

Abstract
The Pd-catalyzed coupling of malonate nucleophiles with alkenes bearing tethered aryl or alkenyl triflates is described. These alkene difunctionalization reactions afford malonate-substituted cyclopentanes that contain fused aryl or cycloalkenyl rings. The transformations generate a five-membered ring, two C–C bonds, and provide products bearing up to three stereocenters with good chemical yield and generally high diastereoselectivity.
Graphical Abstract

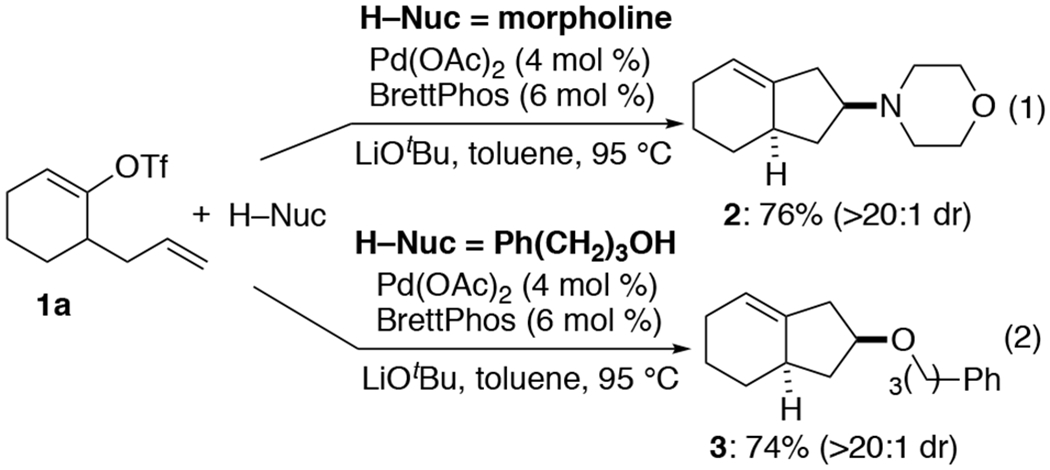
In recent years, transition metal-catalyzed alkene difunctionalization reactions that form two carbon-carbon bonds have emerged as powerful tools for the construction of substituted alkanes, heterocycles, and carbocycles.1 Our group has described a new series of Pd-catalyzed alkene difunctionalization reactions that involve the coupling of amine,2 alcohol/phenol,3 or indole1i nucleophiles with alkenes tethered to aryl or alkenyl triflates (Scheme 1). The transformations generate functionalized cyclopentane derivatives in good yield with high diastereoselectivity. For example, treatment of 1a with morpholine in the presence of Pd(OAc)2, BrettPhos, and LiOtBu afforded 2 in 76% yield with >20:1 dr (Scheme 1, eq 1), and similar conditions effected the coupling of 1a with 3-phenylpropanol to provide 3 in 74% yield and >20:1 dr (Scheme 1, eq 2).
Scheme 1.
Prior Studies
We sought to explore the use of malonates as carbon centered nucleophiles in alkene aryl-alkylation or alkenyl-alkylation variants of these transformations. Malonate anions are good nucleophiles with basicity that is comparable to that of phenoxides, so the feasibility of the transformation seemed high. Importantly, the resulting malonate-substituted cyclopentane products could be further elaborated through manipulation of the malonate and/or functionalization of the double bond in alkenyl-triflate derived products. Moreover, malonate substituted cyclopentanes have found prior utility as synthetic intermediates.4,5 The results of our preliminary studies are described in this manuscript.
In initial experiments, we examined the coupling of 2-allyllphenyltriflate 4a with diethyl malonate using conditions that previously were effective in analogous reactions of amines or alcohol nucleophiles.2b,3 We were gratified to find those conditions, which employed a Pd(OAc)2/BrettPhos catalyst, LiOtBu as base, toluene solvent, and a temperature of 95 °C also performed well with malonates. The desired product 5a was obtained in 93% isolated yield on a 0.2 mmol scale, and upon scaleup to 1 mmol the product was obtained in comparable but slightly lower (84%) isolated yield (eq 3).
