

Summary
The 2014–2016 West Africa Ebola virus (EBOV) outbreak coupled with the most recent outbreaks in Central Africa underscore the need to develop effective treatment strategies against EBOV. Although several therapeutic options have shown great potential, developing a wider breadth of countermeasures would increase our efforts to combat the highly lethal EBOV. Here we show that human cathelicidin antimicrobial peptide (AMP) LL-37 and engineered LL-37 AMPs inhibit the infection of recombinant virus pseudotyped with EBOV glycoprotein (GP) and the wild-type EBOV. These AMPs target EBOV infection at the endosomal cell-entry step by impairing cathepsin B-mediated processing of EBOV GP. Furthermore, two engineered AMPs containing D-amino acids are particularly potent in blocking EBOV infection in comparison with other AMPs, most likely owing to their resistance to intracellular enzymatic degradation. Our results identify AMPs as a novel class of anti-EBOV therapeutics and demonstrate the feasibility of engineering AMPs for improved therapeutic efficacy.
Subject Areas: Drugs, Molecular Biology, Viral Microbiology
Graphical Abstract
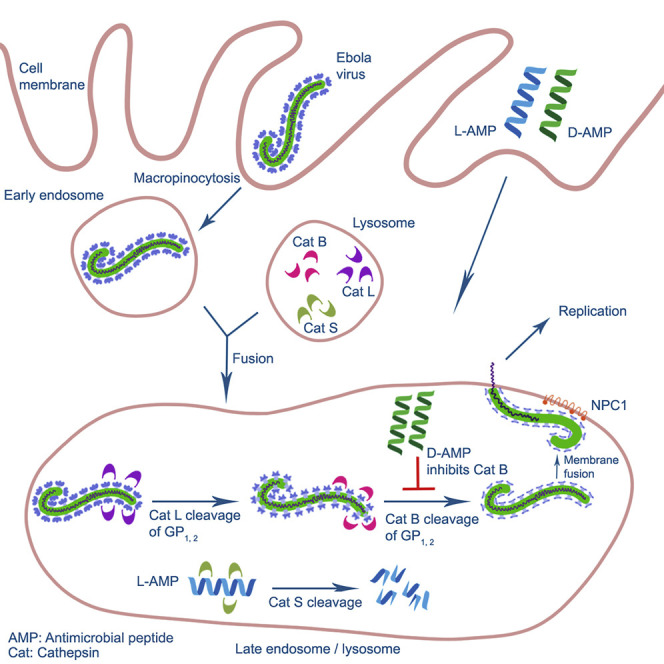
Highlights
-
•
Cathelicidin-derived antimicrobial peptides (AMPs) potently inhibit EBOV infection
-
•
D-form AMPs are more resistant to proteolytic cleavage than L-form AMPs in the cell
-
•
AMPs prevent cathepsin B-mediated processing of EBOV GP1, 2
Drugs; Molecular Biology; Viral Microbiology
Introduction
Ebola virus (EBOV) is a nonsegmented negative-sense RNA virus belonging to the ebolavirus genus in the family Filoviridae. These viruses are highly pathogenic and cause Ebola virus disease (EVD), previously referred to as Ebola hemorrhagic fever with case fatality rates up to 90% (Feldmann and Geisbert, 2011, Feldmann and Kiley, 1999). During 2014–2016, West Africa experienced the largest EBOV outbreak in history that resulted in over 28,000 cases and 11,000 deaths (Coltart et al., 2017). Alarmingly, the most recent EBOV-Kivu outbreak in the Democratic Republic of the Congo (DRC) has a reported 3,224 confirmed and probable cases of EVD and 2,152 deaths as of October 16, 2019 and does not currently show signs of abating (https://www.who.int/csr/don/02-may-2019-ebola-drc/en/). Currently, there are no approved vaccines or therapeutics to prevent or treat EVD; this fact combined with these unprecedented outbreaks in recent years underscores the urgent need to develop a wide variety of effective treatment strategies.
The current armamentarium of treatment options against EBOV primarily consists of small molecules and immunotherapeutics (Edwards and Basler, 2019, King et al., 2019). Of the reported small molecules that demonstrate anti-EBOV activity, the nucleoside analogs BXC4430, GS-5734, and Favipriravir have shown anti-EBOV activity in in vivo infection models (Bixler et al., 2018, Warren et al., 2014, Warren et al., 2016). Similarly, numerous reports have demonstrated that monoclonal antibodies (MAbs) are effective at inhibiting infection (Brannan et al., 2019, Furuyama et al., 2016, Howell et al., 2017, King et al., 2019, Marzi et al., 2012, Pettitt et al., 2013, Qiu et al., 2012, Audet et al., 2014). These MAbs target the EBOV glycoprotein (GP), which acts as an attachment factor, binding the host receptor Neimann-Pick C1 (NPC1) and mediating viral-host cell membrane fusion within the endosomal compartment (Davey et al., 2017, Carette et al., 2011, Cote et al., 2011, Hunt et al., 2012). Prior to receptor engagement, GP is cleaved by host cathepsins (Cat) B and L (Chandran et al., 2005, Schornberg et al., 2006). Cleavage of GP by CatB is required for GP-NPC1 binding and subsequent endosomal fusion (Miller et al., 2012). As a result, targeting GP endosomal processing and receptor binding can serve as an effective method of EBOV inhibition. Indeed, inhibitors of CatB/L or blockade of GP-NPC1 binding have shown efficacy against EBOV infection (Carette et al., 2011, Chandran et al., 2005, Elshabrawy et al., 2014, Herbert et al., 2015, Schornberg et al., 2006, Zhang et al., 2018).
Antimicrobial peptides (AMPs, also called host defense peptides) serve as an essential component of the innate immune system, in part, owing to their pleiotropic functions in microbial killing, inflammation, and wound healing (Nakatsuji and Gallo, 2012). The two major AMP families in mammalian cells are the defensins and the cationic cathelicidin peptides (Lehrer and Ganz, 2002a, Lehrer and Ganz, 2002b, Zanetti et al., 1995). Although there are multiple defensin genes, there is only one known cathelicidin gene in humans (Lehrer and Ganz, 2002a, Sorensen et al., 1997). The human cathelicidin, hCAP-18, is abundantly expressed in neutrophils, monocytes, and epithelial cells of skin and mucosal membranes (Agerberth et al., 2000, Sorensen et al., 1997). hCAP-18 is stored as an inactive precursor and upon stimulation is processed to generate the active peptide LL-37 (Sorensen et al., 2001). Interestingly, besides its broad anti-bacterial property, LL-37 can also inhibit several viruses, including Influenza A virus, human rhinovirus, human adenovirus, and human immunodeficiency virus (Bergman et al., 2007, Currie et al., 2013, Uchio et al., 2013, Sousa et al., 2017). Recently, it has been demonstrated that LL-37-derived AMPs inhibit Zika virus infection, although the mechanisms are not well defined (He et al., 2018). Nevertheless, these studies suggest LL-37 AMPs can serve as viable anti-viral therapeutics.
