

Background: SCCRO3/DCUN1D3 is proposed to function as a tumor suppressor. However, the mechanisms are not well defined.
Results: SCCRO3 inhibits SCCRO-promoted nuclear translocation and neddylation of cullins by sequestering them to the cell membrane.
Conclusion: SCCRO3 putatively functions as a tumor suppressor by antagonizing the activity of SCCRO.
Significance: Understanding how SCCRO and its family members cooperatively regulate cullin neddylation is important for neddylation-targeted cancer therapy.
Keywords: Head and Neck Cancer, Lung Cancer, Oncogene, Tumor Suppressor Gene, Ubiquitin
Abstract
The activity of cullin-RING type ubiquitination E3 ligases is regulated by neddylation, a process analogous to ubiquitination that culminates in covalent attachment of the ubiquitin-like protein Nedd8 to cullins. As a component of the E3 for neddylation, SCCRO/DCUN1D1 plays a key regulatory role in neddylation and, consequently, cullin-RING ligase activity. The essential contribution of SCCRO to neddylation is to promote nuclear translocation of the cullin-ROC1 complex. The presence of a myristoyl sequence in SCCRO3, one of four SCCRO paralogues present in humans that localizes to the membrane, raises questions about its function in neddylation. We found that although SCCRO3 binds to CAND1, cullins, and ROC1, it does not efficiently bind to Ubc12, promote cullin neddylation, or conform to the reaction processivity paradigms, suggesting that SCCRO3 does not have E3 activity. Expression of SCCRO3 inhibits SCCRO-promoted neddylation by sequestering cullins to the membrane, thereby blocking its nuclear translocation. Moreover, SCCRO3 inhibits SCCRO transforming activity. The inhibitory effects of SCCRO3 on SCCRO-promoted neddylation and transformation require both an intact myristoyl sequence and PONY domain, confirming that membrane localization and binding to cullins are required for in vivo functions. Taken together, our findings suggest that SCCRO3 functions as a tumor suppressor by antagonizing the neddylation activity of SCCRO.
Introduction
Ubiquitination regulates the activity of proteins involved in diverse and essential cellular processes, including transcription, differentiation, signal transduction, cell cycle progression, and apoptosis (1). Although ubiquitination serves as the primary signal targeting protein for degradation at the proteasome, it can modify protein function in many other ways (2). Ubiquitination results from the sequential activity of activating (E1), conjugating (E2), and ligating (E3) enzymes (3, 4), with substrate-derived signals (e.g. misfolding, mutation, and post-translational modifications (i.e. phosphorylation)) serving as initiators of the cascade (5). E3s provide specificity and serve as the rate-limiting step in ubiquitination. Thus, factors regulating assembly of multiprotein complexes to constitute functional E3 ligases are the primary regulators of ubiquitination activity. For cullin-RING ligases (CRLs),3 the largest class of mammalian ubiquitination E3s, neddylation of cullin serves as the key signal for assembly of the E3 complex (6, 7). Neddylation is a process analogous to ubiquitination, in which a tripartite cascade resulting in covalent modification of the cullin family of proteins by the ubiquitin-like protein Nedd8 is regulated by activity of the neddylation E3 (8–10). We and others identified SCCRO/DCUN1D1 and showed that it functions as a regulatory component in the neddylation E3 (11–15). SCCRO promotes neddylation in three ways: 1) it binds to cullin-ROC1 complexes in the cytoplasm and promotes their nuclear translocation, 2) it enhances recruitment of E2∼Nedd8 (Ubc12∼Nedd8) thioester to the complex, and 3) it optimizes the orientation of proteins in the complex to allow efficient transfer of Nedd8 from the E2 to the cullin substrates.
SCCRO is not required for neddylation in in vitro assays (14), but studies in yeast and Caenorhabditis elegans suggest that SCCRO activity is essential for neddylation, because targeted inactivation of SCCRO results in lethality (12). In contrast, SCCRO knock-out mice are viable, likely because of compensation by the SCCRO paralogues, which are exclusively present in higher organisms (Ref. 14).4 Bioinformatics analysis shows that SCCRO has four paralogues in mammals that can be classified into three subgroups on the basis of their phylogeny and N-terminal sequences: SCCRO and SCCRO2 (DCUN1D2) contain an ubiquitin-associated domain, SCCRO3 (DCUN1D3) contains a myristoyl sequence, and SCCRO4 (DCUN1D4) and SCCRO5 (DCUN1D5) contain a nuclear localization signal in the N terminus. Although it has been suggested that each of the SCCRO family members promotes neddylation, the precise in vivo contributions remain to be defined (11–18). Interestingly, N-terminal motifs either directly or indirectly regulate subcellular localization of all SCCRO paralogues. The ubiquitin-associated domain of SCCRO and SCCRO2 regulates its nuclear localization (19). We recently showed that the function of SCCRO5 as an oncogene requires its nuclear localization signal (16). Given the importance of nuclear localization in the neddylation function of SCCRO family members, the presence of a myristoyl sequence in SCCRO3 that localizes it to the membrane raises questions about its in vivo activities (17). Moreover, in contrast to the other SCCRO family members, SCCRO3 is reported to function as a tumor suppressor (20). In this study, we sought to determine the mechanisms underlying SCCRO3 contributions to neddylation and human cancer pathogenesis. We show that by sequestering cullins to the membrane to prevent their nuclear translocation, SCCRO3 inhibits SCCRO-promoted neddylation and, consequently, CRL-promoted ubiquitination. Its inhibitory effects on SCCRO activity endow SCCRO3 with putative tumor suppressor function. The high prevalence of reduced SCCRO3 expression in multiple tumor types suggests that it plays a significant role in human cancer pathogenesis.
EXPERIMENTAL PROCEDURES
Tumor Tissue, Cell Lines, Antibodies, and Plasmids
Primary tumor specimens and adjacent normal tissue from head and neck, lung, oral, ovarian, and thyroid neoplasms were collected from patients undergoing surgical resection after informed consent was obtained and in accordance with institutional guidelines. Cell lines H1299, NIH-3T3, U2OS, and HeLa were obtained from American Type Culture Collection (Manassas, VA). 16HBE was a gift from Dr. Alan Hall (Memorial Sloan-Kettering Cancer Center). The following antibodies were used: anti-DCUN1D3 (Abnova, Taipei City, Taiwan); anti-Cul1 and Alexa Fluor 488 Phalloidin (Invitrogen); anti-Cul3 (BD Biosciences, San Jose, CA); anti-ROC1 (Spring Bioscience, Pleasanton, CA); anti-Ubc12 (Rockland, Gilbertsville, MA); anti-CAND1 and anti-GST (Upstate, Lake Placid, NY); anti-tubulin (Sigma-Aldrich); anti-HA (Covance, Princeton, NJ); anti-Aurora B (BD Biosciences); anti-actin and anti-RhoA (Santa Cruz Biotechnology, Santa Cruz, CA); Cy3-conjugated anti-Myc and FITC-conjugated anti-HA (Jackson ImmunoResearch Laboratories, West Grove, PA); anti-GAPDH (Millipore, Billerica, MA); and secondary antibodies conjugated to horseradish peroxidase (Santa Cruz Biotechnology, Santa Cruz, CA), with dilutions according to the manufacturers' specifications. Anti-SCCRO (rabbit polyclonal) antibody was produced and used as described previously (21).
All DNA constructs were generated using a standard PCR-ligation technique and verified by automated sequencing. Proteins were expressed as GST fusions in Escherichia coli strains BL21 (DE3) (Novagen, Madison, WI), were induced overnight at 18 °C with the addition of 1 mm isopropyl β-d-thiogalactopyranoside, and were purified by passage through glutathione-Sepharose 4B beads (GE Healthcare), followed by thrombin cleavage as required. APPBP1/Uba3, Ubc12, and Nedd8 were obtained from a commercial source (Boston Biochem, Cambridge, MA). The human SCCRO3 plasmid was obtained from a commercial source (Clontech). The human PUM2 construct was a gift from Dr. Judith Kimble (Howard Hughes Medical Institute investigator, University of Wisconsin, Madison). SCCRO and SCCRO3 shRNA lentiviral constructs were obtained from Sigma-Aldrich through the high throughput screening core facility at Memorial Sloan Kettering Cancer Center (see Table 1 for sequences). SCCRO3 cDNAs were cloned into pGEX-4T-3 (GE Healthcare), pBABE (Addgene, Cambridge, MA), or pCMV-HA (BD Biosciences, San Jose, CA) vectors per the manufacturers' protocols. Transfections were performed with Lipofectamine 2000 (Invitrogen), in accordance with the manufacturer's protocols. Stable overexpression and shRNA knockdown clones were selected in the presence of puromycin. An NIH-3T3 cell line stably expressing SCCRO was derived as described previously (21).
TABLE 1.
