

Abstract
The broadly active glutamine antagonist 6-diazo-5-oxo-L-norleucine (DON) has been studied for sixty years as a potential anticancer therapeutic. Clinical studies of DON in the 1950s using low daily doses suggested antitumor activity, but later phase I and II trials of DON given intermittently at high doses were hampered by dose-limiting nausea and vomiting. Further clinical development of DON was abandoned. Recently the recognition that multiple tumor types are glutamine dependent has renewed interest in metabolic inhibitors such as DON. Here we describe the prior experience with DON in humans. Evaluation of past studies suggests that the major impediments to successful clinical use included unacceptable gastrointestinal (GI) toxicities, inappropriate dosing schedules for a metabolic inhibitor, and lack of targeted patient selection. To circumvent GI toxicity, prodrug strategies for DON have been developed to enhance delivery of active compound to tumor tissues, including the CNS. When these prodrugs are administered in a low daily dosing regimen, appropriate for metabolic inhibition, they are robustly effective without significant toxicity. Patients whose tumors have genetic, metabolic, or imaging biomarker evidence of glutamine dependence should be prioritized as candidates for future clinical evaluations of novel DON prodrugs, given either as monotherapy or in rationally-directed pharmacologic combinations.
Keywords: glutamine, prodrug, DON, glutaminase, cancer
Preclinical studies:
Glutamine metabolism as a therapeutic target in cancer
Glutamine is the most abundant amino acid in blood, with serum concentration of several hundred micromolar. Several recent reviews detail glutamine utilization in multicellular organisms (1–3). Rapidly proliferating healthy cells (GI epithelium, lymphocytes) or cells under physiologic stress have increased demand for glutamine. Glutamine is transported into cells by one of multiple amino acid transporters (e.g. ASCT2, BOAT2), several of which are thought to be upregulated in cancer cells (2). Once inside the cell, glutamine is utilized in multiple metabolic processes (Figure 1): 1) it is hydrolyzed to glutamate and ammonia by glutaminase (‘glutaminolysis’) (4); 2) it is subject to glutamine amidotransferases which catalyze the use of its amido group as a building block for nucleosides, amino acids, and hexosamine sugars (2); and 3) it is used directly to charge tRNA for protein synthesis. Glutamate, produced from glutamine by glutaminase and glutamine amidotransferase activities, may be further metabolized to alpha ketoglutarate and provide a carbon skeleton source for the mitochondrial tricarboxylic acid cycle (TCA cycle). In addition to generating reducing equivalents and high-energy intermediates through mitochondrial respiration, the TCA cycle also exports carbon skeletons for synthesis of lipids, nucleotides, and reducing equivalents, particularly in rapidly proliferating cells (4,5). Glutamine-derived glutamate is also involved in the synthesis of the reducing equivalent glutathione, vital to maintaining cellular redox status.
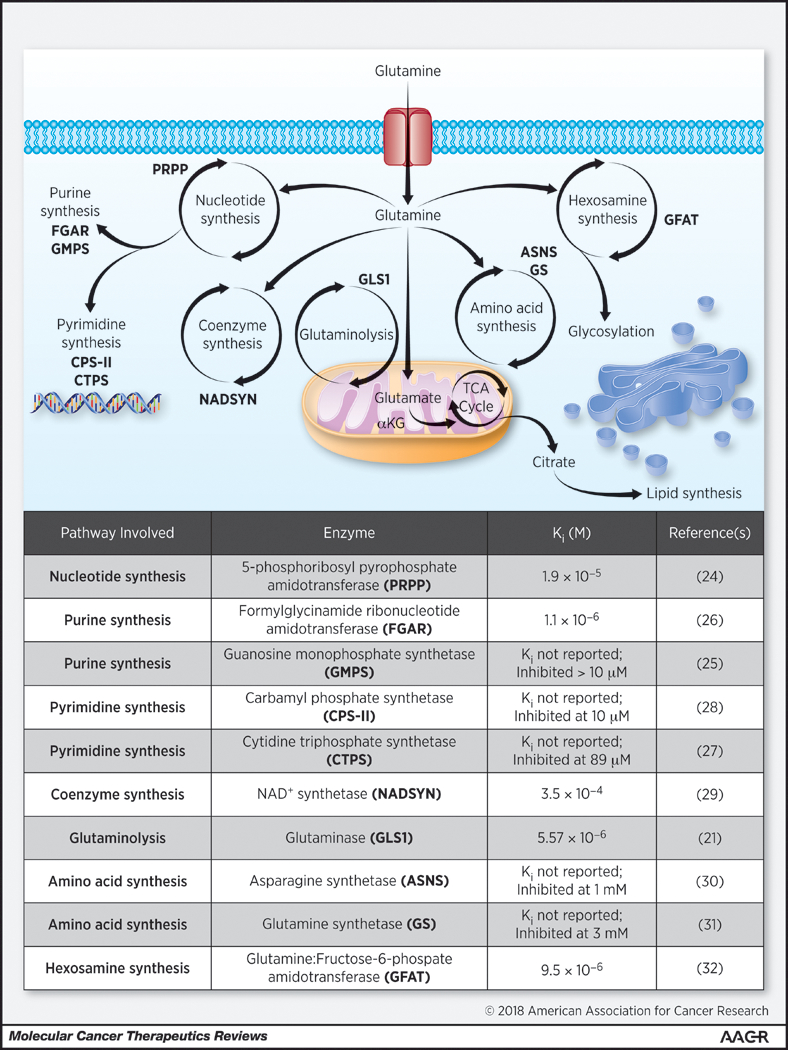
Figure 1: Cellular Glutamine Utilization Targeted by DON.
Top: Illustration depicting major glutamine utilizing pathways in mammalian cells with target enzymes (pink abbreviations) known to be inhibited by DON. Bottom: List of known pathways and enzymes affected by DON with established Ki values where available.
Many tumors become largely dependent on glutamine to provide carbon and nitrogen building blocks needed for proliferation. Warburg noted in the 1920s that in the presence of adequate oxygen, tumors increase glucose uptake and ferment much of it to lactate (6). In cancer model systems, Eagle and colleagues first demonstrated tumor cells in culture require supplementation with exogenous glutamine for efficient proliferation (7). It was subsequently shown that when deprived of glutamine tumor cells undergo apoptosis (8). As interest in cancer metabolism has grown, glutamine utilization by cancer cells and its genetic regulation have become areas of intense interest (1–3). The most well-characterized oncogene to regulate glutamine metabolism is MYC (9), which enhances glutaminase expression, upregulates glutamine transporters, and enhances glutamine utilization in energy production and biosynthesis (1). Other pro-tumorigenic regulators such as KRAS and mTOR, as well as tumor suppressors (p53, VHL) have also been associated with alterations in glutamine metabolism (5,10).