In the present study, we show that human LL-37 and in particular, two engineered LL-37 variants function as inhibitors for EBOV infection. AMPs are known to have both direct and indirect antimicrobial effects; therefore, we explored the mechanism of action. We found that these AMPs impaired early events during EBOV infection but had no effect on virus replication. Further analysis revealed that AMPs impaired CatB-mediated cleavage of EBOV GP. Taken together, these data demonstrate that engineered AMPs are potent inhibitors of EBOV entry.
Results
Engineered AMPs Inhibit EBOV Pseudovirion Infection of Cell Lines and Human Primary Macrophages
Since EBOV is a highly lethal agent, EBOV-GP pseudotyped viruses based on attenuated vectors have been widely used in research laboratories. Here we used a recombinant vesicular stomatitis virus (rVSV) expressing EBOV-GP and green fluorescence protein (rVSV-EBOVgp-GFP, in short, VSV-eGP) as an experimental system to test the effects of AMPs on EBOV-GP-mediated infection. Initially, we screened 20 AMPs including the parent human LL-37 and its engineered variants. We found that two engineered LL-37 variant peptides 17BIPHE2 (in short 17BI) and GI-20d were most potent in inhibiting VSV-eGP infection (data not shown). Therefore, we decided to focus on these two peptides and the relevant control peptides in this study (listed in Figure 1 ). The parent LL-37 peptide was included as a baseline control. Peptides GF-17 and GI-20 containing the native sequences for 17BI and GI-20d, respectively, were derived from the major antimicrobial region of LL-37 (Li et al., 2006, Wang et al., 2008). Peptide 17BI was engineered with three D-form amino acids and two biphenylalanines based on the sequence of GF-17 (Wang et al., 2014). GI-20d is a novel peptide engineered with all D-form amino acids with identical sequence to GI-20. Peptide RI-10 is an inactive LL-37 derivative with only 10 amino acids, serving as a negative control (Wang, 2008).
Figure 1.

Amino Acid Sequences of Engineered AMPs
All the engineered AMPs are C-terminally amidated and derived from the sequence of human native LL-37 (hLL-37). 17BI is a short name for 17BIPHE2 as previously published.
HeLa cells were infected with VSV-eGP viruses together with individual AMPs (at 5 μM) or an EBOV neutralizing mAb 13C6 as a positive control. At 20 h post infection, cells were harvested for flow cytometry analysis to determine percentages of cells positive for GFP as the measurement of VSV-eGP infection. Parent LL-37 and engineered AMPs (except the negative control RI-10) displayed different degrees of inhibitory effects on VSV-eGP infection (Figure 2 A). Peptides 17BI and GI-20d were the most potent in inhibiting VSV-eGP infection with over 80% inhibition at 5 μM, whereas peptides LL-37, GF-17, and GI-20 led to 20%–44% inhibition under the same condition (Figure 2A). All these peptides showed dose-dependent inhibition of VSV-eGP infection in HeLa cells (Figure 2B).
Figure 2.

Engineered AMPs Inhibit the Infection of Pseudo-EBOV Virion in Cell Lines and Human Primary Macrophages
(A) AMPs inhibited pseudo-EBOV (VSV-eGP) infection in HeLa cells. VSV-eGP viruses were preincubated with an EBOV-neutralizing monoclonal antibody 13C6 (at 10 μg/mL, as a positive control) or individual AMPs (at 5 μM) for 30 min at 37°C in PBS buffer (pH 7.4) and then added to HeLa cells. After 20 h of culture, HeLa cells were harvested for flow cytometry analysis of GFP expression (percentages of GFP-positive cells represent percentages of cells infected with VSV-eGP).
(B) AMPs inhibited pseudo-EBOV (VSV-eGP) infection in HeLa cells in a dose-dependent manner. VSV-eGP viruses were added to HeLa cells together with monoclonal antibody 13C6 (at 5, 10, or 20 μg/mL) or individual AMPs (at 2.5, 5, or 10 μM). After 20 h of culture, HeLa cells were harvested for flow cytometry analysis to measure viral infection. ∗∗∗, p < 0.001 by two-way ANOVA test.
(C) AMPs inhibit pseudo-EBOV (VSV-eGP) infection in human primary macrophages. Human monocytes were freshly purified from blood and differentiated to macrophages by culturing the cells in media containing M-CSF. Macrophages were then infected with VSV-eGP in the presence of mAb 13C6 (at 5, 10, or 50 μg/mL) or AMPs (at 2.5, 5, or 10 μM). Cells were analyzed by flow cytometry at 20 h post infection. ∗, p < 0.05; ∗∗, p < 0.01 by two-way ANOVA test.
(D) A summary of the efficacy and toxicity of the AMPs in HeLa cells and human primary macrophages. Cells were infected with VSV-eGP together with AMPs at different concentrations ranging from 0.1 to 50 μM and analyzed by flow cytometry at 20 h post infection to evaluate the IC50 (half maximal inhibitory concentration). To measure cytotoxicity, cells were treated alone with the AMPs at different concentrations for 20 h. Cell viability was measured by MTT cell proliferation assay to determine the TC50 (half maximal toxicity concentration).
