

Abstract
Gene expression is regulated through the activity of transcription factors and chromatin modifying proteins acting on specific DNA sequences, referred to as cis-regulatory elements. These include promoters, located at the transcription initiation sites of genes, and a variety of distal cis-regulatory modules (CRMs), the most common of which are transcriptional enhancers. Because regulated gene expression is fundamental to cell differentiation and acquisition of new cell fates, identifying, characterizing, and understanding the mechanisms of action of CRMs is critical for understanding development. CRM discovery has historically been challenging, as CRMs can be located far from the genes they regulate, have few readily-identifiable sequence characteristics, and for many years were not amenable to high-throughput discovery methods. However, the recent availability of complete genome sequences and the development of next-generation sequencing methods has led to an explosion of both computational and empirical methods for CRM discovery in model and non-model organisms alike. Experimentally, CRMs can be identified through chromatin immunoprecipitation directed against transcription factors or histone post-translational modifications, identification of nucleosome-depleted “open” chromatin regions, or sequencing-based high-throughput functional screening. Computational methods include comparative genomics, clustering of known or predicted transcription factor binding sites, and supervised machine-learning approaches trained on known CRMs. All of these methods have proven effective for CRM discovery, but each has its own considerations and limitations, and each is subject to a greater or lesser number of false-positive identifications. Experimental confirmation of predictions is essential, although shortcomings in current methods suggest that additional means of validation need to be developed.
Keywords: cis-regulatory module, CRM, enhancer, gene expression, transcriptional regulatory elements, transcription factor binding site, regulatory genomics, chromatin immunoprecipitation, model organisms, machine-learning
INTRODUCTION
Transcriptional regulation is a fundamental feature of development. Genes need to be transcribed at the right time, in the right amount, and in the right cells in order for development to proceed correctly. Inappropriate regulation of even a single gene can have dramatic consequences (witness, for instance, the severe dysmorphologies resulting from regulatory mutations in Hox genes1). In animals, a significant portion of gene regulation results from the interaction of transcription factors (TFs) with specific cis-regulatory DNA sequences. For genes transcribed by RNA polymerase II (PolII), regulatory elements include both the promoter, which is situated at the transcription start site (TSS) and binds PolII and a set of core transcription factors, and more distal sequences, which can range from immediately upstream of the promoter to hundreds of kilobases away from the TSS. We discuss here only the distal regulatory elements, collectively referred to as cis-regulatory modules (CRMs). (For several excellent recent reviews of CRMs, see2–6). CRMs tend to be organized in a modular fashion, with each controlling a discrete subset of a gene’s overall expression pattern (Fig. 1A). They are typically a few hundred base pairs in length and can be located 5′, 3′, intronically, or even exonically relative to their target genes. CRMs as a class thus include transcriptional enhancers, and the two terms are often used interchangeably despite the fact that in the majority of cases, the regulatory sequences have not truly been shown to meet the formal requirement that an enhancer act without regard to orientation, distance, or placement (5′/3′) relative to its target gene.7 In keeping with this common usage, we will mainly use the term CRM here to mean enhancer-like positive regulatory sequences and will focus on discovery of these elements, except where otherwise stated. It is important to note, however, the existence of other types of cis-regulatory sequences, including negatively-acting silencers, locus control regions, insulator elements, and others.8
Figure 1. cis-Regulatory Modules.
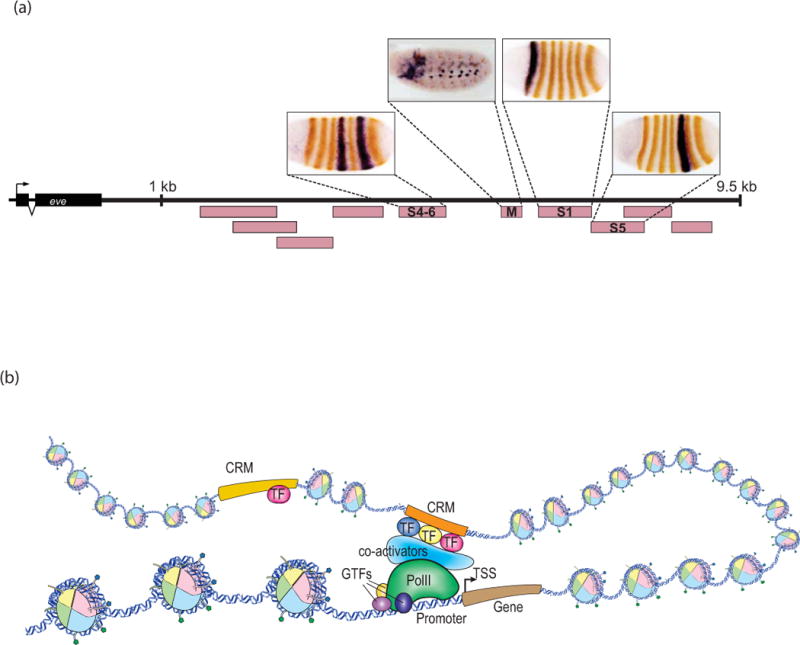
(a) Modular nature of CRMs. The region downstream of the Drosophila even skipped (eve) gene has numerous CRMs (pink boxes), each of which controls a different portion of the gene’s expression pattern. Reporter gene expression directed by individual CRMs (black) is shown superimposed on Eve protein expression (brown). During the early blastoderm stages, individual stripes are regulated by separate CRMs (S1, S4–6, S5), as is later embryonic expression in the somatic musculature (M). Expression from other CRMs including those in the 5′ flanking region are not pictured. Photos courtesy of James Jaynes and Miki Fujioka. (b) Generalized mechanisms of CRM function. Active CRMs (orange), bound by multiple transcription factors (TF), contact their associated promoter by DNA looping. Either through direct contact or via bridging interactions from coactivators, the CRMs help to recruit and/or stabilize RNApolII and the general transcription factors (GTFs). TSS, transcription start site.
The broad outlines of CRM function are well established, although many details remain to be understood (Fig. 1B). In essence, CRMs serve as a scaffold for the assembly of specific combinations of TFs, which in turn recruit various co-activators and co-repressors (many of which are chromatin-modifying enzymes) and nucleosome-remodeling complexes.9 These enhancer complexes are brought into proximity of their target promoters via DNA looping (and/or additional mechanisms), where they serve to recruit or stabilize interactions with PolII and the general transcription factors.3, 4, 9 CRMs may also play an active role in the release of engaged but paused PolII from the promoter to allow productive transcription elongation.3, 4, 9
The rise of genomic profiling methods (i.e., methods that can interrogate gene expression, protein-DNA interactions, chemical modification of DNA, and so forth on a genome-wide scale in a single experiment) has revealed several distinct properties of CRMs. Nucleosomes flanking CRM sequences are characterized by a number of histone modifications, most notably monomethylation of lysine 4 of histone H3 (H3K4me1) and acetylation of histone H3 lysine 27 (H3K27ac). Although these histone modifications mark CRM locations, their functional significance has yet to be determined.10 Also in need of mechanistic explanation is the finding that many active CRMs are themselves transcribed into RNA. Initially noted as a general enhancer feature in a broad survey of Drosophila CRMs,11 enhancer RNAs (“eRNAs”) appear to be widespread and have been implicated in a number of mechanisms including recruitment of cohesin (important for enhancer-promoter looping), mediating chromatin accessibility to allow TF binding, and interacting with the Mediator complex to stimulate transcription.12, 13 However, it remains unclear if there is a single or multiple types of eRNAs and what the various functions of these transcripts will ultimately be revealed to be.
Despite their crucial role in regulating gene expression, CRMs remain poorly annotated in sequenced genomes, and the vast majority of CRMs are yet to be characterized. The reasons for this are several and stem in part from the fact that the number of CRMs likely outweighs the number of genes by at least several fold (as many genes are known to have multiple CRMs). Whereas other functional elements such as genes and promoters have long been amenable to medium- and high-throughput assays, especially since the development of microarray and next-generation sequencing methods, genome-scale assays for CRM discovery have only recently become feasible. Until a few years ago, CRMs could be defined only through low-throughput functional assays—primarily, reporter gene assays (Figure 2)—that demonstrated the ability of a given sequence fragment to affect transcription. The problem of CRM discovery has been exacerbated by the fact that unlike protein-coding regions, which have recognizable sequence-level features such as open reading frames and codon-usage biases, no similar properties are known for CRMs. Unlike promoters, which by definition lie immediately 5′ to the gene and which are often characterized by a limited number of well-defined sequence motifs, CRMs are constrained neither in location nor by motif. Thus, while reasonably effective computational methods have been developed for gene-finding and promoter identification,14, 15 in silico approaches to CRM discovery have until recently enjoyed only limited success.
Figure 2. Reporter Genes.
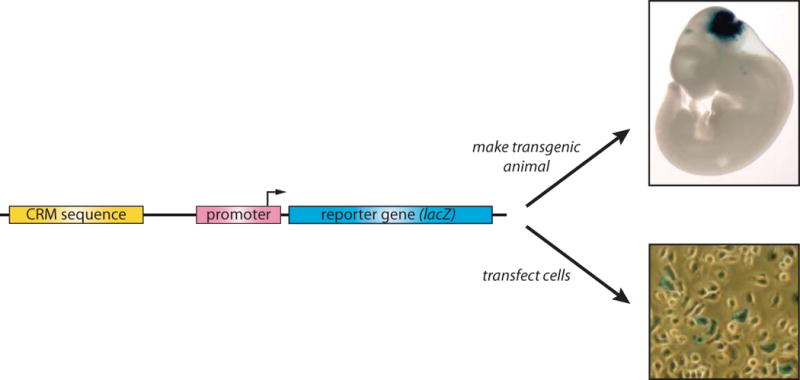
The “gold-standard” test for CRM function is the reporter gene assay, in which a putative CRM sequence is cloned upstream of a minimal promoter-reporter cassette sequence that on its own has little or no transcription. The reporter gene can be any gene whose expression is easily assayed. Current common reporters include luciferase, ß-galactosidase (the E. coli lacZ gene), and fluorescent proteins such as the A. victoria green fluorescent protein (GFP) and its derivatives. lacZ and the fluorescent protein genes are particularly suitable for use as in vivo reporters as they are readily assayed in whole animals or histological sections, whereas luciferase provides high sensitivity in cell culture assays. The recent availability of affordable next-generation sequencing has enabled the development of methods using DNA barcodes or even the CRM sequence itself as a reporter (see main text). While high-throughput, these approaches however lose the valuable ability possessed by visible reporter genes to spatially localize domains of CRM activity. Mouse embryo photo courtesy of VISTA Enhancer Browser,153 cell culture photo courtesy of Satrijat Sinha.
The development of genomic and epigenomic technologies, however, has dramatically changed the outlook for CRM discovery. Next-generation sequencing methods now enable high-throughput functional CRM discovery assays, and the vastly increasing amounts of available data, including large-scale libraries of transcription factor binding site (TFBS) motifs, collections of annotated, validated CRMs, and extensive epigenetic data of many kinds across many cell types, are making accurate computational CRM discovery an attainable goal. In this review, we provide a guide to this changing landscape of CRM discovery. We describe both experimental and computational methods for identifying CRMs and highlight some of the benefits and disadvantages of each approach. We stress the need for in vivo validation of CRM predictions, but note the limitations of current methods.
IDENTIFYING TRANSCRIPTION FACTOR BINDING SITES
The basic units of function for CRMs are transcription factor binding sites (TFBSs), and many CRM discovery approaches, both empirical and computational, therefore begin with an attempt to characterize TFBS sequences. These regions are typically 6–20 base pairs long. Although TFs bind to DNA in a sequence-specific fashion, almost all TFs bind a degenerate recognition sequence; that is, they recognize a range of similar but not identical sequences (Fig. 3a). Collectively, this family of binding site sequences is referred to as a binding site “motif” and can be represented textually as a consensus sequence (Fig. 3b), graphically as a sequence logo (Fig. 3c) and mathematically as a position weight matrix or “PWM” (Fig. 3e; see also discussion in Box 1). A large number of both experimental16 and computational17, 18 approaches have been developed to determine motifs for specific transcription factors, or to identify putative regulatory motifs for unknown factors in long DNA sequences, including entire genomes.
Figure 3. Transcription factor binding site motifs.

A TFBS motif describes the sequences to which a TF can bind, and can be represented in various ways, each with its own advantages and disadvantages. (a) A subset of sequences to which the Drosophila TF Paired binds in a bacterial one-hybrid assay, drawn from FlyFactorSurvey.35 The simplest representation is as a single text string consensus sequence (b). In the consensus sequence, a single base is shown when it occurs in more than half of the binding site sequences and at least twice as much as the next most frequently occurring base at that position; otherwise, degenerate symbols are used.186 The example in (b) has H = {A, C, T} in the first column and Y = {C,T} in the final position. Consensus sequences have the advantage of being simple to portray and easy to search for, but convey limited information about the range of individual sequences comprising the motif. (c) A better sense of nucleotide variability at each position is seen with a motif logo.187 Logos can be derived from a position frequency matrix (d), which totals the presence of each base at each position and which can also be used to develop position weight matrices (PWMs) such as the logodds-adjusted matrix in (e).188 PWMs reflect the probability distributions of the four possible nucleotides at each location and relate closely to the binding energy of TFs to the DNA motifs.189 PWMs lend themselves well to sophisticated sequence-search algorithms and are the basis for most bioinformatics approaches to TFBS detection.18, 190–192
Box 1. Strong and Weak Motifs.
Determining if a genomic sequence is a TFBS is a non-trivial task, regardless of whether one is searching empirically or computationally. In the former case, a decision must be made whether or not a binding event is sufficiently strong to qualify as specific binding rather than experimental noise; in the latter, it must be determined whether the sequence is a close enough match to a known TFBS motif. Motifs are often considered either “good” or “bad” depending on their information content, i.e., the extent to which they show sequence degeneracy, especially in the central or “core” region of the motif. For most search algorithms, motifs with less degeneracy will match genomic sites with higher scores. However, equating higher-scoring matches with more meaningful biological results is a risky proposition. The importance of low-affinity DNA binding by TFs has been demonstrated in many circumstances,26, 108, 164–166 quite aside from the fact that binding affinities in vivo can be modulated by cooperative interactions with other proteins and by alterations in local DNA conformation. A recent study demonstrated that “good” PWMs performed worse than “bad” ones in being able to predict accurately the full range of sequences bound in protein-binding microarray experiments, using algorithms specifically designed for that task. The authors conclude, counter to the conventional wisdom, that “information content has little to do with the accuracy and utility of a motif.”167 This presents a clear conundrum for researchers in determining a satisfactory balance between sensitivity and specificity in TFBS identification using common motif scanning algorithms, with important implications for choosing methods for CRM discovery (see text).