Primer sequences for SCCRO3 methylation analysis and SCCRO3 mutations and sequences for SCCRO and SCCRO3 knockdown
| Sequence type | Primer |
|---|---|
| DNA methylation primer sequences | |
| Methylated(s) | 5′-TGGCGATGATATCGAGTCGTTTC-3′ |
| Methylated(as) | 5′-GACTCCAAACGCGAAAAACTAACG-3′ |
| Unmethylated(as) | 5′-AAAACACCAACTCCAAACACAAAAAAC-3′ |
| Unmethylated(s) | 5′-GTTGGTGGTGATGATATTGAGTTGTTTT-3′ |
| cDNA cloning primer sequences | |
| DCUN1D3 G2A | 5′-ATGGCCCAGTGTGTCACCAAGTGTAAG-3′ |
| DCUN1D3 D241N | 5′-CATGTTCCAAGTGTTCCGGGAGATGCCCTT-3′ |
| DCUN1D3 A265R | 5′-AAAGAGACTTGGCCACCGCTCATCTTCACT-3′ |
| DCUN1D3 D271N | 5′-CCACAAAGGTGTTAAAGAGACTTGGCC-3′ |
| DCUN1D3 deletion 1–26 | 5′-CGGAATTCCGCCGCCACCATGAAGTCACAT-3′ |
| shRNA primer sequences | |
| SCCRO3 shRNA1 | 5′-CCGGCACTTGGAACATGTTCCTTAACTCGAGTTAAGGAACATGTTCCAAGTGTTTTTTG-3′ |
| SCCRO3 shRNA2 | 5′-CCGGCCTTAACTTCACTCAGGTGATCTCGAGATCACCTGAGTGAAGTTAAGGTTTTTTG-3′ |
| SCCRO shRNA1 | 5′-CCGGGCTTCTAGTCTCTTTACTGTTCTCGAGAACAGTAAAGAGACTAGAAGCTTTTTTG-3′ |
| SCCRO shRNA2 | 5′-CCGGGCCTGTTCTTATTGATGACTTCTCGAGAAGTCATCAATAAGAACAGGCTTTTTTG-3′ |
Real Time Reverse Transcriptase-PCR
Total RNA was extracted with TRIzol reagent (Invitrogen) and repurified using the RNeasy Mini spin column (Qiagen). Gene-specific primers were designed using the Primer3 program and were purchased from Operon Technologies (Alameda, CA) (for primer sequences, see Table 2). The relative quantitative analysis of SCCRO3 mRNA expression, normalized to GAPDH, was performed using the 7900HT Sequence Detector System (Applied Biosystems, Foster City, CA). PCR cycling conditions for all samples were as follows: denaturation (94 °C for 5 min) and amplification repeated for 40 cycles (94 °C for 15 s, Tm for 20 s, and 72 °C for 20 s). Real time PCR assays were conducted in duplicate for each sample, and each PCR experiment included two nontemplate control wells. A standard curve for serial dilutions of cDNA of head and neck cancer cell lines MDA686 and MDA1186 was similarly generated. The comparative threshold cycle method was used to calculate the SCCRO3/DCUN1D3-like gene expression ratio in each sample relative to the value observed in the control standard curve, using GAPDH as a control for normalization among samples. Melt curve analysis was performed after amplification. The acquisition temperature was set at 1–2 °C below the Tm of the specific PCR product. The relative quantification of a target gene, compared with that of a reference gene (GAPDH rRNA), was performed as described previously (21).
TABLE 2.
Real time RT-PCR primer sequences
| Gene | Primer |
|
|---|---|---|
| Forward | Reverse | |
| SCCRO1/DCUN1D1 | 5′-CTGGAGGACACCAACATG-3′ | 3′-TTCACTAGATTGTGTGAAGATC-5′ |
| SCCRO2/DCUN1D2 | 5′-GTTCACCTCCATTTCTCAATGTG-3′ | 3′-CTTAGAAATGGCTGTTGCGT-5′ |
| SCCRO3/DCUN1D3 | 5′-CACAGAATTTCGAGTGCTG-3′ | 3′-TGCACTTATTGCTTTGCAG-5′ |
| SCCRO4/DCUN1D4 | 5′-CTGGCAAATATTCATAAGATCTACC-3′ | 3′-AAGACCGCAGACTTCCTG-5′ |
| SCCRO5/DCUN1D5 | 5′-TGCGCTCACAGTTGAATGATATTTCGTC-3′ | 3′-CAGTGGCCATGTCCTCCCAAGCAG-5′ |
| GAPDH | 5′-GCACCACCAACTGCTTAG-3′ | 3′-CATGGACTGTGGTCATGAG-5′ |
DNA Sequencing, Mutational Analysis, and DNA Methylation Analysis
Putative exonic regions of the SCCRO3 gene (NCBI Human Genome Build 36.1) were broken into 61 amplicons of 500 bp or less, and specific primers were designed using Primer3. Sequence reactions were run on an ABI PRISM 3730xl sequencing apparatus (Applied Biosystems). PCR was performed in 384-well plates, in a Duncan DT-24 water bath thermal cycler, with 10 ng of whole genome-amplified DNA (Repli-G Midi; Qiagen) as template, using a touchdown PCR protocol with HotStart Taq (Kapa Biosystems, Cape Town, South Africa). Templates were purified using AMPure (Agencourt Biosciences, Beverly, MA). The purified PCRs were split in two and sequenced bidirectionally with M13 forward and reverse primer and Big Dye Terminator kit (version 3.1; Applied Biosystems) at Agencourt Biosciences. Dye terminators were removed using the CleanSEQ kit (Agencourt Biosciences). Mutations were detected using an automated detection pipeline at the Memorial Sloan-Kettering Cancer Center Bioinformatics Core. Bidirectional reads and mapping tables (to link read names with sample identifiers, gene names, read direction, and amplicon) were subjected to a quality control filter, which excludes reads that have an average Phred score of <10 for bases 100–200. Passing reads were assembled against the SCCRO3 reference sequence, containing all coding and UTR exons, including 5 kb upstream and downstream of the gene, using command line Consed 16.0 (22). Assemblies were passed on to Polyphred 6.02b (23) and Polyscan 3.0 (24), generating lists of putative mutations. Putative mutation calls were normalized to “+” genomic coordinates and annotated using the Genomic Mutation Consequence Calculator (25). The resulting list was loaded into a Postgres database, along with select assembly details for each mutation call. To reduce the number of false positives generated by the mutation detection software packages, only point mutations supported by at least one bidirectional read pair and at least one sample mutation called by Polyphred were considered, and only the putative mutations annotated as having nonsynonymous coding effects and that occurred within 11 bp of an exon boundary or had a conservation score >0.699 were included in the final candidate list. Indels called by any method were manually reviewed and included in the candidate list if found to hit an exon. All putative mutations were confirmed by a second PCR and sequencing reaction, in parallel with amplification and sequencing of matched normal-tissue DNA.
For DNA methylation analysis, genomic DNA was extracted from primary lung tumors by use of All Prep DNA/RNA (Qiagen). Bisulfite conversion of genomic DNA was performed using the EZ DNA methylation kit (Zymo Research, Irvine, CA). One microgram of genomic DNA was used. Methylation-specific PCR was performed using primers designed for methylated and unmethylated promoter sequences using the program MSPPrimer. Methylation-specific PCR was performed as previously described (26). All PCR products were analyzed by electrophoresis on a 2.5% agarose gel. Primers are listed in Table 1.
GST Pulldown Assay
GST-tagged proteins were bound to glutathione-Sepharose beads (GE Healthcare) by gentle rocking at 4 °C for 30 min. The beads were washed four times with EBC buffer (50 mm Tris-HCl, pH 7.5, 2.5 mm MgCl2, 150 mm NaCl, and 0.5% Nonidet P-40) at 20× bead volume. The beads were incubated with 500 μg of HeLa cell lysate or purified proteins, as indicated, at 4 °C for 1 h, followed by three washes with EBC buffer at 20× bead volume. Bound proteins were eluted by the addition of 6× Laemmli buffer or nonreducing Laemmli buffer (when thioester bonds were involved), were resolved on SDS-PAGE gels, and were analyzed by Western blot.
Thioester Reactions
Reactions were performed at room temperature in buffer (50 mm Tris-HCl, pH 7.6, 50 mm NaCl, 10 mm MgCl2, and 0.5 mm DTT) with 4 mm ATP, 80 nm APPBP1/Uba3, 800 nm Ubc12, and 9 μm Nedd8. After 10 min, which was previously determined to be the optimal reaction time (15), reactions were quenched by the addition of EDTA (to a final concentration of 50 mm) and were purified on a G-50 micro spin desalting column (GE Healthcare). Next, 13.1 μl of reaction product containing 10 pmol of Ubc12 was added to 1 nmol of GST-SCCRO binding assay with a gradient of purified protein as indicated. The wash and detection sequences were as described above. The presence of Ubc12∼Nedd8 thioester complexes was verified by Western blot analysis for Ubc12. Ponceau S stain was performed as a loading control.
