

ABSTRACT
Zika virus (ZIKV), a mosquito-transmitted flavivirus responsible for sporadic outbreaks of mild and febrile illness in Africa and Asia, reemerged in the last decade causing serious human diseases, including microcephaly, congenital malformations, and Guillain-Barré syndrome. Although genomic and phylogenetic analyses suggest that genetic evolution may have led to the enhanced virulence of ZIKV, experimental evidence supporting the role of specific genetic changes in virulence is currently lacking. One sequence motif, VNDT, containing an N-linked glycosylation site in the envelope (E) protein, is polymorphic; it is absent in many of the African isolates but present in all isolates from the recent outbreaks. In the present study, we investigated the roles of this sequence motif and glycosylation of the E protein in the pathogenicity of ZIKV. We first constructed a stable full-length cDNA clone of ZIKV in a novel linear vector from which infectious virus was recovered. The recombinant ZIKV generated from the infectious clone, which contains the VNDT motif, is highly pathogenic and causes lethality in a mouse model. In contrast, recombinant viruses from which the VNDT motif is deleted or in which the N-linked glycosylation site is mutated by single-amino-acid substitution are highly attenuated and nonlethal. The mutant viruses replicate poorly in the brains of infected mice when inoculated subcutaneously but replicate well following intracranial inoculation. Our findings provide the first evidence that N-linked glycosylation of the E protein is an important determinant of ZIKV virulence and neuroinvasion.
IMPORTANCE The recent emergence of Zika virus (ZIKV) in the Americas has caused major worldwide public health concern. The virus appears to have gained significant pathogenicity, causing serious human diseases, including microcephaly and Guillain-Barré syndrome. The factors responsible for the emergence of pathogenic ZIKV are not understood at this time, although genetic changes have been shown to facilitate virus transmission. All isolates from the recent outbreaks contain an N-linked glycosylation site within the viral envelope (E) protein, whereas many isolates of the African lineage virus lack this site. To elucidate the functional significance of glycosylation in ZIKV pathogenicity, recombinant ZIKVs from infectious clones with or without the glycan on the E protein were generated. ZIKVs lacking the glycan were highly attenuated for the ability to cause mortality in a mouse model and were severely compromised for neuroinvasion. Our studies suggest glycosylation of the E protein is an important factor contributing to ZIKV pathogenicity.
KEYWORDS: Zika virus, E protein glycosylation, attenuation, neuroinvasion
INTRODUCTION
Following isolation of Zika virus (ZIKV) in 1947 from a rhesus monkey in Uganda (1), only a few cases of human infections were reported in Africa and Asia in the next 60 years (2–4). However, the virus reemerged during the last decade, causing outbreaks on Yap Island of the Federated States of Micronesia in 2007 (5) and in French Polynesia and other South Pacific islands in 2013 and 2014 (6, 7). The virus was subsequently detected for the first time in Brazil in 2015 (8), causing infections in epidemic proportions, and spread rapidly to many countries in the Americas and other parts of the world. The virus exists as two major lineages, African and Asian (2, 9, 10). Sequence analyses have revealed that the Asian lineage ZIKV is responsible for the recent outbreaks (4, 10). Typically, the majority of ZIKV infections in humans are asymptomatic, but approximately 20% of the infections are associated with mild, self-limiting symptoms of flu-like febrile illness that are resolved within days. However, the most alarming observations from the recent outbreaks were the increased incidences of Guillain-Barré syndrome (GBS), an immune system-mediated autoimmune disease of the peripheral nervous system, and congenital malformations, including microcephaly, fetal growth restriction, and other disorders that have been collectively termed congenital ZIKV syndrome (CZS) (3, 4, 11–13).
ZIKV is a mosquito-transmitted flavivirus in the family Flaviviridae. Its ∼10.8-kb positive-sense RNA genome encodes a single polyprotein that is processed by viral and host proteases to generate three structural (capsid [C], premembrane [prM], and envelope[E]) and seven nonstructural (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) proteins. Like those of closely related flaviviruses, such as dengue (DENV), West Nile (WNV), yellow fever (YFV), and Japanese encephalitis (JEV) viruses, the structural proteins are involved in virion formation, attachment, and entry into host cells (14), whereas the nonstructural proteins participate in genome replication, virion assembly, and evasion of host antiviral responses (15–17). The recent expansion of the geographic range and the severity of the virus-induced diseases in humans suggest genetic evolution of ZIKV (2, 9, 18–21). Adaptive evolution resulting from accumulation of genetic changes and/or stochastic introduction of the virus into large naive human populations have been proposed to account for ZIKV emergence with pathogenic potential (2, 4, 18, 22). Comparative genomic and phylogenetic analyses have identified amino acid substitutions (18, 21, 23) that may contribute to the increased rate of transmission and/or pathogenicity. However, formal investigation of the genetic changes that are responsible for the pathogenic traits of the virus is currently lacking. Only one recent study shows that the enhanced infectivity of ZIKV in the Aedes aegypti mosquito vector resulting from a mutation in the NS1 protein could potentially facilitate transmission and contribute to the spread of the virus from Asia to the Americas (24).
In the present study, we developed a reverse genetic system for ZIKV to address the role of a specific sequence motif, 153VNDT156, containing an N-linked glycosylation site (underlined) within the E protein, in pathogenic properties of the virus. The VNDT motif is absent in many of the African ZIKV isolates but present in all isolates from the recent outbreaks (20) and may play a critical role in viral pathogenicity. By genetic manipulation of the viral genome, we recovered mutant ZIKVs either lacking the VNDT motif or with a single amino acid substitution (N154A) in the glycosylation site and showed that the mutant viruses are highly attenuated in their ability to cause morbidity and mortality in the mouse model and are compromised for neuroinvasion. Our results provide evidence for a critical role of N-linked glycosylation of the E protein in ZIKV virulence and neuroinvasion.
RESULTS
Infectious-clone-derived ZIKV mimics the in vitro growth and in vivo pathogenic attributes of the parental virus.
We sought to establish a reverse genetics system for ZIKV by constructing a full-length cDNA clone of the strain MR766 virus (here called MR). Four cDNA fragments (A to D) (Fig. 1A) spanning the entire viral genome were amplified by reverse transcription (RT)-PCR and initially cloned in the pBR322 vector. We consistently observed deletion of 1 or 2 nucleotides at positions around 2800 or 3500 in fragment A (within the NS1 coding region) of all (about 20) examined clones. Attempts to repair the deletion at one site resulted in deletion at the other site, indicating sequence instability around this region, as has been reported (25, 26). Strategies such as insertion of introns into the viral genome (25, 26) or maintaining the genome in multiple small fragments (27–31) have been used to overcome the inherent instability of the ZIKV and other flavivirus genomes. Here, using a novel linear vector (32) (Fig. 1A) with bacterial transcription terminators flanking the cloning site, we generated stable full-length cDNA clones of the ZIKV genome. When transfected into Vero cells, in vitro transcripts from a full-length cDNA clone with correct sequence (pJ-rMR) resulted in specific detection of E protein, whereas no such signal was detected in cells transfected with RNA from a construct encoding an inactive polymerase (pJ-rMR/Pol−; GDD→AAA mutation in NS5) (Fig. 1B). The transfected cells produced infectious ZIKV with a maximum titer at 5 days posttransfection (Fig. 1C). After at least 10 successive passages of the plasmid (extraction and retransformation) in bacterial culture, the plasmid was found to be stable and maintained the viral sequences, leading to recovery of infectious virus (data not shown).
FIG 1.