Genomic-era technologies such as microfluidics, microarrays, and next-generation sequencing enable high-throughput determination of the binding motif for a given TF, and include methods such as MARE (Mechanically induced trapping of molecular interactions (MITOMI)19 for the analysis of regulatory elements),20, 21 SELEX-seq,22,23 protein-binding microarrays (PBMs),24 and bacterial one-hybrid (B1H) assays.25 A significant feature shared by these methods is that they comprehensively sample the “sequence space”—all possible DNA sequences—and thus provide data on weak as well as strong binding sites. This is important as the strongest binding is not always the most functionally relevant binding, as demonstrated for instance by the critical role played by low-affinity binding sites in generating the appropriate readout of the Hedgehog morphogen gradient during Drosophila wing disc development26 (see Box 1).
A substantial advance in TFBS discovery has been the ability to interrogate what sequences are bound in vivo through chromatin immunoprecipitation (ChIP) coupled with genome-tiling microarrays (ChIP-chip27) or more commonly now, next-generation sequencing (ChIP-seq)28, and conceptually similar approaches such as DamID (DNA adenine methyltransferase identification).29 This has been of major importance as it is clear that in vivo binding does not always correlate with in vitro binding capability, presumably due to any of a number of factors including incomplete motif definition, chromatin accessibility, and interactions with other TFs. In ChIP-seq, TFs are physically crosslinked to their binding sites in vivo using formaldehyde fixation. The TF-DNA complexes are isolated, sheared into small chromatin fragments, and co-precipitated using antibodies against the TF of interest. The DNA is then isolated by reversing the crosslinking and sequenced, thus identifying the regions that had been bound by the TF. Since the regions obtained from ChIP (up to a few hundred base pairs) are larger than the TFBSs themselves, additional computational analysis is then used to discover the individual TFBSs within these regions. The motif-finding issue can be overcome by using methods such as ChIP-exo30 or the more sensitive ChIP-nexus,31 in which an exonuclease trims the DNA to give a higher resolution in TFBS mapping.
Extensive application of these methods has led to the generation of TFBS motifs for the bulk of the currently annotated TFs in the major model organisms including yeast, worms, flies, mice, and humans. Several resources are available for accessing these data, including the curated motif databases JASPAR32 and TRANSFAC33, the UniPROBE34 database of protein-binding microarray results, and the Drosophila-specific FlyFactorSurvey.35 These resources can be used to assemble libraries of TFBS motifs for use as input into computational CRM discovery algorithms, as discussed below.
CRM DISCOVERY
Identifying CRMs presents a more challenging task than discovering TFBS motifs, individual motif instances in the genome, or even verified instances of in vivo TF binding. Not all predicted TFBSs are bound, and not all TF binding can be directly linked to regulating gene expression.36, 37 Thus, mere presence of a TFBS cannot be taken as evidence that a sequence is part of a CRM, and explicit CRM-discovery approaches must be used.
For roughly two decades following the initial description of transcriptional enhancers in 1981,38 CRM discovery was confined primarily to the low-throughput approach of testing successive sequence fragments for regulatory activity using reporter gene assays (Fig. 2; Fig. 4a,d). However, with the advent of fully-sequenced genomes around the turn of the 21st century, methods for computational CRM prediction were developed that greatly accelerated the pace of discovery. These were followed a few years later by ChIP and other chromatin profiling approaches (e.g., DNase-seq, FAIRE; see below, “Chromatin Accessibility”) that could predict CRMs on a genomic scale with what generally has been believed (although in many cases not demonstrated) to be fairly high accuracy. All of these methods rely on one or more of a small set of strategies rooted in current understanding and assumptions about CRM biology: sequence conservation, the presence of a TF combinatorial “code” (Box 2), and chromatin modification or conformation. The last few years have seen the development of approaches for high-throughput relatively unbiased genome-scale functional screening as well as methods that attempt to directly capture CRM-gene interactions (reviewed by6, 39). The following sections review each of these families of approaches, focusing on a few representative methods for each class. We divide our discussion into empirical (Fig. 4) and computational (Fig. 5) approaches although in reality the two are often intertwined, as many experimental approaches require at least some computational analysis, and most computational methods rely to some extent on empirically-derived data to serve as input.
Figure 4. Experimental methods for CRM discovery.
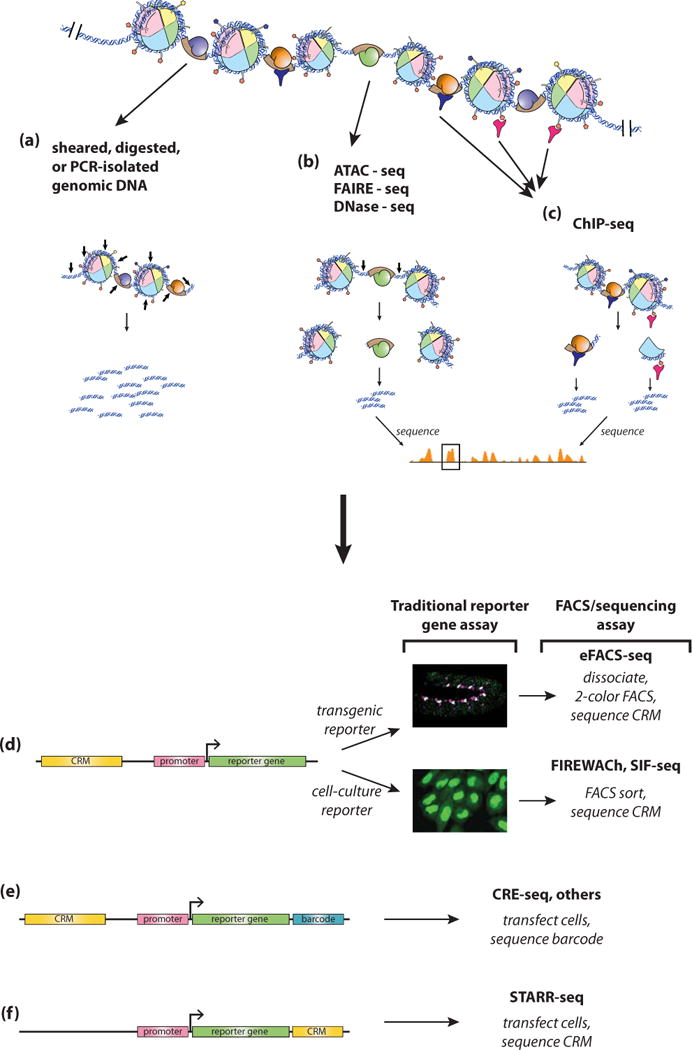
(a) Genomic DNA to be tested for CRM function can be isolated in an unbiased way through shearing or digestion (small arrows), or in a more directed way by PCR amplification. The fragments are then tested for regulatory activity through one of several assays (d-f). (b) CRMs can also be predicted through assays for accessible chromatin, in which “open” chromatin regions (small arrows) can be distinguished from regions of less accessible chromatin. (c) An additional method used for CRM discovery is ChIP-seq directed against histone modifications (pink) or one or more TFs (blue). For both chromatin accessibility and ChIP-seq assays, predicted CRM regions identified by next-generation sequencing (boxed orange peak in b, c) can be cloned and validated by the assays in panels d-f. (d) Cloned sequences can be tested individually by traditional reporter gene assays in transgenic animals or cells (middle), or in a higher-throughput fashion following FACS sorting and next-generation sequencing. (e) Alternatively, reporter constructs can be built to contain unique sequence “barcodes” which can then be matched to the associated CRMs subsequent to RNA-seq analysis. (f) In STARR-seq, the CRM serves as its own reporter, allowing for direct identification following RNA-seq analysis.
Box 2. Transcriptional Codes.
The notion that CRMs regulate gene expression through interpretation of a combinatorial code in the form of a defined set of TFs, whose activities are integrated by the CRM when they are bound together on the DNA, has been instrumental in facilitating both empirical and computational CRM discovery. In the context of developmental gene regulatory networks,168, 169 these transcriptional codes often take the form of a mix of signal-induced TFs, composed of the nuclear effectors of a surprisingly small coterie of signaling pathways,170 and tissue-specific TFs already active in the cells as a result of their developmental history (see Figure). This is likely to be a major mechanism by which cell-type specificity is conferred on what would otherwise be fairly generic inductive signals.171, 172
An important corollary to the transcriptional code concept is the idea of the “gene battery.” Britten and Davidson adapted this term of Morgan’s over four decades ago to refer to a group of genes that are coordinately expressed as a result of their regulatory regions responding to the same transcription factor inputs.173 In molecular terms, a gene battery is a group of genes that are co-expressed by virtue of having CRMs composed of a similar cohort of TF binding sites. The CRMs associated with genes in a battery are usually not identical in terms of either number or arrangement of TFBSs, and a given CRM will not necessarily contain binding sites for all of the TFs. Nevertheless, the relatedness of CRMs regulating co-expressed genes in terms of TF binding underlies many current approaches to CRM discovery.
An important unresolved question about transcriptional codes is to what extent the order and spacing of individual TFs—what is sometimes referred to as CRM “grammar”—plays a role in determining CRM function. Unfortunately, this likely depends on the particular CRM in question. CRMs that form a tight “enhanseosome” structure, epitomized by the mammalian IFNβ enhancer,174 appear to require a highly constrained arrangement of TFBSs, whereas at the other extreme “billboard”-type CRMs175 can tolerate extensive reshuffling of binding sites. Many CRMs—quite probably most—appear able to support rearrangement of some, but not all, TFBSs.108, 176 Until this issue is more fully understood, knowledge of transcriptional codes can aid in CRM discovery, but cannot ensure accurate prediction of CRMs and their regulatory functions.
Figure. Transcriptional codes for developmental CRMs.
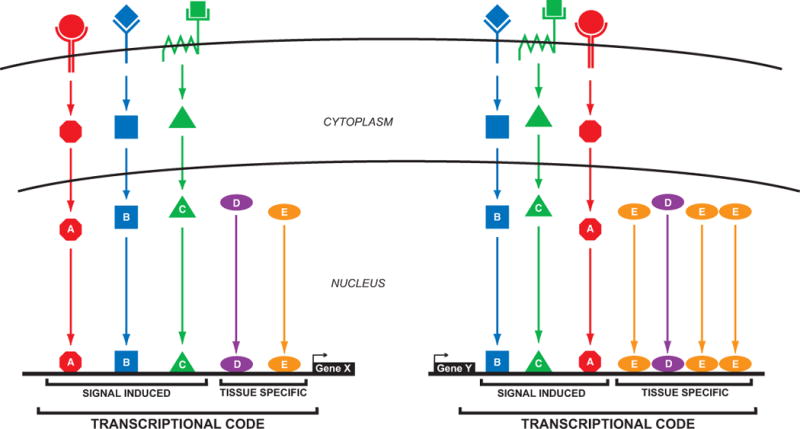
TFs downstream of intercellular signaling pathways (A, B, C) mix with tissue-specific TFs (D, E) to form a “transcriptional code” to activate gene transcription. CRMs for two genes are pictured. Both respond to the same transcriptional code, but the arrangement of the TFBSs is different between the two, and the Gene Y CRM (right) has gained additional binding sites for TF “E”.
Figure 5. Computational approaches to CRM discovery.
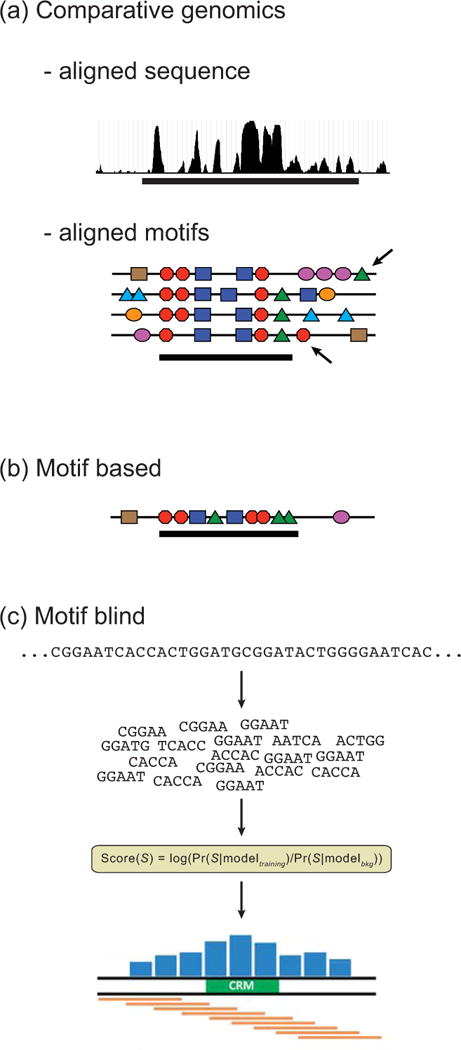
Computational methods for CRM discovery fall into three basic classes. (a) Comparative genomics methods find regions of conservation between two or more species, either by sequence alignment (“aligned sequence”, shown here as a PhastCons score193 over multiple species) or by alignment of TFBS motifs (“aligned motifs”). A horizontal bar indicates predicted CRMs. Note that a method based on alignment of motifs may miss important unaligned compensatory sites (arrows). (b) Motif-based methods identify clusters of TFBS motifs, usually with some foreknowledge of which TFBSs are expected for the CRMs being sought (the “transcriptional code”). Here, a tight cluster of multiple red octagonal, blue square, and green triangle motifs predicts the CRM (horizontal bar). (c) Motif-blind methods rely on statistical models of the DNA sequence rather than identification of motifs. Regions of the genome that receive high scores based on a particular model are predicted as CRMs (green box).
Empirical approaches to CRM Discovery
ChIP-based methods
The ability to obtain in vivo profiles of TF binding via ChIP allows for identification of CRMs based on appropriately clustered TF binding at a single locus. ChIP-based CRM discovery strategies directed against a defined combination of TFs, or even a single TF, have been effective in a number of studies, including, for example, identifying CRMs involved in Drosophila dorsal-ventral patterning,40 Drosophila mesoderm development,41 and human hematopoiesis.42 This approach is most appropriate when the “transcriptional code” (see Box 2) is known, as it is then apparent which combinations of bound TFs should be searched for. However, this information is currently available for only a minority of developmental systems.