Tumor glutamine dependence has been targeted with selective glutaminase inhibitors with some success. Several allosteric inhibitors including BPTES (bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide), compound 968, and CB-839 (Calithera) have shown robust activity in cell culture experiments and promising single agent preclinical activity (11–13). CB-839 has proceeded into clinical studies. Although target engagement was clearly observed (14), single agent antitumor activity was minimal; combination trials are now underway with promising initial results (15,16).
Perhaps a reason for the lack of robust clinical effect of selective glutaminase inhibitors is that glutamine metabolism in tumors is more complex than initially hypothesized. Tumor cells are highly adaptable and alter nutrient uptake and metabolic networks to resist single agent glutaminase inhibition (17,18). Therefore short term cell culture and preclinical studies may not adequately predict the metabolic response of tumors with longer term drug exposure. Additionally, in vitro studies rarely account for the effects of stromal cells or the microenvironment on nutrient availability to tumor. Indeed, it was recently shown that cells in the microenvironment of several tumor types upregulate glutamine production, thereby enabling tumor cells to escape glutaminase inhibition (19). All of these studies recommend combination therapy as a means to improve efficacy and avoid tumor resistance to single-agent glutaminase inhibition or a broader approach to inhibition of glutamine utilization.
DON broadly inhibits glutamine-utilizing enzymes
6-diazo-5-oxo-norleucine (DON) (Figure 2A) is the best-studied broadly active glutamine antagonist, having multiple supporting biochemical, preclinical and clinical evaluations. DON was originally isolated from fermentation broth of a Streptomyces in the 1950s (20). Biochemical studies on DON identified a two-step, mechanism-based mode of inhibition across multiple glutamine-utilizing enzymes. First, DON binds competitively to the glutamine active site, then a covalent adduct is formed irreversibly inhibiting the enzyme (21). Importantly DON’s diazoketone group is stable under physiological conditions because of the electron-withdrawing carbonyl group stabilizing the diazo dipole. As a result, DON acts as a reactive electrophile only when protonated at the α-position under certain conditions (e.g. in the proximity of the active-site serine residue in glutaminase), triggering the release of nitrogen (N2) (22). Thus DON serves as a selective mechanism-based inactivator of glutamine-utilizing reactions rather than a non-specific reactive intermediate. DON inhibits glutamine-utilizing enzymes including glutaminase at low micromolar levels (21) as well as multiple glutamine amidotransferases (23) involved in de novo purine and pyrimidine synthesis (24–28), coenzyme synthesis (29), amino acid synthesis (30,31), and hexosamine production (32) (Figure 1). The kinetics of inhibition and inactivation have been described for some, though not all, of DON’s target enzymes. At far higher concentrations DON also serves as a substrate and an inhibitor of several amino acid transporters and transglutaminases (28), as well as a number of amino acid synthesis reactions more relevant in prokaryotic systems (33).
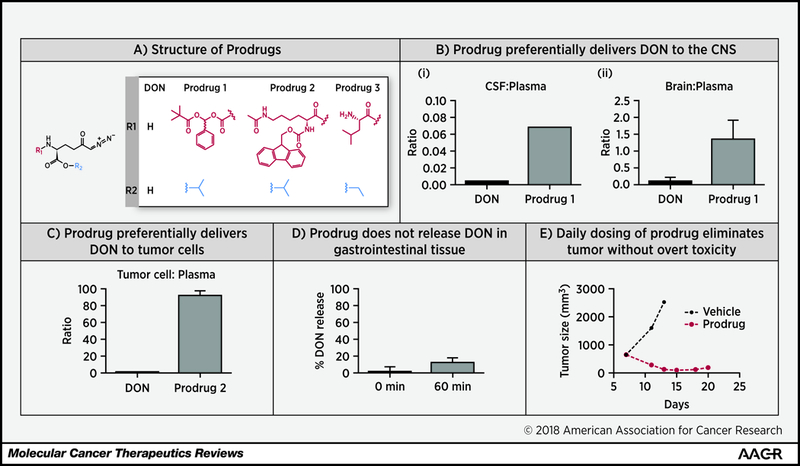
Figure 2: DON prodrugs enhance DON tissue-to-plasma ratios and possess potent antitumor activity without overt toxicities.
(A) Structures of DON Prodrugs. (B) Prodrug 1 preferentially delivers DON to the CNS. DON and Prodrug 1 were dosed by IV infusion (1.6 mg/kg equivalent doses for 1h) in swine and DON was quantified in plasma, CSF and brain samples. Prodrug 1 delivered DON preferentially to CSF (i) and brain (ii) resulting in 15-fold enhanced CSF/plasma ratio and 9 fold enhanced brain/plasma ratio when compared directly with equimolar doses of DON. Adapted with permission from reference (58). Copyright 2017 American Chemical Society. (C) Prodrug 2 preferentially delivers DON to P493B tumor cells. In head-to-head comparison, Prodrug 2 shows a 90-fold enhanced tumor cell/plasma ratio versus DON. (D) Prodrug 2 does not release significant DON in gastrointestinal tissue. Prodrug 2 was incubated with pig gastrointestinal homogenate for 60 minutes. At 60 minutes, DON liberation from Prodrug 2 was limited to 20%. (E) Daily dosing of Prodrug 3 eliminates EL4 engrafted tumors without weight loss or overt toxicity. 8–10 week old male C57BL/6 mice (6–7 per treatment group) were injected subcutaneously into the right flank with 1×106 EL4 cells on day 0, and after five days of growth the mice had measurable tumors. On day 6 after implantation, treatment was initiated with either vehicle or DON Prodrug (1 mg/kg p.o. daily × 5 days followed by 0.3 mg/kg, p.o.). Tumors were measured every other day with calipers and volumes calculated by the formula V=(LxW2)/2. DON prodrug eliminated tumor in all mice, while mice treated with vehicle succumbed to tumors by day 13.