In Vivo and in Vitro Neddylation Assay
For in vivo neddylation, cell lysates were directly subjected to immunoblotting for cullin(s). In vitro neddylation assays were performed, as described previously (15), using HeLa lysate (as a source of cullin-ROC1) and purified SCCRO/SCCRO3 proteins or lysate from U2OS cells transfected with HA-SCCRO3 and Myc-Cul1. The reaction mixture also contained recombinant Nedd8 (2 μm), E1 (5 nm), E2 (5 μm), and ATP (4 mm).
Cell Morphologic Analysis
Cells were seeded in chamber slides (Lab-Tek; Thermo-Fisher Scientific, Rochester, NY), incubated in appropriate medium for 24 h at 37 °C with 5% CO2, washed with PBS, and photographed using phase contrast microscopy at 4- and 10× magnification (Olympus IX71).
Immunofluorescence Analysis
H1299 cells, 16HBE cells, and U2OS cells transfected with the indicated plasmids were seeded in chamber slides or 6-well plates with cover glass and left overnight at 37 °C in 5% CO2. Twenty-four hours after seeding or after transfection, cells were washed (PBS) and fixed in 4% formaldehyde for 10 min or were fixed with methanol acetone for 20 min at −20 °C. Fixed cells were permeabilized with 0.5% Triton X-100 (Spectrum Chemicals, Gardena, CA) for 5 min, were stained with Alexa Fluor 488 Phalloidin for 30 min at room temperature or incubated in blocking buffer (1% bovine serum albumin in PBS-T) for 30 min, were washed (PBS), and were stained overnight in a humid chamber at 4 °C with fluorochrome-conjugated antibodies. The cells were washed three times (PBS), were counterstained with DAPI (Vector Laboratories, Burlingame, CA), were washed an additional three times (PBS), were covered by ProLong Gold Antifade Reagent (Invitrogen), and were examined with Leica and Olympus IX71 inverted confocal microscopes fitted with appropriate fluorescence filters at 20× or 63× magnification. For the subcellular localization assay, percentages of nuclear and nonnuclear Cul1 were calculated on the basis of 200 transfected cells for each condition.
Cell Motility and Transformation Assays
For scratch assays, cells were plated and grown until they formed a confluent monolayer. Cells were then starved of serum for 24 h and treated with mitomycin C for 90 min to arrest cell proliferation, using a previously published protocol (27). The monolayer was scratched in a straight line using a 20-μl pipette tip. We checked these cells at intervals to ensure that cells were migrating across the scratch rather than broad front advancement resulting from cell proliferation. The cells were washed with PBS and then photographed using a phase contrast microscope (Olympus IX71) at 4× magnification. The Petri dishes were placed at 37 °C in humidified air with 5% CO2 for 24 or 36 h, depending on the cell line, and were then photographed in the same location. For soft agar assays, vector, SCCRO3, or mutant DNA was stably transfected into H1299 cells, as well as into NIH-3T3 cells stably transfected with SCCRO, and the cells were plated in 0.35% agarose-coated 6-well plates, at densities of ∼1000 and ∼5000 cells in agarose. After incubation for 2–6 weeks, the cells were stained with 0.005% crystal violet (Sigma-Aldrich) and photographed using a dissecting microscope (Leica Stereoscope MZ FL-III). Colony counts were obtained using Image J software (National Institutes of Health, Bethesda, MD).
Statistical Analysis
Descriptive statistics were used to summarize study data. Qualitative (Fisher's exact test) and quantitative (Mann-Whitney U test) comparisons were performed using nonparametric assays. Statistical significance was defined as a two-tailed p ≤ 0.05. All statistical analyses were performed using a commercially available statistical software package (SPSS, Chicago, IL, or SAS, Durham, NC).
RESULTS
SCCRO3 Expression Is Decreased in Human Tumors
We and others have shown that amplification and overexpression of SCCRO are common in many different types of human cancer and that they underlie its function as an oncogene (21, 28–30). To determine whether SCCRO3 is also involved in cancer pathogenesis, we assessed its expression in lung, oral, ovarian, and thyroid neoplasms and matched normal tissue by real time PCR (Fig. 1A). Expression of SCCRO mRNA was increased in many different tumor types studied, which is consistent with its known oncogenic activity (Fig. 1B). In contrast, SCCRO3 mRNA levels were decreased in a wide range of cancers, including in 58% of lung squamous cell carcinomas (n = 40), 38% of lung adenocarcinomas (n = 40), 85% of lung neuroendocrine tumors (n = 54), 21% of oral squamous cell carcinomas (n = 39), 20% of ovarian carcinomas (n = 40), and 27% of thyroid tumors (n = 56). We further subclassified tumors to assess those with an increase of SCCRO only, a decrease of SCCRO3 only, or both (Fig. 1B). Interestingly, there was a trend toward mutual exclusivity, with 127 cases having either an increase of SCCRO or a decrease of SCCRO3 expression and only 31 tumors having both. Co-dysregulation of SCCRO and SCCRO3 was most common among lung squamous cell carcinomas and neuroendocrine carcinomas. It remains to be determined whether co-dysregulation of SCCRO paralogues increases oncogenicity. Given that SCCRO and SCCRO5 function as oncogenes, the decreased expression of SCCRO3 in human cancers is somewhat unexpected, but it is supported by previously published analyses of liver, bladder, and renal tumors (16, 20, 31).
FIGURE 1.

SCCRO3 expression in human tumors has an inverse relationship with PUM2 levels. A, box plot showing fold decrease in SCCRO3 mRNA levels as measured by real time RT-PCR in lung adenocarcinoma (Adeno), squamous cell carcinoma (SCC), neuroendocrine carcinomas (NE), oral squamous cell carcinoma (OSCC), ovarian carcinoma (CA), and thyroid tumor samples, compared with those in matched normal tissue. B, results from quantitative real time PCR analysis showing percentage of cases with SCCRO overexpression and decreased SCCRO3 expression in the indicated tumor types. Columns 4–6 represent the numbers of cases with an increase of SCCRO only, with a decrease of SCCRO3 only, and with both an increase of SCCRO and a decrease of SCCRO3. C, bisulfate-treated DNA from three representative neuroendocrine lung tumor samples (and matched normal samples) with decreased SCCRO3 mRNA expression was PCR-amplified using SCCRO3 promoter-specific primer pairs for methylated (M) and unmethylated (U) DNA. PCR product was obtained (lanes 3, 7, and 11) only in template DNA from normal tissue, using primer pair (M). D, Western blot analysis of lysates from lung adenocarcinoma or neuroendocrine carcinomas (T) and matched histologically normal tissue (N) showing inverse correlation between PUM2 protein levels and SCCRO3 mRNA expression in tumors by real time PCR analysis (shown below the blot). E, Western blot analysis of lysates from untransfected (WT), empty vector (EV), or HA-PUM2 transfected 16HBE cells probed with anti-HA (top panel), anti-SCCRO3 (second panel), anti-SCCRO (third panel), and anti-α-tubulin (loading control; bottom panel) antibodies, showing a decrease in SCCRO3 but no change in SCCRO levels in cells expressing HA-PUM2 (lane 3). F, Western blot analysis of lysates from H1299 cells showing a decrease in PUM2 and an increase in SCCRO3 levels in cells infected with virus expressing shRNA against PUM2 (shRNA1 and shRNA2) compared with uninfected cells or those infected with a virus expressing scrambled shRNA. G, real time RT-PCR analysis for SCCRO3 expression on RNA extracted from experiments in E (left two panels) and F (right panel). Actinomycin D (AMD) was used to inhibit transcription after transfection.
PUM2 Regulates SCCRO3 Levels in Human Tumors
To determine the cause of the decreased expression of SCCRO3 in human cancers, we assessed published genomic screening data. This did not identify any tumor types with recurrent chromosomal losses involving the SCCRO3 locus at 16p12.3 (32–34). The absence of chromosomal losses at 16p12.3 was validated by our analysis of results from array comparative genomic hybridization and/or single-nucleotide polymorphism investigations performed on thyroid, head and neck, lung, and neuroendocrine tumors at our institution (data not shown). These findings suggest that genomic loss is not a cause of the decreased expression of SCCRO3 in human cancers. To determine whether expression of SCCRO3 is altered by mutations, we performed exon sequencing on DNA extracted from 216 thyroid, oral, and lung tumors with decreased expression of SCCRO3 mRNA, as well as on matched normal tissue. Although several single-nucleotide polymorphisms were detected (NCBI numbers rs1858901, rs34248677, rs35094690, and rs7187522), no mutations were identified in the coding region of SCCRO3 in these tumors (data not shown). Analysis of large scale cancer genomics data sets using the cBioPortal revealed the presence of several mutations in SCCRO3. The prevalence of mutation (highest prevalence in melanoma; 2.5% in Broad data set and 2% in TCGA data set) was significantly lower than the prevalence of a decrease of expression of SCCRO3, suggesting that additional mechanisms may be responsible for changes in expression.
Analysis of the SCCRO3 promoter sequence identified multiple CpG islands, prompting us to investigate DNA methylation as a mechanism of SCCRO3 silencing (35). Methylation was assessed by PCR using the bisulfate method, with matched normal tissue serving as controls. Hypermethylation of the SCCRO3 promoter region was not detected in any of the samples tested (Fig. 1C). These findings suggest that the decreased expression of SCCRO3 is likely not caused by changes in transcription.