An intriguing discovery has been the presence of “HOT” (highly-occupied target) regions, in which unexpectedly high numbers of different TFs are observed to bind, in a range of organisms from C. elegans to Drosophila to human.43–47 A detailed study in Drosophila demonstrated that 94% (102/108) of tested HOT regions functioned as CRMs,48 yet these regions are actually depleted for the binding site motifs of many of the bound factors, implying non-specific binding. Moreover, much of the TF binding has been suggested to be non-functional, as there is poor correlation between cells in which the CRMs are active and cells in which the bound TFs are expressed.48 Other studies also suggest widespread non-functional binding of TFs throughout the genome,36, 49 although it has been proposed that at least some weak, widespread TF binding does have regulatory function.50 One possibility is that the HOT regions represent stretches of unusually accessible chromatin that are particularly amenable to non-specific TF binding. (A second possibility is that the HOT regions are merely artifacts of the ChIP procedure.51, 52) These findings underscore the somewhat counterintuitive idea that while TF binding or TFBS presence often factors importantly in CRM prediction, the identified TFs and TFBSs may not be the ones that are functional in the discovered regulatory modules.53
A related CRM discovery strategy is to use ChIP–seq to identify the in vivo binding sites of transcriptional coactivators which are present at large numbers of CRMs, such as the acetyltransferase p300/CBP.54 Although coactivators do not directly bind the DNA, they are retained in complex with sequence-specific TFs and DNA at active CRMs following formaldehyde cross-linking and are thus amenable to ChIP. This approach has the benefit that sets of relevant transcription factors do not need to be known a priori, with the disadvantage that focusing on a generic coactivator does not allow for preferential discovery of CRMs active in a specific tissue type. However, some extent of cell specificity can be achieved by performing the assays using a homogeneous cell line or isolated tissue. ChIP directed against p300 has been used in this fashion to identify mouse and human CRMs, the former in a tissue-specific fashion by performing the ChIP on dissected embryonic tissues.55–58 Transgenic reporter gene validation assays place the false-positive prediction rate (i.e., sequences selected as CRMs but failing to show activity in the reporter gene assays) in the range of 10%-40%, indicating that the method is not an infallible predictor of CRM function; nevertheless, these are considered strong success rates among methods for both empirical and computational CRM-discovery.
Chromatin “signatures” as means to identify active CRMs
Another variant of ChIP-based methods is to target the various post-translational modifications (PTMs) seen on the tails of histones in the nucleosomes flanking CRMs, such as high levels of H3K4me1 and H3K27ac (Fig. 4c, right-hand and center arrows).10, 59, 60 A growing number of studies have used this approach as a primary method for CRM discovery, including large-scale undertakings such as the ENCODE and modENCODE projects and the NIH Roadmap Epigenomics Mapping Consortium.61–64 Several studies have interpreted the combinations of histone PTMs further, breaking out CRM categories such as “active” versus “poised” enhancers based on the presence or absence of particular modifications.56, 59
Unfortunately, there has been only limited validation performed relative to the very large number of CRM predictions that have been made based on histone PTM profiles, leading to a somewhat circular logic when new modifications are examined: CRMs are predicted based on a certain set of histone PTMs, but then that same, still unvalidated set is used to evaluate whether or not a new modification is enriched in regulatory sequences. Bonn et al.65 performed a retrospective analysis of histone-PTM-based CRM discovery by comparing a tissue-specific ChIP data set to a large set of well-characterized CRMs known from reporter gene analysis in Drosophila. They found that although there are clear associations of modifications such as H3K4me1 and H3K27ac with active CRMs, these and other PTMs are not dispositive; substantial numbers of CRMs contained various numbers, or even none at all, of the six chromatin marks they profiled. Another study that profiled multiple histone modifications, this time in human T cells, similarly found that multiple different combinations of marks could be found at characterized enhancer regions.66 This suggests that significant caution should be taken before using histone PTMs as a sole method for CRM discovery.
Because functional genomic elements, including CRMs, are associated with multiple histone PTMs and other chromatin features, several machine learning approaches (e.g., hidden Markov models, dynamic Bayesian networks) have been developed to segment the chromosome into domains based on patterns of chromatin marks. Examples include ChromHMM67 and Segway.68 These algorithms combine multiple histone PTMs to divide a user-supplied genome into different “chromatin states” which are then assigned to classes based on correlation to known functions including “strong enhancers,” “weak enhancers,” “insulators,” “transcribed,” and others. Segmentation approaches are an intuitively appealing way of integrating the ever-growing amounts of genomic data to identify functional genomic regions, but recent validation experiments raise serious questions about their utility for accurate CRM discovery. A test of over 2000 sequences using a high-throughput functional reporter assay (CRE-seq; see below, “Function-based Methods”) showed that although there was a clear bias for regions defined by ChromHMM and Segway as “enhancers” or “weak enhancers” to activate gene expression, a full three-quarters of such sequences fail to show regulatory activity.69 While these validation experiments are subject to the same caveats as all reporter gene assays (see below, “Outstanding Issues”), the results suggest strongly that the “histone code” is not yet sufficiently understood to enable CRM discovery with accuracies approaching those obtained from more direct methods such as ChIP for p300 or specific TFs, or for effective computational CRM prediction algorithms such as those by Kantorovitz et al.70 and Narlikar et al.71 (see below, “Computational Approaches”).
Chromatin Accessibility
Chromatin accessibility72—the degree to which DNA is wrapped in nucleosomes—is an important aspect of gene regulation, likely due to the inability of many TFs to bind nucleosomal DNA.73 Active CRMs are therefore regions of nucleosome-depleted “open” chromatin74 and can be identified on a genome-wide scale through a variety of methods (Fig. 4b). DNase-seq,75 which is sensitive enough to resolve individual TFBSs,76 makes use of the enhanced susceptibility of open chromatin to enzymatic cleavage by DNase I in a genome-wide, next-generation sequencing-based extension of traditional DNase I footprinting and hypersensitivity assays. Alternatively, FAIRE (Formaldehyde Assisted Isolation of Regulatory Elements) separates nucleosome-containing from nucleosome-free DNA using formaldehyde crosslinking followed by phenol extraction.77, 78 The histone-bound nucleosomal fraction stays in the organic phase while the nucleosome-free open chromatin partitions into the aqueous phase, from where it can be recovered and sequenced. Although simpler to preform than DNase-seq, FAIRE tends to have high signal-to-noise ratios, and it lacks the resolution to identify individual TFBSs. A powerful new approach, ATAC-seq,79 appears to combine the high-resolution of DNase-seq (it can resolve TFBSs) with the simplicity of FAIRE and can be performed on up to five orders of magnitude fewer cells. ATAC-seq takes advantage of the preference for a Tn5 transposon derivative to insert at higher rates in accessible chromatin, using a modification of the “tagmentation” method already optimized for preparing genomic sequencing libraries for use on Illumina next-generation sequencing platforms.80 This elegant method thus generates tagged DNA fragments directly usable for amplification and next-generation sequencing in a single, simple step, with preference for open chromatin locations.
Function-based Methods
Next-generation sequencing-based technologies have also fostered the development of new high-throughput function-based methods for enhancer discovery (reviewed by39). Several of these, such as CRE-seq,81 work by adding DNA “barcodes” to reporter constructs (Fig. 4e).82–86 The reporter plasmids are then transfected into cells or injected in vivo in large batches (hundreds to thousands), and the barcode-containing reporter transcripts are quantified using deep sequencing. The number of reads per barcode reflects the abundance of the respective reporter transcript and thus the activity of the corresponding candidate CRM. Other methods, such as SIF-seq (Site-specific Integration Fluorescence-activated cell sorting followed by sequencing)87 and FIREWAch (Functional Identification of Regulatory Elements Within Active Chromatin),88 avoid the need for barcodes—an often technically-challenging step—by testing large pools of potential regulatory sequences simultaneously in a fluorescent reporter-gene assay and using fluorescently activated cell sorting (FACS) to separate out the cells containing functional regulatory sequences (Fig. 4d). Next-generation sequencing then determines the identities of the sequences driving the reporter-gene expression in the sorted cells. Enhancer-FACS-seq was developed for identification of Drosophila CRMs and uses two-color FACS-based filtering to detect developmentally relevant, tissue-specific enhancers active in developing Drosophila embryos.89 One color is used to register reporter gene activity and the other to mark cell types of interest, allowing for selection and sequencing of only those cells which are both the desired cell-type and which have a functional regulatory sequence driving the reporter gene (Fig. 4d). The approach thus dispenses with the need for time-consuming and labor-intensive screening of individual enhancer constructs in transgenic animals and allows instead for simultaneous testing of multiple pooled putative regulatory sequences, although full characterization of identified CRMs still requires subsequent generation of a new transgenic line. STARR-seq90 requires neither barcodes nor fluorescent reporters and FACS, but rather works by inserting putative CRMs downstream of a minimal promoter such that each sequence serves double-duty as both CRM and reporter (Fig. 4f). Millions of these constructs can then be transfected into cells, and the strength of each regulatory sequence is determined by its abundance in subsequent RNA-seq analysis. A major advantage to all of these function-based screening methods is that they are largely unbiased: although the need to construct libraries of potential CRM sequences still prevents fully comprehensive coverage of the entire genome, choices of candidate CRMs do not need to be constrained by preconceived ideas about TF binding, histone modification, evolutionary conservation, and the like. At the same time, however, these methods are still subject to some of the same limitations as traditional reporter gene assays, in that the putative CRM sequences are tested outside of their native genomic context (see below, “Outstanding Issues”).
Computational approaches to CRM Discovery
Computational methods comprise a vital component of the CRM-discovery arsenal.91–93 In the decade between the initial sequencing of the major model organism genomes and the widespread availability of next-generation sequencing, computational prediction was the only reasonable alternative to one-gene-at-a-time, reporter-gene-based analysis. Even with the development of the high-throughput empirical methods discussed above, however, computational CRM discovery remains an important complement to experimental approaches. Despite rapidly decreasing costs, high-throughput empirical methods remain expensive and technically challenging and to be comprehensive, must be performed at multiple developmental stages, in multiple cell types, and under various growth conditions. Experimental methods furthermore depend on the availability of reagents and technologies—e.g. antibodies, cell lines, or methods for efficient transgenesis—that may not exist for non-model or emerging-model organisms. Computational CRM prediction can provide a rapid and low-cost screening step for identifying an enriched set of candidate CRMs to be followed up with in vivo validation assays, and can also help to refine results from chromatin profiling and other experimental approaches.
Computational methods for CRM discovery fall broadly into three classes, depending on the types of data they require (Fig. 5). Comparative genomics (Fig. 5a) relies on identifying regions of conserved non-coding DNA sequences across related species. Motif-based methods (Fig. 5b) search for short (e.g. 500bp) genomic regions containing clusters of TFBSs. “Motif-blind” approaches (Fig. 5c) require no a priori knowledge of TFs or TFBSs and are based instead on statistical properties of the sequence itself. These categories are not mutually exclusive, and methods combining multiple approaches often perform strongly.
Comparative genomics approaches
Functionally important genomic sequences are under more evolutionary constraint than sequences with less-vital functions. This fact has frequently been exploited for CRM discovery, with varying levels of success. Among factors that need to be considered are the evolutionary distances between species being compared and what tools are being used to assess conservation.94 As more and more species become sequenced, it has become apparent that the answer to the former question varies greatly, not only depending on the species under study but even for individual CRMs within a species: some CRMs are highly conserved throughout genera, families, or beyond, whereas others may show conservation only when compared to close sister species, if at all.11, 94, 95
Several studies have tested the ability to discriminate CRMs from non-CRMs based on sequence conservation, with mixed results. Li et al.11 showed that while Drosophila CRMs are more highly conserved than randomly selected non-coding sequences when compared over eight sequenced Drosophilids, the distributions are highly overlapping and unlikely to lead to accurate prediction if merely assessing overall percentage of conserved bases. Similar results were obtained in a recent study of orthologous sequences from five fly species assayed functionally by STARR-seq.96 In contrast, a windowed version of the PhastCons conservation score was able to achieve reasonable prediction of CRMs using a set of sequences similar to that analyzed by Li et al.,11 although performance was less encouraging on other data sets.97
Broadly speaking, predictions based on analysis of genomic regions surrounding developmentally important regulators and/or based on extreme conservation (for example, conserved from humans to fish98–103) have been reasonably effective, with validation rates averaging somewhat over 50%, whereas more unbiased studies of less-deeply conserved sequences have led to low rates of validation (e.g. < 20%).104 As extreme conservation of CRMs appears to be the exception—only ~5% of mammalian CRMs, for instance, fall into this category105—comparative genomics as a sole criterion for CRM discovery is not recommended. An additional limitation is that sequence conservation obviously is poorly suited for discovering newly-evolved regulatory modules, or those that may have been gained or lost in a lineage-specific fashion.106 This may be a not-uncommon phenomenon. Although purifying selection causes CRMs to evolve at a relatively slower pace compared to DNA without important function, CRMs can appear de novo when random mutations create clusters of TFBS or when deletion brings formerly separated TFBSs into proximity, and can be lost through the same processes by disruption of key TFBSs or TFBS pairings.107
Conservation of CRM content—the individual TFBSs which constitute a CRM—may be more important than conservation of the overall CRM sequence. Sequence conservation has been most effective when mixed with TFBS identification, for instance by using only TFBSs that are conserved in additional species as input to a motif-based CRM discovery algorithm (see below, “Motif-based approaches”). One difficulty in assessing CRM conservation on the TFBS level is that while individual TFBSs may be conserved, their sequence degeneracy can make the conservation difficult to detect through standard nucleotide-level alignment. Further complicating the issue is that the order and arrangement of TFBSs can change substantially even over fairly short evolutionary timescales (e.g.108, 109) (Fig. 5a). A seminal study by Ludwig et al.110 clearly demonstrated the phenomenon of TFBS turnover—TFBS loss in one location with compensatory TFBS gain in another—via fusion of the 5′ half of the eve_stripe2 CRM from D. melanogaster with the 3′ half of the orthologous CRM from D. pseudoobscura. Despite the fact that the two native sequences are easily alignable and function identically in transgenic D. melanogaster, most of the known TFBSs within them are incompletely conserved, and the spacing between them is varied. The result is that the chimeric CRM is completely non-functional. Recent large-scale functional analyses suggest that this is a common CRM feature.96 For this reason, comparative genomics for CRM discovery has proven particularly useful when used in the context of alignment-free sequence comparison frameworks,111 with or without concomitant TFBS identification, rather than in more standard alignment-based sequence conservation approaches (Figure 6).
Figure 6. TFBS conservation in aligned vs. alignment-free settings.