Because human macrophages are a primary target during natural EBOV infection, we next examined whether these AMPs inhibit VSV-eGP infection in human primary macrophages (Bray and Geisbert, 2005, Gupta et al., 2007). CD14+ monocytes were freshly purified from human blood samples and differentiated to macrophages by treatment with macrophage-colony-stimulating factor (M-CSF) (Murray et al., 2014). Macrophages were then infected with VSV-eGP together with mAb 13C6 or AMPs. Similar to the results in HeLa cells, all the AMPs (except the RI-10 negative control) displayed dose-dependent inhibition of VSV-eGP infection in macrophages (Figure 2C). Peptides 17BI and GI-20d were the most potent and almost completely inhibited VSV-eGP infection in macrophages at 10 μM (Figure 2C). We next determined the half-maximal concentration of inhibition (IC50) and half-maximal concentration of cell toxicity (TC50) of these AMPs in HeLa cells and primary macrophages. All the AMPs displayed lower IC50 concentrations in HeLa cells than in macrophages, although they also displayed slightly lower TC50 concentrations in HeLa cells than in macrophages (Figure 2D). Among the parent LL-37 and its engineered variants, peptide GI-20d had the most favorable therapeutic window in both HeLa cells and primary macrophages (with an IC50 of 0.99 μM and TC50 of 18.8 μM in HeLa cells and an IC50 of 2.2 μM and TC50 of more than 30 μM in macrophages). Peptide 17BI also had a favorable therapeutic window with an IC50 of 0.71 μM and TC50 of 13.2 μM in HeLa cells and an IC50 of 5.6 μM and TC50 of more than 30 μM in macrophages (Figure 2D). We also examined the effects of AMPs on VSV-eGP infection in Vero cells, a cell line commonly used for propagating viruses and in analyzing virus infection. Similarly, the parent LL-37 and its engineered variants (except the negative control RI-10) displayed dose-dependent inhibition of VSV-eGP infection in Vero cells (Figures S1A and S1B). In agreement with our findings in HeLa cells and primary macrophages, 17BI and GI-20d were the most potent peptides in inhibition of VSV-eGP infection in Vero cells. Both peptides almost completely inhibited VSV-eGP infection at concentrations ≥5 μM in Vero cells (Figures S1A and S1B).
Taken together, our data show that engineered LL-37 variant peptides 17BI and GI-20d are potent inhibitors of EBOV pseudovirion infection in HeLa and Vero cells as well as in human primary macrophages.
Engineered AMPs Inhibit Wild-Type EBOV-Kiwit Infection
Given the observed potency of AMP's within our surrogate EBOV system, we next tested whether these AMPs have similar effects on wild-type (WT) EBOV infection. HeLa cells were pre-incubated with AMPs at different concentrations for 2 h and then infected with EBOV-Kiwit. At 24 h post infection, cells were fixed, permeabilized, and immuno-stained with an anti-EBOV GP antibody followed by a fluorescence-conjugated secondary antibody to identify infected cells. In agreement with our EBOV pseudovirion studies, engineered peptides 17BI and GI-20d displayed much higher potency in blocking the infection of WT EBOV than the peptides containing only L-amino acids, GF-17 and GI-20 (Figure 3 A). Peptides 17BI and GI-20d had an IC50 of 3 and 1.6 μM, respectively, whereas GF-17 and GI-20 both had an IC50 of about 20 μM (Figure 3B). Interestingly, these AMPs did not significantly affect Marburg virus (MARV) infection under the same conditions, demonstrating the specificity of AMPs in inhibiting EBOV infection (Figure S2). The inhibitory effects of engineered AMPs on WT EBOV infection further support the finding that engineered AMPs may be a new class of anti-EBOV countermeasures and highlights the consistency between WT EBOV and the surrogate systems.
Figure 3.

Engineered AMPs Inhibit Wild-Type EBOV Infection
HeLa cells were preincubated with individual AMPs at different concentrations (16 serial dilutions from 50 μM) for 2 h at 37°C and then infected with EBOV (Zaire-Kiwit) at MOI of 4. At 24 h post infection, cells were fixed, permeabilized, and immuno-stained with anti-EBOV GP antibodies followed by fluorescence-conjugated secondary antibody to identify the infected cells.
(A) Representative immunostaining images (red: nuclear staining; green: anti-EBOV GP staining).
(B) A summary of IC50 of the AMPs.
Engineered AMPs Inhibit EBOV Cell Entry but Not Viral Replication
To determine potential mechanisms responsible for the inhibitory effects of LL-37-derived AMPs on EBOV infection, we next examined whether these AMPs targeted EBOV cell entry by comparing the effects of pretreatment or delayed AMP treatment of cells prior to viral infection. As previously observed (Figure 2C), AMPs 17BI and GI-20d, but not LL-37, GF-17, or GI-20, reduced VSV-eGP infection in human primary macrophages with over 40% inhibition at 5 μM when added at the time of infection (Figure 4 A). Pre-treatment of macrophages with 17BI and GI-20d for 4 h prior to viral infection blocked VSV-eGP infection with similar efficacy (Figure 4A). However, delayed treatment of macrophages with 17BI and GI-20d peptides resulted in a time-dependent reduction of effects, with approximately 20% of inhibition when added at 4 h post infection and a complete loss of inhibition when added at 8 h post infection (Figure 4A). These data suggested that engineered AMP peptides may target the early cell entry stage of EBOV infection. Similar results were observed for Vero cells (Figure S1C).
Figure 4.

Engineered AMPs Inhibit EBOV Cell Entry but Do Not Affect EBOV Replication in a Mini-Genome System
(A) Engineered AMPs targeted pseudo-EBOV (VSV-eGP) at the early stage of infection. Human monocyte-derived primary macrophages were treated with 5 μM of individual AMPs at 4 h before VSV-eGP infection (-4 h), at the same time with VSV-eGP (0 h), or at 4 or 8 h after VSV-eGP infection (+4 h or +8 h, respectively). Macrophages were harvested for flow cytometry analysis of GFP levels at 20 h post infection. ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001 by two-way ANOVA test.
(B) Engineered AMPs inhibit the cell entry of Ebola virus-like particles (VLPs). Ebola VLPs were produced by co-expressing the EBOV matrix protein, VP40 (fused to β-lactamase), and the EBOV GP. Vero cells were pre-treated with AMPs (at 2.5, 5, and 10 μM) or cathepsin B inhibitor CA-074 methyl ester (at 50 μM as a positive control) for 1 h at 37°C and then infected with Ebola VLPs. At 4 h post infection, a membrane-permeable β-lactamase substrate (CCF-2AM) was added to the culture and incubated for 1 h at room temperature. The release of β-lactamase by VLPs into the cytoplasm of cells (which is the result of successful processing and cell entry of VLPs from the endosomes) was measured by fluorescence emission of CCF-2AM substrate within 4 h of infection. ∗, p < 0.05; ∗∗, p < 0.01 by two-way ANOVA test.