 |
(3) |
Given this initial success, we briefly surveyed the reactivity of several different aryl triflate substrates. As shown in Scheme 2, transformations of terminal alkene substrates 4b-d proceeded smoothly to afford 5b-d in good chemical yield. Substrate 4d that contains an allylic methyl group was converted to trans disubstituted product 5d with high (>20:1) diastereoselectivity. However, reactions of substrates 4e-g that contain 1,2-disubstituted alkenes were much more challenging.6 The binding of the more sterically hindered alkene to the Pd-center is less favorable, and these substrates are also prone to base-mediated isomerization of the allyl group to a 2-propenyl group. In addition, the sluggish reactivity of these substrates led to other competing side reactions, such as base-mediated cleavage of the triflate to yield the corresponding phenoxide. After some experimentation, we found that decreasing the reaction temperature to 65 °C diminished the amount of alkene isomerization, and when the amount of diethyl malonate and base were increased to 3.6 and 2.2 equiv respectively, products 5e-g were obtained in modest yield and excellent (>20:1) diastereoselectivity.
Scheme 2. Reactions of Aryl Triflatesa,b,c.

aConditions: 1.0 equiv 4, 1.2 equiv diethyl malonate, 1.4 equiv LiOtBu, 4 mol % Pd(OAc)2, 6 mol % BrettPhos, toluene (0.1 M), 95 °C, 16 h. Reactions were conducted on a 0.1-0.25 mmol scale. bDiastereomeric ratios were determined by 1H NMR analysis. Diastereomeric ratios were identical for crude reaction mixtures and isolated compounds unless otherwise noted. cYields are average isolated yields of two or more experiments. dThe reaction was conducted using (BrettPhos)Pd(allyl)(Cl) in place of Pd(OAc)2. eThe reaction was conducted using 2.2 equiv of LiOtBu and 3.6 equiv of diethyl malonate, a substrate concentration of 0.8 M, and a reaction temperature of 65 °C.
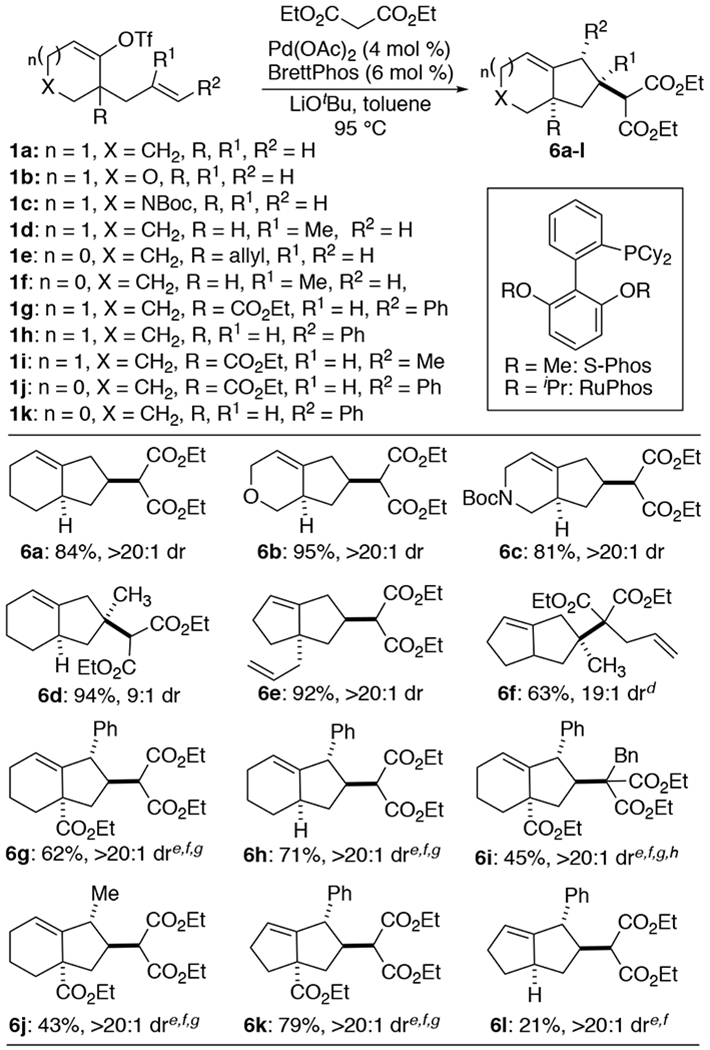
As shown in Scheme 3, the reactions are also effective using alkenyl triflate substrates 1a-k. The substrates can easily be prepared from the corresponding cyclic ketones via allylation followed by enol triflate formation.7 Transformations of terminal alkene substrates 1a-f proceeded smoothly under our standard conditions to afford 6a-f in good yield and generally high diastereoselectivity. The presence of heteroatoms in the starting material is well tolerated (6b-c), as was the presence of a methyl group at the internal position of the alkene (6d and 6f). In addition, malonates bearing an allyl (6f) or benzyl substituent (6i) were also effective coupling partners, although use of smaller ligands (RuPhos or S-Phos) was required.
