of the right eye and inserted an implant in the right orbit. The histopathological examination revealed classical pathological features of retinoblastoma with the presence of a classical rosette pattern (Figure 5). Immunohistochemistry was performed for exact diagnosis and was positive for glial fibrillary acidic protein (GFAP), Vimentin, S-100, p53, Ki-67, bcl-2 and negative for neuron specific enolase (NSE), pRb. The histopathological diagnosis was retinoblastoma.
Figure 4. Cytological analysis of the anterior chamber puncture fluid showing neoplastic cells forming loosely cohesive clusters. Note the scant cytoplasm, the high nuclear to cytoplasmic ratio (staining by hematoxylin-eosin; original magnification, ×400).

Figure 5. A: The tumor was present in the anterior segment and implanted into the iris and ciliary body with neovascularization on the anterior surface of the iris (staining by hematoxylin-eosin; original magnification, ×40). B: The tumor was found to have invaded the optic nerve head without any extension to the lamina cribrosa and optic nerve (staining by hematoxylin-eosin; original magnification, ×40). C: The presence of a classical Homer-Wright rosette pattern (staining by hematoxylin-eosin; original magnification, ×100).

He was transferred to the department of internal medicine and received chemotherapy after ocular surgery. We followed up with the patient at approximately 6 months after surgery, but it is too early to evaluate for the absence of recurrence or metastasis.
DISCUSSION
Retinoblastoma is the most common childhood intraocular malignant tumor. It may occur at any age but occurs most commonly in younger children, usually before the age of two years[1]. More than 90% of cases are diagnosed before 5 years of age, and presentation of retinoblastoma in adults is rare[2]. Although retinoblastoma uncommonly occurs after the age of 20 years, cases in adults have been reported[3]-[11]. Based on epidemiologic studies of several investigators, retinoblastoma affects approximately 1 in 16,000 to 18,000 births, resulting in an incidence of 7000 to 8000 new annual cases worldwide[12]. In 60% of the cases, the disease is unilateral and the median age at diagnosis is two years. Retinoblastoma is bilateral in approximately 40% of cases with a median age at diagnosis of one year. All bilateral and multifocal unilateral forms are hereditary, and in nonhereditary cases, retinoblastoma is always unilateral and unifocal[1]. In our case, this twenty-year-old young man presented with a single tumor in the right eye.
Pathogenesis
There are a number of proposed hypotheses to explain the late presentation of retinoblastomas. One of the hypotheses is that persistence of rare embryonal retinal cells may lead to malignant transformation later in life. Another suggestion is that retinoblastomas may arise from previously undiagnosed spontaneously regressed/arrested retinoblastomas which have been reactivated[13]. The “two-hit” mechanism also explains rare cases of late retinoblastomas[14],[15]. In patients a germline inactivated RB1 allele present in all cells in the body and somatic loss of the second allele in the retinal cells, multiple retinoblastomas arise in both eyes, usually at a young age, whereas in patients both RB1 alleles are inactivated somatically in a single developing retinal progenitor cell, a single tumor usually develops in only one eye and often at a later age, as in our case.
Clinical Presentation
Retinoblastoma can present in many different ways, with the most frequent clinical manifestations including leukocoria and strabismus, especially during childhood[16]. Leukocoria is the result of an altered pupillary red reflex in the eye and usually occurs when there is a large tumor present; it can also occur with smaller tumors associated with retinal detachment. This sign is initially only visible at certain angles and under certain light conditions. Strabismus results from a loss of central vision in one or both eyes. This sign is often overlooked when physicians may not examine the ocular fundus. Some other signs including iris rubeosis, hypopyon, hyphema, buphthalmia, orbital cellulitis, and exophthalmia may be observed.
Retinoblastoma in adult always present with atypical manifestations, such as decreased vision, floaters, and pain. It rarely presents with classic manifestations. These atypical presentations such as decreased vision and floaters can be seen in this case, although he is very young, and their case report is extremely rare. This case explains the late diagnosis and increased risk of death in adult retinoblastoma well[17].