(C) Engineered AMPs do not affect EBOV replication in a mini-genome system. The EBOV mini-genome plasmids (encoding a firefly luciferase reporter gene flanked by the leader and trailer sequences from EBOV genome) were co-transfected into HeLa cells with four supporting plasmids encoding EBOV replication complex components NP, L, VP35, and VP30 and a plasmid encoding Renilla luciferase (for normalization of transfection efficiency). The ratio of firefly to Renilla luciferase represented the relative levels of EBOV mini-genome replication. ∗∗∗, p < 0.001 by one-way ANOVA test.
We next used Ebola virus-like particles (VLPs) to directly test the effects of AMPs on EBOV cell entry. Ebola VLPs were produced by co-expressing the EBOV matrix protein VP40 (fused to β-lactamase) and the EBOV GP. The VLPs possess a structure and biochemical composition similar to the WT EBOV, but unlike the WT virus, lack genetic materials for virus replication. The activities of cytoplasmic β-lactamase thus represent the relative levels of productive cell entry of Ebola VLPs. Vero cells were pre-incubated with AMPs or a cathepsin B (CatB)-specific inhibitor (as a positive control) followed by Ebola VLP infection. CatB inhibitor and peptides 17BI and GI-20d displayed dose-dependent inhibition of cytoplasmic β-lactamase activity, providing direct evidence that engineered AMPs inhibit productive EBOV cell entry (Figure 4B).
After entering the cytoplasm, EBOV undergoes replication to amplify its genetic material in order to produce progeny virions. In the absence of WT virus, replication can be studied using an EBOV mini-genome system (Cressey et al., 2017). We next examined the effects of AMPs on EBOV replication using this system. The EBOV minigenome plasmids (encoding firefly luciferase reporter gene flanked by the leader and trailer sequences from EBOV genome) were co-transfected into HeLa cells with four supporting plasmids encoding EBOV replication complex components NP, L, VP35, and VP30 and a plasmid encoding Renilla luciferase (for normalization of transfection efficiency). The ratio of firefly to Renilla luciferase represented the relative levels of EBOV mini-genome replication. None of the tested AMPs (at 5 μM) led to a significant change of the ratio of firefly to Renilla luciferase, whereas the positive control Ribavirin significantly reduced the ratio (Figure 4C). Our data suggest that LL-37 and its engineered variants do not affect EBOV replication in the mini-genome system.
Collectively, the results suggest that engineered LL-37 variant peptides, 17BI and GI-20d, block EBOV cell entry but not viral replication.
LL-37 and Engineered AMPs Block CatB-Dependent Processing of EBOV-GP
Productive EBOV cell entry and infection requires the processing and cleavage of EBOV-GP by cathepsins within the endosome. We therefore next tested whether AMPs block CatB- and CatL-mediated cleavage of EBOV-GP. CatB was pre-incubated with AMPs or a known CatB inhibitor (CA-074 methyl ester, as a positive control) in a pH 5.0 buffer before the addition of EBOV GP protein. The reaction mixture was then subjected to western blot analysis to analyze EBOV GP proteolytic cleavage. As expected, EBOV GP (about 110 kD) was cleaved by CatB and generated a 19-kD product in a denaturing gel (Figure 5 A). CatB inhibitor and all the tested AMPs (except the negative control RI-10) effectively blocked the processing of EBOV-GP by CatB (Figure 5A). However, none of the tested AMPs had any effects on the processing of EBOV-GP by CatL under the same conditions (Figure 5B).
Figure 5.

LL-37 and Engineered AMPs Block the Cleavage of EBOV GP by CatB, but Not by CatL
(A) LL-37 and engineered AMPs blocked CatB-dependent cleavage of EBOV GP. CatB was pre-incubated with the CatB inhibitor (CA-074 methyl ester at 50 μM, as a positive control) or AMPs (at 10 μM) in 100 mM sodium acetate buffer pH 5.0 at 37°C for 30 min. Ebola GP protein was added to the reaction and incubated at 37°C for 1 h. The reaction mixtures were boiled in SDS-PAGE loading buffer and applied to denatured SDS PAGE followed by western blot with anti-Ebola GP mAb 13C6 (to detect the 110 kD full-length GP) and rabbit anti-Ebola GP polyclonal antibodies (to detect the 19 kD cleaved GP product).
(B) None of the tested AMPs blocked CatL-dependent cleavage of EBOV GP. The same reaction was performed as in (A) except using CatL instead of CatB.
Using standard CatB and CatL substrates, we next tested whether the AMPs block the intrinsic activity of CatB and CatL. All the tested AMPs (except RI-10 negative control) inhibited CatB-mediated cleavage of Z-Arg-Arg-AMC in a dose-dependent manner (Figure S3A). Engineered peptide 17BI is the most effective in inhibiting CatB activity among the tested peptides with an IC50 of 6 μM (Figure S3B). However, none of the peptides had inhibition effects on CatL-mediated cleavage of Z-Phe-Arg-AMC (Figure S3C). The data demonstrated that LL-37 and engineered AMPs directly inhibit the intrinsic enzymatic activity of CatB, but not that of CatL.
AMPs Composed of L-Amino Acids Are Susceptible to Degradation by Cathepsin S (CatS) and Blockage of CatS Enhances AMP Inhibition of EBOV Infection
Notably, the parent LL-37 and derivatives GF-17 and GI-20 had efficacy similar to engineered variants 17BI and GI-20d in their inhibition of CatB-mediated EBOV GP (eGP) cleavage in vitro; however, their efficacy in inhibiting EBOV infection in cells was much lower than the variants containing D-amino acids. We postulated that AMPs composed of L-amino acids are more susceptible to degradation by proteolytic enzymes inside cells, thus reducing their availability to block CatB in vivo. To test this hypothesis, we examined whether the AMPs were susceptible to CatS cleavage in an in vitro reaction. CatS was used because it is the most abundant cathepsin in the endosome/lysosome of macrophages and LL-37 has been shown to be an excellent substrate of CatS (Shi et al., 1992, Andrault et al., 2015). All the AMPs composed of L-amino acids only (LL-37, GF-17, GI-20, and RI-10) were completely degraded by CatS treatment in the reaction, whereas AMPs containing D-amino acids (17BI and GI-20d) were resistant to CatS cleavage (Figure 6 A). The data indicate that AMPs composed of L-amino acids are susceptible to degradation by CatS upon entering cells. We then hypothesized that blockage of CatS may stabilize these AMPs inside cells, thus making them available to inhibit CatB-mediated EBOV entry. To test the hypothesis, Vero cells were pre-treated with a CatS inhibitor for 2 h, followed by the addition of AMPs and VSV-eGP. CatS inhibitor treatment significantly improved the efficacy of LL-37, GF-17, and GI-20 by 4- to 10-fold in their inhibition of VSV-eGP infection (Figure 6B). Notably, CatS inhibitor also improved the efficacy of 17BI and GI-20d by 1.5- to 2-fold and CatS inhibitor treatment alone resulted in 30% inhibition of VSV-eGP infection, likely due to the known ability of the inhibitor to cross-inhibit CatB.