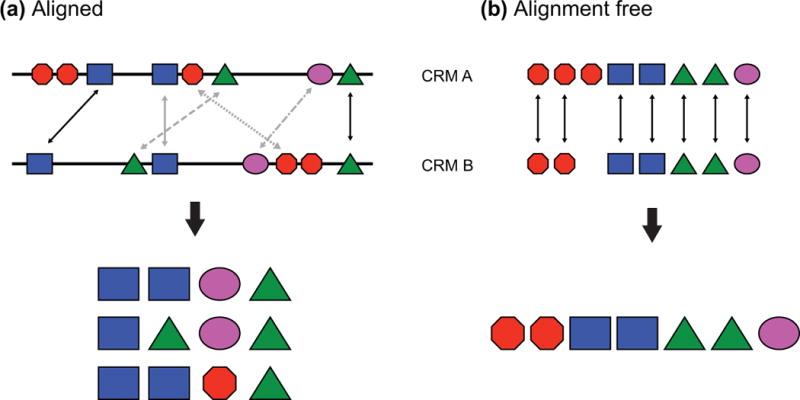
Each colored polygon represents a binding site. (a) When considering conservation based on sequence alignment only a fraction of the binding sites are seen to be conserved (4/8 for CRM A, 4/7 for CRM B), and several different alignments can be proposed. Arrows represent aligned sites, with gray arrows indicating alternative alignments. Note that choosing the proper alignment is significant, as the identities of the conserved sites are sensitive to the chosen alignment; in this example, presence of the sites represented by the purple oval and the red octagon depends on alignment choice. (b) In an alignment-free setting, TFBSs are identified and considered conserved if they appear in both sequences, regardless of how they are ordered. Using this approach, 7/8 sites from CRM A and all seven sites from CRM B are conserved. Moreover, the full complement of different sites is conserved, with merely a small reduction in the number of sites represented by the red octagon. The same principle applies to nucleotide-based (rather than motif-based) alignments, where subsequence (k-mer) composition can be substituted for motifs (see text).
Motif-based approaches
Just as ChIP-seq can be used to localize sequences which bind the TFs comprising a particular transcriptional code empirically and thus predict that they function as CRMs, computational identification of the TFBSs corresponding to a transcriptional code can be used for motif-based computational CRM prediction (“find the binding sites, find the enhancer”)(Fig. 5b). Motif-based CRM prediction was pioneered by Wasserman and Fickett,112 who trained a logistic regression classifier on a set of human muscle gene upstream sequences for which they had first determined a set of five co-occurring TFBSs. Although its discovery impact was limited by the fact that at the time there was no fully-sequenced genome on which to apply the model, this important study demonstrated the effectiveness of the transcriptional code approach to CRM discovery, the utility of using training sets of co-expressed genes to determine the transcriptional code in a statistically-sound, unbiased way, and the value of combining conserved-sequence data with motif-based predictions.
Genome-scale CRM predictions by several groups rapidly followed publication of the Drosophila genome in early 2000.113–117 The earliest implementations avoided the supervised, statistical approach of Wasserman and Fickett112 and used the extensive available knowledge of the CRMs (and their constituent TFBSs) involved in early fly pattern formation to simply search for clusters of TFBSs using PWM-based motif representations in sliding windows of a fixed size (e.g. 500 bp). Windows with matches to the motifs making up the presumed transcriptional code were considered predicted CRMs. The number of matches to each individual TFBS and whether or not a window needed to contain a match to each TFBS was based on the nature of the transcriptional code model and on assumptions about homotypic versus heterotypic TFBS clustering (see Box 3). Regardless of whether they were based on transcriptional code models derived from many known CRMs and stipulating dense TFBS clustering,113 or on a single CRM example with limited TFBS clustering,114 all of these analyses successfully discovered novel Drosophila CRMs. However, false-positive prediction rates—prediction of CRMs that subsequently failed to regulate reporter gene expression in in vivo validation assays—were high. As additional genomes were sequenced, it became possible to incorporate measures of sequence conservation into the searches, either over the entire length of the predicted CRM or just for the identified TFBSs. Both Berman et al.118 and Halfon et al.53 used comparisons to D. pseudoobscura sequence to filter the results of earlier, D. melanogaster-only CRM predictions, but although success rates improved, the gains were not dramatic.
Box 3. TFBS clustering.
CRMs are composed of multiple TFBSs. A cluster of binding sites for different TFs is referred to as a “heterotypic” cluster, whereas a series of sites for the same TF is called a “homotypic” cluster.177 Many CRMs are a mix of homotypic and heterotypic sites; that is, they contain multiple instances of multiple TFBSs (see Figure). Heterotypic clustering is an expected CRM attribute in keeping with the view that CRMs integrate a combinatorial transcriptional code. Many of the first and most well-characterized CRMs were those that regulate gene expression in the early, blastoderm-stage Drosophila embryo, which have a high degree of homotypic clustering. This helped to establish an oft-asserted view that homotypic clustering is a general feature of all CRMs,178 which seemed to be borne out as additional CRMs were identified through early computational discovery methods. However, this was at least partly due to ascertainment bias: since the methods functioned by looking for homotypic motif clusters,113, 115 that is what they found. Less-effective performance of these methods in mammalian systems subsequently led to the suggestion that mammalian CRMs were fundamentally different from insect CRMs in that they lacked significant homotypic clustering.179 However, neither of these views has stood up well to analysis of larger, unbiased collections of CRMs. Li et al.11 demonstrated that the Drosophila blastoderm CRMs have an atypically high degree of homotypic clustering that differentiates them from the majority of fly CRMs. Conversely, Gotea et al.180 studied a large collection of mammalian CRMs and were able to distinguish homotypic clustering in a substantial fraction. It thus appears that homotypic TFBS clustering is a common but not predominant feature of both fly and mammalian CRMs. Whether or not it has functional relevance or is simply an evolutionary artifact of binding site evolution and turnover has been widely debated.11, 181–185
Figure. Degrees of homotypic TFBS clustering.
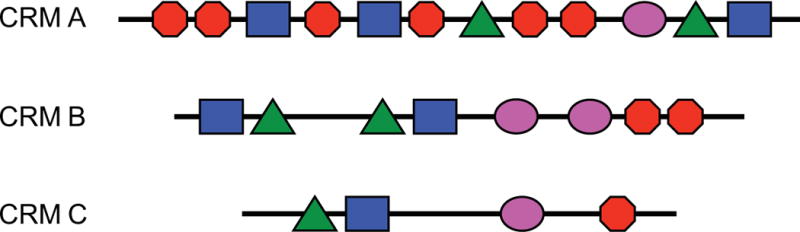
The three pictured CRMs each have a different level of homotypic TFBS clustering ranging from high (CRM A) to low (CRM B) to none (CRM C). All three CRMs have an identical degree of heterotypic site clustering. TFBS are represented by colored polygons.
Subsequent and more sophisticated implementations of the motif clustering approach returned to statistical classification and machine learning methods and in particular to probabilistic models such as Hidden Markov Models (HMMs)119; examples include Ahab,117 Cluster-Buster,120 PFR-searcher,121 and Stubb.122 HMMs, first implemented for CRM discovery by Crowley et al.,123 use a number of parameters such as TFBSs, clustering requirements, TF organization, etc. as “hidden states” to predict the sequence that is most likely to be a putative CRM as compared to a background sequence of non-CRM DNA. Many of these approaches again incorporate sequence conservation (e.g.101, 118, 121, 124, 125), either by limiting searching to conserved regions or through including sequence conservation as a state in the HMM. In a comparison of CRM discovery methods performed by Su et al.,97 Cluster-Buster120 was a top performer when only a single genome was considered, while MorphMS126 was superior when considering sequence conservation. The limitations of assessing conservation via whole-genome alignment is highlighted by the fact that MorphMS consistently outscored the similar StubbMS124; the primary difference between the two algorithms is that StubbMS relies on column-based sequence alignment followed by TFBS identification, whereas MorphMS uses a unified probabilistic approach that considers the evolution of TFBS sequences and finds motifs and alignments simultaneously. The PhylCRM algorithm127 similarly models binding site evolution over a set of aligned genomes prior to performing motif clustering, but has not been compared directly to related methods.
Despite the development of these elegant CRM discovery algorithms, motif-based searches still tend to suffer from a lack of specificity and give a large number of false positives, and methods relying primarily on motif clustering are consistently outperformed when compared directly with motif-agnostic methods.70, 97, 128 This somewhat disappointing performance of motif-based algorithms is likely due to several factors. There are few CRMs for which the entire set of bound TFs or relevant TFBSs is known, meaning that choices of which TFs to consider may often omit relevant factors. Conversely, not all motifs found within CRMs are functionally important, including those that may have been used as input for successful motif-based CRM discovery.53 TFBS motifs themselves are degenerate, and the motifs for many TFs are incompletely characterized, especially as pertaining to altered binding due to interactions with other TFs or local DNA conformations.129, 130 Overall, TFBS prediction is notoriously error-prone,37 with significant issues in terms of selecting an appropriate balance between sensitivity and specificity of results (see Box 1), and it is likely that this error rate propagates directly into motif-based CRM prediction efforts. Nevertheless, in those cases where the constituent TFBSs are well-defined and well-modeled, motif-based CRM discovery can be an effective approach.
Motif-blind approaches
Most often, a detailed transcriptional code is not known and TFBS data are incomplete. This, combined with the generally limited performance of motif-clustering methods, suggests a need for CRM discovery methods that do not rely on prior knowledge of TFBS motifs. CisModule131 (and the related MultiModule132) address this issue by attempting to learn both motifs and CRMs simultaneously from the input sequences. CisModule has shown strong performance in some settings, particularly for its motif finding phase.131, 133 However, in several comparisons it has been outperformed by other methods, both by those which rely on known motifs97 and by methods that do not rely on first predicting TFBSs.128 A more recent algorithm, Imogene, similarly learns motifs first and then uses these to search for CRMs.134 Imogene uses a training set of known CRMs, complemented with orthologous regions from related species (as in135) for the motif-learning phase, and in cross-validation tests has shown strong predictive performance.
Some of the most effective CRM discovery approaches in current use completely bypass TFBS identification, with its various attendant shortcomings, and predict CRMs based solely on DNA sequence (Fig. 5c). These “motif blind”70 methods rely on alignment-free sequence comparison measures and have been applied in both unsupervised and supervised settings.70, 128, 135–137 Although the former do not require any a priori knowledge of CRM sequences, the latter, which require a training set of known CRMs, have proven the most successful. The supervised motif-blind methods determine statistical features of the CRM sequences in the training set as compared to those of a background set of non-CRM sequences, and then search the genome for sequence windows with similar attributes (Fig. 7). This paradigm has been explored in depth by the Sinha and Halfon groups, who have developed “SCRMshaw,” a set of methods that use various machine learning algorithms to identify sequence “words” or “k-mers” (i.e., short DNA subsequences) over-represented in a training set of known CRMs that regulate gene expression in a related pattern.70, 135 Note that like motif-based methods, this approach still relies on the idea of a common transcriptional code through which similarly-expressed genes are regulated by CRMs that bind a similar complement of TFs. However, here the k-mers stand as proxies for the TFBSs, which are not explicitly considered by the algorithm and do not need to be known or modeled. Sequence conservation is taken into account by adding sequences orthologous to the training CRMs drawn from aligned related genomes, allowing conserved k-mers to acquire higher weights without requiring a specific model of TFBS evolution and without unduly penalizing CRMs that are not highly conserved (as conservation is not required for consideration of a sequence or subsequence). The SCRMshaw supervised motif-blind method has been applied to both Drosophila and mouse with in vivo validation revealing successful CRM discovery rates averaging about 80% (100% for some training sets), and SCRMshaw has consistently scored as well or better than motif-based methods applied in similar settings.70 Outperformance of motif-based approaches likely stems from the fact that all subsequences are considered, so unknown/unidentified motifs are not ignored, and from the reduction in error achieved by not relying on often inaccurate motif prediction steps.
Figure 7. Supervised motif-blind CRM discovery.
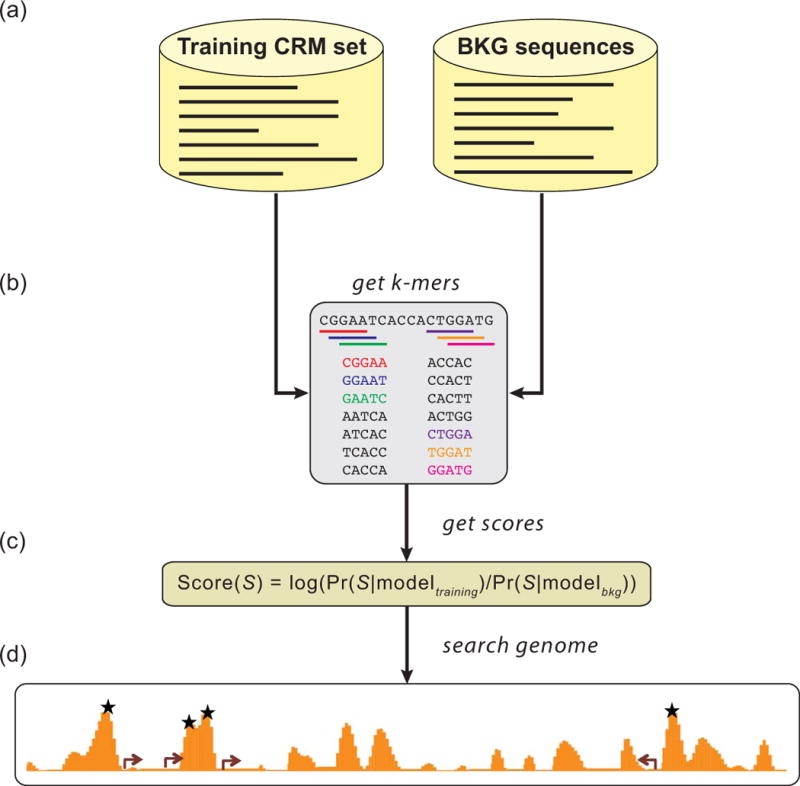
(a) A set of CRMs with related activity (e.g., midbrain, heart, wing, muscle) is selected as a training set, and a set of similarly-sized non-CRMs as a background (BKG) set. The training set can also include orthologous sequences from related species. (b) The k-mer profile of the sequence sets is obtained and used to train one of several statistical models. (c) The score for a given sequence S is the log-likelihood ratio of the models for the positive (“training”) and negative (“background”) sets on S. (d) Overlapping sequence windows are scored throughout the genome. High-scoring windows (stars) are predicted CRMs.