We next investigated the presence of post-transcriptional factors that could affect SCCRO3 levels in human cancer. Proteins that decrease mRNA stability have emerged as key regulators of gene expression in normal and cancerous cells (36). Recent studies identified PUM2 as a SCCRO3 mRNA-binding protein, raising the possibility that it affects translation (37). PUM2 is a member of the PUF family of proteins, which bind to sequence elements in the 3′ UTR of target mRNAs to affect stability and/or translation of the message. Immunoblotting analysis showed that PUM2 protein levels inversely correlated with levels of SCCRO3 mRNA in tumor and matched normal tissue and cancer cell lines, suggesting that an association between PUM2 levels and decreased SCCRO3 expression exists in human cancers (Fig. 1D). To determine whether PUM2 directly affects SCCRO3 levels, we expressed PUM2 in 16HBE cells, an immortalized benign human bronchial epithelial cell line that has low levels of PUM2 and high levels of endogenous SCCRO3. Western blotting analysis of lysates from 16HBE cells transiently transfected with HA-tagged PUM2 showed a decrease in levels of SCCRO3, but not SCCRO (Fig. 1E, lane 3). Moreover, knockdown of PUM2 using shRNA in H1299 cells (a human non-small cell lung carcinoma cell line that has high levels of PUM2 and low levels of SCCRO3) resulted in an increase in levels of SCCRO3, but not SCCRO (Fig. 1F, lanes 3 and 4). To begin to elucidate how PUM2 controls SCCRO3 expression, we assessed SCCRO3 mRNA levels, using real time PCR, in 16HBE cells overexpressing PUM2 and H1299 cells after siRNA knockdown of PUM2. We found that overexpression of PUM2 reduced the levels of SCCRO3 mRNA, whereas knockdown of PUM2 increased its levels relative to controls (Fig. 1G). These findings suggest that PUM2 regulates SCCRO3 at the mRNA level. Next, to determine whether PUM2 regulates SCCRO3 mRNA at the level of transcription or whether it affects its stability, we treated PUM2-transfected 16HBE cells with actinomycin D to stop transcription. Inhibition of transcription in 16HBE cells resulted in a significantly greater decrease in SCCRO3 mRNA levels in PUM2, compared with vector-transfected cells, suggesting that PUM2 affects SCCRO3 mRNA stability (Fig. 1G). Taken together, these findings suggest that SCCRO3 mRNA is a target of PUM2-mediated decay, which may explain the decreased SCCRO3 expression in at least some human tumors.
SCCRO3 Interacts with CAND1, Cullins, and ROC1 via Its PONY Domain
To begin to define the role of SCCRO3 in cancer pathogenesis, we first assessed its biochemical activities. Previous studies have shown that SCCRO interacts with proteins involved in neddylation, including CAND1, cullins, ROC1, and Ubc12 via its PONY domain (11, 12, 15, 38). Given the presence of a highly conserved PONY domain in SCCRO3, we assessed its binding to components of the neddylation E3. Immunoblots of products from GST-SCCRO3 pulldown assays of HeLa lysates showed that SCCRO3 interacts with CAND1, Cul1, Cul3, and ROC1 (Fig. 2A, lane 3). To define the regions involved in binding, we created mutations in residues in the PONY domain of SCCRO3 (corresponding to the DAD patch) that are required for binding of SCCRO to neddylation components (SCCRO3D241N, SCCRO3A265R, SCCRO3D271N, SCCRO3A265R/D271N, and SCCRO3D241N/A265R/D271N [SCCRO3DAD]), as well as in the N-terminal myristoyl sequence (SCCRO3Δ1–26 and SCCRO3G2A) (11). GST pulldown assays of HeLa lysates showed that the SCCRO3-DAD patch mutants (Fig. 2A, lanes 4–8), but not the N-terminal mutants (Fig. 2A, lanes 9 and 10), lost binding to CAND1, cullins, and ROC1. These findings confirm that, as with SCCRO, the interaction of SCCRO3 with neddylation components requires its PONY domain.
FIGURE 2.

SCCRO3 does not function as a component of the neddylation E3. A, Western blot analysis of the pulldown products of GST-SCCRO, GST-SCCRO3, or GST-SCCRO3 mutants from HeLa extracts probed with indicated antibodies, which showed GST-SCCRO (control; lane 1) binds to all the indicated proteins. SCCRO3 and its N-terminal mutants, but not its C-terminal mutants, bind to CAND1, Cul1, Cul3, and ROC1. Neither SCCRO3 nor its mutants showed binding to Ubc12. The levels of the various GST-tagged proteins used in the pulldown experiment were confirmed by probing a Western blot with anti-GST antibody (bottom panel). B, Western blot analysis probing for Ubc12 on a thioester reaction, which showed generation of Ubc12∼Nedd8 thioester (left panel, left lane, 30-kDa band) and a specific reduction of thioester bonds with the addition of DTT (left panel, right lane), and Western blot analysis of the products of GST, GST-SCCRO, and GST-SCCRO3 pulldown assays from the same Ubc12 thioester reaction mixture, which showed a preferred interaction of GST-SCCRO with Ubc12∼Nedd8 (right panel, lane 2) and no interaction of GST or GST-SCCRO3 with Ubc12∼Nedd8 or free Ubc12 (right panel, lane 3). Coomassie Blue staining showing levels of GST and GST-tagged proteins (bottom panel). C, Western blot analysis of a thioester reaction, showing a generation of UBC12∼Nedd8 thioester (left panel, left lane, 30-kDa band) and its reduction with the addition of DTT (left panel, right lane), and Western blot analysis of the products of GST-SCCRO pulldown assays (1 nmol GST-SCCRO per assay) from the same Ubc12 thioester reaction (right panel) supplemented with 10 nmol of untagged SCCRO or SCCRO3 protein as indicated, which showed addition of 10× free SCCRO but SCCRO3 blocked binding between of UBC12 to GST-SCCRO. Ponceau S staining of the blot showed equal loading. D, Western blot analysis of the products of an in vitro neddylation reaction with HeLa lysate (as a source of cullin-ROC1) supplemented with a concentration gradient of either SCCRO (upper panel) or SCCRO3 (lower panel) purified proteins, which showed a dose-dependent increase in Cul3 neddylation with addition of SCCRO but not SCCRO3.
SCCRO3 Does Not Bind to Ubc12
Unlike SCCRO, SCCRO3 had no interactions with Ubc12 detected in GST pulldown assays of HeLa lysates (Fig. 2A). These findings contrast results from Meyer-Schaller et al. (17), who found that GST-Ubc12 binds to bacterially purified His-SCCRO3. We were also able to detect binding between GST-SCCRO3 and Ubc12 using purified proteins, but only when very high concentrations of protein were used for GST pulldown, raising the possibility that the observed interactions are nonspecific (data not shown). Selective interaction between E3s and E2s charged with ubiquitin or ubiquitin-like protein, rather than free E2s, is a key mechanism for maintenance of reaction processivity. To determine whether binding to Ubc12 is functionally relevant, we assessed whether SCCRO3 maintains the reaction processivity paradigms that are conserved in all E3s for ubiquitin and ubiquitin-like protein in their interactions with E2s. We previously showed that SCCRO binds to Ubc12∼Nedd8 thioester with a much higher affinity than to free Ubc12, which is consistent with its function as an E3 (11, 15, 39). GST pulldown assays of the products of thioester reactions containing roughly equal quantities of free Ubc12 and Ubc12∼Nedd8 thioester, followed by immunoblotting, showed that whereas GST-SCCRO preferentially bound to Ubc12∼Nedd8 thioester, GST-SCCRO3 did not bind to either Ubc12 or Ubc12∼Nedd8 under identical conditions (Fig. 2B). To confirm these differences in binding efficiency, we performed the GST-SCCRO pulldown in identical experiments, adding either bacterially derived, thrombin-cleaved SCCRO or SCCRO3 to the reaction mix before the pulldown. The addition of a 10-fold molar excess of SCCRO, but not SCCRO3, blocked pulldown of Ubc12∼Nedd8 by GST-SCCRO, confirming that SCCRO binds to Ubc12∼Nedd8 with much higher efficiency than SCCRO3 does (Fig. 2C). These observations suggest that SCCRO3 does not conform to the conserved processivity paradigms for E3s in the ubiquitin and ubiquitin-like protein pathway, and they raise questions about its function as an E3 in neddylation.