Scheme 3. Reactions of Alkenyl Triflatesa,b,c.
aConditions: 1.0 equiv 1, 1.2 equiv diethyl malonate, 1.4 equiv LiOtBu, 4 mol % Pd(OAc)2, 6 mol % BrettPhos, toluene (0.1 M), 95 °C, 16 h. Reactions were conducted on a 0.1-0.25 mmol scale. bDiastereomeric ratios were determined by 1H NMR analysis. Diastereomeric ratios were identical for crude reaction mixtures and isolated compounds unless otherwise noted. cYields are average isolated yields of two or more experiments. dThe reaction was conducted using RuPhos as ligand. eThe reaction was conducted using 2.2 equiv of LiOtBu and 3.6 equiv of diethyl malonate, a substrate concentration of 0.8 M. fThe reaction was conducted using S-Phos as ligand. gThe reaction was conducted using Pd(acac)2 in place of Pd(OAc)2. hThe reaction was conducted in xylenes solvent at 110 °C.
Reactions of alkenyl triflate substrates bearing tethered internal alkenes proved to be even more challenging than the corresponding reactions of aryl triflates. Efforts to employ either the standard conditions, or the conditions that were effective with aryl triflates, led to very low yields of desired products (< 5%), along with significant amounts of a side product resulting from reduction of the triflate group. We reasoned that a smaller phosphine ligand might lead to improved results, as complexation of the more sterically hindered 1,2-disubstituted alkenes to the metal center is more difficult. As such, we explored the use of different phosphine ligand palladium sources,7 and after some experimentation found that the combination of Pd(acac)2 and either S-Phos or RuPhos provided the best results for most internal alkene substrates (1g-k). In all cases, products bearing three stereocenters were generated with >20:1 diastereoselectivity, with products resulting from anti-addition of the alkenyl group and the malonate to the double bond. Not surprisingly, the coupling of a substituted malonate with a disubstituted alkene proceeded in lower yield. However, the transformation of substrate 1i bearing an alkenyl methyl group proceeded in considerably lower yield than the analogous reaction of 1h, which contains an alkenyl phenyl substituent.
Substrates 1g, and 1h, which differ in the nature of their R substituent (H vs CO2Et), were transformed to 6g and 6h respectively, in comparable yield. However, the outcome of related cyclopentane-derived substrates 1j and 1k was much different, as 1j was converted to 6k in 79% yield, but the coupling of 1k with diethyl malonate afforded 6l in only 21% yield. The difference in the reactivity of 1j and 1k is likely due to a Thorpe-type effect,8 but the difference in reactivity of cyclohexene substrate 1h and cyclopentane substrate 1k is presumably a result of ring strain in the transition state for the formation of bicyclo[3.3.0]octane product 6l.