A variety of other supervised CRM discovery methods have been developed over the years, in particular classification-based methods employing Support Vector Machines (SVMs; e.g. kmerSVM,138, 139 KIRMES140). In general, these methods perform well when the training sets are comprehensive, with a definite trend toward motif-blind approaches trumping motif-aware approaches, although for many methods evaluation has been based on limited in-silico cross-validation and not on direct in vivo testing of predicted CRM sequences. So far, the strongest reported success rates for validated discovery of novel CRMs are those obtained from the SCRMshaw motif-blind pipeline (although only very limited mammalian CRM discovery has been attempted with this method70). However, direct comparisons between methods is difficult other than in cases where the same training data and assessment criteria are used, and further evaluation along the lines of that performed by Su et al.97 will be necessary to determine the most effective strategies.
Integrated approaches
As greater numbers of CRMs are discovered, the opportunities for supervised learning approaches improve. An effective method developed by Narlikar et al.71 and subsequently dubbed “CLARE” (Cracking the Language of Regulatory Elements)141 returns to the linear regression approach first put forward by Wasserman and Fickett112 by taking advantage of the vastly increased CRM and TFBS data that have become available in the intervening years. Like other supervised methods, CLARE takes as input a training set of CRMs with common activity and a set of non-CRM control sequences. The sequences are then searched for (1) the presence of TFBSs, using available motif libraries for all known (vertebrate) TFs; (2) novel overrepresented motifs, using a Gibbs-sampling de novo motif discovery approach; and (3) overrepresented k-mers, using Markov chain discrimination. These results are then passed to the LASSO linear regression algorithm to develop a classifier to predict CRMs from genomic sequence (Fig. 8). In cross-validation tests this method outperformed strictly motif-based methods such as Cluster-Buster,120 Stubb,122 and CisModule,131 and achieved a success rate of 62% in in vivo validations in transgenic zebrafish, putting it on par with ChIP-based empirical CRM discovery methods.
Figure 8. CLARE: Cracking the Language of Regulatory Elements.
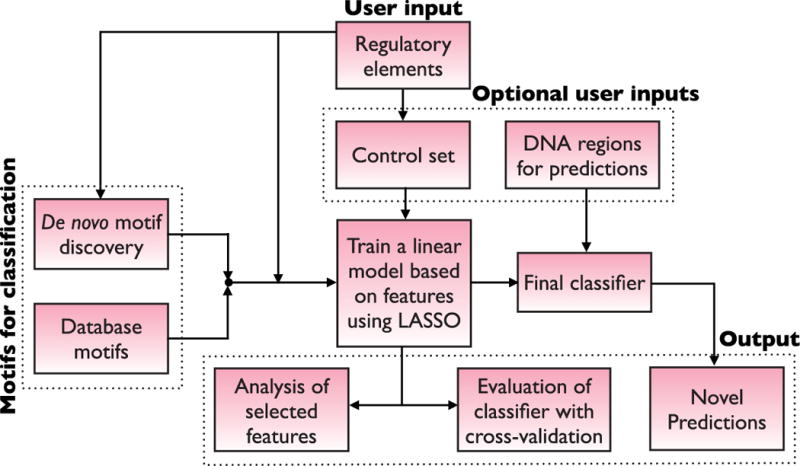
Flowchart of the CLARE method. Figure from Taher et al. (2012),141 © Oxford University Press, used with permission.
EnhancerFinder142 is another example of a supervised CRM prediction pipeline that assesses various genomic features of a CRM training set including sequence conservation, k-mer counts, and p300 binding and histone modifications from ChIP-seq data. These data are integrated using a machine-learning method known as multiple kernel learning and used to predict CRMs genome-wide. A second phase of the algorithm builds classifiers to attempt to determine the tissues in which the CRMs are active. The latter aspect is noteworthy, as CRM discovery in general has proven a much easier prospect than accurate prediction of CRM tissue specificity.70, 135 In cross-validation testing EnhancerFinder outperformed CLARE when tested on the same training data.
i-CisTarget,143 on the other hand, is a promising integrative unsupervised approach, i.e., one that does not require a training set of known CRMs. Similar to previous unsupervised methods,121, 128 the input is a set of co-expressed genes or, more uniquely, a set of genomic regions drawn, for example, from a set of ChIP-seq peaks. i-CisTarget then calculates, for a predefined set of sequences around these loci (akin to what Ivan et al.128 refer to as the “control region” for each gene), enrichment scores for motifs from a TFBS motif library as well as for a variety of features based on ChIP and other genomic data. Output consists of a list of enriched features and predicted CRMs. The related cisTargetX,144 which uses motif data only, has been effective in discovering previously unknown CRMs (subsequently confirmed in vivo) as has the conceptually similar PhylCRM/Lever method.127 To what extent integrating genomic features in addition to TFBS motifs will improve CRM discovery for these approaches is not yet certain. A significant drawback to unsupervised approaches like i-CisTarget is their restriction to a “control region” in proximity to the genes of interest, which precludes their ever being able to identify CRMs that lie outside the analyzed sequences. On the other hand, they do not require training sets of known CRMs, which are not available for all regulatory networks and cannot be compiled for organisms in which few CRMs have yet been identified.
Cross-species CRM prediction
Supervised CRM discovery for regulatory networks currently lacking any known CRMs poses a difficult challenge in terms of acquiring the requisite training data, but supervised CRM discovery for organisms with few characterized CRMs has recently been shown to be an obtainable goal as long as there are sufficient CRM data available for a related species. Kazemian et al.145 used training sets composed of Drosophila CRMs to successfully undertake CRM discovery in other insects, including the Hymenopteran species Apis mellifera (honeybee) and Nasonia vitripennis (jewel wasp), which diverged from flies roughly 350 million years ago.146 At this evolutionary distance, which in terms of molecular divergence likely exceeds that between humans and pufferfish,147 noncoding regions are essentially unalignable, preventing the application of any alignment-based CRM discovery approaches. However, the SCRMshaw motif-blind pipeline showed strong CRM prediction performance based on Drosophila training data, with in vivo validation providing an approximately 75% prediction success rate as inferred from reporter gene assays in transgenic flies. Thus the underlying transcriptional codes involved in many developmental processes appear to be conserved enough to be identified through alignment-free comparison, despite the unalignable nature of the orthologous regulatory sequences.
ASSIGNING CRMS TO GENES
As CRM discovery moves from targeted analyses of individual loci to large-scale prediction by either empirical or computational means, a significant problem becomes identifying the target gene (or genes) which a CRM is regulating. Many studies, for the sake of simplicity, use “the closest active gene” criterion to assign CRMs to target genes. However, there are abundant examples in which such assignments do not hold true, and a recent large-scale study by Kvon et al.148 that analyzed over 7000 Drosophila transgenic reporter constructs suggests that 30% or more of CRMs target a gene other than one of their two immediate neighbors. Therefore, assigning CRM target genes based on proximity is a risky assumption in the absence of in vivo spatio-temporal expression data for both gene and CRM activity. CRM-gene contacts can be identified using the chromosome conformation capture (3C) assay and its many higher-throughput variants (4C, 5C, Hi-C, ChIA-PET).149 However, these assays have limited resolution, particularly in identifying short-range contacts, and interpretation of results is not always straightforward. For instance, known CRMs are frequently observed making multiple contacts,150, 151 but whether this means that they are regulating more than a single target gene is not known. CRMs also contact other CRMs,150 raising the possibility that at least some of the observed interactions may reflect proximity induced by localization of multiple distinct active CRM-promoter pairs to the same nuclear region (e.g., the same transcription factory152), rather than co-regulation by a single CRM. Accurate CRM-gene assignment therefore remains an important area in need of further development, as the number of “orphan” CRMs continues to increase rapidly.
RESOURCES FOR TRAINING DATA
The most effective computational methods we have discussed here rely on the availability of collections of known CRMs and/or on libraries of TFBS motifs. Indeed, the increased efficacy of CRM discovery in recent years is due at least in part to the much greater amounts of input data that are currently available. There are several resources for motif data, as discussed previously. Options for CRM training data are more limited. For vertebrates, the main available resource is the VISTA Enhancer Browser.153 This exceptional resource contains in vivo validated CRM data—sequences and images—for over 2100 sequence fragments assayed in transgenic mouse embryos, over half of which show regulatory activity. Most of the data are for CRMs predicted from ChIP-seq analysis of p300-bound regions from mouse embryonic day 11.5 limb, forebrain, and midbrain tissues, although it also includes regions obtained from comparative genomic analysis across multiple vertebrate species. The main drawback to the Enhancer Browser is that all analysis is performed at a single embryonic stage, so that any activity of the tested sequences at other timepoints is unknown. Also, the reporter gene activity is described in very broad anatomical terms, limiting the specificity of tissue-specific training sets that can be compiled from the data. Nevertheless, this ongoing project provides a powerful source of training data to facilitate vertebrate CRM discovery.
The other major source of CRM training data is REDfly, the Regulatory Element Database for Drosophila.154 REDfly takes a biocuration approach, seeking to annotate all of the verified Drosophila CRMs that have been reported in the literature. This makes REDfly, with more than 5500 CRMs based on analysis of over 11,600 reporter constructs, the broadest and most unbiased available collection of CRMs for any metazoan. REDfly also curates known TFBSs, which are cross-referenced with the CRMs for easy identification of TFBSs that lie within a CRM. One strength of REDfly as a resource for CRM discovery is its extensive array of search functions. Regulatory activity is described using the Drosophila anatomy ontology,155 which allows for tissue-specific CRM datasets of different granularity to be assembled. CRMs can be filtered by size, genomic location, and position relative to target genes (e.g., upstream, downstream, intronic). Overlapping regions between multiple CRMs are automatically calculated to suggest minimal CRM sequences and the regulatory activity of these inferred CRMs. The core REDfly CRM annotations are provided to FlyBase,156 making Drosophila the only model organism whose genome annotation provides comprehensive coverage of validated CRMs and providing direct integration with other Drosophila genomic and genetic data.
OUTSTANDING ISSUES IN REGULATORY ELEMENT DISCOVERY
We have focused our discussion of CRM discovery here on enhancers and similar positive-acting regulatory sequences. Effective methods for large-scale discovery of other types of cis-regulatory sequences are still in need of development. Although ChIP-based experimental or motif-based computational methods can (and have) been applied to discovery of silencers and insulators, a lack of good functional validation assays leaves prediction of these elements open to question, and high-quality, verified collections such as the CRM collections contained in REDfly and the VISTA Enhancer Browser are not available.
Even for enhancers, there are considerable gaps. Empirical assays require purified tissue or defined cell types, and complete coverage in even a single organism is still a long way off. Although computational methods can address this in part, and even help discover CRMs in organisms for which there is little or no experimental data,145 requirements for training data or motif libraries remain a significant limitation. While motif data are becoming more comprehensive, motif-based methods tend to suffer from lower accuracy than motif-blind approaches and are most effective for motif-dense, heavily homotypically-clustered CRMs, which comprise only a subset of regulatory regions. For both empirical and computational approaches, most of the current methods assume that CRMs are relatively compact, binding-site rich DNA segments such as CRMs A and B in Box 3. However, as we and others have argued previously, other CRM architectures may exist in which TFBSs are spread over long regions of the DNA or in which multiple separated regions act as a composite CRM to regulate gene expression.157, 158
It is important to stress that the majority of methods discussed in this review—both computational and experimental—are merely predictive, and biological validation is essential before assigning a definitive regulatory function to a genomic region. Although reporter gene assays remain the gold standard for CRM validation due to their overall efficacy and relative ease of use, they possess a number of shortcomings that are worth bearing in mind. In particular, the potential for false-negative results—true CRMs which fail to activate reporter gene expression—is high and can stem from a number of factors. One, a sequence could have regulatory activity at a time or in a cell type not assayed. This is especially true for assays performed in cell culture, where only a single cell type is tested, but holds as well for in vivo assays, where it is unusual for all life-cycle stages to be assessed. Two, it is well known that certain CRMs will only work properly when paired with the correct promoter.159 Thus, an incompatible CRM-promoter pairing will cause a true CRM to fail to activate gene expression. Three, reporter gene assays cannot detect negative regulatory elements (e.g., transcriptional silencers), meaning that only positive-acting enhancer-like CRMs will be detected. Four, typical reporter gene assays place the CRMs in close proximity to the promoter, which is in contrast to the native genomic positioning of most regulatory sequences. Although the classical definition of an “enhancer” stipulates distance independence, there are clear examples in which CRM-promoter distance has important effects.160, 161 Finally, most reporter gene assays take place in an artificial, non-native genomic context where they might be influenced by other nearby CRMs, promoters, or chromatin configurations.162
Alternatives to the standard reporter gene assay include direct deletion or mutation of a putative CRM by manipulating large, native-like regions using bacterial artificial chromosomes (BACs) or using genome engineering methods to alter the endogenous locus in cultured cells or in vivo. Although the latter has long been prohibitively expensive, time-consuming, and/or challenging in most model organisms, recent successes with CRISPR/Cas9 genome engineering in multiple systems163 raise the possibility that direct tests of putative CRMs in a completely native chromosomal context may soon become routine. Deleting or mutating sequences in their native locus would provide a vital complement to reporter gene assays and greatly facilitate the identification and study of silencers, insulators, and other regulatory features.
CONCLUDING REMARKS
One of the vital but daunting challenges confronting us today is to annotate the regulatory genomes of the rapidly-growing number of sequenced organisms. Despite the Herculean nature of this task, dramatic strides forward are continuing to be made. Genome-scale approaches fueled by advances in next-generation sequencing have already led to the prediction and in many cases validation of thousands of new CRMs in all of the major model organisms, including humans. Decreasing costs coupled with the ability to apply genomic assays to increasingly small numbers of cells—in some cases even single cells—will allow for detection of cell-type-specific CRMs and TF binding events and help to remove the potentially confounding effect of having experimental results reflect an average of genetic and epigenetic events within multiple different cell types. Genome engineering methods such as the CRISPR/Cas9 system will open up heretofore non-model organisms to experimental analysis and will enable new assays to better detect additional types of regulatory elements beyond just enhancers. Computational methods, which have matured greatly over the last dozen years, can now predict with growing accuracy CRMs in both model and non-model organisms with comparable success rates, an ability that will continue to grow as the corpus of regulatory genomic data for key model organisms increases. In this era of rapid genome sequencing, it is important to recognize that in order to take advantage of the availability of genomics to improve our understanding of development, evolution, and disease, characterization of regulatory sequences is arguably as necessary as identification of the genes themselves. Fortunately, the methods and studies reviewed in this article suggest that the outlook for regulatory element discovery has never been brighter.
