

Abstract
Background
Dilated cardiomyopathy (DCM), characterized by progressive left ventricular enlargement and systolic dysfunction, is the most common type of cardiomyopathy and a leading cause of heart failure and cardiac death. Accumulating evidence underscores the critical role of genetic defects in the pathogenesis of DCM, and >250 genes have been implicated in DCM to date. However, DCM is of substantial genetic heterogeneity, and the genetic basis underpinning DCM remains elusive in most cases.
Methods and Results
By genome‐wide scan with microsatellite markers and genetic linkage analysis in a 4‐generation family inflicted with autosomal‐dominant DCM, a new locus for DCM was mapped on chromosome 15q13.1–q13.3, a 4.77‐cM (≈3.43 Mbp) interval between markers D15S1019 and D15S1010, with the largest 2‐point logarithm of odds score of 5.1175 for the marker D15S165 at recombination fraction (θ)=0.00. Whole‐exome sequencing analyses revealed that within the mapping chromosomal region, only the mutation in the KLF13 gene, c.430G>T (p.E144X), cosegregated with DCM in the family. In addition, sequencing analyses of KLF13 in another cohort of 266 unrelated patients with DCM and their available family members unveiled 2 new mutations, c.580G>T (p.E194X) and c.595T>C (p.C199R), which cosegregated with DCM in 2 families, respectively. The 3 mutations were absent from 418 healthy subjects. Functional assays demonstrated that the 3 mutants had no transactivation on the target genes ACTC1 and MYH7 (2 genes causally linked to DCM), alone or together with GATA4 (another gene contributing to DCM), and a diminished ability to bind the promoters of ACTC1 and MYH7. Add, the E144X‐mutant KLF13 showed a defect in intracellular distribution.
Conclusions
This investigation indicates KLF13 as a new gene predisposing to DCM, which adds novel insight to the molecular pathogenesis underlying DCM, implying potential implications for prenatal prevention and precision treatment of DCM in a subset of patients.
Keywords: biological assay, dilated cardiomyopathy, functional genomics, KLF13, molecular genetics, transcriptional factor
Subject Categories: Genetics, Cardiomyopathy, Heart Failure
Nonstandard Abbreviations and Acronyms
- DCM
dilated cardiomyopathy
- WES
whole‐exome sequencing
Clinical Perspective.
What Is New?
By genome‐wide screening with microsatellite markers and genetic linkage analysis in a 4‐generation family suffering from autosomal‐dominant dilated cardiomyopathy (DCM), a new locus for DCM was mapped on chromosome 15q13.1–q13.3.
A novel mutation in the KLF13 gene, c.430G>T (p.E144X), was identified by whole‐exome sequencing analysis to cosegregate with DCM in the family within the mapping chromosomal region; sequencing analyses of KLF13 in another cohort of 266 unrelated patients with DCM and their available family members revealed 2 novel mutations, c.580G>T (p.E194X) and c.595T>C (p.C199R), which cosegregated with DCM in 2 families, respectively.
The 3 mutants had no transactivation on the target genes ACTC1 and MYH7, alone or together with GATA4, and the 3 mutations showed a diminished ability to bind the promoters of ACTC1 and MYH7; the E144X‐mutant KLF13 showed a defect in intracellular distribution.
What Are the Clinical Implications?
The discovery of a new DCM‐causative gene has potential implications for antenatal prophylaxis and precise management of DCM.
The DCM‐associated genetic data may be used for prognostic risk stratification of patients with DCM.
Dilated cardiomyopathy (DCM), defined by left ventricular enlargement with systolic dysfunction at the exclusion of other usual clinically detectable causes responsible for impaired global contractile function, is the most prevalent form of myocardial disease in humans, with an estimated prevalence of up to 1 in 250 individuals worldwide. 1 , 2 , 3 Notably, there is a significant sex difference in the incidence of DCM, with an overall combined mean male:female sex ratio of 2.5:1. 4 DCM confers a significantly enhanced risk for heart failure, ventricular and supraventricular dysrhythmias, cardiac transplantation, and death. 5 , 6 , 7 , 8 , 9 Factually, DCM is the second most prevalent cause of heart failure next to coronary heart disease, accounting for 30% to 40% of all heart failures, and the most common indication for cardiac transplantation worldwide, with an average 5‐year survival rate of approximately 50% after diagnosis of DCM, though a significant improvement of heart function has been achieved by evidence‐based therapy for heart failure. 4 , 10 , 11 , 12 , 13 , 14 It was reported that in female patients the annual incidence of DCM‐related death was about 2%, whereas in male patients, the annual incidence of DCM‐causative death was nearly 4%, and in the United States alone, DCM contributed to ≈46 000 hospitalizations and ≈10 000 deaths annually. 3 , 15 Obviously, DCM causes substantial morbidity and mortality as well as medical care cost, imposing an enormous socioeconomic burden on humans. Despite the clinical significance, the molecular pathogenesis underlying DCM remains incompletely understood.