Construction of an infectious clone of ZIKV MR766 and recovery and characterization of the clone-derived virus. (A) The viral genome with the encoded proteins is shown at the top. The solid circle at the 5′ end represents the cap. The genomic RNA from purified virions was amplified by RT-PCR with specific primers to generate four cDNA fragments (A, B, C, and D), which were assembled into a full-length clone using naturally occurring restriction enzyme sites shown above the fragments. The green rectangular box immediately upstream of fragment A represents the bacteriophage T7 RNA polymerase promoter (ϕ10). A unique BssHII site was incorporated immediately following the viral sequences in fragment D to linearize the plasmid prior to in vitro transcription. The blue lines on the left and right sides of the boxed regions represent sequences from pBR322. The entire genome, along with some sequence of pBR322 released by digestion with ScaI and NruI, was cloned in the pJAZZ-OC linear vector (bottom) at the unique SmaI site in the multiple cloning site of the vector. telL and telR, left and right telomeric sequence from coliphage N15; TelN, protelomerase gene of coliphage N15; repA, replication factor gene and origin of replication of coliphage N15; cB, repressor protein of coliphage N15; Camr, chloramphenicol resistance gene. The approximate positions of transcriptional terminators, T1 (T7 early transcription terminator), T2 (transcription terminator from the E. coli rrnB gene), and T3 (bidirectional E. coli tonB-P14 transcription terminator) are shown. The arrows indicate the direction of the coding sequence for repA and Camr. (B) In vitro transcripts from a full-length infectious clone (pJ-rMR) or from a clone encoding the catalytically inactive polymerase (pJ-rMR/Pol−) were transfected into Vero cells, and after 4 days, the cells were fixed and examined by immunofluorescent staining for E protein using 4G2 antibody. Mock, cells treated with the transfection reagent only. The top row shows phase-contrast images; the bottom row shows immunofluorescent images. (C) Infectious virus recovered from the transfected Vero cells at various days posttransfection as determined by plaque assay on Vero cells. The dashed line represents the limit of detection. (D) Silent nucleotide substitutions (shown in red) were introduced into rMR sequences as genetic tags, resulting in elimination of Bsu36I naturally present in the pMR and creation of AatII and BspEI sites in the rMR. The numbers on the left and right of the sequences are the nucleotide positions in the viral genome. Amino acid residues encoded by these sequences (in blue) are shown in the middle. (E) Confirmation of the presence of the genetic tag. A 1,822-bp DNA fragment was amplified by RT-PCR using the genomic RNA from the pMR or rMR virus and primers (Table 2) and digested with the restriction enzymes shown. Upon digestion of the PCR-amplified product with Bsu36I or AatII, two DNA fragments of 1,112 bp and 710 bp were generated. (F) Multistep growth of the pMR and rMR viruses in Vero and C6/36 cell lines. The graphs show mean values with error bars representing SD from three independent experiments. An unpaired Student t test (two-tailed) was used to determine significance. ns, nonsignificant.
To distinguish the infectious-clone-derived recombinant MR virus (rMR) from the parental MR virus (pMR), we had introduced four silent nucleotide substitutions, resulting in the elimination of a naturally occurring Bsu36I site and creation of new AatII and BspEI sites (Fig. 1D). We confirmed that the AatII site was present in the rMR but not in the pMR, whereas the Bsu36I site was present in the pMR but not in the rMR (Fig. 1E). The rMR grew to similar or slightly (but not significantly) lower titers than the pMR in Vero and C6/36 cell lines (Fig. 1F), suggesting that the infectious-clone-derived virus exhibits growth kinetics similar to those of the pMR.
We next examined the in vivo pathogenic attributes of the rMR compared to the pMR. Several mouse models have been established to study ZIKV pathogenesis in immunocompetent, as well as immunodeficient, animals (for a review, see reference 33). Immunodeficient mice lacking the type I interferon (IFN) signaling pathways are highly susceptible to ZIKV infection (34–39). A129 mice genetically lacking the IFN-α/β receptor undergo significant body weight loss, show clinical disease with limb paralysis, and succumb to infection. Using this model, following subcutaneous (s.c.) inoculation, mice infected with the pMR or the rMR began to exhibit body weight loss 4 days postinfection (dpi). The infected mice continued to lose weight, and at 6 dpi, weight loss was significantly higher (Fig. 2A). One or two mice from each infected group died at 6 dpi, whereas all the mice either died or were moribund and euthanized at 7 dpi (Fig. 2B). The weight loss or survival of mice infected with the pMR or the rMR were similar. The viral genome copy numbers in the plasma of animals infected with the rMR were lower than in those infected with the pMR at 3 dpi but were comparable at 4 to 6 dpi (Fig. 2C). Infectious virus titers in the plasma at 3 dpi or 6 dpi were slightly lower but statistically nonsignificant for the two groups of infected animals (Fig. 2D), indicating that the clone-derived virus exhibited similar in vivo growth kinetics during the infection period examined. Infectious-virus titers in tissues such as brain, spleen, or liver at 6 dpi were also not significantly different between the two groups (Fig. 2E). Overall, the results suggest that the rMR recapitulates the growth and pathogenic properties of the pMR.
FIG 2.

Pathogenic properties of the pMR and rMR viruses in mice. (A and B) Weight loss and mortality of A129 mice inoculated s.c. with 1,000 PFU of the pMR (n = 6) or rMR (n = 6) virus. PBS (n = 5) was used as a negative control. (C) Genome copies in the plasma of animals described in panels A and B at 3, 4, 5, and 6 dpi. (D) Plasma from blood collected at 3 dpi or 6 dpi from pMR- or rMR-inoculated mice was quantitated for infectious virus. (E) Infectious-virus loads in the brain, spleen, and liver in mice inoculated with the pMR or the rMR. Two-way ANOVA (Tukey's multiple-comparison test) (A), multiple t tests (C and D), and an unpaired Student's t test, two tailed (E) were used to determine significance. ****, P < 0.0001; *, P < 0.05; ns, nonsignificant.
ZIKVs encoding nonglycosylated E protein exhibit growth properties similar to those of the virus with the glycosylated protein.
We examined available sequences (n = 330) of the E protein of ZIKV in GenBank, and four major types of polymorphism were noteworthy (Table 1) around the sequence VNDT. Several African isolates of the virus lack or have mutations in the VNDT motif, whereas the vast majority of the Asian and all American isolates contain the motif (20). Furthermore, the MR virus also exhibits polymorphism in the sequences around the VNDT motif. Because of the lack of detailed knowledge on the passage history of the MR virus, it is unclear how the polymorphism developed in this prototypic ZIKV over time. The motif, located in a loop, which we refer to as the “glycan loop” (residues 146-SQHSGMIVNDTGHETDE-162), is in close proximity to the E protein dimer interface and the fusion loop (40) of the adjacent E protein (Fig. 3A). The glycan loop, which is located in domain I (D-I) of the E protein (40), is 6 to 9 amino acids longer in ZIKVs than in several (but not all) other flaviviruses. Glycosylation at the N154 site in ZIKV E protein has been proposed to play an important role(s) in virus growth, pathogenicity, or disease severity (41), although experimental evidence supporting this contention is lacking at this time. In related flaviviruses, this modification was found to influence virus growth, stability, and virulence (42–46).
TABLE 1.
Amino acid sequence polymorphism around the VNDT motif in ZIKV isolates
| Type | Polymorphism(s) | Sequence (148–162)a | No. with polymorphism/total |
||
|---|---|---|---|---|---|
| African | Asian | American | |||
| I | ΔVNDT | HSGMI....GHETDE | 5/61 | 0/41 | 0/228 |
| II | ΔIVNDT, ΔTGHETD, ΔTGHETDE | HSGM........TDE | 8/61 | 1/41 | 0/228 |
| III | VNDI | HSGMIVNDIGHETDE | 24/61 | 2/41 | 0/228 |
| IV | VNDT | HSGMIVNDTGHETDE | 24/61 | 38/41 | 184/228b |
Of the total number of sequences (n = 330) examined, 264 were from full-length genomes and 66 from partial E-coding sequences currently available in GenBank. The African sequences represented 17 full-length genomes and 44 partial E sequences; Asian sequences represented 32 full-length genomes and 9 partial E sequences; American sequences represented 215 full-length genomes and 13 partial E sequences. Amino acid sequences from 148 to 162 of E are shown. VNDT is in bold-face with the glycosylation site underlined. The periods represent sequence not present in the isolates.
Forty-four sequences from the American isolates had incomplete sequence information.
FIG 3.

Characterization of the mutant viruses lacking the glycosylation site in the E protein. (A) Three-dimensional structure of dimeric E protein of ZIKV (Protein Data Bank [PDB] code 5IRE). The boxed region in the top diagram is enlarged below, showing the glycan moiety, glycan loop, fusion loop, and dimer interface. (B) Amino acid sequence of the rMR and the mutant (m1MR and m2MR) viruses around the VNDT motif. (C) Examination of the glycosylation status of the E protein in cells infected with the rMR, m1MR, or m2MR virus. Vero cells infected with the viruses (MOI = 1) were radiolabeled with Expre35S35S protein-labeling mix, immunoprecipitated with 4G2 antibody, digested with endo H or PNGase F, analyzed by SDS-PAGE, and detected by fluorography. Relative migration of molecular mass markers (in kilodaltons) is shown on the left. (D) Multistep growth of the rMR, m1MR, and m2MR in Vero and C6/36 cells. The graphs show mean values, with error bars representing standard deviations from the results of three independent experiments. An unpaired Student t test (two-tailed) was used to determine significance. ns, nonsignificant.