Acknowledgments
We thank John Nyquist for help with illustrations, Jim Jaynes, Miki Fujioka, and Satrajit Sinha for providing images, and Michael Buck for comments on the manuscript. We apologize to all those whose valuable contributions to the field we were unable to acknowledge due to space limitations. Work in the Halfon laboratory is supported by the National Institutes of Health (R01 GM85233), the U. S. Department of Agriculture (2011-04656), and the National Science Foundation (DBI-1355511).
RELATED ARTICLES
- Cho KW. Enhancers. Wiley Interdiscip Rev Dev Biol. 2012;1(4):469–478. doi: 10.1002/wdev.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadonaga JT. Perspectives on the RNA polymerase II core promoter. Wiley Interdiscip Rev Dev Biol. 2012;1(1):40–51. doi: 10.1002/wdev.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noordermeer D, Duboule D. Chromatin looping and organization at developmentally regulated gene loci. Wiley Interdiscip Rev Dev Biol. 2013;2(5):615–630. doi: 10.1002/wdev.103. [DOI] [PubMed] [Google Scholar]
References
- 1.Pick L, Heffer A. Hox gene evolution: multiple mechanisms contributing to evolutionary novelties. Ann N Y Acad Sci. 2012;1256:15–32. doi: 10.1111/j.1749-6632.2011.06385.x. [DOI] [PubMed] [Google Scholar]
- 2.Smith E, Shilatifard A. Enhancer biology and enhanceropathies. Nat Struct Mol Biol. 2014;21:210–219. doi: 10.1038/nsmb.2784. [DOI] [PubMed] [Google Scholar]
- 3.Bulger M, Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011;144:327–339. doi: 10.1016/j.cell.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maston GA, Landt SG, Snyder M, Green MR. Characterization of enhancer function from genome-wide analyses. Annu Rev Genomics Hum Genet. 2012;13:29–57. doi: 10.1146/annurev-genom-090711-163723. [DOI] [PubMed] [Google Scholar]
- 5.Ong CT, Corces VG. Enhancers: emerging roles in cell fate specification. EMBO Rep. 2012;13:423–430. doi: 10.1038/embor.2012.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spitz F, Furlong EE. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet. 2012;13:613–626. doi: 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- 7.Schaffner W. Encyclopedia of Molecular Biology. John Wiley & Sons, Inc; 2002. Enhancer. [Google Scholar]
- 8.Maston GA, Evans SK, Green MR. Transcriptional regulatory elements in the human genome. Annu Rev Genomics Hum Genet. 2006;7:29–59. doi: 10.1146/annurev.genom.7.080505.115623. [DOI] [PubMed] [Google Scholar]
- 9.Weake VM, Workman JL. Inducible gene expression: diverse regulatory mechanisms. Nat Rev Genet. 2010;11:426–437. doi: 10.1038/nrg2781. [DOI] [PubMed] [Google Scholar]
- 10.Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell. 2013;49:825–837. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li L, Zhu Q, He X, Sinha S, Halfon MS. Large-scale analysis of transcriptional cis-regulatory modules reveals both common features and distinct subclasses. Genome Biol. 2007;8:R101. doi: 10.1186/gb-2007-8-6-r101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lam MT, Li W, Rosenfeld MG, Glass CK. Enhancer RNAs and regulated transcriptional programs. Trends Biochem Sci. 2014;39:170–182. doi: 10.1016/j.tibs.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mousavi K, Zare H, Koulnis M, Sartorelli V. The emerging roles of eRNAs in transcriptional regulatory networks. RNA Biol. 2014;11:106–110. doi: 10.4161/rna.27950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bajic VB, Tan SL, Suzuki Y, Sugano S. Promoter prediction analysis on the whole human genome. 2004;22:1467–1473. doi: 10.1038/nbt1032. [DOI] [PubMed] [Google Scholar]
- 15.Wang Z, Chen Y, Li Y. A brief review of computational gene prediction methods. Genomics Proteomics Bioinformatics. 2004;2:216–221. doi: 10.1016/S1672-0229(04)02028-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jolma A, Taipale J. Methods for Analysis of Transcription Factor DNA-Binding Specificity In Vitro. Subcell Biochem. 2011;52:155–173. doi: 10.1007/978-90-481-9069-0_7. [DOI] [PubMed] [Google Scholar]
- 17.MacIsaac KD, Fraenkel E. Practical strategies for discovering regulatory DNA sequence motifs. PLoS Comput Biol. 2006;2:e36. doi: 10.1371/journal.pcbi.0020036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zambelli F, Pesole G, Pavesi G. Motif discovery and transcription factor binding sites before and after the next-generation sequencing era. Brief Bioinform. 2013;14:225–237. doi: 10.1093/bib/bbs016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maerkl SJ, Quake SR. A systems approach to measuring the binding energy landscapes of transcription factors. Science. 2007;315:233–237. doi: 10.1126/science.1131007. [DOI] [PubMed] [Google Scholar]
- 20.Gubelmann C, Waszak SM, Isakova A, Holcombe W, Hens K, Iagovitina A, Feuz JD, Raghav SK, Simicevic J, Deplancke B. A yeast one-hybrid and microfluidics-based pipeline to map mammalian gene regulatory networks. Mol Syst Biol. 2013;9:682. doi: 10.1038/msb.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hens K, Feuz JD, Isakova A, Iagovitina A, Massouras A, Bryois J, Callaerts P, Celniker SE, Deplancke B. Automated protein-DNA interaction screening of Drosophila regulatory elements. Nat Methods. 2011;8:1065–1070. doi: 10.1038/nmeth.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Slattery M, Riley T, Liu P, Abe N, Gomez-Alcala P, Dror I, Zhou T, Rohs R, Honig B, Bussemaker HJ, et al. Cofactor binding evokes latent differences in DNA binding specificity between Hox proteins. Cell. 2011;147:1270–1282. doi: 10.1016/j.cell.2011.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jolma A, Kivioja T, Toivonen J, Cheng L, Wei G, Enge M, Taipale M, Vaquerizas JM, Yan J, Sillanpaa MJ, et al. Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Res. 2010;20:861–873. doi: 10.1101/gr.100552.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berger MF, Philippakis AA, Qureshi AM, He FS, Estep PW, 3rd, Bulyk ML. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat Biotechnol. 2006;24:1429–1435. doi: 10.1038/nbt1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Noyes MB, Meng X, Wakabayashi A, Sinha S, Brodsky MH, Wolfe SA. A systematic characterization of factors that regulate Drosophila segmentation via a bacterial one-hybrid system. Nucleic Acids Res. 2008;36:2547–2560. doi: 10.1093/nar/gkn048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parker DS, White MA, Ramos AI, Cohen BA, Barolo S. The cis-regulatory logic of Hedgehog gradient responses: key roles for gli binding affinity, competition, and cooperativity. Sci Signal. 2011;4:ra38. doi: 10.1126/scisignal.2002077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buck MJ, Lieb JD. ChIP-chip: considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments. Genomics. 2004;83:349–360. doi: 10.1016/j.ygeno.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 28.Mardis ER. ChIP-seq: welcome to the new frontier. Nat Methods. 2007;4:613–614. doi: 10.1038/nmeth0807-613. [DOI] [PubMed] [Google Scholar]
- 29.van Steensel B, Henikoff S. Identification of in vivo DNA targets of chromatin proteins using tethered dam methyltransferase. Nat Biotechnol. 2000;18:424–428. doi: 10.1038/74487. [DOI] [PubMed] [Google Scholar]
- 30.Rhee HS, Pugh BF. ChIP-exo method for identifying genomic location of DNA-binding proteins with near-single-nucleotide accuracy. Curr Protoc Mol Biol. 2012 doi: 10.1002/0471142727.mb2124s100. Chapter 21: Unit 21 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He Q, Johnston J, Zeitlinger J. A novel ChIP-exo method reveals genome-wide in vivo transcription factor binding footprints influenced by local DNA sequence. Nat Biotechnol. 2014 doi: 10.1038/nbt.3121. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley-Hunt R, Arenillas DJ, Buchman S, Chen CY, Chou A, Ienasescu H, et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2014;42:D142–147. doi: 10.1093/nar/gkt997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robasky K, Bulyk ML. UniPROBE, update 2011: expanded content and search tools in the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Res. 2011;39:D124–128. doi: 10.1093/nar/gkq992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu LJ, Christensen RG, Kazemian M, Hull CJ, Enuameh MS, Basciotta MD, Brasefield JA, Zhu C, Asriyan Y, Lapointe DS, et al. FlyFactorSurvey: a database of Drosophila transcription factor binding specificities determined using the bacterial one-hybrid system. Nucleic Acids Res. 2011;39:D111–117. doi: 10.1093/nar/gkq858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fisher WW, Li JJ, Hammonds AS, Brown JB, Pfeiffer BD, Weiszmann R, MacArthur S, Thomas S, Stamatoyannopoulos JA, Eisen MB, et al. DNA regions bound at low occupancy by transcription factors do not drive patterned reporter gene expression in Drosophila. Proc Natl Acad Sci U S A. 2012;109:21330–21335. doi: 10.1073/pnas.1209589110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wasserman WW, Sandelin A. Applied bioinformatics for the identification of regulatory elements. Nat Rev Genet. 2004;5:276–287. doi: 10.1038/nrg1315. [DOI] [PubMed] [Google Scholar]
- 38.Banerji J, Rusconi S, Schaffner W. Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell. 1981;27:299–308. doi: 10.1016/0092-8674(81)90413-x. [DOI] [PubMed] [Google Scholar]
- 39.Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet. 2014;15:272–286. doi: 10.1038/nrg3682. [DOI] [PubMed] [Google Scholar]
- 40.Zeitlinger J, Zinzen RP, Stark A, Kellis M, Zhang H, Young RA, Levine M. Whole-genome ChIP-chip analysis of Dorsal, Twist, and Snail suggests integration of diverse patterning processes in the Drosophila embryo. Genes Dev. 2007;21:385–390. doi: 10.1101/gad.1509607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zinzen RP, Girardot C, Gagneur J, Braun M, Furlong EE. Combinatorial binding predicts spatio-temporal cis-regulatory activity. Nature. 2009;462:65–70. doi: 10.1038/nature08531. [DOI] [PubMed] [Google Scholar]
- 42.Cheng Y, King DC, Dore LC, Zhang X, Zhou Y, Zhang Y, Dorman C, Abebe D, Kumar SA, Chiaromonte F, et al. Transcriptional enhancement by GATA1-occupied DNA segments is strongly associated with evolutionary constraint on the binding site motif. Genome Res. 2008;18:1896–1905. doi: 10.1101/gr.083089.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Negre N, Brown CD, Ma L, Bristow CA, Miller SW, Wagner U, Kheradpour P, Eaton ML, Loriaux P, Sealfon R, et al. A cis-regulatory map of the Drosophila genome. Nature. 2011;471:527–531. doi: 10.1038/nature09990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rhee HS, Pugh BF. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 2011;147:1408–1419. doi: 10.1016/j.cell.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moorman C, Sun LV, Wang J, de Wit E, Talhout W, Ward LD, Greil F, Lu XJ, White KP, Bussemaker HJ, et al. Hotspots of transcription factor colocalization in the genome of Drosophila melanogaster. Proc Natl Acad Sci U S A. 2006;103:12027–12032. doi: 10.1073/pnas.0605003103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science. 2010;330:1775–1787. doi: 10.1126/science.1196914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yip KY, Cheng C, Bhardwaj N, Brown JB, Leng J, Kundaje A, Rozowsky J, Birney E, Bickel P, Snyder M, et al. Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcription-related factors. Genome Biol. 2012;13:R48. doi: 10.1186/gb-2012-13-9-r48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kvon EZ, Stampfel G, Yanez-Cuna JO, Dickson BJ, Stark A. HOT regions function as patterned developmental enhancers and have a distinct cis-regulatory signature. Genes Dev. 2012;26:908–913. doi: 10.1101/gad.188052.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li XY, MacArthur S, Bourgon R, Nix D, Pollard DA, Iyer VN, Hechmer A, Simirenko L, Stapleton M, Luengo Hendriks CL, et al. Transcription factors bind thousands of active and inactive regions in the Drosophila blastoderm. PLoS Biol. 2008;6:e27. doi: 10.1371/journal.pbio.0060027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cao Y, Yao Z, Sarkar D, Lawrence M, Sanchez GJ, Parker MH, MacQuarrie KL, Davison J, Morgan MT, Ruzzo WL, et al. Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev Cell. 2010;18:662–674. doi: 10.1016/j.devcel.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park D, Lee Y, Bhupindersingh G, Iyer VR. Widespread misinterpretable ChIP-seq bias in yeast. PLoS One. 2013;8:e83506. doi: 10.1371/journal.pone.0083506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Teytelman L, Thurtle DM, Rine J, van Oudenaarden A. Highly expressed loci are vulnerable to misleading ChIP localization of multiple unrelated proteins. Proc Natl Acad Sci U S A. 2013;110:18602–18607. doi: 10.1073/pnas.1316064110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Halfon MS, Zhu Q, Brennan ER, Zhou Y. Erroneous attribution of relevant transcription factor binding sites despite successful prediction of cis-regulatory modules. BMC Genomics. 2011;12:578. doi: 10.1186/1471-2164-12-578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Blow MJ, McCulley DJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. ChIP-Seq identification of weakly conserved heart enhancers. Nat Genet. 2010;42:806–810. doi: 10.1038/ng.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.May D, Blow MJ, Kaplan T, McCulley DJ, Jensen BC, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, et al. Large-scale discovery of enhancers from human heart tissue. Nat Genet. 2012;44:89–93. doi: 10.1038/ng.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 61.modEncode Consortium. Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, Landolin JM, Bristow CA, Ma L, et al. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science. 2010;330:1787–1797. doi: 10.1126/science.1198374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Encode Project Consortium. Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488:116–120. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bonn S, Zinzen RP, Girardot C, Gustafson EH, Perez-Gonzalez A, Delhomme N, Ghavi-Helm Y, Wilczynski B, Riddell A, Furlong EE. Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat Genet. 2012;44:148–156. doi: 10.1038/ng.1064. [DOI] [PubMed] [Google Scholar]