Figure 6.

AMPs Composed of L-Amino Acids Are Susceptible to Degradation by CatS and Blockage of CatS Enhances AMP Inhibition of EBOV Infection
(A) AMPs composed of L-amino acids were susceptible to degradation by CatS. AMPs (5 μM) were incubated with or without CatS (8 μg/mL) in a sodium acetate buffer (pH 5.0) for 60 min at 37°C. The reaction mixture was boiled in SDS sample buffer and loaded onto 4%–20% gradient gel, followed by staining with Coomassie blue.
(B) Blockage of CatS enhanced AMP inhibition of pseudo-EBOV infection. Vero cells were infected with VSV-eGP in the absence or presence of CatB inhibitor (50 μM) as a positive control, CatS inhibitor (75 μM), AMPs (5 μM), or the combination of CatS inhibitor (75 μM) and AMPs (5 μM). After 20–24 h of culture, cells were harvested and applied to flow cytometry analysis.
Collectively, our data suggest that LL-37 AMPs are susceptible to enzymatic degradation inside cells and engineered AMPs with D-amino acids are more resistant to such degradation, thus more effective at inhibiting EBOV infection.
Discussion
In this report, we demonstrated that engineered AMPs based on human cathelicidin LL-37 inhibit EBOV infection. Mechanistic studies revealed that these AMPs act as CatB inhibitors to block the endosomal processing of EBOV GP, thus preventing virus entry. Our results identify engineered AMPs as a potential novel class of anti-EBOV therapeutics, providing an additional avenue for combating the highly lethal EBOV.
Human cathelicidin LL-37 has several limitations as a therapeutic molecule. First, it is relatively long with 37 amino acids. Second, it can be rapidly degraded by proteases. Third, it can lose activity under certain conditions. The design of engineered LL-37 aimed to overcome these limitations. Different active regions in LL-37 have been identified in previous structure-function studies (Sigurdardottir et al., 2006, Nell et al., 2006, Braff et al., 2005, Nagaoka et al., 2002, Li et al., 2006). We have chosen GF-17 as a template for further peptide engineering because GF-17 is the most active against methicillin-resistant Staphylococcus aureus (MRSA) among active peptides discovered previously (Wang et al., 2014). In another study, we found that GI-20 (with three amino acid extension at the N-terminus of GF-17) had increased activity in inhibiting HIV-1 (Wang et al., 2008). Based on GF-17 and GI-20, respectively, 17BI and GI-20d were designed with incorporation of D-amino acids to enhance their biostability.
Interestingly, 17BI and GI-20d are found to be more effective in inhibiting EBOV infection than the parent LL-37 and other derivative peptides. Previous studies have shown that the 17BI peptide is more resistant to chymotrypsin, bacterial V8 protease, and fungal protease K degradation than GF-17 and other peptides composed of L-amino acid (Wang et al., 2014). Here we show that peptides 17BI and GI-20d are more resistant to degradation by CatS, a cysteine proteinase abundant in the lysosomes of macrophages, dendritic cells, and some epithelial cells (Shi et al., 1992, Shi et al., 1994). In addition, pretreatment of cells with a CatS inhibitor significantly enhanced the efficacy of parent LL-37 and its derivative AMPs compose of L-amino acids in inhibiting EBOV infection (Figure 6). These results suggest the important role of CatS in mediating the instability of LL-37-based AMPs inside cells. A previous study has reported that LL-37 is an excellent substrate for CatS with seven potential cleavage sites (Andrault et al., 2015). Therefore, we anticipate that targeted engineering of AMPs to eliminate CatS cleavage sites will produce more stable versions of AMPs that may display enhanced efficacy in inhibiting EBOV infection.
Our in vitro study showed that LL-37 and its derived AMPs were inhibitors of CatB, but not CatL, using both EBOV GP and the standard substrates for each enzyme. These results are in contrast to a previous study that claimed LL-37 was a selective inhibitor of CatL but not CatB (Andrault et al., 2015). To address any potential experimental differences, we additionally confirmed our study results using two independent sources of CatB and CatL (one source provided cathepsins purified from the liver and the other source provided cathepsins in recombinant form). In all cases, these confirmatory experiments agreed with our original results (data not shown) and the commercially available CatB- and CatL-specific inhibitors performed as expected in our assays (Figure S3).
Although endosomal cysteine proteases are required for cell entry of all filovirus family members, different viruses have distinct protease preferences (Misasi et al., 2012). CatB is required for EBOV cell entry, whereas several studies suggest that MARV cell entry does not require CatB, although it is not clear what proteases are required (Gnirss et al., 2012, Misasi et al., 2012, Sanchez, 2007). In agreement of those studies, we found that none of our tested AMPs significantly inhibited MARV infection (Figure S2). The two D-amino acid-containing AMPs (17BI and GI-20d) that display potent inhibition of EBOV infection only have minimal inhibition of MARV infection (with an IC50 of 41 and 25 μM, respectively). In future studies, it will be interesting to test whether these AMPs also inhibit the infection of other viruses where viral cell entry involves CatB, such as other filovirus family members (e.g., Bundibugyo virus) (Misasi et al., 2012) and Middle East Respiratory Syndrome (MERS) and Severe Acute Respiratory Syndrome (SARS) coronaviruses (Elshabrawy et al., 2014).
Notably, all the LL-37 AMPs are less efficient in inhibiting EBOV infection in macrophages than in HeLa and Vero cells (Figures 2 and S1). Two potential mechanisms may explain the observation. First, as professional antigen-presenting cells, macrophages likely contain an abundance of proteases such as cathepsin S; therefore, AMP peptides are less available in macrophages to inhibit viral infection. Second, it is known that primary macrophages are more refractory to EBOV infection compared with tissue-culture cell lines (i.e., Vero, HeLa). As such, EBOV macrophage infection requires a 10-fold higher MOI compared with cell lines. Therefore, it is likely that an increase in the amounts of AMPs is needed to achieve similar degrees of inhibition. Finally, it should be noted that our data are consistent with findings that EBOV neutralizing antibodies, such as 13C6, demonstrate less potency with higher IC50 values in primary macrophages relative to cell lines.