Diagnosis
When the appearance of the tumor is classical, the diagnosis can be made by ophthalmoscopy. The lesion appears as a white tumor with angiomatous dilatation of the vessels. However, in many questionable cases, as in our patient, the ophthalmologist does not have the luxury of direct visualization of the fundus and must rely on imaging modalities. In this case, the diagnostic difficulty was due to the misleading result of the B-scan, in which the echogenicity was stronger in the intraocular mass than the vitreous. Ultrasonography cannot help distinguish retinoblastoma from Coats' disease, which showed massive subretinal exudation from leaking telangiectatic retinal blood vessels[18]. Since Coats' disease occurs most commonly in young males, just like our patient, it leaded us to the wrong orientation of diagnosis. However, the highly reflective lesion in the mass could be interpreted as a calcification, which is featured in retinoblastoma. MRI assessed the local extension in our case. The mass of retinoblastoma should have a signal equivalent to or a slightly more intense than that of the vitreous on T1-weighted sequences, with a relatively low-intensity signal on T2-weighted sequences, which was consistent with our case. CT is the preferred imaging modality to detect intraocular calcifications. In our case, the CT showed a slightly higher density in the intraocular mass than the vitreous body, but the calcification was not observed. Therefore, even with all imaging investigations, the diagnosis cannot be clear in this case.
As leukocoria is not specific to retinoblastoma, atypical manifestations of retinoblastoma can pose diagnostic challenges. Toxocariasis and Coats' disease make up more than 45% of cases in children with leukocoria[19]. Persistence of the primitive vitreous, retinal dysplasia, congenital cataract, and retinopathy of prematurity may also simulate retinoblastoma. The ultrasonography in this case strongly mimicked typical findings in Coats' disease, including the lack of calcification and dilated talangiectatic vessels with leakage. Shields et al[20] have suggested that the caliber and distribution of retinal blood vessels can help distinguish Coats' disease from retinoblastoma. However, fluorescein angiography could not be performed in this case because the right lens was opaque. Cytological analysis can also be used as a diagnostic tool. Coats' disease can be suspected when a subretinal aspirate reveals foamy machophages frequently containing coarse, oval melanin pigments. Inflammatory cells, especially lymphocytes, are also noted.
In clinically uncertain cases, cytological analysis can be very helpful in making a definite diagnosis. The puncture fluid was very thick, with a significant amount of white floccular exudates, as shown by the slit-lamp examination. The presence of anterior chamber particles, called pseudo-hypopyon, is very highly suggestive of AC invasion by the tumor and is never seen in Coat's disease. The cytological smears demonstrated the typical morphological features of retinoblastoma, including undifferentiated cells on a background of abundant necrotic debris. Single or clusters of undifferentiated cells were characterized by a high nuclear/cytoplasmic ratio (N:C). The nuclear size is at least twice as large as in neuronal cells. The cytoplasm was scant and ill defined[21],[22]. The specimen of enucleation confirmed the diagnosis. The tumor was composed of undifferentiated cells along dilated blood vessels with a large ischemic necrosis around it. Homer-Wright rosettes were present. The tumor invaded the optic nerve head without reaching the level of surgical transsection. The tumor had also started to invade the anterior segment and implanted into the iris and ciliary body. Immunohistochemical staining was negative for neuroendocrine markers (neuron-specific enolase, NSE) as well as for the melanocytic marker (S-100). The proliferation index marker Ki-67 stained 90% of the atypical cells' nuclei, in contrast to non-neoplastic retinal neuronal cells, which was negative for the marker. This reinforced the impression that these atypical cells were neoplastic rather than disorganized and fragmented benign retinal tissue. Immunihistochmistry was negative for the protein produced by the RB gene (pRb). In the absence of pRB, cells continuously replicate, which is a property of cancer[23].
Generally, preoperative cytological analysis is not routinely performed in retinoblastomas to minimize the theoretical risk of tumor seeding in the needle tract[24]. Occasionally, cytological analysis is used as a diagnostic tool either when the diagnosis is clinically uncertain, or when the patient insists on pathologic verification of the clinical diagnosis prior to treatment[25]. Despite the rare use of cytological analysis of intraocular tumors, the current literature does not show an increased risk of tumor recurrence post-enucleation following cytological analysis[21].