SCCRO3 Does Not Augment Cullin Neddylation in Vitro
Given the limited binding of SCCRO3 to Ubc12∼Nedd8, we questioned whether SCCRO3 can augment cullin neddylation. In vitro neddylation reactions were performed using HeLa cell lysate (as a source of cullin-ROC1 substrate) supplemented with recombinant Nedd8, APPBP1/Uba3 (E1), Ubc12 (E2), ATP, and varying amounts of either purified SCCRO or SCCRO3 (Fig. 2D). As expected, SCCRO augmented cullin neddylation in a dose-dependent manner. Under identical conditions, SCCRO3 was associated with a minimal increase of neddylated Cul3. The changes in the levels of neddylated Cul3 were independent of the dose of SCCRO3, suggesting that the observed changes may not reflect the physiological activity of SCCRO3. Combined, these findings suggest that SCCRO3 does not promote neddylation of cullins with the same efficiency as SCCRO.
SCCRO3 Competes with SCCRO for Cul1 Localization
Recent work from our laboratory suggests that subcellular localization plays an important role in regulating cullin neddylation (14). To determine whether SCCRO3 affects cullin localization, HA-tagged SCCRO, SCCRO3, or selected SCCRO3 mutants were co-transfected with Myc-tagged Cul1 into U2OS cells. Localization of proteins expressed from transgenes was assessed by immunofluorescence with FITC-conjugated anti-HA and Cy3-conjugated anti-Myc antibodies. We found that Cul1 and SCCRO3 were co-localized to the plasma membrane (Fig. 3, A, second row, and B) and SCCRO3DAD was localized to the membrane without Cul1, whereas neither SCCRO3G2A nor Cul1 localized to the membrane when co-transfected (Fig. 3A, third and fourth rows). These findings show that SCCRO3 promotes localization of Cul1 to the membrane and that this requires both an intact myristoyl sequence and a PONY domain.
FIGURE 3.

SCCRO3 competes with SCCRO for subcellular localization of cullins. A, representative images from fluorescence microscopy (scale bar represents 20 μm) showing U2OS cells co-transfected with Myc-Cul1, the indicated HA-tagged constructs and varying concentrations of untagged SCCRO (fifth and sixth rows) stained with FITC-conjugated anti-HA (first column) and Cy3-conjugated anti-Myc antibody (second column) or DAPI (third column). Merged images are shown (fourth column). Myc-Cul1 co-localized with HA-SCCRO (first row) in the nucleus and HA-SCCRO3 at the membrane (second row). HA-SCCRO3G2A loses membrane localization, and Myc-Cul1 is primarily nuclear in these cells (third row). HA-SCCRO3DAD retains membrane localization, but HA-Cul1 does not co-localize with it (fourth row). Co-transfection of increasing concentrations of SCCRO with HA-SCCRO3 results in progressive translocation of Myc-Cul1 from the membrane to the nucleus (fifth and sixth rows). B, graph showing subcellular distribution of HA-Cul1 from experiment above at varying concentrations of SCRRO and SCCRO3. C, representative fluorescence microscopic images of U2OS cells transfected with scrambled shRNA, or shRNA against SCCRO or SCCRO3 stained for Cul1 using antibody against Cul1 (first column) and DAPI (second column) and merged (third column). Knockdown of SCCRO resulted in localization of a fraction of Cul1 to the cell membrane (second row). Knockdown of SCCRO3 increased the proportion of Cul1 in the nucleus (third row). D, Western blot analysis of lysates from U2OS cells expressing shRNA against SCCRO (shRNA1 and shRNA2) showing reduced levels of SCCRO (third row), but not SCCRO3 (fourth row), and a decrease in the proportion of neddylated Cul1 and Cul3. E, Western blot analysis of lysates from U2OS cells expressing shRNA against SCCRO3 (shRNA1 and shRNA2) showing decreased SCCRO3, but not SCCRO expression, and an increase in the proportion of neddylated Cul1 and Cul3. The numbers below the blots are the ratios of neddylated to nonneddylated cullins.
Given our previous findings showing that SCCRO-promoted nuclear translocation of Cul1 is required for neddylation in vivo, we next questioned whether SCCRO3 competes with SCCRO for binding and localization of Cul1. To address this question, we assessed the localization of Myc-Cul1 while varying the ratio of SCCRO to SCCRO3 by shRNA knockdown or transgene expression in U2OS cells. As expected, we found that SCCRO promoted nuclear translocation, resulting in its co-localization with Cul1 in the nucleus (Fig. 3, A, top row, and B). Knockdown of SCCRO using shRNA cells led to localization of Cul1 at the cell membrane (Fig. 3, C and D). In contrast, knockdown of SCCRO3 led to an increase of Cul1 in the nucleus (Fig. 3, C and E). Note that shRNA knockdown of SCCRO3 did not result in removal of all Cul1 from the membrane, likely because of incomplete knockdown of SCCRO3. When SCCRO3 and SCCRO were co-expressed at equal levels in U2OS cells, Cul1 primarily localized to the membrane. Increasing the level of SCCRO expression relative to SCCRO3 expression in U2OS cells resulted in a dose-dependent increase in nuclear translocation of Cul1 (Fig. 3, A, last two rows, and B). These findings suggest that SCCRO3 antagonizes the SCCRO-promoted nuclear translocation of Cul1.
SCCRO3 Antagonizes SCCRO-promoted Cullin Neddylation
Given that SCCRO3 antagonizes SCCRO-promoted nuclear translocation, we questioned whether this activity also affects cullin neddylation in vivo. U2OS cells were transfected with SCCRO3, SCCRO3G2A, or SCCRO3DAD, alone or in combination with varying amounts of SCCRO, and the levels of neddylated Cul1 in vivo were assessed by immunoblotting. Transfection of vector alone, SCCRO3, SCCRO3G2A, or SCCRO3DAD did not enhance Cul1 neddylation beyond basal levels (Fig. 4A, lanes 1–4). Expression of SCCRO increased levels of neddylated Cul1. Co-expression of SCCRO3, but not SCCRO3G2A or SCCRO3DAD, reduced the level to which SCCRO enhanced Cul1 neddylation (Fig. 4, A, lanes 6 and 7, and B). The inhibitory effects of SCCRO3 on Cul1 neddylation could be rescued by increasing SCCRO expression in a dose-dependent manner (Fig. 4A, lanes 2, 5, and 6). These results suggest that SCCRO3 antagonizes SCCRO-mediated cullin neddylation in vivo.
FIGURE 4.
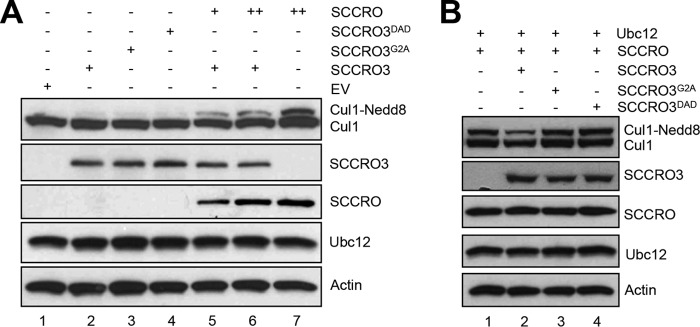
SCCRO3 antagonizes SCCRO-promoted cullin neddylation. A, Western blot analysis of the products of lysates of U2OS cells transfected with the SCCRO, SCCRO3, and/or SCCRO3 mutants as indicated. Expression of SCCRO3 or its mutants had no effect on levels of neddylated Cul1 (lanes 2–4). Expression of SCCRO increased the fractions of neddylated Cul1 (lane 7). Co-expression of SCCRO3 decreased the extent to which SCCRO increased levels of neddylated Cul1 (lane 6). The effect of SCCRO3 on neddylated Cul1 levels could be partially rescued by increasing SCCRO expression levels (lanes 2, 5, and 6). B, Western blot analysis on lysates from U2OS cells co-transfected with Ubc12 and the indicated SCCRO/SCCRO3 constructs. Co-expression of SCCRO3 (compare lane 1 with lane 2), and not SCCRO3 mutants, inhibits SCCRO-augmented Cul1 neddylation (compare lane 2 with lanes 3 and 4).
Putative Tumor Suppressor Function of SCCRO3 Requires Its PONY Domain and Myristoyl Sequence
Because SCCRO is known to function as an oncogene, we questioned whether the putative tumor suppressor activity of SCCRO3 involves its antagonism of SCCRO neddylation activity. To confirm that SCCRO3 functions as a tumor suppressor, we assessed effects of its knockdown in 16HBE cells. Knockdown of SCCRO3 by virally delivered shRNA resulted in a decrease in levels of SCCRO3 protein (but not SCCRO) relative to levels in 16HBE cells infected with virus expressing scrambled shRNA controls (Fig. 5A). Microscopic evaluation showed 16HBE cells in which SCCRO3 was stably knocked down changed from epitheloid to spindle-shaped. These cells also showed alteration in their pattern of growth, spreading widely across the plate rather than growing in islands, as seen in parental 16HBE cells or those infected with scrambled shRNA control (Fig. 5B). Scratch assays showed that cells in which SCCRO3 was knocked down had more directional motility than scrambled shRNA-infected cells (Fig. 5C). Staining for actin, using Alexa Fluor 488 Phalloidin, showed a reduction in stress fibers in SCCRO3 knockdown cells, compared with scrambled shRNA-infected cells (Fig. 5D).
FIGURE 5.