- 66.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ernst J, Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods. 2012;9:215–216. doi: 10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hoffman MM, Buske OJ, Wang J, Weng Z, Bilmes JA, Noble WS. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat Methods. 2012;9:473–476. doi: 10.1038/nmeth.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kwasnieski JC, Fiore C, Chaudhari HG, Cohen BA. High-throughput functional testing of ENCODE segmentation predictions. Genome Res. 2014 doi: 10.1101/gr.173518.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kantorovitz MR, Kazemian M, Kinston S, Miranda-Saavedra D, Zhu Q, Robinson GE, Gottgens B, Halfon MS, Sinha S. Motif-blind, genome-wide discovery of cis-regulatory modules in Drosophila and mouse. Dev Cell. 2009;17:568–579. doi: 10.1016/j.devcel.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Narlikar L, Sakabe NJ, Blanski AA, Arimura FE, Westlund JM, Nobrega MA, Ovcharenko I. Genome-wide discovery of human heart enhancers. Genome Res. 2010;20:381–392. doi: 10.1101/gr.098657.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bell O, Tiwari VK, Thoma NH, Schubeler D. Determinants and dynamics of genome accessibility. Nat Rev Genet. 2011;12:554–564. doi: 10.1038/nrg3017. [DOI] [PubMed] [Google Scholar]
- 73.Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gross DS, Garrard WT. Nuclease hypersensitive sites in chromatin. Annu Rev Biochem. 1988;57:159–197. doi: 10.1146/annurev.bi.57.070188.001111. [DOI] [PubMed] [Google Scholar]
- 75.Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132:311–322. doi: 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, Thurman RE, Neph S, Kuehn MS, Noble WS, et al. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods. 2009;6:283–289. doi: 10.1038/nmeth.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007;17:877–885. doi: 10.1101/gr.5533506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Giresi PG, Lieb JD. Isolation of active regulatory elements from eukaryotic chromatin using FAIRE (Formaldehyde Assisted Isolation of Regulatory Elements) Methods. 2009;48:233–239. doi: 10.1016/j.ymeth.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Adey A, Morrison HG, Asan, Xun X, Kitzman JO, Turner EH, Stackhouse B, MacKenzie AP, Caruccio NC, Zhang X, et al. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol. 2010;11:R119. doi: 10.1186/gb-2010-11-12-r119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kwasnieski JC, Mogno I, Myers CA, Corbo JC, Cohen BA. Complex effects of nucleotide variants in a mammalian cis-regulatory element. Proc Natl Acad Sci U S A. 2012;109:19498–19503. doi: 10.1073/pnas.1210678109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nam J, Davidson EH. Barcoded DNA-tag reporters for multiplex cis-regulatory analysis. PLoS One. 2012;7:e35934. doi: 10.1371/journal.pone.0035934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Patwardhan RP, Hiatt JB, Witten DM, Kim MJ, Smith RP, May D, Lee C, Andrie JM, Lee SI, Cooper GM, et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol. 2012;30:265–270. doi: 10.1038/nbt.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Melnikov A, Murugan A, Zhang X, Tesileanu T, Wang L, Rogov P, Feizi S, Gnirke A, Callan CG, Jr, Kinney JB, et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol. 2012;30:271–277. doi: 10.1038/nbt.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sharon E, Kalma Y, Sharp A, Raveh-Sadka T, Levo M, Zeevi D, Keren L, Yakhini Z, Weinberger A, Segal E. Inferring gene regulatory logic from high-throughput measurements of thousands of systematically designed promoters. Nat Biotechnol. 2012;30:521–530. doi: 10.1038/nbt.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.White MA, Myers CA, Corbo JC, Cohen BA. Massively parallel in vivo enhancer assay reveals that highly local features determine the cis-regulatory function of ChIP-seq peaks. Proc Natl Acad Sci U S A. 2013;110:11952–11957. doi: 10.1073/pnas.1307449110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dickel DE, Zhu Y, Nord AS, Wylie JN, Akiyama JA, Afzal V, Plajzer-Frick I, Kirkpatrick A, Gottgens B, Bruneau BG, et al. Function-based identification of mammalian enhancers using site-specific integration. Nat Methods. 2014;11:566–571. doi: 10.1038/nmeth.2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Murtha M, Tokcaer-Keskin Z, Tang Z, Strino F, Chen X, Wang Y, Xi X, Basilico C, Brown S, Bonneau R, et al. FIREWACh: high-throughput functional detection of transcriptional regulatory modules in mammalian cells. Nat Methods. 2014;11:559–565. doi: 10.1038/nmeth.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gisselbrecht SS, Barrera LA, Porsch M, Aboukhalil A, Estep PW, 3rd, Vedenko A, Palagi A, Kim Y, Zhu X, Busser BW, et al. Highly parallel assays of tissue-specific enhancers in whole Drosophila embryos. Nat Methods. 2013;10:774–780. doi: 10.1038/nmeth.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Arnold CD, Gerlach D, Stelzer C, Boryn LM, Rath M, Stark A. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science. 2013;339:1074–1077. doi: 10.1126/science.1232542. [DOI] [PubMed] [Google Scholar]
- 91.Aerts S. Computational strategies for the genome-wide identification of cis-regulatory elements and transcriptional targets. Curr Top Dev Biol. 2012;98:121–145. doi: 10.1016/B978-0-12-386499-4.00005-7. [DOI] [PubMed] [Google Scholar]
- 92.Haeussler M, Joly JS. When needles look like hay: how to find tissue-specific enhancers in model organism genomes. Dev Biol. 2011;350:239–254. doi: 10.1016/j.ydbio.2010.11.026. [DOI] [PubMed] [Google Scholar]
- 93.Van Loo P, Marynen P. Computational methods for the detection of cis-regulatory modules. Brief Bioinform. 2009;10:509–524. doi: 10.1093/bib/bbp025. [DOI] [PubMed] [Google Scholar]
- 94.Miller W, Makova KD, Nekrutenko A, Hardison RC. Comparative genomics. Annu Rev Genomics Hum Genet. 2004;5:15–56. doi: 10.1146/annurev.genom.5.061903.180057. [DOI] [PubMed] [Google Scholar]
- 95.Hardison RC. Conserved noncoding sequences are reliable guides to regulatory elements. Trends Genet. 2000;16:369–372. doi: 10.1016/s0168-9525(00)02081-3. [DOI] [PubMed] [Google Scholar]
- 96.Arnold CD, Gerlach D, Spies D, Matts JA, Sytnikova YA, Pagani M, Lau NC, Stark A. Quantitative genome-wide enhancer activity maps for five Drosophila species show functional enhancer conservation and turnover during cis-regulatory evolution. Nat Genet. 2014;46:685–692. doi: 10.1038/ng.3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Su J, Teichmann SA, Down TA. Assessing computational methods of cis-regulatory module prediction. PLoS Comput Biol. 2010;6:e1001020. doi: 10.1371/journal.pcbi.1001020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Loots GG, Locksley RM, Blankespoor CM, Wang ZE, Miller W, Rubin EM, Frazer KA. Identification of a coordinate regulator of interleukins 4, 13, and 5 by cross-species sequence comparisons. Science. 2000;288:136–140. doi: 10.1126/science.288.5463.136. [DOI] [PubMed] [Google Scholar]
- 99.Johnson DS, Davidson B, Brown CD, Smith WC, Sidow A. Noncoding regulatory sequences of Ciona exhibit strong correspondence between evolutionary constraint and functional importance. Genome Res. 2004;14:2448–2456. doi: 10.1101/gr.2964504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nobrega MA, Ovcharenko I, Afzal V, Rubin EM. Scanning human gene deserts for long-range enhancers. Science. 2003;302:413. doi: 10.1126/science.1088328. [DOI] [PubMed] [Google Scholar]
- 101.Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, Minovitsky S, Dubchak I, Holt A, Lewis KD, et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature. 2006;444:499–502. doi: 10.1038/nature05295. [DOI] [PubMed] [Google Scholar]
- 102.Shin JT, Priest JR, Ovcharenko I, Ronco A, Moore RK, Burns CG, MacRae CA. Human-zebrafish non-coding conserved elements act in vivo to regulate transcription. Nucleic Acids Res. 2005;33:5437–5445. doi: 10.1093/nar/gki853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Woolfe A, Goodson M, Goode DK, Snell P, McEwen GK, Vavouri T, Smith SF, North P, Callaway H, Kelly K, et al. Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 2005;3:e7. doi: 10.1371/journal.pbio.0030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Attanasio C, Reymond A, Humbert R, Lyle R, Kuehn MS, Neph S, Sabo PJ, Goldy J, Weaver M, Haydock A, et al. Assaying the regulatory potential of mammalian conserved non-coding sequences in human cells. Genome Biol. 2008;9:R168. doi: 10.1186/gb-2008-9-12-r168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.King DC, Taylor J, Zhang Y, Cheng Y, Lawson HA, Martin J, ENCODE groups for Transcriptional Regulation and Multispecies Sequence Analysis. Chiaromonte F, Miller W, Hardison RC. Finding cis-regulatory elements using comparative genomics: Some lessons from ENCODE data 10.1101/gr.5592107. Genome Res. 2007;17:775–786. doi: 10.1101/gr.5592107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hardison RC, Taylor J. Genomic approaches towards finding cis-regulatory modules in animals. Nat Rev Genet. 2012;13:469–483. doi: 10.1038/nrg3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rubinstein M, de Souza FS. Evolution of transcriptional enhancers and animal diversity. Philos Trans R Soc Lond B Biol Sci. 2013;368:20130017. doi: 10.1098/rstb.2013.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Swanson CI, Schwimmer DB, Barolo S. Rapid evolutionary rewiring of a structurally constrained eye enhancer. Curr Biol. 2011;21:1186–1196. doi: 10.1016/j.cub.2011.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Junion G, Spivakov M, Girardot C, Braun M, Gustafson EH, Birney E, Furlong EE. A transcription factor collective defines cardiac cell fate and reflects lineage history. Cell. 2012;148:473–486. doi: 10.1016/j.cell.2012.01.030. [DOI] [PubMed] [Google Scholar]
- 110.Ludwig MZ, Bergman C, Patel NH, Kreitman M. Evidence for stabilizing selection in a eukaryotic enhancer element. Nature. 2000;403:564–567. doi: 10.1038/35000615. [DOI] [PubMed] [Google Scholar]
- 111.Vinga S, Almeida J. Alignment-free sequence comparison–a review. Bioinformatics. 2003;19:513–523. doi: 10.1093/bioinformatics/btg005. [DOI] [PubMed] [Google Scholar]
- 112.Wasserman WW, Fickett JW. Identification of regulatory regions which confer muscle-specific gene expression. J Mol Biol. 1998;278:167–181. doi: 10.1006/jmbi.1998.1700. [DOI] [PubMed] [Google Scholar]
- 113.Berman BP, Nibu Y, Pfeiffer BD, Tomancak P, Celniker SE, Levine M, Rubin GM, Eisen MB. Exploiting transcription factor binding site clustering to identify cis-regulatory modules involved in pattern formation in the Drosophila genome. Proc Natl Acad Sci U S A. 2002;99:757–762. doi: 10.1073/pnas.231608898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Halfon MS, Grad Y, Church GM, Michelson AM. Computation-based discovery of related transcriptional regulatory modules and motifs using an experimentally validated combinatorial model. Genome Res. 2002;12:1019–1028. doi: 10.1101/gr.228902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Markstein M, Markstein P, Markstein V, Levine MS. Genome-wide analysis of clustered Dorsal binding sites identifies putative target genes in the Drosophila embryo. Proc Natl Acad Sci U S A. 2002;99:763–768. doi: 10.1073/pnas.012591199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rebeiz M, Reeves NL, Posakony JW. SCORE: a computational approach to the identification of cis-regulatory modules and target genes in whole-genome sequence data. Site clustering over random expectation. Proc Natl Acad Sci U S A. 2002;99:9888–9893. doi: 10.1073/pnas.152320899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rajewsky N, Vergassola M, Gaul U, Siggia ED. Computational detection of genomic cis-regulatory modules applied to body patterning in the early Drosophila embryo. BMC Bioinformatics. 2002;3:30. doi: 10.1186/1471-2105-3-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Berman BP, Pfeiffer BD, Laverty TR, Salzberg SL, Rubin GM, Eisen MB, Celniker SE. Computational identification of developmental enhancers: conservation and function of transcription factor binding-site clusters in Drosophila melanogaster and Drosophila pseudoobscura. Genome Biol. 2004;5:R61. doi: 10.1186/gb-2004-5-9-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Eddy SR. What is a hidden Markov model? Nat Biotechnol. 2004;22:1315–1316. doi: 10.1038/nbt1004-1315. [DOI] [PubMed] [Google Scholar]
- 120.Frith MC, Li MC, Weng Z. Cluster-Buster: Finding dense clusters of motifs in DNA sequences. Nucleic Acids Res. 2003;31:3666–3668. doi: 10.1093/nar/gkg540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Grad YH, Roth FP, Halfon MS, Church GM. Prediction of similarly acting cis-regulatory modules by subsequence profiling and comparative genomics in Drosophila melanogaster and D.pseudoobscura. Bioinformatics. 2004;20:2738–2750. doi: 10.1093/bioinformatics/bth320. [DOI] [PubMed] [Google Scholar]
- 122.Sinha S, van Nimwegen E, Siggia ED. A probabilistic method to detect regulatory modules. Bioinformatics. 2003;19(Suppl 1):i292–301. doi: 10.1093/bioinformatics/btg1040. [DOI] [PubMed] [Google Scholar]
- 123.Crowley EM, Roeder K, Bina M. A statistical model for locating regulatory regions in genomic DNA. J Mol Biol. 1997;268:8–14. doi: 10.1006/jmbi.1997.0965. [DOI] [PubMed] [Google Scholar]
- 124.Sinha S, Schroeder MD, Unnerstall U, Gaul U, Siggia ED. Cross-species comparison significantly improves genome-wide prediction of cis-regulatory modules in Drosophila. BMC Bioinformatics. 2004;5:129. doi: 10.1186/1471-2105-5-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Blanchette M, Bataille AR, Chen X, Poitras C, Laganiere J, Lefebvre C, Deblois G, Giguere V, Ferretti V, Bergeron D, et al. Genome-wide computational prediction of transcriptional regulatory modules reveals new insights into human gene expression. Genome Res. 2006;16:656–668. doi: 10.1101/gr.4866006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sinha S, He X. MORPH: probabilistic alignment combined with hidden Markov models of cis-regulatory modules. PLoS Comput Biol. 2007;3:e216. doi: 10.1371/journal.pcbi.0030216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Warner JB, Philippakis AA, Jaeger SA, He FS, Lin J, Bulyk ML. Systematic identification of mammalian regulatory motifs’ target genes and functions. Nat Methods. 2008;5:347–353. doi: 10.1038/nmeth.1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ivan A, Halfon MS, Sinha S. Computational discovery of cis-regulatory modules in Drosophila without prior knowledge of motifs. Genome Biol. 2008;9:R22. doi: 10.1186/gb-2008-9-1-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gordan R, Shen N, Dror I, Zhou T, Horton J, Rohs R, Bulyk ML. Genomic regions flanking E-box binding sites influence DNA binding specificity of bHLH transcription factors through DNA shape. Cell Rep. 2013;3:1093–1104. doi: 10.1016/j.celrep.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Siggers T, Duyzend MH, Reddy J, Khan S, Bulyk ML. Non-DNA-binding cofactors enhance DNA-binding specificity of a transcriptional regulatory complex. Mol Syst Biol. 2011;7:555. doi: 10.1038/msb.2011.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhou Q, Wong WH. CisModule: de novo discovery of cis-regulatory modules by hierarchical mixture modeling. Proc Natl Acad Sci U S A. 2004;101:12114–12119. doi: 10.1073/pnas.0402858101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhou Q, Wong WH. Coupling hidden Markov models for the discovery of cis-regulatory modules in multiple species. The Annals of Applied Statistics. 2007:36–65. [Google Scholar]