In early preclinical cancer models, DON inhibited the growth of multiple cancer cell lines in culture (34), and prevented tumor growth and increased survival several murine cancer models including murine sarcomas, carcinomas, and leukemias (28), (35). Of note the most effective dosing regimens in some early rodent studies were daily low dose therapy (36). Based on these data there was interest in taking DON into human studies as an antitumor agent.
Clinical Studies:
1956 – 1962: Low daily dosing of DON shows anti-tumor activity
The first-in-human study of DON was published in 1957. Magill and colleagues evaluated DON as a single agent in 63 patients (37). The majority of patients had advanced inoperable solid tumors including lung, breast, colon, and genitourinary tumors; a smaller number had leukemia or lymphoma. DON was given as a single agent by either intravenous, intramuscular, or oral routes at doses ranging from 0.2–0.6 mg/kg/day once daily (IV/IM) or 0.2–1.1 mg/kg/day (oral) (conversion 7.4 – 40.7 mg/m2/dose) (Table 1). Several smaller groups of patients also received multiple smaller daily doses (e.g. 0.2 mg/kg/dose q4–6 hours) or less frequent infusions at higher mg/kg dosing (1.6 mg/kg IV q4 days). Duration of DON treatment varied based on toxicities but three-quarters of patients were treated for two or more weeks.
Table 1:
Clinical Experience with DON in Humans
| Early Studies (1957–1962) at low daily dosing | ||||
|---|---|---|---|---|
| Patients enrolled Disease Type |
Dosing Regimen(s) | Toxicities | Outcomes | Reference |
| 63 patients Refractory tumors: majority breast/lung |
Majority dosed at 0.2–1.1 mg/kg/day daily IV, IM, or PO | Mucositis (83%) Diarrhea (48%) Nausea/vomiting (30%) |
7/63 patients with partial response | (37) |
| 10 patients in DON cohort Metastatic testicular cancer |
10 to 15 mg daily in combination (with 6-MP or an alkylator) | Not reported | 1/10 patients with decreased urine tumor markers | (39) |
| 41 patients Hodgkins lymphoma, lymphosarcoma, bronchogenic carcinoma, melanoma |
0.2 mg/kg PO daily x 30 days | Stomatitis Diarrhea Vomiting |
47% Hodgkin’s lymphoma lesions decreased by >/= 20% | (38) |
| 71 pediatric patients Untreated acute leukemia |
0.25 mg/kg DON PO daily + 2.5 mg/kg 6MP daily for 28 days | Mucositis (85%) GI symptoms (28%) Leukopenia (60%) |
30/71 patients with complete remission |
(40) |
| Phase II Studies (1980s-2000s) at high intermittent dosing | ||||
| 23 patients Advanced lung cancer |
160 mg/m2/dose IV Three consecutive days every 3 weeks |
Nausea and vomiting Diarrhea Leukopenia/Thrombocytopenia |
No responses | (48) |
| 23 patients (14 evaluable) Advanced colorectal carcinoma |
200 mg/m2/dose IV Twice weekly for four doses every 3 weeks |
Nausea and vomiting Thrombocytopenia | Progressive disease: 11/14 patients | (50) |
| 30 patients Advanced colorectal carcinoma |
160 mg/m2/dose IV Three consecutive days every 3 weeks |
Nausea/vomiting Rare CNS toxicity (diplopia/ataxia/visual hallucinations) Hyperbilirubinemia |
Progressive disease: 13/30 Stable disease: 16/30 |
(51) |
| 98 patients, 41 treated with DON Advanced sarcomas |
50 mg/m2/dose IV Five consecutive days every 4 weeks |
Nausea/vomiting Diarrhea Myelosuppression |
No objective responses | (49) |
| 55 patients Advanced refractory solid tumors |
140 mg/m2/dose IVs Twice weekly every 3 weeks + once weekly 120 IU/m2s PEGylated glutaminase |
Fatigues Nausea and vomitings Diarrhea |
Progression free at 5 months: 5/17s Partials response: 1/17 |
(52) |
Magill’s early study was not designed or powered to show therapeutic efficacy and clinical trial terminology was less standardized than it is today; nonetheless there were hints of activity among the patients treated (Table 1). Seven of 47 (15%) patients who had at least two weeks’ duration of DON exposure were reported to have partial response to treatment. These included two breast cancer patients and two bronchogenic carcinoma patients with decrease in metastases. An advanced sigmoid adenocarcinoma patient, a uterine cancer patient, and an advanced Hodgkin’s lymphoma patient also experienced transient partial responses of metastatic disease. Four additional patients (two breast tumor and two “other carcinoma” patients) showed reversal of hypercalcemia of malignancy in response to DON. In twenty-four patients there was neither demonstrable regression nor progression of the disease, and DON therapy was deemed to have “no effect” on this subset. Twelve patients had disease progression.
Following the report by Magill, several other groups evaluated daily dosing of DON in humans against a variety of malignant diseases (Table 1) (38–40). Included among these was one pediatric study, in which children acute leukemia were treated with the combination of DON (daily 0.25 mg/kg) and 6-mercaptopurine (6-MP). Forty two percent of patients achieved complete remission with the combination therapy, an advantage over 6-MP alone.
1980s-2000s: Higher intermittent dosing of DON does not show efficacy
In the 1980s interest in the antitumor effects of DON were rekindled by its robust activity against several human tumor xenografts in the newly introduced nude mouse model. DON was reported to be effective against human lung, colon, and breast tumor xenografts, inducing tumor regression at high intermittent dosing regimens (25–100 mg/kg IP every 4 days) (41). Mechanistic studies showed DON treatment led to decreased cell proliferation and S phase blocks (42). Animal toxicity studies were performed by the National Cancer Institute using azotomycin, which is metabolized to DON. These studies established target organs of the gastrointestinal tract and bone marrow; effects on the heart, kidneys, and liver became more pronounced with increased drug exposure (43), (20). Since the DON highest exposures could be achieved with intermittent doses and this dosing regimen was effective at inducing tumor regression in the nude mouse model, an intermittent dosing paradigm was employed in further clinical trials.
Five phase I dose escalation studies of DON were carried out and published in the 1980s (43–47). Patients received DON intravenously in doses ranging from 50 mg/m2/dose to 600 mg/m2/dose (1.35 – 16.2 mg/kg equivalent) generally two to three times weekly every three to four weeks. Four studies examined adult patients with a range of treatment refractory cancers, and one study focused on a pediatric population. Among the five phase I studies, no objective responses to therapy were noted in any adult solid tumor group with measureable disease. Among the pediatric patients treated with DON as a single agent, a handful of both hematologic and solid tumor patients had studies suggesting stable disease to partial responses (43).