SCCRO3 has tumor suppressor activity that requires its myristoyl sequence and PONY domain. A, Western blot analysis of lysates from 16HBE cells showing a decrease in SCCRO3 levels in cells infected with shRNA against SCCRO3 (shRNA1 and shRNA2). Western blot for tubulin is shown as a loading control. B, dark field microscopic image of 16HBE cells expressing shRNA against SCCRO3 (shRNA1 and shRNA2) showing a change from epitheloid to spindle-shaped and decreased clumping, compared with cells expressing scrambled shRNA. C, scratch assay on the same cells showing enhanced migration in cells with SCCRO3 knocked down relative to those transfected with scrambled shRNA. D, indirect immunofluorescence analysis of 16HBE cells infected with shRNA against SCCRO3 and stained with Alexa Fluor 488 Phalloidin for actin, which showed a reduction in stress fibers. E, representative images form microscopic analysis of H1299 cells stably transfected with the indicated constructs. Empty vector (EV) transected cells dispersed throughout the plate. SCCRO3 transfected cells (second panel) shows cells maintained intercellular contact and grown in nests. Cells expressing SCCRO3G2A or SCCRO3DAD showed no change in growth characteristics relative to empty vector transfected cells. F, scratch assay on H1299 cells transfected with the indicated constructs showing decreased migration in cells transfected with SCCRO3 and no change in those transfected with SCCRO3G2A, SCCRO3DAD, or empty vector (EV). G, graph showing the average numbers of cells that migrated into wounded areas from experiments above. H, representative images form fluorescence microscopy of H1299 cells transfected with the indicated constructs and stained with Alexa Fluor 488 Phalloidin showing development of actin containing stress fibers in SCCRO3 relative to empty vector transected cells, but not SCCRO3G2A or SCCRO3DAD transfected cells. All scale bars represent 20 μm.
To begin to determine the mechanisms underlying the effects of SCCRO3 on transformation, we performed structure-function studies in H1299 cells. We stably transfected H1299 cells with empty vector, HA-SCCRO3, HA-SCCRO3G2A, or HA-SCCRO3DAD and confirmed that transgenes were expressed at approximately equal levels by Western blot (data not shown). Microscopic analysis showed that SCCRO3-transfected cells acquired an epitheloid shape and grew in nests (Fig. 5E, second row). In contrast, vector-, SCCRO3G2A-, and SCCRO3DAD-transfected cells were spindle-shaped and were scattered throughout the plate, resembling features seen in untransfected H1299 cells (Fig. 5E, first, third, and fourth rows). Scratch assays showed that migration across the scratch was markedly reduced in H1299 cells expressing SCCRO3, compared with that in vector-, SCCRO3G2A-, and SCCRO3DAD-transfected cells (Fig. 5, F and G). Staining with phalloidin showed rearrangement of the actin cytoskeleton, with stress fibers developing in SCCRO3-transfected cells but not in vector- or SCCRO3 mutant-transfected cells (Fig. 5H). To determine SCCRO3 oncogenic activity, we performed soft agar assays that showed reduced colony formation in SCCRO3-transfected but not SCCRO3G2A- or SCCRO3DAD-transfected H1299 clones (Fig. 6, A and B). These observations suggest that SCCRO3 functions as a putative tumor suppressor that requires membrane localization and binding to cullins.
FIGURE 6.
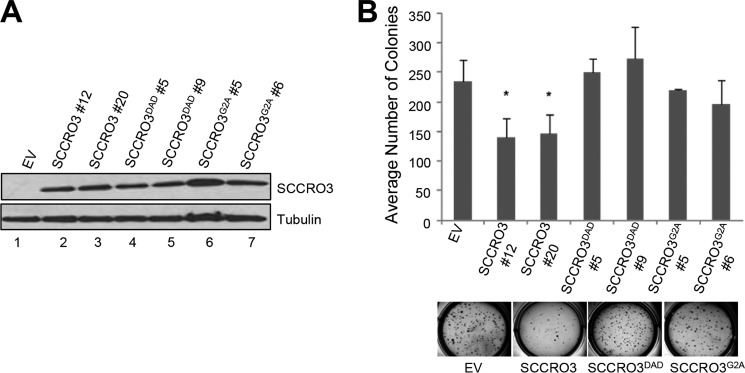
SCCRO3 has tumor suppressor activity that requires its myristoyl sequence and PONY domain. A, Western blot analysis of lysates from H1299 cells stably transfected with the indicated SCCRO3 constructs, probed with anti-SCCRO3 antibody showing equivalent expression of constructs in selected clones. B, results from soft agar assay on the same H1299 clones showing decreased colony formation in cells expressing SCCRO3, relative to clones expressing SCCRO3 mutants or vector alone (average number of colonies per well of a 6-well plate ± S.D.; p = 0.03 for SCCRO3#12 and p = 0.03 for SCCRO3#20, compared with vector). EV, empty vector.
Given the significant impact of mutation on SCCRO3 function, we critically assessed mutations occurring in human cancers. Interestingly, we found mutations in both the myristoyl sequence (SCCRO3G2S) and the PONY domain, including SCCRO3S239F, which occurred with a loss of the wild-type allele. The presence of naturally occurring mutations in human tumors that can abrogate SCCRO3 function supports the role of SCCRO3 as a putative tumor suppressor as well as the requirement of the PONY domain and myristoyl sequence for this activity.
SCCRO3 Antagonizes SCCRO in Vivo Function
To determine whether SCCRO3 tumor suppressor activity results from inhibition of SCCRO oncogeneic activity, we assessed whether SCCRO3 inhibits SCCRO-promoted transformation in vivo. To do this, we established NIH-3T3 clones that stably expressed SCCRO. We found that in contrast to vector-transected NIH-3T3 cells, SCCRO-expressing NIH-3T3 cells formed colonies in soft agar. SCCRO-expressing NIH-3T3 cells were then co-transfected with HA-tagged SCCRO3, SCCRO3G2A, or SCCRO3DAD (Fig. 7A). Co-transfection of SCCRO3 induced morphological changes in SCCRO-expressing NIH-3T3 cells, from a spindle to an epitheloid shape. The pattern of growth was also changed, with cells growing in clusters rather than spreading throughout the plate, as seen in the parental line. No change in shape or growth pattern was seen in SCCRO-expressing NIH-3T3 cells transfected with SCCRO3G2A or SCCRO3DAD (Fig. 7B). Scratch assays showed reduced directional migration of NIH-3T3 cells co-expressing SCCRO and SCCRO3 but not of cells co-expressing SCCRO and SCCRO3G2A or SCCRO3DAD (Fig. 7C). Moreover, colony formation in soft agar was significantly lower in SCCRO-expressing NIH-3T3 cells transfected with SCCRO3 than in those transfected with SCCRO3G2A or SCCRO3DAD (Fig. 7D). Combined, these findings suggest that SCCRO3 tumor suppressor activity involves inhibition of SCCRO-promoted oncogenesis.
FIGURE 7.

SCCRO3 antagonizes SCCRO-induced transformation. A, Western blot analysis of lysates of NIH-3T3 cells stably expressing SCCRO co-transfected with HA-SCCRO3, HA-SCCRO3-G2A, HA-SCCRO3-DAD, or vector alone and probed for the HA tag, which showed equal expression of transgenes. B, representative dark field microscopic images showing SCCRO3 transfected cells maintaining contact and growing in nests, whereas those stably co-expressing SCCRO with empty vector (EV) or SCCRO3G2A or SCCRO3DAD detached and began to disperse across the plate. C, scratch assay on the same set of cells at times 0 h and at 14 h showing a decrease in migration across the scratch for SCCRO expressing cells co-transfected with SCCRO3 relative to empty vector, but not those co-transfected with SCCRO3G2A or SCCRO3DAD. D, graph showing the average numbers of cells that migrated into wounded areas from the experiments above. E, graph showing results from soft agar assays on the same set of cells showing a decrease in colony formation in cell co-transfected with SCCRO3 relative to those co-transfected with empty vector. No change in colony formation was seen with co-transfection of SCCRO3G2A or SCCRO3DAD (showing the average number per high power field [HPF] ± S.D.; p = 0.002, compared with vector).