Epidemiological investigations demonstrate that DCM is frequently associated with miscellaneous structural heart diseases, systemic comorbidities, and other environmental predisposing factors, including valvular heart disease, coronary heart disease, tachycardia‐mediated cardiomyopathy, viral myocarditis, neuromuscular disease, essential hypertension, autoimmune disorder, endocrine homeostatic imbalance, inadequate epigenetic modifier, nutritional deficiency, hemochromatosis, systematic amyloidosis, alcohol abuse, and exposure to cardiotoxic chemicals. 3 , 4 , 11 , 16 However, there is accumulating evidence highlighting the pivotal role of genetic defects in the pathogenesis of DCM, especially for familial DCM, which is considered when ≥2 first‐degree relatives have been diagnosed with DCM or have experienced sudden cardiac death with ages ≤35 years. 1 , 2 , 3 , 4 Familial DCM, accounting for approximately 20% to 50% of all DCM, is predominantly inherited in an autosomal‐dominant mode, although other inheritance patterns, encompassing autosomal‐recessive, X‐linked recessive, and mitochondrial inheritance, are also reported. 17 To date, DCM‐causing variations in >250 genes have been implicated with the development of DCM, of which the vast majority code for sarcomere proteins, nuclear envelope proteins, cytoskeleton proteins, Z‐band proteins, intercalated disc proteins, ion channel proteins, gap junction channel proteins, RNA‐binding proteins, mitochondrial proteins, and transcriptional factor proteins. 1 , 2 , 3 , 4 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 , 49 , 50 , 51 , 52 , 53 , 54 , 55 Additionally, genome‐wide association studies have led to the discovery of many new common variants involved in DCM, in addition to novel rare variants involved in DCM. 33 Nevertheless, the genetic architecture of DCM is highly complex and diverse owing to pronounced genetical heterogeneity, and the genetic determinants underpinning DCM in a large proportion of cases remain to be identified. 56
METHODS
The supporting data are available within the article, its online supplemental files, and upon reasonable request. Data analysis came from our institutional laboratories, including the cardiovascular research laboratory and central laboratory of Shanghai Fifth People's Hospital, Fudan University, Shanghai, China.
Recruitment of Study Participants
For the current research, a 43‐member Chinese family spanning 4 generations with a high incidence of DCM was identified, and the available family members were recruited. Another cohort of 266 probands with DCM and their available family members were enlisted. A total of 418 unrelated healthy individuals, who were matched with the patients for ethnicity, sex, and age, were enrolled as controls. This cohort of DCM patients with DCM and their family members was specifically included for the purpose of this study, and the same for the healthy individuals. They were recruited from the inpatient and outpatient individuals of the Shanghai Fifth People's Hospital. All research participants underwent a comprehensive clinical assessment, encompassing a thorough review of personal, familial, and medical histories, detailed physical examination, standard 12‐lead electrocardiogram, transthoracic echocardiogram, and routine laboratory tests. All patients with DCM also experienced coronary artery angiography and exercise performance tests, but cardiovascular magnetic resonance imaging, coronary computed tomography angiography, and endomyocardial biopsy were performed only when indicated. DCM was diagnosed as described elsewhere: a left‐ventricular end‐diastolic diameter >117% of the predicted value corrected for body surface area and age and a left‐ventricular ejection fraction <45% and/or a fractional shortening <25%, in the absence of abnormal loading conditions, coronary artery disease, toxin exposure, and other systemic diseases. 2 , 57 The inclusion criteria for the patients with DCM were consistent with the diagnosis of DCM. 2 The exclusion criteria included peripartum DCM and secondary DCM caused by other cardiovascular diseases, systemic diseases, or known environmental risk factors, including rheumatic heart disease, ischemic heart disease, viral myocarditis, primary hypertension, metabolic disorder, and exposure to toxic chemicals. The current investigation involving human participants complies with the tenets of the Declaration of Helsinki. All study procedures used in this research were approved by the Medical Ethics Committee of Shanghai Fifth People's Hospital, Fudan University, Shanghai, China (approval number 2020–011). Before recruitment into the study, all study participants signed informed consent. Approximately 6 mL of whole blood sample was collected from each study subject.