The subsequent phase II studies of DON at intermittent high doses carried out between the 1980s and 2000s generally had disappointing outcomes (Table 1) (48–51). Four of these studies used DON as a single agent in advanced or refractory adult solid tumors. These studies used maximum tolerated dosing regimens based on the phase I trials ranging from 50 mg/m2/dose daily for five days every three weeks, given to sarcoma patients, to 200 mg/m2/dose twice weekly for two weeks with one week off, given to colorectal cancer patients. There were few clinical responses even in the more well-defined oncology populations, with the exception of data published as an abstract more recently on concurrent therapy with DON and PEGylated glutaminase (52). Based on these collective experiences, it was concluded that intermittent high doses were ineffective and too toxic (see below), and DON was abandoned as an anticancer therapy.
Low daily dosing of DON: local gastrointestinal toxicities and evidence for target engagement
In studies from the 1950s employing daily doses of DON, the most common toxicities were direct GI toxicity to the gastrointestinal mucosa (Table 1). Mucositis was observed in up to 83% patients, in spite of preventive treatments, and diarrhea in 48% patients (37). Nausea and vomiting (30% patients), GI bleeding (16% patients), and abdominal pain (8% patients) also occurred. One-third of patients also had hematologic changes (leukopenia/thrombocytopenia) but these included patients with underlying bony metastases or disease involving the marrow. Mucositis and diarrhea were more severe in patients receiving multiple daily divided doses compared to patients receiving the same total dose in one daily infusion, though sample sizes in the divided dose regimens were small. Similar side effects were reported across the early studies using low daily DON doses (37–40). In the DON and 6-MP combination study the improved outcomes with combination therapy were offset by an increase in toxicity, especially mucositis and other gastrointestinal symptoms.
When DON was administered on intermittent high dose schedules, nausea and vomiting were the major dose limiting toxicities (Table 1) (48–52) – the stomatitis/mucositis/diarrhea observed on earlier daily dosing regimens was less frequently observed even though the total dose administered was much greater. Moderate to severe nausea and vomiting was observed in one study using DON doses between 300–550 mg/m2 given by rapid IV infusion (45). In these patients, DON peak plasma concentration (Cmax) was found to be over 20 micrograms/ml (45). Longer infusion times of high intermittent doses of DON in two additional studies resulted in peak plasma concentrations measured at less than 10 micrograms/ml and slightly milder side effects (46). By contrast, low daily dosing of DON in earlier studies (0.2–0.3 mg/kg IV) resulted in peak plasma concentrations of only 400–500 nanograms/ml measured by an indirect microbiological assay (37). This dose schedule primarily resulted in oral mucositis and some diarrhea, with less nausea and vomiting reported.
Taken together, these observations suggest that low daily or divided dose exposures produced low plasma concentrations and a characteristic local GI toxicity, while high intermittent doses lead to more typical chemotherapy-induced nausea and vomiting. Of note, at the time many of the clinical studies on DON were done, 5HT3 receptor antagonists were not routinely in clinical practice as antiemetics, although several of the phase II studies and a pediatric phase I study pretreated patients with prochlorperazine or chlorpromazine and observed decreased severity of nausea and vomiting (48,51), (43). Other non-dose limiting toxicities of DON observed in the high intermittent dosing studies include reversible myelosuppression and transient hypocalcemia (43,44,47).
Our review of the clinical observations suggests that the low dosing paradigm of the early studies was sufficient to inhibit glutamine utilization. In the trials using low daily dosing stomatitis and diarrhea were consistently observed, reflecting local glutamine starvation of the gastrointestinal mucosa and inhibition of cell cycle in rapidly proliferating intestinal epithelial cells. Of note, these side effects are not reported with CB-839, the glutaminase inhibitor currently in human trials (14) suggesting that full blockade of glutamine utilization is dependent on enzymes beyond glutaminase. Evidence for low dose DON inhibition of glutamine amidotransferases was directly observed in humans in a study of DON for gout (53). In this study, DON was administered at low daily intramuscular doses of only 5 to 20 mg and inhibited glutamine utilization as measured by radiotracer. These doses were more than an order of magnitude lower than those used in subsequent cancer trials in the 1980s (45,50,51), supporting the notion that high intermittent dosing was not an optimal schedule to achieve blockade of glutamine metabolism with DON.
Targeting cancer metabolism may be complementary to classic cytotoxic approaches and many inhibitors of “metabolic targets” are in preclinical studies or in early clinical trials (54). As is suggested for DON optimal dosing of metabolic inhibitors will differ compared from the cytotoxic agents typically used with intermittent dosing. Constant target inhibition with overall low dose therapy is thought to be important for avoiding resistance mechanisms that can occur more readily when agents are started and stopped intermittently. Currently approved metabolic inhibitors are dosed clinically on a daily to BID basis, allowing for constant target inhibition at doses below the maximally tolerated dose (55).
Strategies for Clinical Redevelopment
Development of a targeted DON prodrug to improve therapeutic index
Given DON’s breadth of activity and promising efficacy in treating glutamine dependent tumors, particularly with daily low doses, it is of clinical importance to identify a strategy to deliver it preferentially to the tumor while minimizing its peripheral exposure and thus toxicity to non-tumor tissues. Prodrugs have proven to be a valuable strategy to alter tissue distribution and improve delivery of active agents to target tissues (56). We have developed a dual prodrug strategy to mask DON’s carboxylic acid and amino groups with addition of promoieties (Figure 2A). The resulting prodrugs circulate intact and inert in plasma and preferentially release DON at the target tissue, permitting dose reductions and improved therapeutic index. We have developed two types of DON prodrugs – those that preferentially deliver DON to brain and those that preferentially deliver DON to peripheral tumors.