- 133.Johnson DS, Zhou Q, Yagi K, Satoh N, Wong W, Sidow A. De novo discovery of a tissue-specific gene regulatory module in a chordate. Genome Res. 2005;15:1315–1324. doi: 10.1101/gr.4062605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rouault H, Santolini M, Schweisguth F, Hakim V. Imogene: identification of motifs and cis-regulatory modules underlying gene co-regulation. Nucleic Acids Res. 2014;42:6128–6145. doi: 10.1093/nar/gku209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kazemian M, Zhu Q, Halfon MS, Sinha S. Improved accuracy of supervised CRM discovery with interpolated Markov models and cross-species comparison. Nucleic Acids Res. 2011;39:9463–9472. doi: 10.1093/nar/gkr621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Arunachalam M, Jayasurya K, Tomancak P, Ohler U. An alignment-free method to identify candidate orthologous enhancers in multiple Drosophila genomes. Bioinformatics. 2010;26:2109–2115. doi: 10.1093/bioinformatics/btq358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sosinsky A, Honig B, Mann RS, Califano A. Discovering transcriptional regulatory regions in Drosophila by a nonalignment method for phylogenetic footprinting 10.1073/pnas.0701614104. PNAS. 2007;0701614104 doi: 10.1073/pnas.0701614104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lee D, Karchin R, Beer MA. Discriminative prediction of mammalian enhancers from DNA sequence. Genome Res. 2011;21:2167–2180. doi: 10.1101/gr.121905.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fletez-Brant C, Lee D, McCallion AS, Beer MA. kmer-SVM: a web server for identifying predictive regulatory sequence features in genomic data sets. Nucleic Acids Res. 2013;41:W544–556. doi: 10.1093/nar/gkt519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schultheiss SJ, Busch W, Lohmann JU, Kohlbacher O, Ratsch G. KIRMES: kernel-based identification of regulatory modules in euchromatic sequences. Bioinformatics. 2009;25:2126–2133. doi: 10.1093/bioinformatics/btp278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Taher L, Narlikar L, Ovcharenko I. CLARE: Cracking the LAnguage of Regulatory Elements. Bioinformatics. 2012;28:581–583. doi: 10.1093/bioinformatics/btr704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Erwin GD, Oksenberg N, Truty RM, Kostka D, Murphy KK, Ahituv N, Pollard KS, Capra JA. Integrating diverse datasets improves developmental enhancer prediction. PLoS Comput Biol. 2014;10:e1003677. doi: 10.1371/journal.pcbi.1003677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Herrmann C, Van de Sande B, Potier D, Aerts S. i-cisTarget: an integrative genomics method for the prediction of regulatory features and cis-regulatory modules. Nucleic Acids Res. 2012;40:e114. doi: 10.1093/nar/gks543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Aerts S, Quan XJ, Claeys A, Naval Sanchez M, Tate P, Yan J, Hassan BA. Robust target gene discovery through transcriptome perturbations and genome-wide enhancer predictions in Drosophila uncovers a regulatory basis for sensory specification. PLoS Biol. 2010;8:e1000435. doi: 10.1371/journal.pbio.1000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kazemian M, Suryamohan K, Chen JY, Zhang Y, Samee MA, Halfon MS, Sinha S. Evidence for deep regulatory similarities in early developmental programs across highly diverged insects. Genome Biol Evol. 2014 doi: 10.1093/gbe/evu184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wiegmann BM, Trautwein MD, Kim JW, Cassel BK, Bertone MA, Winterton SL, Yeates DK. Single-copy nuclear genes resolve the phylogeny of the holometabolous insects. BMC Biol. 2009;7:34. doi: 10.1186/1741-7007-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zdobnov EM, Bork P. Quantification of insect genome divergence. Trends Genet. 2007;23:16–20. doi: 10.1016/j.tig.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 148.Kvon EZ, Kazmar T, Stampfel G, Yanez-Cuna JO, Pagani M, Schernhuber K, Dickson BJ, Stark A. Genome-scale functional characterization of Drosophila developmental enhancers in vivo. Nature. 2014 doi: 10.1038/nature13395. [DOI] [PubMed] [Google Scholar]
- 149.Noordermeer D, Duboule D. Chromatin looping and organization at developmentally regulated gene loci. Wiley Interdiscip Rev Dev Biol. 2013;2:615–630. doi: 10.1002/wdev.103. [DOI] [PubMed] [Google Scholar]
- 150.Ghavi-Helm Y, Klein FA, Pakozdi T, Ciglar L, Noordermeer D, Huber W, Furlong EE. Enhancer loops appear stable during development and are associated with paused polymerase. Nature. 2014;512:96–100. doi: 10.1038/nature13417. [DOI] [PubMed] [Google Scholar]
- 151.Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cook PR. A model for all genomes: the role of transcription factories. J Mol Biol. 2010;395:1–10. doi: 10.1016/j.jmb.2009.10.031. [DOI] [PubMed] [Google Scholar]
- 153.Visel A, Minovitsky S, Dubchak I, Pennacchio LA. VISTA Enhancer Browser–a database of tissue-specific human enhancers. Nucleic Acids Res. 2007;35:D88–92. doi: 10.1093/nar/gkl822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gallo SM, Gerrard DT, Miner D, Simich M, Des Soye B, Bergman CM, Halfon MS. REDfly v3.0: toward a comprehensive database of transcriptional regulatory elements in Drosophila. Nucleic Acids Res. 2011;39:D118–123. doi: 10.1093/nar/gkq999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Costa M, Reeve S, Grumbling G, Osumi-Sutherland D. The Drosophila anatomy ontology. J Biomed Semantics. 2013;4:32. doi: 10.1186/2041-1480-4-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.St Pierre SE, Ponting L, Stefancsik R, McQuilton P. FlyBase 102–advanced approaches to interrogating FlyBase. Nucleic Acids Res. 2014;42:D780–788. doi: 10.1093/nar/gkt1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Frankel N. Multiple layers of complexity in cis-regulatory regions of developmental genes. Dev Dyn. 2012;241:1857–1866. doi: 10.1002/dvdy.23871. [DOI] [PubMed] [Google Scholar]
- 158.Halfon MS. (Re)modeling the transcriptional enhancer. Nat Genet. 2006;38:1102–1103. doi: 10.1038/ng1006-1102. [DOI] [PubMed] [Google Scholar]
- 159.Juven-Gershon T, Kadonaga JT. Regulation of gene expression via the core promoter and the basal transcriptional machinery. Dev Biol. 2010;339:225–229. doi: 10.1016/j.ydbio.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Swanson CI, Evans NC, Barolo S. Structural rules and complex regulatory circuitry constrain expression of a Notch- and EGFR-regulated eye enhancer. Dev Cell. 2010;18:359–370. doi: 10.1016/j.devcel.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kwon D, Mucci D, Langlais KK, Americo JL, DeVido SK, Cheng Y, Kassis JA. Enhancer-promoter communication at the Drosophila engrailed locus. Development. 2009;136:3067–3075. doi: 10.1242/dev.036426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Atkinson TJ, Halfon MS. Regulation of gene expression in the genomic context. Comput Struct Biotechnol J. 2014;9:e201401001. doi: 10.5936/csbj.201401001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Harrison MM, Jenkins BV, O’Connor-Giles KM, Wildonger J. A CRISPR view of development. Genes Dev. 2014;28:1859–1872. doi: 10.1101/gad.248252.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.John S, Marais R, Child R, Light Y, Leonard WJ. Importance of low affinity Elf-1 sites in the regulation of lymphoid-specific inducible gene expression. J Exp Med. 1996;183:743–750. doi: 10.1084/jem.183.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Tanay A. Extensive low-affinity transcriptional interactions in the yeast genome. Genome Res. 2006;16:962–972. doi: 10.1101/gr.5113606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jaeger SA, Chan ET, Berger MF, Stottmann R, Hughes TR, Bulyk ML. Conservation and regulatory associations of a wide affinity range of mouse transcription factor binding sites. Genomics. 2010;95:185–195. doi: 10.1016/j.ygeno.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Weirauch MT, Cote A, Norel R, Annala M, Zhao Y, Riley TR, Saez-Rodriguez J, Cokelaer T, Vedenko A, Talukder S, et al. Evaluation of methods for modeling transcription factor sequence specificity. Nat Biotechnol. 2013;31:126–134. doi: 10.1038/nbt.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Carroll SB, Grenier JK, Weatherbee SD. From DNA to diversity : molecular genetics and the evolution of animal design. 2. Malden, MA: Blackwell Pub.; 2005. [Google Scholar]
- 169.Davidson EH. The regulatory genome : gene regulatory networks in development and evolution. Burlington, MA: San Diego: Academic; 2006. [Google Scholar]
- 170.Barolo S, Posakony JW. Three habits of highly effective signaling pathways: principles of transcriptional control by developmental cell signaling. Genes Dev. 2002;16:1167–1181. doi: 10.1101/gad.976502. [DOI] [PubMed] [Google Scholar]
- 171.Halfon MS, Carmena A, Gisselbrecht S, Sackerson CM, Jimenez F, Baylies MK, Michelson AM. Ras pathway specificity is determined by the integration of multiple signal-activated and tissue-restricted transcription factors. Cell. 2000;103:63–74. doi: 10.1016/s0092-8674(00)00105-7. [DOI] [PubMed] [Google Scholar]
- 172.Mann RS, Carroll SB. Molecular mechanisms of selector gene function and evolution. Current Opinion in Genetics & Development. 2002;12:592–600. doi: 10.1016/s0959-437x(02)00344-1. [DOI] [PubMed] [Google Scholar]
- 173.Britten RJ, Davidson EH. Gene regulation for higher cells: a theory. Science. 1969;165:349–357. doi: 10.1126/science.165.3891.349. [DOI] [PubMed] [Google Scholar]
- 174.Ford E, Thanos D. The transcriptional code of human IFN-beta gene expression. Biochim Biophys Acta. 2010;1799:328–336. doi: 10.1016/j.bbagrm.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 175.Arnosti DN, Kulkarni MM. Transcriptional enhancers: Intelligent enhanceosomes or flexible billboards? J Cell Biochem. 2005;94:890–898. doi: 10.1002/jcb.20352. [DOI] [PubMed] [Google Scholar]
- 176.Liu F, Posakony JW. Role of architecture in the function and specificity of two Notch-regulated transcriptional enhancer modules. PLoS Genet. 2012;8:e1002796. doi: 10.1371/journal.pgen.1002796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lifanov AP, Makeev VJ, Nazina AG, Papatsenko DA. Homotypic regulatory clusters in Drosophila. Genome Res. 2003;13:579–588. doi: 10.1101/gr.668403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Arnone MI, Davidson EH. The hardwiring of development: organization and function of genomic regulatory systems. Development. 1997;124:1851–1864. doi: 10.1242/dev.124.10.1851. [DOI] [PubMed] [Google Scholar]
- 179.Hallikas O, Palin K, Sinjushina N, Rautiainen R, Partanen J, Ukkonen E, Taipale J. Genome-wide prediction of mammalian enhancers based on analysis of transcription-factor binding affinity. Cell. 2006;124:47–59. doi: 10.1016/j.cell.2005.10.042. [DOI] [PubMed] [Google Scholar]
- 180.Gotea V, Visel A, Westlund JM, Nobrega MA, Pennacchio LA, Ovcharenko I. Homotypic clusters of transcription factor binding sites are a key component of human promoters and enhancers. Genome Res. 2010;20:565–577. doi: 10.1101/gr.104471.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.He X, Duque TS, Sinha S. Evolutionary origins of transcription factor binding site clusters. Mol Biol Evol. 2012;29:1059–1070. doi: 10.1093/molbev/msr277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hertel KJ, Lynch KW, Maniatis T. Common themes in the function of transcription and splicing enhancers. Curr Opin Cell Biol. 1997;9:350–357. doi: 10.1016/s0955-0674(97)80007-5. [DOI] [PubMed] [Google Scholar]
- 183.Lusk RW, Eisen MB. Evolutionary mirages: selection on binding site composition creates the illusion of conserved grammars in Drosophila enhancers. PLoS Genet. 2010;6:e1000829. doi: 10.1371/journal.pgen.1000829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Papatsenko DA, Makeev VJ, Lifanov AP, Regnier M, Nazina AG, Desplan C. Extraction of functional binding sites from unique regulatory regions: the Drosophila early developmental enhancers. Genome Res. 2002;12:470–481. doi: 10.1101/gr.212502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Crocker J, Potter N, Erives A. Dynamic evolution of precise regulatory encodings creates the clustered site signature of enhancers. Nat Commun. 2010;1:99. doi: 10.1038/ncomms1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.D’Haeseleer P. What are DNA sequence motifs? Nat Biotechnol. 2006;24:423–425. doi: 10.1038/nbt0406-423. [DOI] [PubMed] [Google Scholar]
- 187.Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Hertz GZ, Stormo GD. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics. 1999;15:563–577. doi: 10.1093/bioinformatics/15.7.563. [DOI] [PubMed] [Google Scholar]
- 189.Stormo GD. DNA binding sites: representation and discovery. Bioinformatics. 2000;16:16–23. doi: 10.1093/bioinformatics/16.1.16. [DOI] [PubMed] [Google Scholar]
- 190.Stormo GD. Modeling the specificity of protein-DNA interactions. Cold Spring Harb Symp Quant Biol. 2013;1:115–130. doi: 10.1007/s40484-013-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.GuhaThakurta D. Computational identification of transcriptional regulatory elements in DNA sequence 10.1093/nar/gkl372. Nucl Acids Res. 2006;34:3585–3598. doi: 10.1093/nar/gkl372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Pavesi G, Mauri G, Pesole G. In silico representation and discovery of transcription factor binding sites. Brief Bioinform. 2004;5:217–236. doi: 10.1093/bib/5.3.217. [DOI] [PubMed] [Google Scholar]
- 193.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]