Genome‐Wide Screening With Microsatellite Markers and Genetic Linkage Analysis
Genomic DNA was extracted from blood leukocytes using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI). Genome‐wide screening for genotyping in Family 1 with DCM was implemented with the ABI PRISM Linkage Mapping Set (Applied Biosystems, Foster City, CA), using 386 polymorphic microsatellite markers spaced at a mean resolution of ≈10 cM from chromosomes 1 to 22. Multiplex amplification of markers was conducted using the AmpliTaq Gold DNA Polymerase (Applied Biosystems) by polymerase chain reaction (PCR) under the T100 Thermal Cycler (Bio‐Rad, Hercules, CA). Genotyping was carried out on the ABI PRISM 3130XL Genetic Analyzer (Applied Biosystems) as per the manufacturer's protocol. Genetic linkage analysis was made with the software Statistical Analysis for Genetic Epidemiology (http://darwin.cwru.edu/sage/), under an autosomal‐dominant mode of inheritance with the allele frequency and penetrance rate of DCM being set at 0.1% and 95%, respectively. 58 A 2‐point logarithm of odds (LOD) score between each marker and the disease locus was calculated as described previously. 59 To finely map the chromosomal region determined by the marker D15S165 with an initial supportive LOD score, 5 additional markers (D15S1002, D15S1019, D15S3976, D15S1010, and D15S144) near the region were genotyped to delimit the critical chromosomal interval by refining the recombination boundaries. The distances between loci and marker order were derived from the Généthon genetic map. 58 A haplotype map of Family 1 with DCM was constructed with the software Cyrillic v2.1.3 (Cherwell Scientific, Oxford, UK) to reveal the shared genomic regions among affected members from the DCM family and confine the recombination boundaries.
Sequencing Analysis of the Candidate Genes Within the Mapped Chromosomal Region
Whole‐exome sequencing (WES) and bioinformatics analyses in 3 affected family members (II‐5, III‐11, and III‐16) and 2 unaffected family members (II‐6 and III‐14) from Family 1 were completed as described elsewhere. 59 , 60 , 61 , 62 Briefly, for each family member selected to undergo WES, 3 μg of genomic DNA were used for construction of a whole exome library, which was captured with the SureSelect Human All Exon V6 Kit (Agilent Technologies, Santa Clara, CA) following the manufacturer's manual. The captured exome libraries were sequenced on the Illumina HiSeq 2000 Genome Analyzer (Illumina, San Diego, CA), with the HiSeq Sequencing Kit (Illumina) according to the manufacturer's protocol. Raw image data were processed by the Pipeline software to call bases and generate the set of sequence reads. Sequence reads were mapped to the human referential genome (hg19, GRCh37) through the Burrows‐Wheeler Aligner software. Sequence variants were called via the GATK software. When a potential DCM‐causative variation was identified by WES analysis within the mapped chromosomal region, Sanger sequencing analysis in the whole family with DCM was fulfilled to confirm it. Once a gene possessing an identified DCM‐causing variation was discovered, Sanger sequencing analysis of the gene was carried out in another cohort of 266 index patients with DCM, the available family members of the index patients harboring an identified DCM‐causing variation, and 418 unrelated healthy subjects. For a verified deleterious mutation, such population genetics databases as the UK Biobank (https://www.ukbiobank.ac.uk/), the Single Nucleotide Polymorphism database (https://www.ncbi.nlm.nih.gov/snp/), and the Genome Aggregation Database (http://gnomad.broadinstitute.org/) were consulted to check its novelty.