We were particularly interested in delivering DON to the CNS as many brain tumors (e.g. gliomas, medulloblastoma) have amplification of the MYC or MYCN oncogenes, potentially rendering them glutamine dependent (57). The brain-targeted prodrugs, exemplified by Prodrug 1 (Figure 2A) were designed to introduce bulky hydrophobic groups to enhance the cLogP and cellular permeability of DON, while maintaining plasma stability such that the peripheral exposure to DON would be limited (11,58). Following infusion in swine, Prodrug 1 delivered DON preferentially to CSF and brain, resulting in a 15-fold enhanced CSF/plasma ratio and 9-fold enhanced brain/plasma ratio when compared directly with equimolar doses of DON (Figure 2B, I and II) (58). A second class of prodrugs was designed to enhance tumor delivery by utilizing promoieties which are cleaved by enzymes enriched in tumor. This approach is exemplified by Prodrug 2, designed to take advantage of HDAC and Cathepsin L overexpression by tumors (59–61). When directly compared to DON, Prodrug 2 shows a greater than 50-fold enhanced tumor cell/plasma ratio (Figure 2C). Importantly, when incubated with GI tissue homogenates, Prodrug 2 did not release significant amounts of DON (Figure 2D), confirming that DON delivery is preferential to tumor cells and suggesting that the prodrug approach should decrease gut exposure to DON, and thereby reduce GI toxicity.
Low daily dosing of DON prodrug provides efficacy without toxicity
We have evaluated the in vivo tolerability and efficacy of DON prodrugs using a low daily dosing paradigm, reminiscent of the original studies in humans (37) and similar to dosing paradigms employed with other metabolic inhibitors (55). Mice bearing EL-4 lymphoma were treated with vehicle or DON prodrug (1 mg/kg p.o. x 5 days followed by 0.3 mg/kg, p.o. x 9 days). As clearly demonstrated, Prodrug 3 (62) robustly induced tumor elimination while mice succumbed to vehicle-treated tumors (Figure 2E). No overt side effects or weight changes were observed with prodrug treatment, in agreement with the observation that DON prodrugs do not release significant DON in gut tissue (Figure 2D).
DON prodrugs may also avoid the resistance to glutaminase inhibition and metabolic adaptation to single agent therapy that has been a concern in trials of the selective glutaminase inhibitor CB-839. In contrast, DON broadly inhibits glutamine utilization including glutaminase, glutamine amidotransferases (used in de novo purine and pyrimidine synthesis, coenzyme synthesis, and hexosamine synthesis), and glutamine synthetase (Figure 1). Thus DON prodrug inhibition may be more difficult for tumor and stromal cells to adapt to and circumvent. DON’s breadth of inhibition, tolerability at low daily doses, and potential utility in combinations can now be exploited with the prodrug approach in further clinical oncology studies.
The way forward: evaluating novel DON prodrugs administered at daily low doses in patients with glutamine dependent tumors
With the growing appreciation of the complexity of targeting glutamine metabolism in cancers and the promising preclinical results with the novel DON prodrugs described above, we believe that clinical studies of these DON prodrugs are warranted. Dosing in humans should be trialed at low daily doses – the prodrug doses that we have tested in mice of 0.3 to 1 mg/kg are equivalent to less than 0.1 mg/kg in humans, which is below the lower limit of tolerable doses evaluated in the original 1957 human study (37,63). Unlike the early DON clinical trials however, modern DON prodrug clinical studies should aim to include oncology patients whose tumors have the best chance of benefiting from therapy targeting tumor glutamine dependence.
We propose that investigations of DON prodrugs be prioritized in three clinical areas. First, many CNS tumors are in need of new therapeutic options that cross the blood-brain barrier. Preclinical evidence suggests that subtypes of brain tumors (e.g. glioblastoma, group III medulloblastoma) have MYC or MYCN amplification, are glutamine-avid, thus should benefit therapeutically from glutamine antagonists (64,65). Brain-targeted DON prodrugs have been shown to deliver DON preferentially to the CSF to enhance drug activity in a tissue usually inaccessible to chemotherapy. In this way brain-targeted DON prodrugs offer an advantage when compared to CB-839, which is reported to have limited CNS availability (66).
Second, non-CNS tumors with evidence of glutamine dependence are good candidates for treatment with DON prodrugs. Multiple means exist to identify such populations. Mutations in genes established to drive glutamine dependence (e.g. MYC or MYCN, KRAS) as well as more recently identified genetic mutations affecting glutamine utilization (e.g. VHL, KEAP1, ASS1) can be used for rational patient selection (67–72). Alternatively, or in combination, new technologies to evaluate tumor metabolism in vivo may be employed. Positron emission-tomography (PET) based imaging with 18F-glutamine, evaluation of the tumor environment with magnetic resonance spectroscopy, and metabolomics or tracer profiling of tumor biopsy at diagnosis can identify increased glutamine uptake or dependence in vivo (64,65,73–75). These techniques may also prove valuable in clinical trials to establish makers of blockade of glutamine utilization after DON prodrug administration.
Finally, we propose that DON prodrugs be evaluated in combination with either standard-of-care therapies or in rationally-directed new pairings. For example, in an early clinical trial, DON was successful when combined with 6-MP in pediatric acute leukemias (1,40). In addition DON prodrugs should be combined with newer targeted therapies. Glutamine metabolism promotes tumor resistance to mTOR inhibitors in certain tumor contexts (54,64); the combination of a DON prodrug and an mTOR inhibitor will be interesting to study. Finally, it is known that aberrant glutamine metabolism by a tumor can lead to more wide ranging effects on the local microenvironment or even whole organism that play into the broader effects of cancer (3,19). Therefore, DON prodrugs should be evaluated for synergy with tumor cell non-autonomous therapeutics, including immunotherapy agents.
By inhibiting a wide range of metabolic pathways utilizing glutamine that are critically upregulated in cancer cells, DON has been shown to be a selective but broadly targeted therapy. In order to deliver the active drug to compartments like the CNS and minimize GI exposure, we have synthesized and conducted thorough preclinical investigations of targeted DON prodrugs. Now with an improved understanding of how to dose these agents and what tumor types are best suited for broad inhibition of glutamine metabolism, these novel glutamine antagonists are well positioned for future success in the clinic.
Acknowledgements:
We apologize to authors whose work we could not cite due to space limitations. We thank Eric Raabe, Jonathan Powell, and Frank Smith for critical reading and comments. We thank members of Johns Hopkins Drug Discovery for helpful discussion and figure preparation. We gratefully acknowledge Jennifer Fairman’s assistance with the preparation of Figure 1. This work was supported by NIH T32CA060441 (K.M.L.), an Alleghany Health Network grant (R.R.), and NIH P30MH075673, R01 CA193895 and Alex’s Lemonade Stand Foundation, Children’s Cancer Foundation, Alleghany Health Network, and TEDCO Maryland Innovation Initiative grants (B.S.S.).