To examine the roles of this motif and N-linked glycosylation, we generated mutant viruses by either deleting the VNDT motif (m1MR) or generating a single-amino-acid substitution mutant (m2MR) by altering the asparagine to alanine in the glycosylation site. The sequences of the E proteins of these mutant viruses compared to that of the wild-type (wt) virus (rMR) around this site are shown in Fig. 3B. The E protein synthesized in m1MR- or m2MR-infected cells was not glycosylated, since it was not digested by endo-β-N-acetylglucosaminidase H (endo H) or peptide-N-glycosidase F (PNGase F), whereas it was glycosylated in rMR-infected cells (Fig. 3C). The mutant viruses grew to slightly (but not statistically significantly) lower titers than the rMR in Vero cells and C6/36 cells (Fig. 3D). Additionally, the glycan-deficient mutant viruses grew to titers similar to those of the rMR virus in at least three other cell lines (HTR-8, a human trophoblast cell line; NTERA-2, a human testicular cell line; and SH-SY5Y, a human neuroblastoma cell line) that are the targets of ZIKV infection in humans (data not shown). These results indicate that the deletion of the VNDT motif or the loss of a glycosylation site in the E protein does not adversely affect in vitro growth of the mutant viruses. The mutant viruses were also found to be stable following multiple passages in Vero and C6/36 cells (data not shown).
The glycosylation mutant ZIKVs are attenuated in vivo.
To elucidate the influence of glycosylation of the E protein on ZIKV pathogenicity, A129 mice were inoculated s.c. with the rMR, m1MR, or m2MR virus. The rMR-infected mice began to show signs of illness and weight loss at 4 dpi. All the mice in this group showed enhanced clinical symptoms and by 6 dpi exhibited significantly high overall clinical scores (Fig. 4A) and weight loss (Fig. 4B). The mice were either moribund and euthanized for tissue sample collection at 6 dpi or succumbed to infection at 7 dpi (Fig. 4C). In contrast, none of the m1MR- or m2MR-infected mice exhibited clinical symptoms, and they appeared healthy at 6 dpi (Fig. 4A). The m1MR-infected mice showed slight weight loss at 7 dpi but began to gain weight after that (Fig. 4B), whereas m2MR-infected mice did not exhibit body weight loss. All the animals except one (dead at 8 dpi) in the m1MR-infected group appeared healthy at 8 dpi. A number of randomly chosen animals from the m1MR- and m2MR-infected groups were also sacrificed at 6 dpi to examine virus replication in tissues. About 25% of the remaining mice in the m1MR-infected group began showing paralysis of one or both hind limbs at 9 dpi, but interestingly, a majority of them recovered within 2 days. Importantly, over 75% of the m1MR-infected animals did not show any clinical symptoms for prolonged periods of up to 2 months, when the animals were eventually euthanized. Likewise, none of the animals in the m2MR-infected group showed any clinical symptoms for up to 2 weeks of observation. Viral-genome copy numbers in the plasma were higher for rMR-infected animals at 3 to 5 dpi than those in m1MR- or m2MR-infected animals but were not significantly different for all three groups at 6 dpi (Fig. 4D). Since the m1MR and m2MR viruses replicated to slightly lower titers under in vitro growth conditions (Fig. 3D), it is possible that the diminished viral load in the plasma of the mutant-virus-infected animals at 3 to 5 dpi could be due to their lower replication potential compared to the rMR. It should be noted that the viral-genome copy numbers in the plasma of m1MR- and m2MR-infected animals were not significantly different from each other at various times examined, indicating that both the mutant viruses replicated to nearly similar levels in the animals. The infectious-virus loads in the plasma of the infected animals in the three groups showed a trend at 3 and 6 dpi similar to that of the genome copy numbers (Fig. 4E). The viral loads in the plasma of m1MR- or m2MR-infected animals were reduced significantly at 7, 10, and 14 dpi compared to 6 dpi (data not shown). These results suggest that the mutant ZIKVs with the deletion of the VNDT motif or lacking the glycan moiety on the E protein were attenuated, as their ability to cause disease and lethality was markedly diminished.
FIG 4.

Mutant viruses are attenuated in mice. (A) Clinical scores at 6 dpi in A129 mice inoculated s.c. with 1,000 PFU of the rMR (n = 8), m1MR (n = 8), or m2MR (n = 6) virus. See Materials and Methods for clinical symptoms and scores. (B and C) Weight loss and mortality of A129 mice inoculated s.c. with 1,000 PFU of the rMR (n = 13), m1MR (n = 23), or m2MR (n = 12) virus. PBS (n = 6) was used as a negative control. (D and E) Viral-RNA copies and infectious-virus titers in the plasma of infected animals at various times postinfection. The data presented in panels B to E are combined data from two independent experiments. Unpaired Student t tests, two-tailed (A) and multiple t tests (D and E) were used to determine significance. ****, P < 0.0001; ***, P < 0.001; ns, nonsignificant.
Replication of the glycosylation mutant ZIKVs is severely restricted in the brain but not in the spleen or liver in infected animals.
To understand pathogenic differences between the rMR and the mutant ZIKVs, we examined tissues of the infected animals collected at 6 dpi. Histopathological examination of brain sections revealed specific differences between the groups. rMR-infected animals showed moderate to severe lesions in the meninges and moderate inflammation in the brain compared to those infected with m1MR or m2MR virus (data not shown). Diffuse lymphocytic infiltrates in the meninges and scattered dark hyperchromatic neurons were prominent in the brain sections from rMR-infected but not m1MR- or m2MR-infected animals (Fig. 5i, iv, and vii). There were mild to moderate levels of karyorrhexic debris and lymphocytic cuffing in the brains of rMR-infected animals but minimal brain inflammation or neuronal pathology in m1MR- or m2MR-infected animals (Fig. 5ii, v, and viii). Interestingly, the spleens of m1MR- or m2MR-infected mice showed significantly higher lymphoid hyperplasia than those of rMR-infected animals (Fig. 5iii, vi, and ix). The rMR-infected group had moderate neutrophil infiltrates throughout the spleen that were absent in the mutant-virus-infected groups (Fig. 5iii, vi, and ix). The histological presentations of livers and kidneys in all three groups of animals were similar.
FIG 5.

Histopathological examination of tissues of animals infected with the wt or mutant virus. H&E staining of sections of brains (meninges and cerebrum) and spleens of animals infected with the rMR, m1MR, or m2MR virus is shown. Representative images from the experiment are shown. Bars, 60 μm in meninges and cerebrum, 30 μm in spleen.
To determine if the histopathological differences were due to the differences in the levels of replication of the wt and mutant viruses, we examined the viral loads in tissues of the animals. Indeed, the rMR replicated in the brains of infected animals robustly, and both numbers of genome copies (Fig. 6A) and levels of infectious virus (Fig. 6B) were significantly higher than those from m1MR- or m2MR-infected animals. Both m1MR and m2MR viruses replicated poorly in the brain at this time. The genome copies and infectious virus titers were about 750- to 1,500-fold and 5,000- to 8,000-fold, respectively, lower than those from the rMR-infected group (Fig. 6A and B). The levels of infectious virus in the spleen were similar among the three groups but were somewhat lower in the livers of the m1MR- and m2MR-infected groups than in those of the rMR-infected group (Fig. 6C). The results suggest that the presence and/or replication of the mutant viruses in the brain is severely restricted.
FIG 6.
The mutant viruses replicate less efficiently in the brains of infected animals. (A and B) Viral-genome copy numbers (A) and infectious-virus titers (B) in the brains of animals infected with the rMR (n = 8), m1MR (n = 8), and m2MR (n = 8) viruses at 6 dpi. (C) Infectious-virus titers in the spleens and livers of animals infected with the rMR, m1MR, and m2MR viruses at 6 dpi. Data from two separate experiments were combined. An unpaired Student t test (two-tailed) was used to determine significance. ****, P < 0.0001; ns, nonsignificant.
The glycosylation mutant ZIKVs are defective in neuroinvasion.