DISCUSSION
The role of SCCRO in the neddylation E3 is to promote assembly and optimize complex orientation to enhance ligase activity, which it achieves by binding to neddylation components via its PONY domain. Although the contributions of SCCRO are important, they are not required for neddylation E3 activity in vitro. The essential contribution of SCCRO to neddylation in vivo is the nuclear translocation of neddylation components, which is regulated by its ubiquitin-associated domain (Ref. 19).5 To compensate for the complexities of the expanded genome, SCCRO has evolved in higher organisms to include four paralogues that have highly conserved PONY domains but variable N-terminal domains. Previous studies have suggested that all SCCRO paralogues can promote neddylation (18). However, because these studies used in vitro assays, they do not account for the contributions of N-terminal domains, which are all directly or indirectly involved in subcellular localization. Given the requirement of nuclear localization of cullins for neddylation, the presence of a myristoyl sequence in SCCRO3 that localizes it in the cytoplasm raises questions about its in vivo function. We found that, in sharp contrast to other SCCRO family members, SCCRO3 does not function as a component of the E3 or promote cullin neddylation. Although SCCRO3 binds to cullins, ROC1, and CAND1 via its PONY domain, it does not bind to Ubc12 at levels that are functionally relevant. Similar to previous reports, in the present study, we detected binding between SCCRO3 and Ubc12 only when high concentrations of purified proteins were used in in vitro pulldown assays (17). However, the affinity of SCCRO3 for Ubc12 is significantly lower than that of SCCRO. These findings are supported by recent work from Monda et al. (18), who showed a Kd of 21 μm for the interaction between SCCRO3 and Ubc12, compared with 2.0 μm for SCCRO and Ubc12. Moreover, unlike SCCRO, SCCRO3 does not conform to the reaction processivity paradigms for E3s, because it did not preferentially bind to Ubc12∼Nedd8. Consistent with this, we found that SCCRO3 does not promote Cul1 neddylation in vivo. The lack of conformity with reaction processivity paradigms, combined with the absence of neddylation-promoting activity, suggests that SCCRO3 does not function as a component of the E3 for neddylation.
What then is the function of SCCRO3? Previous studies have shown that the Kd for binding to Cul1–Cul5 varies between SCCRO paralogues (18). The Kd for binding of Cul1 to SCCRO is 1.8 μm, compared with 1.1 μm for SCCRO3. Consistent with this, when SCCRO and SCCRO3 were co-expressed at relatively equal levels, HA-Cul1 predominately localized to the membrane with SCCRO3. Because the transgenic expression of SCCRO is increased above that of SCCRO3, Cul1 translocates to the nucleus. Moreover, the relative levels of SCCRO and SCCRO3 expression also affected the level of cullin neddylation. SCCRO3 inhibited SCCRO-promoted Cul1 neddylation, which could be overcome by increasing the level of SCCRO expression in vivo. Mutation of either the myristoyl sequence or the PONY domain in SCCRO3 abrogated co-localization and inhibition of Cul1 neddylation. These findings suggest that SCCRO and SCCRO3 compete for binding and localization of cullins. This competition affects neddylation activity and suggests that SCCRO3 functions as an antagonist of SCCRO neddylation activity. As expected, the PONY domain mutant of SCCRO3, which loses binding to the neddylation component, also loses its in vivo functions. That SCCRO3G2A, a mutant that is unable to localize to the membrane but retains the ability to bind to cullins, also loses its effects on SCCRO function suggests that membrane localization is required for SCCRO3 to impart its effects. To confirm these findings, we performed a competition assay by transecting U2OS cells with SCCRO and an increasing amount of SCCRO3G2A. We found that, even at significantly higher expression levels, SCCRO3G2A had no obvious effect on SCCRO-promoted Cul3 neddylation in vivo (data not shown). There are several possible explanations for why the SCCRO3G2A mutant has no effect on SCCRO neddylation and transformation activity. Although the SCCRO3G2A mutant retains binding to neddylation components in the absence of the tether to the membrane, neddylation components can still enter the nucleus (Fig. 3A). Once neddylation components are in the nucleus, the regulatory effects of compartmentalization are lost, allowing neddylation to proceed. It is also possible that co-factors present at the membrane are required for SCCRO3 to inhibit neddylation activity. Our findings need to be reconciled with previous published results, which show that SCCRO3 promotes Cul3 neddylation at the membrane. It remains possible that SCCRO3 has dual activity. Our data and those from Monda et al. (18) clearly show that SCCRO3 has limited binding to Ubc12. As such, SCCRO3 is unlikely to be able to participate in a neddylation reaction that involves Ubc12. However, Monda et al. (18) showed that SCCRO3 binds more efficiently to UBE2F (Kd = 1.1 μm), an alternate E2 in neddylation. Consistent with this finding, the effect of SCCRO3 on Cul5 neddylation was significantly more with the addition of UBE2F to the reaction than with addition of Ubc12. Interestingly, SCCRO3 complemented the neddylation defect of yeast dcn1Δ cells (SCCRO homologue), which does not contain UBE2F, suggesting that SCCRO3 may interact with yeast Ubc12. Combined, these results suggest that SCCRO3 plays a dual role: 1) antagonizing the neddylation activity of the other SCCRO paralogues and 2) promoting neddylation at the membrane by using UBE2F as the E2.
It was previously shown that overexpression of SCCRO3 in HeLa cells leads to inhibition of colony formation in soft agar. UVC irradiation increased expression of SCCRO3 in cancer cell lines, and knockdown of SCCRO3 by siRNA in HeLa cells inhibited UVC-induced cell death (20), suggesting that SCCRO3 functions as a tumor suppressor. We validated SCCRO3 function as a putative tumor suppressor by showing 1) that overexpression of SCCRO3 promotes mesenchymal to epithelial-like changes and is associated with a decrease in oncogenic activity in H1299 cells and 2) that knockdown of SCCRO3 by shRNA in 16HBE cells promotes epithelial to mesenchymal-like changes and is associated with an increase in oncogenic activity. SCCRO3 tumor suppressor activity required both an intact myristoyl sequence and PONY domain, because mutants in these regions abrogate its tumor suppressor activity in 16HBE cells. Moreover, co-expression of SCCRO3 blocked SCCRO-promoted transformation, suggesting that competition between SCCRO and SCCRO3 for cullin substrate has functional implications in vivo.
Consistent with the role of SCCRO3 as a putative tumor suppressor, its expression was decreased in a large proportion of lung, oral, ovarian, and thyroid tumors. Previous reports have shown that its expression is also reduced in liver, bladder, and renal tumors (20), suggesting that SCCRO3 plays a role in the pathogenesis of a broad range of human cancers. In contrast to the other SCCRO family members, changes in SCCRO3 expression in human cancers cannot be explained by copy number changes or by promoter methylation or gene mutation. Screening for the causes of the decreased expression of SCCRO3 in human cancers led us to PUM2, an RNA-binding protein. We found that expression of PUM2 inversely correlates with levels of SCCRO3 expression in human tumors (37). PUM2 is known to bind to SCCRO3 mRNA, and we found that transgenic expression of PUM2 in cells resulted in decreased cellular levels of SCCRO3, but not SCCRO. Similarly, knockdown of PUM2 was associated with increased SCCRO3 expression, suggesting that PUM2 may be a cause of the decreased SCCRO3 expression in many human cancers. Moreover, the presence of mutations in both the PONY domain and myristoyl sequence of SCCRO3 in human tumors suggests that mutation may be the cause for SCCRO3 inactivation in some cases.
Combined, our data clearly show an antagonistic relationship between SCCRO and SCCRO3 in neddylation and oncogenesis. Whether this antagonism is also attributable to competition for binding to targets downstream of CRLs remains to be elucidated. In the broader context, our results suggest that SCCRO family members play independent and overlapping roles in the regulation of cullin neddylation (13). Moreover, CRL targets that mediate the effects of SCCRO and SCCRO3 in oncogenesis have yet to be elucidated.
This work was supported, in whole or in part, by National Institutes of Health/NCI Cancer Center Support Grant P30 CA008748 (to B. S.). This work was also supported in part by grants from the Catherine van Tussenbroek Fonds and the foundation Vrijvrouwe van Renswoude (to C. C. B.); Fulbright, The Dutch Cancer Society, and the Prins Bernhard Culture Foundation (to V. B. W.); and the Clinical Innovator Award from the Flight Attendants Medical Research Institute and a research grant from the Hackers for Hope.
G. Huang, A. J. Kaufman, R. Ryan, L. Huryn, G. Hunnicut, Y. Romin, P. Morris, K. Manova-Todorova, S. Y. Ramanathan, and B. Singh, unpublished data.
G. Huang, C. W. Towe, L. Choi, Y. Yonekawa, C. C. Bommelje, S. Bains, W. Rechler, B. Hao, S. Y. Ramanathan, and B. Singh, unpublished data.
- CRL
- cullin-RING ligase.