We also briefly examined the reactivity of acyclic alkenyl triflate substrate 7. As shown in eq 4, this transformation afforded spirocyclic compound 8 in 82% yield with >20:1 dr. However, preliminary efforts to employ other acyclic alkenyl triflates led to complex mixtures of products; future efforts will be directed towards improving the desired reactivity of these substrates.
 |
(4) |
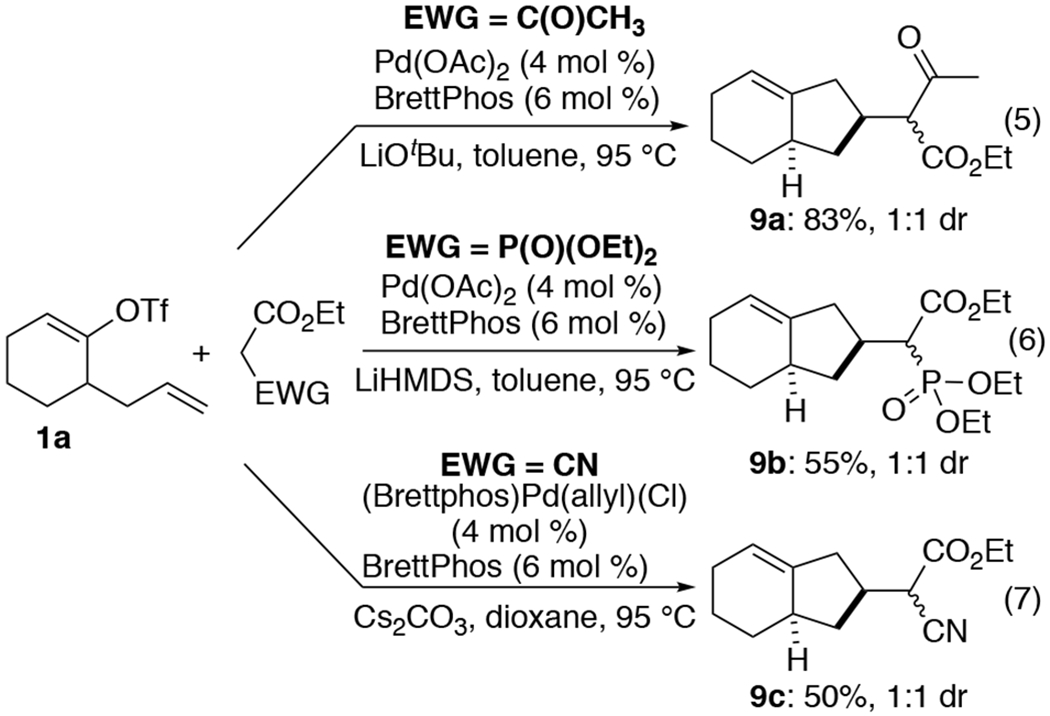
Although we have focused the bulk of our preliminary studies on reactions of diethyl malonate as a nucleophile, other activated methylene nucleophiles can be employed in these transformations. As shown in Scheme 4, the coupling of 1a with ethyl acetoacetate proceeded under standard conditions to afford 9a in 83% yield as a 1:1 mixture of diastereomers epimeric at the stereocenter adjacent to the carbonyl groups (eq 5). Use of triethyl phosphonoacetate provided 9b in a comparable 55% yield and 1:1 dr,9 although use of LiHMDS as base was needed to obtain optimal results (eq 6). The reactivity of ethyl cyanoacetate proved to be more challenging, but after some optimization we found that use of (BrettPhos)Pd(allyl)(Cl) as the precatalyst,10 combined with Cs2CO3 as base and dioxane as solvent provided 9c in 50% yield (eq 7). Although the coupling reactions of 1a with other malonate derivatives provided moderate amounts of the desired products, efforts to combine internal alkene substrates (e.g., 1g) with these nucleophiles were unsuccessful.
Scheme 4.
Reactions of Other Activated Methylene Nucleophiles
The mechanism of these reactions is likely similar to that of related transformations of amine or alcohol nucleophiles.2b,3 As shown in Scheme 5, oxidative addition of the alkenyl (or aryl) triflate to Pd(0) generates intermediate 10, which then undergoes coordination of the alkene to Pd followed by anti-carbopalladation of the alkene through chair-like conformation 11. The resulting intermediate 12 then affords the product 6 via reductive elimination, which also regenerates the active Pd(0) catalyst. The stereochemical outcome of these reactions is consistent with prior models involving minimization of non-bonding interactions in chair-like transition states similar to 11.2,3
Scheme 5.