The current promising treatment options against EBOV primarily consists of nucleoside analog small molecules and immunotherapies using neutralizing antibodies (Edwards and Basler, 2019, King et al., 2019). It will be interesting to test whether the combined use of engineered AMPs with those therapeutic regimens will have additive efficacy, given that distinct inhibition mechanisms are involved. We anticipate that a combination of AMPs with small molecules that interfere with viral replication or with virus-neutralizing antibodies will have improved efficacy against EBOV.
In summary, our study has demonstrated the inhibitory effects of engineered LL-37 AMPs on EBOV cell entry. Future studies will test the therapeutic potential of these AMPs in well-established EBOV animal models. Because the most common route of EBOV infection is through blood or other body fluids, we anticipate that intravenous injection of engineered AMP peptides will be most effective, although topical use of the peptides could be an option to combat EBOV infection through direct skin contact. These animal studies will further confirm that engineered LL-37 AMPs are a novel class of anti-EBOV therapeutics.
Limitations of the Study
Most of the mechanistic studies were performed using EBOV surrogate systems. Follow-up experiments using live virus in animal systems are needed to extend the clinical utility of the findings. The current work was limited to a single filovirus member, future efforts should test against additional filovirus family members.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This study is supported by the Nebraska University (NU) Collaboration Initiatives Systems Biology Seed Grant awarded to K.S. Peptide synthesis is supported by NIH grant R01AI105147 to G.W. All live-virus studies were conducted at USAMRIID within biosafety level 4 containment. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense or U.S. Army.
Author Contributions
Y.Y. performed most of the experiments, analyzed the data, and contributed to manuscript writing. C.L.C. provided VSV-eGP and mAb 13C6, supervised wild-type EBOV and MARV infection experiments, and contributed to experimental design and manuscript writing. G.W. provided antimicrobial peptides and contributed to experimental design and manuscript writing. M.J.M., K.K., J.P.T., Y.L., J.S., A.K.Z., and L.G.L. performed some of the experiments reported in the manuscript. S.B.B. provided Ebola GP proteins and contributed to manuscript writing. S.P.R. supervised EBOV mini-genome replication assay and contributed to experimental design and manuscript writing. S.H.H. supervised the project and contributed to experimental design and manuscript writing. K.S. supervised and managed the project including experimental design, data analysis, manuscript writing, and providing funds for the project.
Declaration of Interests
The authors declare no competing interests.
Published: April 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.100999.
Supplemental Information
References
- Agerberth B., Charo J., Werr J., Olsson B., Idali F., Lindbom L., Kiessling R., Jornvall H., Wigzell H., Gudmundsson G.H. The human antimicrobial and chemotactic peptides LL-37 and alpha-defensins are expressed by specific lymphocyte and monocyte populations. Blood. 2000;96:3086–3093. [PubMed] [Google Scholar]
- Andrault P.M., Samsonov S.A., Weber G., Coquet L., Nazmi K., Bolscher J.G., Lalmanach A.C., Jouenne T., Bromme D., Pisabarro M.T. Antimicrobial peptide LL-37 is both a substrate of cathepsins S and K and a selective inhibitor of cathepsin L. Biochemistry. 2015;54:2785–2798. doi: 10.1021/acs.biochem.5b00231. [DOI] [PubMed] [Google Scholar]
- Audet J., Wong G., Wang H., Lu G., Gao G.F., Kobinger G., Qiu X. Molecular characterization of the monoclonal antibodies composing ZMAb: a protective cocktail against ebola virus. Sci. Rep. 2014;4:6881. doi: 10.1038/srep06881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman P., Walter-Jallow L., Broliden K., Agerberth B., Soderlund J. The antimicrobial peptide LL-37 inhibits HIV-1 replication. Curr. HIV Res. 2007;5:410–415. doi: 10.2174/157016207781023947. [DOI] [PubMed] [Google Scholar]
- Bixler S.L., Bocan T.M., Wells J., Wetzel K.S., Van Tongeren S.A., Dong L., Garza N.L., Donnelly G., Cazares L.H., Nuss J. Efficacy of favipiravir (T-705) in nonhuman primates infected with Ebola virus or Marburg virus. Antivir. Res. 2018;151:97–104. doi: 10.1016/j.antiviral.2017.12.021. [DOI] [PubMed] [Google Scholar]
- Braff M.H., Hawkins M.A., Di Nardo A., Lopez-Garcia B., Howell M.D., Wong C., Lin K., Streib J.E., Dorschner R., Leung D.Y., Gallo R.L. Structure-function relationships among human cathelicidin peptides: dissociation of antimicrobial properties from host immunostimulatory activities. J. Immunol. 2005;174:4271–4278. doi: 10.4049/jimmunol.174.7.4271. [DOI] [PubMed] [Google Scholar]
- Brannan J.M., He S., Howell K.A., Prugar L.I., Zhu W., Vu H., Shulenin S., Kailasan S., Raina H., Wong G. Post-exposure immunotherapy for two ebolaviruses and Marburg virus in nonhuman primates. Nat. Commun. 2019;10:105. doi: 10.1038/s41467-018-08040-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray M., Geisbert T.W. Ebola virus: the role of macrophages and dendritic cells in the pathogenesis of Ebola hemorrhagic fever. Int. J. Biochem. Cell Biol. 2005;37:1560–1566. doi: 10.1016/j.biocel.2005.02.018. [DOI] [PubMed] [Google Scholar]