It is possible that the mutant ZIKVs are either defective in neurotropism or compromised for neuroinvasion when inoculated by the parenteral route (s.c.). To examine this, we inoculated mice with the viruses intracranially (i.c.). This allowed us to bypass the blood-brain barrier (BBB) and examine viral replication and pathology independently of neuroinvasive factors. Following i.c. inoculation, the mice in all three groups appeared healthy for the first 3 days but began to lose weight and show signs of illness at 4 dpi. This was in stark contrast to s.c. inoculation with the mutant ZIKVs, where none of the animals showed any sign of illness. Control mice inoculated with phosphate-buffered saline (PBS) alone did not exhibit any body weight loss or illness. Numbers of viral-genome copies in the plasma at 4 dpi and 5 dpi were higher in the rMR-infected group than in the mutant-virus-infected groups (data not shown), similar to what was observed in s.c. inoculation (Fig. 4D). However, when mice were sacrificed at 6 dpi and tissues were examined, the numbers of genome copies and infectious-virus titers in the mutant-ZIKV-infected groups were significantly higher in the brain (Fig. 7A and B) than those from mice inoculated s.c. with the mutant viruses (Fig. 6A and B). Numbers of genome copies in the brains of the mutant-virus-infected animals inoculated i.c. were about 50- to 80-fold higher than in those inoculated s.c. (1.18 × 1010/g versus 1.39 × 108/g for m1MR virus and 1.3 × 1010/g versus 2.7 × 108/g for m2MR virus), whereas numbers of PFU were nearly 80- to 100-fold higher (4.2 × 105/g versus 4.5 × 103/g for m1MR and 5.4 × 105/g versus 6.8 × 103/g for m2MR virus). The rMR replicated well and to similar levels in the brains of animals inoculated by the two routes. The levels of infectious virus in the spleen were similar between the rMR-infected and the mutant-virus-infected groups but were slightly lower in the liver (Fig. 7C). Furthermore, the levels of replication of the rMR or the mutant viruses in the spleen and liver were similar when inoculated by either route (Fig. 6C and 7C). Lymphocytic meningitis and encephalitis were evident and of comparable severity in all three groups of i.c.-inoculated mice. Mild to moderate lymphoid hyperplasia in the spleen was also observed among the three infected groups. The i.c.-inoculated rMR group was associated with moderate neutrophils in the spleen, similar to the s.c. challenge group, whereas neutrophils were rare in the spleens of the mutant-virus-infected mice inoculated by either route. These studies suggest that the mutant viruses replicate well in the brain when inoculated i.c. but may be compromised in their ability to invade the brain from the peripheral site, as seen with s.c. inoculation.
FIG 7.
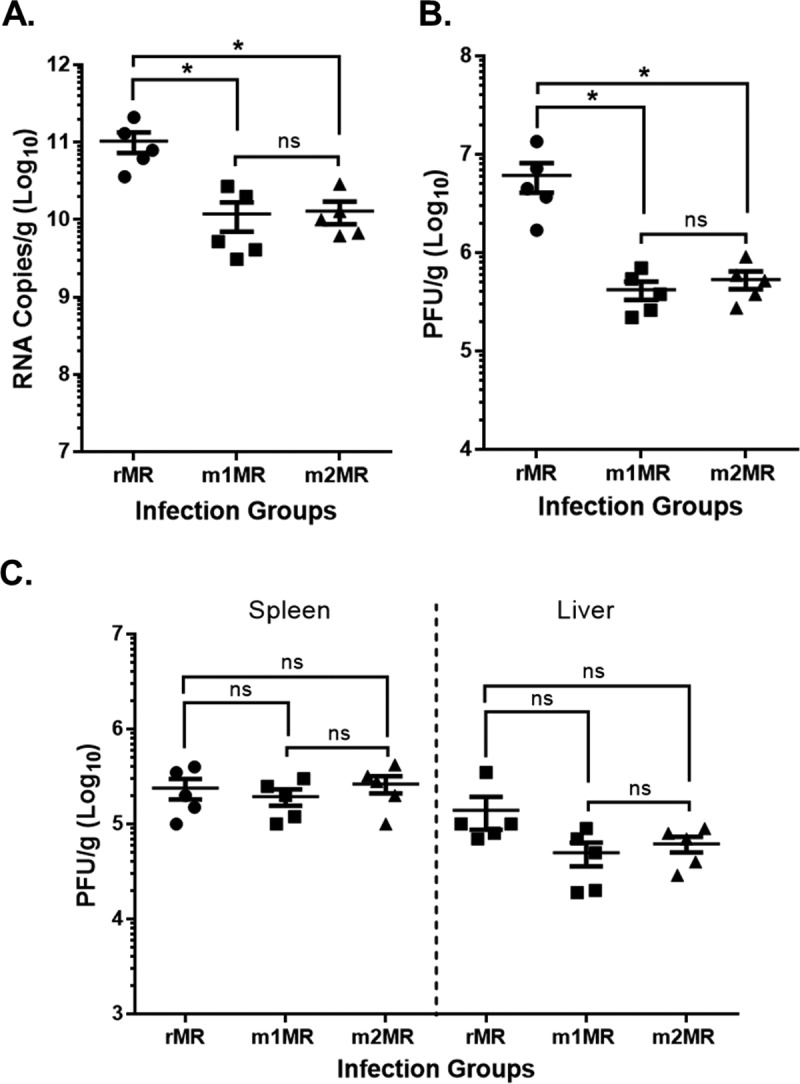
Mutant viruses replicate well in the brain when inoculated i.c. Mice were inoculated i.c. with 1,000 PFU of the rMR (n = 5), m1MR (n = 5), or m2MR (n = 5) virus. (A and B) RNA copies (A) and infectious virus (B) in brains of animals at 6 dpi. (C) Infectious virus in spleens and livers of the same animals at 6 dpi. An unpaired Student t test (two-tailed) was used to determine significance. *, P < 0.05; ns, nonsignificant.
DISCUSSION
With the unprecedented emergence of ZIKV causing diseases, including GBS, microcephaly, and CZS (3, 4, 11–13), attention has focused not only on developing intervention strategies to combat ZIKV diseases, but also on understanding the mechanisms of the virus' emergence and what genetic changes may have contributed to its sudden widespread dissemination and enhanced pathogenicity. Comprehensive genetic, phylogenetic, and evolutionary studies on ZIKV isolates before and during the epidemic have suggested amino acid substitutions in the structural and NS proteins that may contribute to virus transmission, efficient replication, and/or neurotropism (2, 20, 21, 23). In fact, a recent study has shown that a single amino acid substitution (A188V) in the viral NS1 protein is critical for NS1 antigenemia, which in turn facilitates ZIKV acquisition by mosquitoes from viremic animals (24). With the availability of infectious clones of ZIKV (25-29, 47) from historical and contemporary isolates, it will be possible to further examine the specific changes in the viral genome that are responsible for viral transmission, replication, virulence, and tissue tropism.
Here, we investigated the role of the VNDT motif, as well as glycosylation within the motif in the E protein, which appears to exhibit polymorphism in ZIKV isolates. This sequence is located in a loop region in close proximity to the fusion loop as well as the dimer interface, and therefore, glycosylation at this site may impact virus growth, immune evasion, transmission, and/or virulence. VNDT is present in all isolates from the recent outbreaks and absent in several of the African isolates (2, 20) (Table 1), suggesting its potential role in pathogenicity. Additionally, the original African MR766 virus exhibits polymorphism at this site, which could be due to the growth of the virus in the host species from which it was isolated or to the passage of the virus in culture, or both. Given that the passage histories of some of the MR766 isolates whose sequences have been reported in GenBank are unknown, it is unclear how the polymorphism at this site developed. The passage history and animal species from which the virus is isolated could result in deletions, insertions, and/or amino acid substitutions at critical sites in the viral sequences, resulting in polymorphism at those sites, and could potentially lead to increased or decreased transmissibility and pathogenicity. We set out to examine if N-linked glycosylation within the VNDT motif plays any role in ZIKV pathogenicity in an immunocompromised-mouse model. Although infectious clones of MR766 viruses (25, 28, 29), as well as several other ZIKV isolates (26, 27, 47), have been reported, the role of VNDT or glycosylation in this region in pathogenicity has not been explored. A recent study using ectopic expression and pseudoparticle formation with a WNV replicon showed that N-glycosylation of ZIKV E protein is necessary for efficient particle assembly and infectivity (48). The results reported here provide evidence that the deletion of the VNDT motif or a single amino acid substitution within the glycosylation site resulting in the loss of N-glycosylation of the E protein did not significantly affect in vitro or in vivo growth of the virus; however, the pathogenicity of the mutant viruses was significantly compromised.