Treatment
Retinoblastoma has evolved from a deadly childhood cancer to a largely curable cancer within the past 40 years. Current treatment strategies aim to salvage the eye and preserve vision. Currently, the methods available for treatment include laser treatment, cryotherapy, radiotherapy, chemotherapy and enucleation[26]. These treatments are frequently combined for best results. The treatment option chosen for each individual case depends on the overall situation, including threat of metastatic disease, risks for second cancers, systemic status, laterality of the disease, size and location of the tumors, and estimated visual prognosis[1],[27],[28]. Enucleation is the treatment of choice for advanced uniocular retinoblastoma or the worse eye in bilateral cases[26]. In this case, the mass extended anteriorly to the iris and ciliary body with some part of neovascularization in the iris, and there was little hope of salvageable vision, which presented Group E of IRC classification; therefore, enucleation was performed. According to the result of the pathological examination, chemotherapy was administered on this patient after surgery.
In summary, the report of retinoblastoma in this 20-year-old patient mimicking as Coats' disease indicates that, in the presence of uncertain mass lesions in the fundus of an adult, especially young adults, ophthalmologists should maintain a high index of suspicion of retinoblastoma. Ultrasound and CT scans should be the first line of investigations. If diagnostic uncertainty persists, a cytological analysis should be considered.
Footnotes
Foundation items: This work was supported by the National Natural Science Foundation of China (No. 81170785) and a Donation of Beijing Health Systems High-level Health Technology Training Program
REFERENCES
- 1.Aerts I, Lumbroso-Le Rouic L, Gauthier-Villars M, Brisse H, Doz F, Desjardins L. Retinoblastoma. Orphanet J Rare Dis. 2006;1(8):31. doi: 10.1186/1750-1172-1-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karcioglu ZA, Abboud EB, Al-Mesfer SA, Al-Rashed W, Pilapil DH. Retinoblastoma in older children. J AAPOS. 2002;6(1):26–32. doi: 10.1067/mpa.2002.120643. [DOI] [PubMed] [Google Scholar]
- 3.Maghy C. A case of bilateral glioma of the retina in a girl twenty years of age in which the second eye was excised after an interval of nearly eighteen years. Br J Ophthalmol. 1919;3(8):337–340. doi: 10.1136/bjo.3.8.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verhoeff FH. A Retinoblastoma in a Man Aged Forty-eight Years. Trans Am Ophthalmol Soc. 1929;27:176–188. [PMC free article] [PubMed] [Google Scholar]
- 5.Rychener RO. Retinoblastoma in the Adult. Trans Am Ophthalmol Soc. 1948;46:318–326. [PMC free article] [PubMed] [Google Scholar]
- 6.Mehra KS, Hamid S. Retinoblastoma in an adult. Am J Ophthalmol. 1961;52(9):405–406. doi: 10.1016/0002-9394(61)90742-5. [DOI] [PubMed] [Google Scholar]
- 7.Makley TA., Jr Retinoblastoma in a 52-year-old man. Arch Ophthalmol. 1963;69(3):325–327. doi: 10.1001/archopht.1963.00960040331013. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi T, Tamura S, Inoue M, Isayama Y, Sashikata T. Retinoblastoma in a 26-year-old adult. Ophthalmology. 1983;90(2):179–183. doi: 10.1016/s0161-6420(83)34582-6. [DOI] [PubMed] [Google Scholar]
- 9.Nork TM, Millecchia LL, de Venecia GB, Myers FL, Vogel KA. Immunocytochemical features of retinoblastoma in an adult. Arch Ophthalmol. 1996;114(11):1402–1406. doi: 10.1001/archopht.1996.01100140602013. [DOI] [PubMed] [Google Scholar]