- Carette J.E., Raaben M., Wong A.C., Herbert A.S., Obernosterer G., Mulherkar N., Kuehne A.I., Kranzusch P.J., Griffin A.M., Ruthel G. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 2011;477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran K., Sullivan N.J., Felbor U., Whelan S.P., Cunningham J.M. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science. 2005;308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coltart C.E., Lindsey B., Ghinai I., Johnson A.M., Heymann D.L. The Ebola outbreak, 2013-2016: old lessons for new epidemics. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017;372(1721):20160297. doi: 10.1098/rstb.2016.0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote M., Misasi J., Ren T., Bruchez A., Lee K., Filone C.M., Hensley L., Li Q., Ory D., Chandran K., Cunningham J. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature. 2011;477:344–348. doi: 10.1038/nature10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cressey T., Brauburger K., Muhlberger E. Modeling ebola virus genome replication and transcription with minigenome systems. Methods Mol. Biol. 2017;1628:79–92. doi: 10.1007/978-1-4939-7116-9_6. [DOI] [PubMed] [Google Scholar]
- Currie S.M., Findlay E.G., Mchugh B.J., Mackellar A., Man T., Macmillan D., Wang H., Fitch P.M., Schwarze J., Davidson D.J. The human cathelicidin LL-37 has antiviral activity against respiratory syncytial virus. PLoS One. 2013;8:e73659. doi: 10.1371/journal.pone.0073659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey R.A., Shtanko O., Anantpadma M., Sakurai Y., Chandran K., Maury W. Mechanisms of filovirus entry. Curr. Top. Microbiol. Immunol. 2017;411:323–352. doi: 10.1007/82_2017_14. [DOI] [PubMed] [Google Scholar]
- Edwards M.R., Basler C.F. Current status of small molecule drug development for Ebola virus and other filoviruses. Curr. Opin. Virol. 2019;35:42–56. doi: 10.1016/j.coviro.2019.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshabrawy H.A., Fan J., Haddad C.S., Ratia K., Broder C.C., Caffrey M., Prabhakar B.S. Identification of a broad-spectrum antiviral small molecule against severe acute respiratory syndrome coronavirus and Ebola, Hendra, and Nipah viruses by using a novel high-throughput screening assay. J. Virol. 2014;88:4353–4365. doi: 10.1128/JVI.03050-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann H., Geisbert T.W. Ebola haemorrhagic fever. Lancet. 2011;377:849–862. doi: 10.1016/S0140-6736(10)60667-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann H., Kiley M.P. Classification, structure, and replication of filoviruses. Curr. Top. Microbiol. Immunol. 1999;235:1–21. doi: 10.1007/978-3-642-59949-1_1. [DOI] [PubMed] [Google Scholar]
- Furuyama W., Marzi A., Nanbo A., Haddock E., Maruyama J., Miyamoto H., Igarashi M., Yoshida R., Noyori O., Feldmann H., Takada A. Discovery of an antibody for pan-ebolavirus therapy. Sci. Rep. 2016;6:20514. doi: 10.1038/srep20514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnirss K., Kuhl A., Karsten C., Glowacka I., Bertram S., Kaup F., Hofmann H., Pohlmann S. Cathepsins B and L activate Ebola but not Marburg virus glycoproteins for efficient entry into cell lines and macrophages independent of TMPRSS2 expression. Virology. 2012;424:3–10. doi: 10.1016/j.virol.2011.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M., Spiropoulou C., Rollin P.E. Ebola virus infection of human PBMCs causes massive death of macrophages, CD4 and CD8 T cell sub-populations in vitro. Virology. 2007;364:45–54. doi: 10.1016/j.virol.2007.02.017. [DOI] [PubMed] [Google Scholar]
- He M., Zhang H., Li Y., Wang G., Tang B., Zhao J., Huang Y., Zheng J. Cathelicidin-derived antimicrobial peptides inhibit zika virus through direct inactivation and interferon pathway. Front. Immunol. 2018;9:722. doi: 10.3389/fimmu.2018.00722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert A.S., Davidson C., Kuehne A.I., Bakken R., Braigen S.Z., Gunn K.E., Whelan S.P., Brummelkamp T.R., Twenhafel N.A., Chandran K. Niemann-pick C1 is essential for ebolavirus replication and pathogenesis in vivo. MBio. 2015;6 doi: 10.1128/mBio.00565-15. e00565–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell K.A., Brannan J.M., Bryan C., Mcneal A., Davidson E., Turner H.L., Vu H., Shulenin S., He S., Kuehne A. Cooperativity enables non-neutralizing antibodies to neutralize ebolavirus. Cell Rep. 2017;19:413–424. doi: 10.1016/j.celrep.2017.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt C.L., Lennemann N.J., Maury W. Filovirus entry: a novelty in the viral fusion world. Viruses. 2012;4:258–275. doi: 10.3390/v4020258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King L.B., Milligan J.C., West B.R., Schendel S.L., Ollmann Saphire E. Achieving cross-reactivity with pan-ebolavirus antibodies. Curr. Opin. Virol. 2019;34:140–148. doi: 10.1016/j.coviro.2019.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrer R.I., Ganz T. Cathelicidins: a family of endogenous antimicrobial peptides. Curr. Opin. Hematol. 2002;9:18–22. doi: 10.1097/00062752-200201000-00004. [DOI] [PubMed] [Google Scholar]
- Lehrer R.I., Ganz T. Defensins of vertebrate animals. Curr. Opin. Immunol. 2002;14:96–102. doi: 10.1016/s0952-7915(01)00303-x. [DOI] [PubMed] [Google Scholar]
- Li X., Li Y., Han H., Miller D.W., Wang G. Solution structures of human LL-37 fragments and NMR-based identification of a minimal membrane-targeting antimicrobial and anticancer region. J. Am. Chem. Soc. 2006;128:5776–5785. doi: 10.1021/ja0584875. [DOI] [PubMed] [Google Scholar]
- Marzi A., Yoshida R., Miyamoto H., Ishijima M., Suzuki Y., Higuchi M., Matsuyama Y., Igarashi M., Nakayama E., Kuroda M. Protective efficacy of neutralizing monoclonal antibodies in a nonhuman primate model of Ebola hemorrhagic fever. PLoS One. 2012;7:e36192. doi: 10.1371/journal.pone.0036192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller E.H., Obernosterer G., Raaben M., Herbert A.S., Deffieu M.S., Krishnan A., Ndungo E., Sandesara R.G., Carette J.E., Kuehne A.I. Ebola virus entry requires the host-programmed recognition of an intracellular receptor. EMBO J. 2012;31:1947–1960. doi: 10.1038/emboj.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misasi J., Chandran K., Yang J.Y., Considine B., Filone C.M., Cote M., Sullivan N., Fabozzi G., Hensley L., Cunningham J. Filoviruses require endosomal cysteine proteases for entry but exhibit distinct protease preferences. J. Virol. 