Financial Support: K.M.L. is supported by NIH T32CA060441. R.R is supported by an Alleghany Health Network grant. B.S.S. is supported by NIH P30MH075673, R01 CA193895 and Alleghany Health Network, Alex’s Lemonade Stand Foundation, Children’s Cancer Foundation, and TEDCO grants.
Footnotes
Conflict of Interest Disclosure Statement: B.S.S. and R.R. are founders of Dracen Pharmaceuticals, a company pursuing small molecule glutamine antagonists for clinical oncology applications. Other authors declare no conflicts of interest.
References
- 1.Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer 2016;16(10):619–34 doi 10.1038/nrc.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bode BP. Glutamine Transport and Metabolism in Cancer In: Denis DM, editor. Glutamine: Biochemistry, Physiology, and Clinical Applications: CRC Press; 2015. [Google Scholar]
- 3.Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest 2013;123(9):3678–84 doi 10.1172/JCI69600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A 2007;104(49):19345–50 doi 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013;496(7443):101–5 doi 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Warburg O On the Origin of Cancer Cells. Science 1956;123(3191):309–14. [DOI] [PubMed] [Google Scholar]
- 7.Eagle H, Oyama VI, Levy M, Horton CL, Fleischman R. The growth response of mammalian cells in tissue culture to L-glutamine and L-glutamic acid. J Biol Chem 1956;218(2):607–16. [PubMed] [Google Scholar]
- 8.Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol 2007;178(1):93–105 doi 10.1083/jcb.200703099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009;458(7239):762–5 doi 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A 2010;107(16):7455–60 doi 10.1073/pnas.1001006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elgogary A, Xu Q, Poore B, Alt J, Zimmermann SC, Zhao L, et al. Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proc Natl Acad Sci U S A 2016;113(36):E5328–36 doi 10.1073/pnas.1611406113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010;18(3):207–19 doi 10.1016/j.ccr.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell 2015;59(2):298–308 doi 10.1016/j.molcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harding JJ, Telli ML, Munster PN, Le MH, Molineaux C, Bennett MK, et al. Safety and tolerability of increasing doses of CB-839, a first-in-class, orally administered small molecule inhibitor of glutaminase, in solid tumors. Journal of Clinical Oncology 2015;33(15_suppl):2512– doi 10.1200/jco.2015.33.15_suppl.2512. [DOI] [Google Scholar]
- 15.Meric-Bernstam F, Tannir NM, Mier JW, DeMichele A, Telli ML, Fan AC, et al. Phase 1 study of CB-839, a small molecule inhibitor of glutaminase (GLS), alone and in combination with everolimus (E) in patients (pts) with renal cell cancer (RCC). Journal of Clinical Oncology 2016;34(15_suppl):4568– doi 10.1200/JCO.2016.34.15_suppl.4568. [DOI] [Google Scholar]
- 16.DeMichele A, Harding JJ, Telli ML, Munster PN, McKay R, Iliopoulos O, et al. Phase 1 study of CB-839, a small molecule inhibitor of glutaminase (GLS) in combination with paclitaxel (Pac) in patients (pts) with triple negative breast cancer (TNBC). Journal of Clinical Oncology 2016;34(15_suppl):1011– doi 10.1200/JCO.2016.34.15_suppl.1011. [DOI] [Google Scholar]
- 17.Muir A, Danai LV, Gui DY, Waingarten CY, Lewis CA, Vander Heiden MG. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife 2017;6 doi 10.7554/eLife.27713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Biancur DE, Paulo JA, Malachowska B, Quiles Del Rey M, Sousa CM, Wang X, et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nature communications 2017;8:15965 doi 10.1038/ncomms15965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang L, Achreja A, Yeung TL, Mangala LS, Jiang D, Han C, et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab 2016;24(5):685–700 doi 10.1016/j.cmet.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clinical Brochure: DON -- NSC 7365 In: Investigational Drug Branch CTTP, editor. Bethesda, Maryland: 1979. p 1–53. [Google Scholar]
- 21.Thomas AG, Rojas C, Tanega C, Shen M, Simeonov A, Boxer MB, et al. Kinetic characterization of ebselen, chelerythrine and apomorphine as glutaminase inhibitors. Biochem Biophys Res Commun 2013;438(2):243–8 doi 10.1016/j.bbrc.2013.06.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thangavelu K, Chong QY, Low BC, Sivaraman J. Structural basis for the active site inhibition mechanism of human kidney-type glutaminase (KGA). Sci Rep 2014;4:3827 doi 10.1038/srep03827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cervantes-Madrid D, Romero Y, Duenas-Gonzalez A. Reviving Lonidamine and 6-Diazo-5-oxo-L-norleucine to Be Used in Combination for Metabolic Cancer Therapy. Biomed Res Int 2015;2015:690492 doi 10.1155/2015/690492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartman SC. The Interaction of 6-Diazo-5-Oxo-L-Norleucine with Phosphoribosyl Pyrophosphate Amidotransferase. J Biol Chem 1963;238:3036–47. [PubMed] [Google Scholar]
- 25.Kaufman ER. Isolation and characterization of a mutant Chinese hamster cell line resistant to the glutamine analog 6-diazo-5-oxo-l-norleucine. Somatic Cell and Molecular Genetics 1985;11(1):1–10 doi 10.1007/bf01534729. [DOI] [PubMed] [Google Scholar]
- 26.Levenberg B, Melnick I, Buchanan JM. Biosynthesis of the purines. XV. The effect of aza-L-serine and 6-diazo-5-oxo-L-norleucine on inosinic acid biosynthesis de novo. J Biol Chem 1957;225(1):163–76. [PubMed] [Google Scholar]