- 10.Mietz H, Hutton WL, Font RL. Unilateral retinoblastoma in an adult: report of a case and review of the literature. Ophthalmology. 1997;104(1):43–47. doi: 10.1016/s0161-6420(97)30363-7. [DOI] [PubMed] [Google Scholar]
- 11.Berkeley JS, Kalita BC. Retinoblastoma in an adult. Lancet. 1977;2(8036):508–509. doi: 10.1016/s0140-6736(77)91635-x. [DOI] [PubMed] [Google Scholar]
- 12.Houston SK, Murray TG, Wolfe SQ, Fernandes CE. Current update on retinoblastoma. Int Ophthalmol Clin. 2011;51(1):77–91. doi: 10.1097/IIO.0b013e3182010f29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park JJ, Gole GA, Finnigan S, Vandeleur K. Late presentation of a unilateral sporadic retinoblastoma in a 16-year-old girl. Aust N Z J Ophthalmol. 1999;27(5):365–368. doi: 10.1046/j.1440-1606.1999.00217.x. [DOI] [PubMed] [Google Scholar]
- 14.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68(4):820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, Albert DM, Dryja TP. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323(6089):643–646. doi: 10.1038/323643a0. [DOI] [PubMed] [Google Scholar]
- 16.Zhao J, Li S, Shi J, Wang N. Clinical presentation and group classification of newly diagnosed intraocular retinoblastoma in China. Br J Ophthalmol. 2011;95(10):1372–1375. doi: 10.1136/bjo.2010.191130. [DOI] [PubMed] [Google Scholar]
- 17.Shields CL, Shields JA, Shah P. Retinoblastoma in older children. Ophthalmology. 1991;98(3):395–399. doi: 10.1016/s0161-6420(91)32283-8. [DOI] [PubMed] [Google Scholar]
- 18.Junker B, Hansen LL. Coats disease. Ophthalmologe. 2010;107(4):379–388; quiz 389-390. doi: 10.1007/s00347-010-2151-6. [DOI] [PubMed] [Google Scholar]
- 19.Shields JA, Shields CL, Parsons HM. Differential diagnosis of retinoblastoma. Retina. 1991;11(2):232–243. [PubMed] [Google Scholar]
- 20.Shields JA, Shields CL. Differentiation of coats' disease and retinoblastoma. J Pediatr Ophthalmol Strabismus. 2001;38(5):262–266; quiz 302-263. doi: 10.3928/0191-3913-20010901-05. [DOI] [PubMed] [Google Scholar]
- 21.Decaussin M, Boran MD, Salle M, Grange JD, Patricot LM, Thivolet-Bejui F. Cytological aspiration of intraocular retinoblastoma in an 11-year-old boy. Diagn Cytopathol. 1998;19(3):190–193. doi: 10.1002/(sici)1097-0339(199809)19:3<190::aid-dc7>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 22.Orellana ME, Brimo F, Auger M, Galic J, Deschenes J, Burnier MN. Cytopathological diagnosis of adult retinoblastoma in a vitrectomy specimen. Diagn Cytopathol. 2010;38(1):59–64. doi: 10.1002/dc.21135. [DOI] [PubMed] [Google Scholar]
- 23.Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer. 2002;2(12):910–917. doi: 10.1038/nrc950. [DOI] [PubMed] [Google Scholar]
- 24.Karcioglu ZA, Gordon RA, Karcioglu GL. Tumor seeding in ocular fine needle aspiration biopsy. Ophthalmology. 1985;92(12):1763–1767. doi: 10.1016/s0161-6420(85)34105-2. [DOI] [PubMed] [Google Scholar]
- 25.O'Hara BJ, Ehya H, Shields JA, Augsburger JJ, Shields CL, Eagle RC., Jr Fine needle aspiration biopsy in pediatric ophthalmic tumors and pseudotumors. Acta Cytol. 1993;37(2):125–130. [PubMed] [Google Scholar]
- 26.Parulekar MV. Retinoblastoma - current treatment and future direction. Early Hum Dev. 2010;86(10):619–625. doi: 10.1016/j.earlhumdev.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 27.Shields CL, Shields JA. Diagnosis and management of retinoblastoma. Cancer Control. 2004;11(5):317–327. doi: 10.1177/107327480401100506. [DOI] [PubMed] [Google Scholar]
- 28.Chintagumpala M, Chevez-Barrios P, Paysse EA, Plon SE, Hurwitz R. Retinoblastoma: review of current management. Oncologist. 2007;12(10):1237–1246. doi: 10.1634/theoncologist.12-10-1237. [DOI] [PubMed] [Google Scholar]