Proposed Mechanism
In conclusion, we have developed a new Pd-catalyzed alkene difunctionalization reaction between malonate nucleophiles and alkenes bearing tethered alkenyl or aryl triflate electrophiles. The transformations generate two carbon-carbon bonds and provide products that contain up to three stereocenters with good to excellent diastereoselectivity. We have also demonstrated that use of smaller phosphine ligands allows for transformations of substrates bearing 1,2-disubstituted alkenes. Future studies will be directed towards expanding the scope of these transformations to other types of carbanion nucleophiles.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the NIH-NIGMS (GM 124030) for financial support of this work. EMH was partially supported by a University of Michigan Rackham dissertation fellowship.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Experimental procedures, characterization data, and copies of 1H and 13C NMR spectra for all new compounds (PDF). The Supporting Information is available free of charge on the ACS Publications website.
The authors declare no competing financial interest.
REFERENCES
- (1).For a review, see:; (a) Dhungana RK; KC S; Basnet P; Giri R Chem. Rec 2018, 18, 1314. [DOI] [PubMed] [Google Scholar]; For selected recent examples, see; (b) KC S; Basnet P; Thapa S; Shrestha B; Giri R J. Org. Chem 2018, 83, 2920. [DOI] [PubMed] [Google Scholar]; (c) Kuang Y; Wang X; Anthony D; Diao T Chem. Commun 2018, 54, 2558. [DOI] [PubMed] [Google Scholar]; (d) Wang K; Ding Z; Zhou Z; Kong W J. Am. Chem. Soc 2018, 140, 12364. [DOI] [PubMed] [Google Scholar]; (e) Basnet P; Dhungana RK; Thapa S; Shrestha B; KC S; Sears JM; Giri R J. Am. Chem. Soc 2018, 140, 7782. [DOI] [PubMed] [Google Scholar]; (f) Derosa J; Tran VT; Boulous MN; Chen JS; Engle KM J. Am. Chem. Soc 2017, 139, 10657. [DOI] [PubMed] [Google Scholar]; (g) Derosa J; van der Puyl VA; Tran VT; Liu M; Engle KM Chem. Sci 2018, 9, 5278. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Liu Z; Zeng T; Yang KS; Engle KM J. Am. Chem. Soc 2016, 138, 15122. [DOI] [PubMed] [Google Scholar]; (i) Kirsch JK; Manske JL; Wolfe JP J. Org. Chem 2018, 83, 13568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) White DR; Hutt JT; Wolfe JP J. Am. Chem. Soc 2015, 137, 11246. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) White DR; Wolfe JP Chem. Eur. J 2017, 23, 5419. [DOI] [PubMed] [Google Scholar]
- (3).White DR; Herman MI; Wolfe JP Org. Lett 2017, 19, 4311. [DOI] [PubMed] [Google Scholar]
- (4).(a) Iwata C; Kubota H; Yamada M; Takemoto Y; Uchida S; Tanaka T; Imanishi T Tetrahedron Lett. 1984, 25, 3339. [Google Scholar]; (b) Iwata C; Takemoto Y; Kubota H Kuroda T; Imanishi T Tetrahedron Lett. 1985, 26, 3231. [Google Scholar]; (c) Muraoka M; Toshio N; Sugie A; Ono K; Yamamoto M US Patent 4,680,307, 1987.
- (5).Related 2-cyclopentylacetic acid derivatives that would derive from saponification and decarboxylation of the products have also found utility as synthetic intermediates. For representative examples, see:; (a) Taber DF; Paquette CM; J. Org. Chem 2014, 79, 3410. [DOI] [PubMed] [Google Scholar]; (b) Srikrishna A; Beerahah B Tetrahedron Lett. 2007, 48, 2291. [Google Scholar]
- (6).In our prior work, we had demonstrated only one single example of a reaction of a 1,2-disubstituted alkene, which proceeded in low (36%) yield when pyrrolidine was used as nucleophile in a reaction with an alkenyl triflate substrate. See reference 2b.
- (7).See the Supporting Information for complete experimental details.
- (8).Jung ME; Piizzi G Chem. Rev 2005, 105, 1735. [DOI] [PubMed] [Google Scholar]
- (9).This reaction was conducted on a 1.85 mmol scale.
- (10).DeAngelis AJ; Glidner PG; Chow R; Colacot TJ J. Org. Chem 2015, 80, 6794. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.