Construction of Eukaryotic Gene Expression Plasmids
Isolation of total mRNA from discarded human heart tissue and production of cDNA by reverse‐transcription PCR were described previously. 59 The full‐length cDNA of the human KLF13 gene (GenBank accession no. NM_015995.4) was yielded by PCR amplification of the produced cDNA with the PfuUltra High‐Fidelity DNA Polymerase (Stratagene, Santa Clara, CA) and a specific pair of primers (forward primer: 5′‐GAGGAATTCGCGGATGCGCGGCTGACGAC‐3′; reverse primer: 5′‐CTCTCTAGAGCGGCTGCTCATGGCTGTGG‐3′). The yielded KLF13 cDNA was doubly digested with restriction endonucleases EcoRI (NEB, Ipswich, MA) and XbaI (NEB) and then ligated to the plasmid pcDNA3.1 (Invitrogen, Carlsbad, CA) at the EcoRI‐XbaI sites to generate the eukaryotic gene expression plasmid of wild‐type (WT) KLF13‐pcDNA3.1. Each mutant‐type KLF13‐pcDNA3.1 plasmid was generated by site‐targeted mutagenesis of WT KLF13‐pcDNA3.1 with the QuikChange Site‐Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA), as well as a complementary pair of primers, and was confirmed by Sanger sequencing analysis. Similarly, the entire open read frame of the human GATA4 gene (GenBank accession no. NM_001308093.3) was yielded by PCR amplification of the produced cDNA with the PfuUltra High‐Fidelity DNA Polymerase (Stratagene) and a specific pair of primers (forward primer: 5′‐ACTGAATTCGCGTGGCTCCTTGACCTGCG‐3′; reverse primer: 5′‐AGTTCTAGACTCCAAGTCCCAGGTCCGTG‐3′), cut with EcoRI (NEB) and XbaI (NEB), and then inserted to the plasmid pcDNA3.1 (Invitrogen) at the EcoRI‐XbaI sites to construct WT GATA4‐pcDNA3.1. A 680‐bp genomic DNA fragment (from −620 to +60, with the transcriptional start site numbered as +1) of the human ACTTC1 gene (GenBank accession no. NC_000015.10) was amplified from human genomic DNA by PCR with a specific pair of primers (forward primer: 5′‐TCCGCTAGCGAGCCCAGCACCAGCTCTCT‐3′; reverse primer: 5′‐GGACTCGAGCCGGGCGCTGACTCACCGTC‐3′), digested with NheI (NEB) and XhoI (NEB), and inserted into the pGL3‐Basic vector (Promega) to create the ACTC1 promoter‐driven firefly luciferase reporter (ACTC1‐luc). Similarly, a 628‐bp upstream of the translation start site (from −1 to −627) of the human MYH7 gene (GenBank accession no. NC_000014.9) was amplified with a specific pair of primers (forward primer: 5′‐GCAGCTAGCCCATCTCTAAGCTCCATGGT‐3′; reverse primer: 5′‐TGCCTCGAGGGAGCCAGACCTGGTCTCAG‐3′), cut with NheI (NEB) and XhoI (NEB), and subcloned into the pGL3‐Basic vector (Promega) to construct the MYH7 promoter‐driven firefly luciferase reporter (MYH7‐luc).
Cell Culture, Transfection, and Dual‐Luciferase Assays
Hela cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen) containing 10% fetal bovine serum (Invitrogen) in an atmosphere with 5% CO2 at 37 °C. Twenty‐four hours before transient transfection using the Lipofectamine™ 3000 Transfection Reagent (Invitrogen), cells were seeded into 24‐well plates at a density of 1×105 cells per well. Different amounts (ranging from 0.025 to 0.8 μg) of WT KLF13‐pcDNA3.1 were used to assess its dose‐dependent activation of ACTC1‐luc and MYH7‐luc (1.0 μg, respectively). For the analysis of the transactivation of ACTC1 promoter by KLF13, Hela cells were transfected with 0.4 μg of empty pcDNA3.1 or 0.4 μg of WT KLF13‐pcDNA3.1 or 0.4 μg of mutant KLF13‐pcDNA3.1 or 0.2 μg of WT KLF13‐pcDNA3.1 plus 0.2 μg of empty pcDNA3.1 or 0.2 μg of WT KLF13‐pcDNA3.1 plus 0.2 μg of mutant KLF13‐pcDNA3.1, together with 1.0 μg of ACTC1‐luc and 0.04 μg of pRL‐TK (Promega). For the analysis of the transactivation of MYH7 promoter by KLF13, the quantity of WT or mutant KLF13‐pcDNA3.1 or empty pcDNA3.1 were halved whereas the amount of MYH7‐luc or pRL‐TK (Promega) remained the same as described previously. For measurement of the synergistic transcriptional activation between KLF13 and GATA4, the same amount (0.1 μg) of empty pcDNA3.1, WT KLF13‐pcDNA3.1, mutant KLF13‐pcDNA3.1, or GATA4‐pcDNA3.1 was used alone or together, in the presence of 1.0 μg of ACTC1‐luc or MYH7‐luc and 0.04 μg of pRL‐TK (Promega). The pRL‐TK plasmid expressing Renilla luciferase (Promega) was used for the normalization of transfection efficiency. Cells were harvested 48 hours after transfection and the luciferase activity was assayed by using the Dual‐Luciferase Reporter Assay System (Promega). The promoter activity value was expressed as fold activation of firefly luciferase to Renilla luciferase. Experiments were conducted independently 3 times in triplicate for each sample.