- 27.Levitzki A, Stallcup WB, Koshland DE Jr. Half-of-the-sites reactivity and the conformational states of cytidine triphosphate synthetase. Biochemistry 1971;10(18):3371–8. [DOI] [PubMed] [Google Scholar]
- 28.Livingston RB, Venditti JM, Cooney DA, Carter SK. Glutamine antagonists in chemotherapy. Adv Pharmacol Chemother 1970;8:57–120. [DOI] [PubMed] [Google Scholar]
- 29.LaRonde-LeBlanc N, Resto M, Gerratana B. Regulation of active site coupling in glutamine-dependent NAD(+) synthetase. Nature structural & molecular biology 2009;16(4):421–9 doi 10.1038/nsmb.1567. [DOI] [PubMed] [Google Scholar]
- 30.Jayaram HN, Cooney DA, Milman HA, Homan ER, Rosenbluth RJ. Don, conv and donv--I. Inhibition of L-asparagine synthetase in vitro. Biochem Pharmacol 1976;25(14):1571–82. [DOI] [PubMed] [Google Scholar]
- 31.Tiemeier DC, Milman G. Chinese hamster liver glutamine synthetase. Purification, physical and biochemical properties. J Biol Chem 1972;247(8):2272–7. [PubMed] [Google Scholar]
- 32.Walker B, Brown MF, Lynas JF, Martin SL, McDowell A, Badet B, et al. Inhibition of Escherichia coli glucosamine synthetase by novel electrophilic analogues of glutamine--comparison with 6-diazo-5-oxo-norleucine. Bioorg Med Chem Lett 2000;10(24):2795–8. [DOI] [PubMed] [Google Scholar]
- 33.Goto Y, Zalkin H, Keim PS, Heinrikson RL. Properties of anthranilate synthetase component II from Pseudomonas putida. J Biol Chem 1976;251(4):941–9. [PubMed] [Google Scholar]
- 34.Coffey GL, Ehrlich J, Fisher MW, Hillegas AB, Kohberger DL, Machamer HE, et al. 6-Diazo-5-oxo-L-norleucine, a new tumor-inhibitory substance. I. Biologic studies. Antibiot Chemother (Northfield) 1956;6(8):487–97. [PubMed] [Google Scholar]
- 35.Burchenal JH. Antitumor effects of azaserine and DON. Cancer Treat Rep 1979;63(6):1031–2. [PubMed] [Google Scholar]
- 36.Cooney DA, Jayaram HN, Milman HA, Homan ER, Pittillo R, Geran RI, et al. DON, CONV and DONV-III. Pharmacologic and toxicologic studies. Biochem Pharmacol 1976;25(16):1859–70. [DOI] [PubMed] [Google Scholar]
- 37.Magill GB, Myers WP, Reilly HC, Putnam RC, Magill JW, Sykes MP, et al. Pharmacological and initial therapeutic observations on 6-diazo-5-oxo-1-norleucine (DON) in human neoplastic disease. Cancer 1957;10(6):1138–50. [DOI] [PubMed] [Google Scholar]
- 38.A CLINICAL study of the comparative effect of nitrogen mustard and DON in patients with bronchogenic carcinoma, Hodgkin’s disease, lymphosarcoma, and melanoma. J Natl Cancer Inst 1959;22(2):433–9. [PubMed] [Google Scholar]
- 39.Li MC, Whitmore WF Jr., Golbey R, Grabstald H. Effects of combined drug therapy on metastatic cancer of the testis. JAMA 1960;174:1291–9. [DOI] [PubMed] [Google Scholar]
- 40.Sullivan MP, Beatty EC Jr., Hyman CB, Murphy ML, Pierce MI, Severo NC. A comparison of the effectiveness of standard dose 6-mercaptopurine, combination 6-mercaptopurine and DON, and high-loading 6-mercaptopurine therapies in treatment of the acute leukemias of childhood: results of a coperative study. Cancer Chemother Rep 1962;18:83–95. [PubMed] [Google Scholar]
- 41.Ovejera AA, Houchens DP, Catane R, Sheridan MA, Muggia FM. Efficacy of 6-diazo-5-oxo-L-norleucine and N-[N-gamma-glutamyl-6-diazo-5-oxo-norleucinyl]-6-diazo-5-oxo-norleucine against experimental tumors in conventional and nude mice. Cancer Res 1979;39(8):3220–4. [PubMed] [Google Scholar]
- 42.Huber KR, Mayer EP, Mitchell DF, Roberts J. Cell cycle phase perturbations by 6-diazo-5-oxo-L-norleucine and acivicin in normal and neoplastic human cell lines. Br J Cancer 1987;55(6):653–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sullivan MP, Nelson JA, Feldman S, Van Nguyen B. Pharmacokinetic and phase I study of intravenous DON (6-diazo-5-oxo-L-norleucine) in children. Cancer Chemother Pharmacol 1988;21(1):78–84. [DOI] [PubMed] [Google Scholar]
- 44.Earhart RH, Koeller JM, Davis HL. Phase I trial of 6-diazo-5-oxo-L-norleucine (DON) administered by 5-day courses. Cancer Treat Rep 1982;66(5):1215–7. [PubMed] [Google Scholar]
- 45.Kovach JS, Eagan RT, Powis G, Rubin J, Creagan ET, Moertel CG. Phase I and pharmacokinetic studies of DON. Cancer Treat Rep 1981;65(11–12):1031–6. [PubMed] [Google Scholar]
- 46.Rahman A, Smith FP, Luc PT, Woolley PV. Phase I study and clinical pharmacology of 6-diazo-5-oxo-L-norleucine (DON). Invest New Drugs 1985;3(4):369–74. [DOI] [PubMed] [Google Scholar]
- 47.Sklaroff RB, Casper ES, Magill GB, Young CW. Phase I study of 6-diazo-5-oxo-L-norleucine (DON). Cancer Treat Rep 1980;64(12):1247–51. [PubMed] [Google Scholar]
- 48.Eagan RT, Frytak S, Nichols WC, Creagan ET, Ingle JN. Phase II study on DON in patients with previously treated advanced lung cancer. Cancer Treat Rep 1982;66(8):1665–6. [PubMed] [Google Scholar]
- 49.Earhart RH, Amato DJ, Chang AY, Borden EC, Shiraki M, Dowd ME, et al. Phase II trial of 6-diazo-5-oxo-L-norleucine versus aclacinomycin-A in advanced sarcomas and mesotheliomas. Invest New Drugs 1990;8(1):113–9. [DOI] [PubMed] [Google Scholar]
- 50.Lynch G, Kemeny N, Casper E. Phase II evaluation of DON (6-diazo-5-oxo-L-norleucine) in patients with advanced colorectal carcinoma. Am J Clin Oncol 1982;5(5):541–3. [PubMed] [Google Scholar]
- 51.Rubin J, Sorensen S, Schutt AJ, van Hazel GA, O’Connell MJ, Moertel CG. A phase II study of 6-diazo-5-oxo-L-norleucine (DON, NSC-7365) in advanced large bowel carcinoma. Am J Clin Oncol 1983;6(3):325–6. [DOI] [PubMed] [Google Scholar]