Studies with WNV have shown that E protein glycosylation influences particle assembly and infectivity (44), as well as neuroinvasion (43, 46). The results presented here suggest glycosylation of E is an important determinant of ZIKV virulence and neuroinvasion. The data also support the contention that the mutant ZIKVs lacking the glycan cannot breach the BBB and neuroinvade as efficiently as the wt virus. The mechanism(s) by which ZIKV enters the CNS from peripheral sites of inoculation is not known, but it likely does so by disrupting the BBB, which is thought to be a key intrinsic defense against encephalitic viruses (49). The BBB consists of brain microvasculature endothelial cells with specialized tight junctions. One possibility is that ZIKV gains central nervous system (CNS) access either as cell-free virus or through infected blood monocytes, which circumvent the BBB. Alternatively, ZIKV may directly infect the endothelial cells or choroid plexus epithelial cells, disrupt the BBB, and enter the CNS. Since the TAM receptor AXL makes endothelial cells susceptible to ZIKV infection (50–52), access of the mutant ZIKVs encoding nonglycosylated E protein to the CNS might be compromised due to their inability to infect these cells. Since WNV disrupts expression of or signaling by IFN-λ (53) and also uses the Toll-like receptor 3-dependent immune response (54) to gain entry into the CNS, it is possible that ZIKV may employ similar or yet unknown mechanisms to facilitate its entry into the CNS. The mutant ZIKVs may have lost their ability do so. Nevertheless, studies to identify the mechanism(s) by which ZIKV gains entry into the CNS will be important to understand how the mutant viruses are unable to neuroinvade.
The reason(s) for the death of one animal in the m1MR-infected group is unknown but could be due to excessive weight loss by the animal, which impacted the overall weight loss on day 7 in this group (Fig. 4B). It is possible that the mutant virus may have reverted to a virulent phenotype in this particular animal. Future studies examining if the mutant virus reverts to a virulent phenotype in some animals through acquisition of compensatory mutations may shed light on this relatively rare event. Some of the animals infected with the mutant ZIKVs developed transient hind limb paralysis that resolved within days. At this time, the cause of the paralysis and the rapid recovery from the disease is unknown. However, it would be interesting to examine if there is a strong immune response directed against the antigens of Schwann cells, leading to paralysis. The presence of prominent hyperplastic germinal centers in the spleens of the mutant-virus-infected mice suggests robust antibody responses. It is possible that these mice have high overall immunoglobulin levels, some of which may be directed toward Schwann cells, thus causing the paralysis. Whether the paralysis observed in the mice represents a form of GBS seen in some ZIKV-infected humans remains to be investigated. Since the rMR-infected mice succumbed to infection by 7 dpi (before the onset of paralysis), further studies would be necessary to determine whether these mice would have displayed paralysis had they survived longer. In conclusion, our studies presented here suggest that E protein glycosylation is an important determinant of ZIKV pathogenicity and neuroinvasion.
MATERIALS AND METHODS
Ethics statement.
All procedures involving animals were conducted in accordance with the guidelines established in the Guide for Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee at the University of Nebraska—Lincoln (UNL). The animals were housed in the Life Sciences Annex building at the university. Mice were anesthetized with isoflurane prior to inoculation, and all efforts were made to minimize animal suffering.
Cells, viruses, and reagents.
Vero (Cercopithecus aethiops; CCL-81), C6/36 (Aedes albopictus; CRL-1660), SH-SY5Y human neuroblastoma (CRL-2266), HTR-8/SVneo human trophoblast (CRL-3271), and NTERA-2 human embryonal carcinoma (CRL-1973) cells were obtained from the ATCC. The cells were grown and maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% heat-inactivated fetal bovine serum (FBS) and penicillin-streptomycin (PS) in a humidified chamber with 5% CO2 at 37°C, except C6/36 cells, which were maintained at 32°C.
The MR766 strain of ZIKV was obtained from Barbara Johnson at the Centers for Disease Control and Prevention, Fort Collins, CO, USA. We passaged the virus once in Vero cells to prepare a stock of the virus, from which genomic RNA was extracted for construction of the infectious clone. Upon sequencing of some protein-coding regions of the genome, we determined that the virus sequence corresponded to GenBank accession number KU720415. For preparation of the stock virus, Vero cells were infected with MR766 virus at a multiplicity of infection (MOI) of 0.1 PFU per cell. Following adsorption at 37°C for 1 h, the inoculum was removed and replaced with virus growth medium (VGM) (DMEM containing 2% FBS, PS, 20 mM HEPES, 1 mM sodium pyruvate, and nonessential amino acids). The infected cells were incubated at 37°C for 4 days, after which the culture supernatant was collected and clarified, FBS was added to a final concentration of 20%, and the stock virus was stored in small aliquots at −80°C. For multistep growth analysis, cells were infected at an MOI of 0.1 PFU/cell and incubated as described above. Small aliquots of infected cell culture supernatants were collected at various times postinfection, clarified, and stored at −80°C for virus titration at a later time. Virus quantitation was performed by plaque assay on Vero cells. Duplicates of serial 10-fold dilutions of virus in VGM were applied to Vero cell monolayers in 24-well plates. Following virus adsorption, the inoculum was removed, and the cell monolayers were overlaid with medium containing 1% low-gelling-temperature (LGT) agarose in VGM. After incubation for 5 days at 37°C, the cells were fixed in 10% formaldehyde in PBS for 30 min, the agarose plugs were removed, and the monolayers were stained with 0.1% crystal violet in 30% methanol. Plaques were counted manually.
Restriction enzymes, DNA-modifying enzymes, the Q5 High Fidelity PCR kit, the ProtoScript II First Strand cDNA synthesis kit, endo H, and PNGase F were obtained from New England BioLabs (Ipswich, MA). SuperScript II was obtained from Invitrogen (Carlsbad, CA). Anti-flavivirus monoclonal antibody D1-4G2-4-15, which reacts with ZIKV E protein, was obtained from EMD Millipore (Billerica, MA). Secondary antibodies were obtained from Sigma (St. Louis, MO) and Invitrogen. TransIT-mRNA transfection reagent was obtained from Mirus Bio (Madison, WI). Oligonucleotide primers and probes for DNA amplification and quantitative PCR (qPCR) were obtained from Sigma and IDT (Coralville, IA). The mMessage mMachine T7 Ultra transcription kit was from Ambion (Austin, TX). Expre35S35S protein labeling mix was obtained from PerkinElmer (Waltham, MA).
Construction of a full-length cDNA clone and virus recovery.
Viral RNA extracted from culture supernatants of infected Vero cells was used as the template for the generation of infectious clones. Four cDNA fragments were amplified by RT-PCR using specific primers shown in Table 2. The amplified products were digested with the appropriate restriction enzymes and cloned in a stepwise manner into the pBR322 vector. Fragment A, which contained an AatII site along with a T7 RNA polymerase promoter and one extra G nucleotide for efficient transcription initiation by the polymerase at the 5′ end prior to the beginning of the viral sequences and SbfI and NruI sites at the 3′ end, was digested with AatII and NruI and cloned into pBR322 at the same sites. Fragments B, C, and D were likewise cloned with the use of unique restriction enzyme sites naturally present in the viral sequences, as well as additional sites incorporated in the primers. The forward primer for amplification of fragment B contained several silent mutations as genetic tags. Fragment D contained a unique BssHII site immediately following the viral genome to linearize the plasmid for in vitro transcription. Since we detected deletion of one or two nucleotides in fragment A at around nucleotide positions 2800 and 3500, we transferred the full-length assembled clones from the pBR322 vector (by digestion with ScaI and NruI) into the linear plasmid pJAZZ-OC (32) using the unique SmaI site in the multiple cloning region of the plasmid. The linear vector, obtained from Lucigen Corporation (Middleton, WI, USA), employs transcription-free cloning technology due to the presence of multiple transcription terminators flanking the cloning site. This eliminates transcription both into and out of the insert DNA and results in increased stability of the insert DNA in the bacterial host. The important genetic and regulatory elements of the vector are described in the legend to Fig. 1. The plasmid, which contains an inducible origin of replication from phage N15, can be maintained at approximately 2 to 4 copies/cell in the BigEasy TSA host strain (Lucigen Corporation). Further details of the BigEasy TSA host strain can be obtained from Lucigen Corporation or from the corresponding author.
TABLE 2.