2012;86:3284–3292. doi: 10.1128/JVI.06346-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray P.J., Allen J.E., Biswas S.K., Fisher E.A., Gilroy D.W., Goerdt S., Gordon S., Hamilton J.A., Ivashkiv L.B., Lawrence T. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaoka I., Hirota S., Niyonsaba F., Hirata M., Adachi Y., Tamura H., Tanaka S., Heumann D. Augmentation of the lipopolysaccharide-neutralizing activities of human cathelicidin CAP18/LL-37-derived antimicrobial peptides by replacement with hydrophobic and cationic amino acid residues. Clin. Diagn. Lab. Immunol. 2002;9:972–982. doi: 10.1128/CDLI.9.5.972-982.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsuji T., Gallo R.L. Antimicrobial peptides: old molecules with new ideas. J. Invest. Dermatol. 2012;132:887–895. doi: 10.1038/jid.2011.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nell M.J., Tjabringa G.S., Wafelman A.R., Verrijk R., Hiemstra P.S., Drijfhout J.W., Grote J.J. Development of novel LL-37 derived antimicrobial peptides with LPS and LTA neutralizing and antimicrobial activities for therapeutic application. Peptides. 2006;27:649–660. doi: 10.1016/j.peptides.2005.09.016. [DOI] [PubMed] [Google Scholar]
- Pettitt J., Zeitlin L., Kim D.H., Working C., Johnson J.C., Bohorov O., Bratcher B., Hiatt E., Hume S.D., johnson A.K. Therapeutic intervention of Ebola virus infection in rhesus macaques with the MB-003 monoclonal antibody cocktail. Sci. Transl. Med. 2013;5:199ra113. doi: 10.1126/scitranslmed.3006608. [DOI] [PubMed] [Google Scholar]
- Qiu X., Audet J., Wong G., Pillet S., Bello A., Cabral T., Strong J.E., Plummer F., Corbett C.R., Alimonti J.B., Kobinger G.P. Successful treatment of ebola virus-infected cynomolgus macaques with monoclonal antibodies. Sci. Transl. Med. 2012;4:138ra81. doi: 10.1126/scitranslmed.3003876. [DOI] [PubMed] [Google Scholar]
- Sanchez A. Analysis of filovirus entry into vero e6 cells, using inhibitors of endocytosis, endosomal acidification, structural integrity, and cathepsin (B and L) activity. J. Infect. Dis. 2007;196:S251–S258. doi: 10.1086/520597. [DOI] [PubMed] [Google Scholar]
- Schornberg K., Matsuyama S., Kabsch K., Delos S., Bouton A., White J. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J. Virol. 2006;80:4174–4178. doi: 10.1128/JVI.80.8.4174-4178.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G.P., Munger J.S., Meara J.P., Rich D.H., Chapman H.A. Molecular cloning and expression of human alveolar macrophage cathepsin S, an elastinolytic cysteine protease. J. Biol. Chem. 1992;267:7258–7262. [PubMed] [Google Scholar]
- Shi G.P., Webb A.C., Foster K.E., Knoll J.H., Lemere C.A., Munger J.S., Chapman H.A. Human cathepsin S: chromosomal localization, gene structure, and tissue distribution. J. Biol. Chem. 1994;269:11530–11536. [PubMed] [Google Scholar]
- Sigurdardottir T., Andersson P., Davoudi M., Malmsten M., Schmidtchen A., Bodelsson M. In silico identification and biological evaluation of antimicrobial peptides based on human cathelicidin LL-37. Antimicrob. Agents Chemother. 2006;50:2983–2989. doi: 10.1128/AAC.01583-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen O., Arnljots K., cowland J.B., Bainton D.F., Borregaard N. The human antibacterial cathelicidin, hCAP-18, is synthesized in myelocytes and metamyelocytes and localized to specific granules in neutrophils. Blood. 1997;90:2796–2803. [PubMed] [Google Scholar]
- Sorensen O.E., Follin P., Johnsen A.H., Calafat J., Tjabringa G.S., Hiemstra P.S., Borregaard N. Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood. 2001;97:3951–3959. doi: 10.1182/blood.v97.12.3951. [DOI] [PubMed] [Google Scholar]
- Sousa F.H., Casanova V., Findlay F., Stevens C., Svoboda P., Pohl J., Proudfoot L., Barlow P.G. Cathelicidins display conserved direct antiviral activity towards rhinovirus. Peptides. 2017;95:76–83. doi: 10.1016/j.peptides.2017.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchio E., Inoue H., Kadonosono K. Anti-adenoviral effects of human cationic antimicrobial protein-18/LL-37, an antimicrobial peptide, by quantitative polymerase chain reaction. Korean J. Ophthalmol. 2013;27:199–203. doi: 10.3341/kjo.2013.27.3.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G. Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J. Biol. Chem. 2008;283:32637–32643. doi: 10.1074/jbc.M805533200. [DOI] [PubMed] [Google Scholar]
- Wang G., Hanke M.L., Mishra B., Lushnikova T., Heim C.E., Chittezham Thomas V., Bayles K.W., Kielian T. Transformation of human cathelicidin LL-37 into selective, stable, and potent antimicrobial compounds. ACS Chem. Biol. 2014;9:1997–2002. doi: 10.1021/cb500475y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G., Watson K.M., Buckheit R.W., JR. Anti-human immunodeficiency virus type 1 activities of antimicrobial peptides derived from human and bovine cathelicidins. Antimicrob. Agents Chemother. 2008;52:3438–3440. doi: 10.1128/AAC.00452-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren T.K., Jordan R., Lo M.K., Ray A.S., Mackman R.L., Soloveva V., Siegel D., Perron M., Bannister R., Hui H.C. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature. 2016;531:381–385. doi: 10.1038/nature17180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren T.K., Wells J., Panchal R.G., Stuthman K.S., Garza N.L., Van Tongeren S.A., Dong L., Retterer C.J., Eaton B.P., Pegoraro G. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature. 2014;508:402–405. doi: 10.1038/nature13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanetti M., Gennaro R., Romeo D. Cathelicidins: a novel protein family with a common proregion and a variable C-terminal antimicrobial domain. FEBS Lett. 1995;374:1–5. doi: 10.1016/0014-5793(95)01050-o. [DOI] [PubMed] [Google Scholar]
- Zhang X., Liu Q., Zhang N., Li Q.Q., Liu Z.D., Li Y.H., Gao L.M., Wang Y.C., Deng H.B., Song D.Q. Discovery and evolution of aloperine derivatives as novel anti-filovirus agents through targeting entry stage. Eur. J. Med. Chem. 2018;149:45–55. doi: 10.1016/j.ejmech.2018.02.061. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.