- 52.Mueller C, Al-Batran S, Jaeger E, Schmidt B, Bausch M, Unger C, et al. A phase IIa study of PEGylated glutaminase (PEG-PGA) plus 6-diazo-5-oxo-L-norleucine (DON) in patients with advanced refractory solid tumors. 2008. 2533– p. [Google Scholar]
- 53.Grayzel AI, Seegmiller JE, Love E. Suppression of uric acid synthesis in the gouty human by the use of 6-diazo-5-oxo-L-norleucine. J Clin Invest 1960;39:447–54 doi 10.1172/JCI104057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Garber K Cancer anabolic metabolism inhibitors move into clinic. Nat Biotechnol 2016;34(8):794–5 doi 10.1038/nbt0816-794. [DOI] [PubMed] [Google Scholar]
- 55.Tabernero J, Rojo F, Calvo E, Burris H, Judson I, Hazell K, et al. Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: a phase I tumor pharmacodynamic study in patients with advanced solid tumors. J Clin Oncol 2008;26(10):1603–10 doi 10.1200/JCO.2007.14.5482. [DOI] [PubMed] [Google Scholar]
- 56.Wyatt C, Baeten JM. Tenofovir alafenamide for HIV infection: is less more? The Lancet 2015;385(9987):2559–60 doi . [DOI] [PubMed] [Google Scholar]
- 57.Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta neuropathologica 2012;123(4):465–72 doi 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nedelcovych MT, Tenora L, Kim BH, Kelschenbach J, Chao W, Hadas E, et al. N-(Pivaloyloxy)alkoxy-carbonyl Prodrugs of the Glutamine Antagonist 6-Diazo-5-oxo-l-norleucine (DON) as a Potential Treatment for HIV Associated Neurocognitive Disorders. J Med Chem 2017;60(16):7186–98 doi 10.1021/acs.jmedchem.7b00966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jancarik A, Rais R, Alt J, Tenora L, Krecmerova M, Cheng C, et al. Novel lymphoid targeted prodrugs of the glutamine antagonist DON for the treatment of hematological malignancies. The FASEB Journal 2016;30(1_supplement):lb472–lb doi 10.1096/fasebj.30.1_supplement.lb472. [DOI] [Google Scholar]
- 60.Slusher B, Rais R, Tenora L, Majer P, Jancarik A, Nedelcovych M; UNIV JOHNS HOPKINS (UYJO-C) INST ORGANIC CHEM & BIOCHEMISTRY ACAD (ORGA-Non-standard), assignee. New prodrug of a glutamine analog, useful for treating a disease or condition comprising an infection, cancer, an autoimmune disease, an inflammatory disease, and a neurodegenerative or neurological disease patent WO2017023774-A1; WO2017023774-A9. WO2017023774-A1 09 Feb 2017 C07C-227/14 201715 Pages: 208 English WO2017023774-A9 23 Nov 2017 C07C-227/14 201778 English.
- 61.Ueki N, Lee S, Sampson NS, Hayman MJ. Selective cancer targeting with prodrugs activated by histone deacetylases and a tumour-associated protease. Nat Commun 2013;4:2735 doi 10.1038/ncomms3735. [DOI] [PubMed] [Google Scholar]
- 62.Rais R, Jancarik A, Tenora L, Nedelcovych M, Alt J, Englert J, et al. Discovery of 6-Diazo-5-oxo-l-norleucine (DON) Prodrugs with Enhanced CSF Delivery in Monkeys: A Potential Treatment for Glioblastoma. J Med Chem 2016;59(18):8621–33 doi 10.1021/acs.jmedchem.6b01069. [DOI] [PubMed] [Google Scholar]
- 63.Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm 2016;7(2):27–31 doi 10.4103/0976-0105.177703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tanaka K, Sasayama T, Irino Y, Takata K, Nagashima H, Satoh N, et al. Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J Clin Invest 2015;125(4):1591–602 doi 10.1172/jci78239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Venneti S, Dunphy MP, Zhang H, Pitter KL, Zanzonico P, Campos C, et al. Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci Transl Med 2015;7(274):274ra17 doi 10.1126/scitranslmed.aaa1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther 2014;13(4):890–901 doi 10.1158/1535-7163.mct-13-0870. [DOI] [PubMed] [Google Scholar]
- 67.Halbrook CJ, Lyssiotis CA. Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell 2017;31(1):5–19. [DOI] [PubMed] [Google Scholar]
- 68.Kremer JC, Prudner BC, Lange SES, Bean GR, Schultze MB, Brashears CB, et al. Arginine Deprivation Inhibits the Warburg Effect and Upregulates Glutamine Anaplerosis and Serine Biosynthesis in ASS1-Deficient Cancers. Cell Rep 2017;18(4):991–1004 doi 10.1016/j.celrep.2016.12.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab 2012;15(1):110–21 doi 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Qing G, Li B, Vu A, Skuli N, Walton Zandra E, Liu X, et al. ATF4 Regulates MYC-Mediated Neuroblastoma Cell Death upon Glutamine Deprivation. Cancer Cell 2012;22(5):631–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med 2017;23(11):1362–8 doi 10.1038/nm.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xu J, Chen Y, Olopade OI. MYC and Breast Cancer. Genes & Cancer 2010;1(6):629–40 doi 10.1177/1947601910378691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wishart DS. Emerging applications of metabolomics in drug discovery and precision medicine. Nat Rev Drug Discov 2016;15(7):473–84 doi 10.1038/nrd.2016.32. [DOI] [PubMed] [Google Scholar]
- 74.Zhu L, Ploessl K, Zhou R, Mankoff D, Kung HF. Metabolic Imaging of Glutamine in Cancer. J Nucl Med 2017;58(4):533–7 doi 10.2967/jnumed.116.182345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sellers K, Fox MP, Bousamra M 2nd, Slone SP, Higashi RM, Miller DM, et al. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J Clin Invest 2015;125(2):687–98 doi 10.1172/JCI72873. [DOI] [PMC free article] [PubMed] [Google Scholar]