Primers used in this study
| Primer | Sequence (5′–3′)a | Purpose |
|---|---|---|
| 1F | cggatcgacgtcTAATACGACTCACTATAGAGTTGTTGATCTGTGTGAGTCAGACTGCG | Amplification of fragment A |
| 5030R | gagccgtcgcgaGGATCGGAGATCCTGAGGTCCCTGCAGGGTAGT | Amplification of fragment A |
| 5003F | ctctggactacCCTGCAGGGACGTCCGGAAGTCCGATCCTAGACAAATGTGG | Amplification of fragment B |
| 7190R | catcattagcatcgcgaCTCCAAGGTCCCATGCATAAAATGG | Amplification of fragment B |
| 7006F | CATTGATCTGCGGCCAGC | Amplification of fragment C |
| 8860R | gttgattcgcgaTTCTTTGGTGCAGACGCGTGGCCGC | Amplification of fragment C |
| 8584F | CCATGGGAGCTACGAAGC | Amplification of fragment D |
| 10807R | gagacttcgcgagcgcgcAGAAACCATGGATTTCCCCACACCGGCCGCCGAAGTTCGGCGATCTGTGCCTGGC | Amplification of fragment D |
| POL-6126F | CCTCGCTCTATCGGCCTGAGGCCGATAAG | Polymerase mutation |
| POL-9709R | gacactCCTGAGGGCATGTGCAAACCTATCATCGATTGGCTTCACAACGCAGGCAGCTGCACTGACCGCCATTCGTTTG | Polymerase mutation |
| PJAZZ-10222F | GCCCACCCGAAGGTGAG | Glycosylation mutation |
| GLY-1467R | CTATTTTCGTCAGTTTCATATCCAATCATCCCGCTATGC | VNDT deletion |
| N154A | CAGTTTCATATCCTGTATCAGCGACAATCATCCCGCTATG | Glycosylation mutation |
| GLY-3302R | CTTCACTGTGCCATGGC | Glycosylation mutation |
| Tag-3902F | GCTCTTGAAGGTGACTTG | Detection of genetic tag |
| Tag-5723R | CATTTCCGTTTCTCACGC | Detection of genetic tag |
Restriction enzyme sites are underlined. Viral sequences are shown in boldface uppercase letters. Lowercase lightface letters represent random nonspecific sequences in the primers. Uppercase lightface letters represent a T7 RNA polymerase promoter sequence and an extra G (in primer 1F) and vector sequences (in primer PJAZZ-10222F).
Following the transfer of the full-length ZIKV cDNA clone into the pJAZZ-OC vector, we repaired the mutation(s) by fragment exchange using the unique AflII site at position 2993 of the viral genome. Stable full-length clones (pJ-rMR) of correct sequences were recovered using this vector and used subsequently. Fragment A was also subcloned in the linear vector, which was used as the template for mutagenesis to generate the VNDT deletion mutant (m1MR) or the glycosylation mutant (m2MR) by substituting alanine in place of asparagine using the primers (Table 2). A polymerase-defective full-length clone (pJ-rMR/Pol−) was generated using the primers (Table 2) with the full-length clone as the template.
In vitro transcription, RNA transfection, and virus recovery.
An mMessage mMachine T7 Ultra transcription kit from Ambion (Austin, TX) was used for in vitro synthesis of RNA from full-length clones according to the manufacturer's recommendations. Plasmids containing the full-length viral genomes (rMR, Pol−, or mutant constructs) were linearized by digestion with BssHII and then used for in vitro transcription to generate full-length transcripts. Following digestion of the reaction products with RQ1 RNase-free DNase (Promega, Madison, WI, USA), the RNA was recovered and quantitated by spectrometry, as well as by agarose gel electrophoresis. One to 5 μg of RNA was transfected into monolayers of Vero cells in six-well plates using TransIT-mRNA transfection reagent according to the manufacturer's recommendations. The transfected cells were incubated in a CO2 chamber at 37°C and observed for development of cytopathic effects. Aliquots of the culture supernatants were collected at 24-h intervals for plaque assay to determine if infectious virus was recovered from the transfected cells.
Mouse studies and pathogenesis evaluation.
Four-week-old mice deficient in IFN-α/β receptor (A129) were obtained from the Jackson Laboratory, Bar Harbor, ME, USA. After acclimatization for 4 days, the mice were anesthetized as described above and inoculated s.c. with 1,000 PFU of MR766 (pMR), infectious clone-derived recombinant MR766 (rMR), VNDT deletion (m1MR), or glycosylation mutant (m2MR) virus diluted in 100 μl of PBS. For intracranial (i.c.) inoculation, 1,000 PFU of these viruses was diluted in 30 μl of PBS. Following virus inoculation, the mice were monitored twice daily for disease signs. Weight change, clinical symptoms, and survival were recorded daily. Clinical symptoms were scored as follows: 0, normal; 1, ruffled fur; 2, conjunctivitis, lethargy, and hunched posture; 3, paralysis of one hind limb; 4, paralysis of both hind limbs; 5, paralysis of all four limbs; 6, moribund and euthanization; 7, dead. Mice were scored as moribund when they appeared very lethargic, showed limb paralysis, and/or lost greater than 20% of their initial body weight. Blood was collected by retro-orbital bleeding under anesthesia. For tissue sample collection, mice were euthanized by CO2 inhalation.
Histopathological evaluation.
Sections of brain, liver, spleen, and kidney were collected into 10% neutral buffered formalin, processed routinely into paraffin wax, and sectioned at 5 μm. The tissue sections were then stained with hematoxylin and eosin (H&E) for histologic evaluation. The slides were examined by a certified pathologist blinded to the treatment groups. Brain sections were evaluated for meningitis, encephalitis, and neuronal necrosis. Spleens were evaluated for neutrophilic infiltrates, lymphoid karyorrhexis, and follicular hyperplasia. Livers were scored for numbers of inflammatory-cell foci. Any other findings deviating from normal tissues were noted. Severity was scored as follows: 0, normal; 1, minimal; 2, mild; 3, moderate; and 4, severe. Meningoencephalitis scores were combined meningeal and parenchymal scores.
Real-time qPCR.
Mice were euthanized at 6 dpi to collect brain, spleen, and liver samples, with one half used for qPCR and the other half used for H&E staining. Plasma recovered from blood collected at various times postinfection was used for qPCR. All samples for quantitation of viral loads were stored at −80°C until further processing. Tissue samples were weighed and homogenized in 1× DMEM in a Bullet Blender (Next Advance, Inc., Averill Park, NY) using metal beads with the speed setting at 10 for 5 min at 4°C. Homogenized extracts were clarified by centrifugation at 14,000 × g for 15 min at 4°C, and the supernatants were used to perform plaque assays and viral RNA isolation. RNA isolation was performed with a QIAamp viral-RNA extraction kit (Qiagen, Valencia, CA) using 140 μl of tissue homogenate or 10 to 20 μl plasma. We employed a standard curve method for absolute quantification of ZIKV RNA from the samples using a specific real-time qPCR assay. The primer and 6-carboxyfluorescein (FAM) probe sequences were adopted from a published method (55) with in-house modification and optimization. An oligonucleotide corresponding to the E gene sequence at positions 1191 to 1268 was synthesized and used as the template for determination of a standard curve using the primers 5′-GTCGTTGCCCAACACAAG-3′ (forward) and 5′-CCACTAATGTTCTTTTGCAGAC-3′ (reverse) and the probe 5′-/56-FAM (5′ 6-carboxyfluorescein)/AGCCTACCT/ZEN/TGACAAGCAATCAGACACTCAA/3IABkFQ (3′ Iowa black fluorescent quencher)/-3′. Real-time PCR assays were performed in two steps: viral RNA was used for cDNA synthesis by SuperScript II (Thermo Fisher Scientific Inc., MA), and the cDNAs were then used as the templates in qPCR. The cycling conditions used were as follows: initial denaturation at 95°C for 10 min; 35 cycles of 95°C for 30 s, 55.3°C for 30 s, and 72°C for 30 s; and a melt curve from 65°C to 95°C with 0.5°C increments for 5 s. The reactions were run in CFX Connect (Bio-Rad, CA). Viral-genome copy numbers were determined in parallel reactions using RNA samples in duplicate and expressed as copy numbers per gram of tissue or milliliter of plasma.
Immunofluorescence.
For the detection of E protein following transfection with in vitro transcripts, transfected cells were washed three times with cold PBS, fixed in cold methanol-acetone (1:1) for 10 min, and then washed with PBS three times. Primary antibody (4G2) targeting ZIKV E protein was added at a 1:500 dilution and incubated for 1 h at room temperature. The cells were washed three times with PBS, and then Alexa Fluor 488-conjugated secondary antibody was added for 1 h in the dark. Following incubation, the cells were washed three times with PBS and examined under a fluorescence microscope. Images were obtained using the Evos FL Auto Cell Imaging System at the Microscopy Core Facility at the UNL.
Metabolic radiolabeling and immunoprecipitation.
Radiolabeling of viral proteins in infected cells was performed with Expre35S35S protein-labeling mix as described previously (56). The radiolabeled E protein from ZIKV-infected cells was immunoprecipitated with 4G2 antibody, digested with endo H or PNGase F according to the manufacturer's recommendations, examined by electrophoresis in an 8% polyacrylamide gel containing sodium dodecyl sulfate (SDS-PAGE) under reducing conditions, and detected by fluorography as described previously (56).
Statistical analysis.