REFERENCES
- 1. Hershko A., Ciechanover A. (1998) The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 [DOI] [PubMed] [Google Scholar]
- 2. Komander D. (2009) The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 37, 937–953 [DOI] [PubMed] [Google Scholar]
- 3. Pickart C. M. (2001) Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533 [DOI] [PubMed] [Google Scholar]
- 4. Scheffner M., Nuber U., Huibregtse J. M. (1995) Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature 373, 81–83 [DOI] [PubMed] [Google Scholar]
- 5. Hunter T. (2007) The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol. Cell 28, 730–738 [DOI] [PubMed] [Google Scholar]
- 6. Bosu D. R., Kipreos E. T. (2008) Cullin-RING ubiquitin ligases: global regulation and activation cycles. Cell Div. 3, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hotton S. K., Callis J. (2008) Regulation of cullin RING ligases. Annu. Rev. Plant Biol. 59, 467–489 [DOI] [PubMed] [Google Scholar]
- 8. Dye B. T., Schulman B. A. (2007) Structural mechanisms underlying posttranslational modification by ubiquitin-like proteins. Annu. Rev. Biophys. Biomol. Struct. 36, 131–150 [DOI] [PubMed] [Google Scholar]
- 9. Duda D. M., Borg L. A., Scott D. C., Hunt H. W., Hammel M., Schulman B. A. (2008) Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell 134, 995–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pan Z. Q., Kentsis A., Dias D. C., Yamoah K., Wu K. (2004) Nedd8 on cullin: building an expressway to protein destruction. Oncogene 23, 1985–1997 [DOI] [PubMed] [Google Scholar]
- 11. Kurz T., Chou Y. C., Willems A. R., Meyer-Schaller N., Hecht M. L., Tyers M., Peter M., Sicheri F. (2008) Dcn1 functions as a scaffold-type E3 ligase for cullin neddylation. Mol. Cell 29, 23–35 [DOI] [PubMed] [Google Scholar]
- 12. Kurz T., Ozlü N., Rudolf F., O'Rourke S. M., Luke B., Hofmann K., Hyman A. A., Bowerman B., Peter M. (2005) The conserved protein DCN-1/Dcn1p is required for cullin neddylation in C. elegans and S. cerevisiae. Nature 435, 1257–1261 [DOI] [PubMed] [Google Scholar]
- 13. Scott D. C., Monda J. K., Grace C. R., Duda D. M., Kriwacki R. W., Kurz T., Schulman B. A. (2010) A dual E3 mechanism for Rub1 ligation to Cdc53. Mol. Cell 39, 784–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang G., Kaufman A. J., Ramanathan Y., Singh B. (2011) SCCRO (DCUN1D1) promotes nuclear translocation and assembly of the neddylation E3 complex. J. Biol. Chem. 286, 10297–10304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim A. Y., Bommeljé C. C., Lee B. E., Yonekawa Y., Choi L., Morris L. G., Huang G., Kaufman A., Ryan R. J., Hao B., Ramanathan Y., Singh B. (2008) SCCRO (DCUN1D1) is an essential component of the E3 complex for neddylation. J. Biol. Chem. 283, 33211–33220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bommeljé C. C., Weeda V. B., Huang G., Shah K., Bains S., Buss E., Shaha M., Gönen M., Ghossein R., Ramanathan S. Y., Singh B. (2014) Oncogenic function of SCCRO5/DCUN1D5 requires its Neddylation E3 activity and nuclear localization. Clin. Cancer Res. 20, 372–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meyer-Schaller N., Chou Y. C., Sumara I., Martin D. D., Kurz T., Katheder N., Hofmann K., Berthiaume L. G., Sicheri F., Peter M. (2009) The human Dcn1-like protein DCNL3 promotes Cul3 neddylation at membranes. Proc. Natl. Acad. Sci. U.S.A. 106, 12365–12370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Monda J. K., Scott D. C., Miller D. J., Lydeard J., King D., Harper J. W., Bennett E. J., Schulman B. A. (2013) Structural conservation of distinctive N-terminal acetylation-dependent interactions across a family of mammalian NEDD8 ligation enzymes. Structure 21, 42–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu K., Yan H., Fang L., Wang X., Pfleger C., Jiang X., Huang L., Pan Z. Q. (2011) Mono-ubiquitination drives nuclear export of the human DCN1-like protein hDCNL1. J. Biol. Chem. 286, 34060–34070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ma T., Shi T., Huang J., Wu L., Hu F., He P., Deng W., Gao P., Zhang Y., Song Q., Ma D., Qiu X. (2008) DCUN1D3, a novel UVC-responsive gene that is involved in cell cycle progression and cell growth. Cancer Sci. 99, 2128–2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sarkaria I., O-charoenrat P., Talbot S. G., Reddy P. G., Ngai I., Maghami E., Patel K. N., Lee B., Yonekawa Y., Dudas M., Kaufman A., Ryan R., Ghossein R., Rao P. H., Stoffel A., Ramanathan Y., Singh B. (2006) Squamous cell carcinoma related oncogene/DCUN1D1 is highly conserved and activated by amplification in squamous cell carcinomas. Cancer Res. 66, 9437–9444 [DOI] [PubMed] [Google Scholar]
- 22. Gordon D., Abajian C., Green P. (1998) Consed: a graphical tool for sequence finishing. Genome Res. 8, 195–202 [DOI] [PubMed] [Google Scholar]
- 23. Nickerson D. A., Tobe V. O., Taylor S. L. (1997) PolyPhred: automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing. Nucleic Acids Res. 25, 2745–2751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen K., McLellan M. D., Ding L., Wendl M. C., Kasai Y., Wilson R. K., Mardis E. R. (2007) PolyScan: an automatic indel and SNP detection approach to the analysis of human resequencing data. Genome Res. 17, 659–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Major J. E. (2007) Genomic mutation consequence calculator. Bioinformatics 23, 3091–3092 [DOI] [PubMed] [Google Scholar]
- 26. Herman J. G., Graff J. R., Myöhänen S., Nelkin B. D., Baylin S. B. (1996) Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. U.S.A. 93, 9821–9826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ng D. C., Lin B. H., Lim C. P., Huang G., Zhang T., Poli V., Cao X. (2006) Stat3 regulates microtubules by antagonizing the depolymerization activity of stathmin. J. Cell Biol. 172, 245–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Broderick S. R., Golas B. J., Pham D., Towe C. W., Talbot S. G., Kaufman A., Bains S., Huryn L. A., Yonekawa Y., Carlson D., Hambardzumyan D., Ramanathan Y., Singh B. (2010) SCCRO promotes glioma formation and malignant progression in mice. Neoplasia 12, 476–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang J., Qian J., Hoeksema M. D., Zou Y., Espinosa A. V., Rahman S. M., Zhang B., Massion P. P. (2013) Integrative genomics analysis identifies candidate drivers at 3q26–29 amplicon in squamous cell carcinoma of the lung. Clin. Cancer Res. 19, 5580–5590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huang G., Singh B. (2013) Coamplification and cooperation: toward identifying biologically relevant oncogenes. Clin. Cancer Res. 19, 5549–5551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Estilo C. L., O-Charoenrat P., Ngai I., Patel S. G., Reddy P. G., Dao S., Shaha A. R., Kraus D. H., Boyle J. O., Wong R. J., Pfister D. G., Huryn J. M., Zlotolow I. M., Shah J. P., Singh B. (2003) The role of novel oncogenes squamous cell carcinoma-related oncogene and phosphatidylinositol 3-kinase p110α in squamous cell carcinoma of the oral tongue. Clin. Cancer Res. 9, 2300–2306 [PubMed] [Google Scholar]
- 32. Jung S. H., Shin S. H., Yim S. H., Choi H. S., Lee S. H., Chung Y. J. (2009) Integrated analysis of copy number alteration and RNA expression profiles of cancer using a high-resolution whole-genome oligonucleotide array. Exp. Mol. Med. 41, 462–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen M., Ye Y., Yang H., Tamboli P., Matin S., Tannir N. M., Wood C. G., Gu J., Wu X. (2009) Genome-wide profiling of chromosomal alterations in renal cell carcinoma using high-density single nucleotide polymorphism arrays. Int. J. Cancer 125, 2342–2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Desouki M. M., Liao S., Huang H., Conroy J., Nowak N. J., Shepherd L., Gaile D. P., Geradts J. (2011) Identification of metastasis-associated breast cancer genes using a high-resolution whole genome profiling approach. J. Cancer Res. Clin. Oncol. 137, 795–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dobrovic A., Gareau J. L., Ouellette G., Bradley W. E. (1988) DNA methylation and genetic inactivation at thymidine kinase locus: two different mechanisms for silencing autosomal genes. Somat. Cell Mol. Genet. 14, 55–68 [DOI] [PubMed] [Google Scholar]
- 36. Dreyfuss G., Kim V. N., Kataoka N. (2002) Messenger-RNA-binding proteins and the messages they carry. Nat. Rev. Mol. Cell Biol. 3, 195–205 [DOI] [PubMed] [Google Scholar]
- 37. Galgano A., Forrer M., Jaskiewicz L., Kanitz A., Zavolan M., Gerber A. P. (2008) Comparative analysis of mRNA targets for human PUF-family proteins suggests extensive interaction with the miRNA regulatory system. PLoS One 3, e3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang X., Zhou J., Sun L., Wei Z., Gao J., Gong W., Xu R. M., Rao Z., Liu Y. (2007) Structural basis for the function of DCN-1 in protein Neddylation. J. Biol. Chem. 282, 24490–24494 [DOI] [PubMed] [Google Scholar]
- 39. Huang D. T., Miller D. W., Mathew R., Cassell R., Holton J. M., Roussel M. F., Schulman B. A. (2004) A unique E1-E2 interaction required for optimal conjugation of the ubiquitin-like protein NEDD8. Nat. Struct. Mol. Biol. 11, 927–935 [DOI] [PMC free article] [PubMed] [Google Scholar]