Data were analyzed using GraphPad Prism software version 6.0. An unpaired two-tailed Student t test or Mann-Whitney test was performed for pairwise comparisons between the groups to determine significant differences in viral loads (RNA levels and infectious titers). Percent weight change between groups was analyzed by two-way analysis of variance (ANOVA) (Tukey multiple-comparison test). The data are represented as either means and standard errors of the mean (SEM) or means and standard deviations (SD).
Accession number(s).
The sequence of the infectious clone reported here has been deposited in GenBank with accession number KY989511.
ACKNOWLEDGMENTS
We thank Barbara Johnson and Brandy Russell of the Centers for Disease Control and Prevention, Fort Collins, CO, USA, for providing the MR766 isolate. We appreciate the help of Ignacio Correas for statistical analysis.
The research was supported in part by funds from the University of Nebraska—Lincoln.
REFERENCES
- 1.Dick GW, Kitchen SF, Haddow AJ. 1952. Zika virus. I. Isolations and serological specificity. Trans R Soc Trop Med Hyg 46:509–520. doi: 10.1016/0035-9203(52)90042-4. [DOI] [PubMed] [Google Scholar]
- 2.Faye O, Freire CC, Iamarino A, Faye O, de Oliveira JV, Diallo M, Zanotto PM, Sall AA. 2014. Molecular evolution of Zika virus during its emergence in the 20(th) century. PLoS Negl Trop Dis 8:e2636. doi: 10.1371/journal.pntd.0002636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan JF, Choi GK, Yip CC, Cheng VC, Yuen KY. 2016. Zika fever and congenital Zika syndrome: an unexpected emerging arboviral disease. J Infect 72:507–524. doi: 10.1016/j.jinf.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Musso D, Gubler DJ. 2016. Zika virus. Clin Microbiol Rev 29:487–524. doi: 10.1128/CMR.00072-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duffy MR, Chen TH, Hancock WT, Powers AM, Kool JL, Lanciotti RS, Pretrick M, Marfel M, Holzbauer S, Dubray C, Guillaumot L, Griggs A, Bel M, Lambert AJ, Laven J, Kosoy O, Panella A, Biggerstaff BJ, Fischer M, Hayes EB. 2009. Zika virus outbreak on Yap Island, Federated States of Micronesia. N Engl J Med 360:2536–2543. doi: 10.1056/NEJMoa0805715. [DOI] [PubMed] [Google Scholar]
- 6.Cao-Lormeau VM, Roche C, Teissier A, Robin E, Berry AL, Mallet HP, Sall AA, Musso D. 2014. Zika virus, French Polynesia, South Pacific, 2013. Emerg Infect Dis 20:1085–1086. doi: 10.3201/eid2006.140138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Musso D, Nilles EJ, Cao-Lormeau VM. 2014. Rapid spread of emerging Zika virus in the Pacific area. Clin Microbiol Infect 20:O595–O596. doi: 10.1111/1469-0691.12707. [DOI] [PubMed] [Google Scholar]
- 8.Zanluca C, Melo VC, Mosimann AL, Santos GI, Santos CN, Luz K. 2015. First report of autochthonous transmission of Zika virus in Brazil. Mem Inst Oswaldo Cruz 110:569–572. doi: 10.1590/0074-02760150192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haddow AD, Schuh AJ, Yasuda CY, Kasper MR, Heang V, Huy R, Guzman H, Tesh RB, Weaver SC. 2012. Genetic characterization of Zika virus strains: geographic expansion of the Asian lineage. PLoS Negl Trop Dis 6:e1477. doi: 10.1371/journal.pntd.0001477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Enfissi A, Codrington J, Roosblad J, Kazanji M, Rousset D. 2016. Zika virus genome from the Americas. Lancet 387:227–228. doi: 10.1016/S0140-6736(16)00003-9. [DOI] [PubMed] [Google Scholar]
- 11.Lazear HM, Diamond MS. 2016. Zika virus: new clinical syndromes and its emergence in the Western Hemisphere. J Virol 90:4864–4875. doi: 10.1128/JVI.00252-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miner JJ, Diamond MS. 2017. Zika virus pathogenesis and tissue tropism. Cell Host Microbe 21:134–142. doi: 10.1016/j.chom.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coyne CB, Lazear HM. 2016. Zika virus—reigniting the TORCH. Nat Rev Microbiol 14:707–715. doi: 10.1038/nrmicro.2016.125. [DOI] [PubMed] [Google Scholar]
- 14.Lindenbach BD, Murray CL, Thiel RJ, Rice CM. 2013. Flaviviridae, p 712–746. In Knipe DM, Howley PM (ed), Fields virology, 6th ed Lippincott Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 15.Diamond MS, Pierson TC. 2015. Molecular insight into dengue virus pathogenesis and its implications for disease control. Cell 162:488–492. doi: 10.1016/j.cell.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, Schwarz MC, Sanchez-Seco MP, Evans MJ, Best SM, Garcia-Sastre A. 2016. Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe 19:882–890. doi: 10.1016/j.chom.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar A, Hou S, Airo AM, Limonta D, Mancinelli V, Branton W, Power C, Hobman TC. 2016. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep 17:1766–1775. doi: 10.15252/embr.201642627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu Z, Chan JF, Tee KM, Choi GK, Lau SK, Woo PC, Tse H, Yuen KY. 2016. Comparative genomic analysis of pre-epidemic and epidemic Zika virus strains for virological factors potentially associated with the rapidly expanding epidemic. Emerg Microbes Infect 5:e22. doi: 10.1038/emi.2016.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh RK, Dhama K, Malik YS, Ramakrishnan MA, Karthik K, Tiwari R, Saurabh S, Sachan S, Joshi SK. 2016. Zika virus—emergence, evolution, pathology, diagnosis, and control: current global scenario and future perspectives—a comprehensive review. Vet Q 36:150–175. doi: 10.1080/01652176.2016.1188333. [DOI] [PubMed] [Google Scholar]
- 20.May M, Relich RF. 2016. A comprehensive systems biology approach to studying Zika virus. PLoS One 11:e0161355. doi: 10.1371/journal.pone.0161355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang L, Valderramos SG, Wu A, Ouyang S, Li C, Brasil P, Bonaldo M, Coates T, Nielsen-Saines K, Jiang T, Aliyari R, Cheng G. 2016. From mosquitos to humans: genetic evolution of Zika virus. Cell Host Microbe 19:561–565. doi: 10.1016/j.chom.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weaver SC. 2017. Emergence of epidemic Zika virus transmission and congenital Zika syndrome: are recently evolved traits to blame? mBio 8:e02063-16. doi: 10.1128/mBio.02063-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pettersson JH, Eldholm V, Seligman SJ, Lundkvist A, Falconar AK, Gaunt MW, Musso D, Nougairede A, Charrel R, Gould EA, de Lamballerie X. 2016. How did Zika virus emerge in the Pacific Islands and Latin America? mBio 7:e01239-16. doi: 10.1128/mBio.01239-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y, Liu J, Du S, Shan C, Nie K, Zhang R, Li XF, Wang T, Qin CF, Wang P, Shi PY, Cheng G. 2017. Evolutionary enhancement of Zika virus infectivity in Aedes aegypti mosquitoes. Nature 545:482–486. doi: 10.1038/nature22365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwarz MC, Sourisseau M, Espino MM, Gray ES, Chambers MT, Tortorella D, Evans MJ. 2016. Rescue of the 1947 Zika virus prototype strain with a cytomegalovirus promoter-driven cDNA clone. mSphere 1:e00246-16. doi: 10.1128/mSphere.00246-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsetsarkin KA, Kenney H, Chen R, Liu G, Manukyan H, Whitehead SS, Laassri M, Chumakov K, Pletnev AG. 2016. A full-length infectious cDNA clone of Zika virus from the 2015 epidemic in Brazil as a genetic platform for studies of virus-host interactions and vaccine development. mBio 7:e01114-16. doi: 10.1128/mBio.01114-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weger-Lucarelli J, Duggal NK, Bullard-Feibelman K, Veselinovic M, Romo H, Nguyen C, Ruckert C, Brault AC, Bowen RA, Stenglein M, Geiss BJ, Ebel GD. 2017. Development and characterization of recombinant virus generated from a New World Zika virus infectious clone. J Virol 91:e01765-16. doi: 10.1128/JVI.01765-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gadea G, Bos S, Krejbich-Trotot P, Clain E, Viranaicken W, El-Kalamouni C, Mavingui P, Despres P. 2016. A robust method for the rapid generation of recombinant Zika virus expressing the GFP reporter gene. Virology 497:157–162. doi: 10.1016/j.virol.2016.07.015. [DOI] [PubMed] [Google Scholar]
- 29.Widman DG, Young E, Yount BL, Plante KS, Gallichotte EN, Carbaugh DL, Peck KM, Plante J, Swanstrom J, Heise MT, Lazear HM, Baric RS. 2017. A reverse genetics platform that spans the Zika virus family tree. mBio 8:e02014-16. doi: 10.1128/mBio.02014-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rice CM, Grakoui A, Galler R, Chambers TJ. 1989. Transcription of infectious yellow fever RNA from full-length cDNA templates produced by in vitro ligation. New Biol 1:285–296. [PubMed] [Google Scholar]
- 31.Pu SY, Wu RH, Yang CC, Jao TM, Tsai MH, Wang JC, Lin HM, Chao YS, Yueh A. 2011. Successful propagation of flavivirus infectious cDNAs by a novel method to reduce the cryptic bacterial promoter activity of virus genomes. J Virol 85:2927–2941. doi: 10.1128/JVI.01986-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Godiska R, Mead D, Dhodda V, Wu C, Hochstein R, Karsi A, Usdin K, Entezam A, Ravin N. 2010. Linear plasmid vector for cloning of repetitive or unstable sequences in Escherichia coli. Nucleic Acids Res 38:e88. doi: 10.1093/nar/gkp1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morrison TE, Diamond MS. 2017. Animal models of Zika virus infection, pathogenesis, and immunity. J Virol 91:e00009-17. doi: 10.1128/JVI.00009-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, Miner JJ, Diamond MS. 2016. A mouse model of Zika virus pathogenesis. Cell Host Microbe 19:720–730. doi: 10.1016/j.chom.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rossi SL, Tesh RB, Azar SR, Muruato AE, Hanley KA, Auguste AJ, Langsjoen RM, Paessler S, Vasilakis N, Weaver SC. 2016. Characterization of a novel murine model to study Zika virus. Am J Trop Med Hyg 94:1362–1369. doi: 10.4269/ajtmh.16-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aliota MT, Caine EA, Walker EC, Larkin KE, Camacho E, Osorio JE. 2016. Characterization of lethal Zika virus infection in AG129 mice. PLoS Negl Trop Dis 10:e0004682. doi: 10.1371/journal.pntd.0004682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dowall SD, Graham VA, Rayner E, Atkinson B, Hall G, Watson RJ, Bosworth A, Bonney LC, Kitchen S, Hewson R. 2016. A susceptible mouse model for Zika virus infection. PLoS Negl Trop Dis 10:e0004658. doi: 10.1371/journal.pntd.0004658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H, Saucedo-Cuevas L, Regla-Nava JA, Chai G, Sheets N, Tang W, Terskikh AV, Shresta S, Gleeson JG. 2016. Zika virus infects neural progenitors in the adult mouse brain and alters proliferation. Cell Stem Cell 19:593–598. doi: 10.1016/j.stem.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tripathi S, Balasubramaniam VR, Brown JA, Mena I, Grant A, Bardina SV, Maringer K, Schwarz MC, Maestre AM, Sourisseau M, Albrecht RA, Krammer F, Evans MJ, Fernandez-Sesma A, Lim JK, Garcia-Sastre A. 2017. A novel Zika virus mouse model reveals strain specific differences in virus pathogenesis and host inflammatory immune responses. PLoS Pathog 13:e1006258. doi: 10.1371/journal.ppat.1006258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dai L, Song J, Lu X, Deng YQ, Musyoki AM, Cheng H, Zhang Y, Yuan Y, Song H, Haywood J, Xiao H, Yan J, Shi Y, Qin CF, Qi J, Gao GF. 2016. Structures of the Zika virus envelope protein and its complex with a flavivirus broadly protective antibody. Cell Host Microbe 19:696–704. doi: 10.1016/j.chom.2016.04.013. [DOI] [PubMed] [Google Scholar]
- 41.Sirohi D, Chen Z, Sun L, Klose T, Pierson TC, Rossmann MG, Kuhn RJ. 2016. The 3.8 A resolution cryo-EM structure of Zika virus. Science 352:467–470. doi: 10.1126/science.aaf5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chambers TJ, Halevy M, Nestorowicz A, Rice CM, Lustig S. 1998. West Nile virus envelope proteins: nucleotide sequence analysis of strains differing in mouse neuroinvasiveness. J Gen Virol 79:2375–2380. doi: 10.1099/0022-1317-79-10-2375. [DOI] [PubMed] [Google Scholar]
- 43.Beasley DW, Whiteman MC, Zhang S, Huang CY, Schneider BS, Smith DR, Gromowski GD, Higgs S, Kinney RM, Barrett AD. 2005. Envelope protein glycosylation status influences mouse neuroinvasion phenotype of genetic lineage 1 West Nile virus strains. J Virol 79:8339–8347. doi: 10.1128/JVI.79.13.8339-8347.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hanna SL, Pierson TC, Sanchez MD, Ahmed AA, Murtadha MM, Doms RW. 2005. N-linked glycosylation of West Nile virus envelope proteins influences particle assembly and infectivity. J Virol 79:13262–13274. doi: 10.1128/JVI.79.21.13262-13274.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee E, Leang SK, Davidson A, Lobigs M. 2010. Both E protein glycans adversely affect dengue virus infectivity but are beneficial for virion release. J Virol 84:5171–5180. doi: 10.1128/JVI.01900-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shirato K, Miyoshi H, Goto A, Ako Y, Ueki T, Kariwa H, Takashima I. 2004. Viral envelope protein glycosylation is a molecular determinant of the neuroinvasiveness of the New York strain of West Nile virus. J Gen Virol 85:3637–3645. doi: 10.1099/vir.0.80247-0. [DOI] [PubMed] [Google Scholar]
- 47.Shan C, Xie X, Muruato AE, Rossi SL, Roundy CM, Azar SR, Yang Y, Tesh RB, Bourne N, Barrett AD, Vasilakis N, Weaver SC, Shi PY. 2016. An infectious cDNA clone of Zika virus to study viral virulence, mosquito transmission, and antiviral inhibitors. Cell Host Microbe 19:891–900. doi: 10.1016/j.chom.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mossenta M, Marchese S, Poggianella M, Slon Campos JL, Burrone OR. 2017. Role of N-glycosylation on Zika virus E protein secretion, viral assembly and infectivity. Biochem Biophys Res Commun 492:579–586. doi: 10.1016/j.bbrc.2017.01.022. [DOI] [PubMed] [Google Scholar]
- 49.Koyuncu OO, Hogue IB, Enquist LW. 2013. Virus infections in the nervous system. Cell Host Microbe 13:379–393. doi: 10.1016/j.chom.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nowakowski TJ, Pollen AA, Di Lullo E, Sandoval-Espinosa C, Bershteyn M, Kriegstein AR. 2016. Expression analysis highlights AXL as a candidate Zika virus entry receptor in neural stem cells. Cell Stem Cell 18:591–596. doi: 10.1016/j.stem.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu S, DeLalio LJ, Isakson BE, Wang TT. 2016. AXL-mediated productive infection of human endothelial cells by Zika virus. Circ Res 119:1183–1189. doi: 10.1161/CIRCRESAHA.116.309866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Richard AS, Shim BS, Kwon YC, Zhang R, Otsuka Y, Schmitt K, Berri F, Diamond MS, Choe H. 2017. AXL-dependent infection of human fetal endothelial cells distinguishes Zika virus from other pathogenic flaviviruses. Proc Natl Acad Sci U S A 114:2024–2029. doi: 10.1073/pnas.1620558114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lazear HM, Daniels BP, Pinto AK, Huang AC, Vick SC, Doyle SE, Gale M Jr, Klein RS, Diamond MS. 2015. Interferon-lambda restricts West Nile virus neuroinvasion by tightening the blood-brain barrier. Sci Transl Med 7:284ra259. doi: 10.1126/scitranslmed.aaa4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. 2004. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med 10:1366–1373. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- 55.Corman VM, Rasche A, Baronti C, Aldabbagh S, Cadar D, Reusken CB, Pas SD, Goorhuis A, Schinkel J, Molenkamp R, Kummerer BM, Bleicker T, Brunink S, Eschbach-Bludau M, Eis-Hubinger AM, Koopmans MP, Schmidt-Chanasit J, Grobusch MP, de Lamballerie X, Drosten C, Drexler JF. 2016. Assay optimization for molecular detection of Zika virus. Bull World Health Organ 94:880–892. doi: 10.2471/BLT.16.175950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ansari IH, Kwon B, Osorio FA, Pattnaik AK. 2006. Influence of N-linked glycosylation of porcine reproductive and respiratory syndrome virus GP5 on virus infectivity, antigenicity, and ability to induce neutralizing antibodies. J Virol 80:3994–4004. doi: 10.1128/JVI.80.8.3994-4004.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]