Nuclear Protein Isolation and Electrophoretic Mobility Shift Analysis
Nuclear extracts were prepared from Hela cells transfected with 10 μg of WT KLF13‐pcDNA3.1 or mutant KLF13‐pcDNA3.1 by using the NE‐PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific, Rockford, IL) according to the manufacturer's instructions. Oligonucleotide probes used for electrophoretic mobility shift analysis were synthesized, purified, and 5′ end‐labeled with biotin. For the binding reactions, biotinylated probes (0.2 pmol) were incubated with nuclear extracts (5 μg) and the binding buffer (Beyotime Biotechnology, Shanghai, China) for 20 minutes at room temperature. Of note, unlabeled cold probes (20 pmol) were preincubated with the nuclear extracts for 10 minutes. The mixture was fractionated on a 6% nondenaturing polyacrylamide gel in 0.5× Tris‐borate‐EDTA buffer at 100 V for 1 hour, transferred to a nylon membrane (Beyotime Biotechnology) in ice‐cold 0.5× Tris‐borate‐EDTA buffer at 380 mA for 30 minutes, and crosslinked by ultraviolet irradiation for 20 minutes. The biotin‐labeled probe was detected by using the streptavidin‐horseradish peroxidase conjugate and the chemiluminescent substrate (Beyotime Biotechnology) according to the manufacturer's instructions.
Cellular Immunofluorescence Assay of the KLF13 Proteins
Hela cells (5 × 104) grown on glass coverslips in a 12‐well plate were transfected with 0.4 μg of WT or mutant KLF13‐pcDNA3.1. After 48 hours, cells were harvested and fixed with 4% paraformaldehyde for 15 minutes, heated in antigen retrieval buffer at 95 °C for 10 minutes, and permeabilized with 0.5% TritonX‐100 for 20 minutes. Subsequently, cells were blocked with 3% bovine serum albumin at room temperature for 30 minutes and incubated with rabbit anti‐KLF13 polyclonal antibody (Affinity, 1:100) overnight at 4 °C, followed by incubation with the conjugated secondary antibody, goat antirabbit Alexa‐Flour 594 (Affinity, 1:100) for 1 hour at room temperature in the dark. Nuclear staining was conducted by using 4′,6‐diamidino‐2‐phenylindole for 1 minute. Finally, coverslips were mounted with ProLong Glass Antifade Mountants (Thermo Fisher Scientific, Waltham, MA) and sealed with clear nail polish. The images were acquired by using confocal laser‐scanning fluorescence microscopy (Leica, Mannheim, Germany) under an oil objective (Leica).
Statistical Analysis
Continuous variables were presented as mean±SD and compared between the 2 groups using the Student's unpaired t test. Categorical variables were expressed as numbers and percentages and compared between the 2 groups using the Fisher's exact test. A 2‐tailed P value of <0.05 was considered statistically significant.
RESULTS
Demographic and Clinical Features of the Research Participants
As shown in Figure 1, a 43‐member 4‐generation family suffering from autosomal‐dominant DCM (Family 1) was identified from the Chinese Han population, including 40 living family members (19 male members and 21 female members, with ages ranging from 21 to 68 years). The proband of Family 1 (II‐5, Figure 1) was initially diagnosed with DCM at the age of 45 years and received pharmacologic therapy for heart failure. His brother (II‐1, Figure 1) and father (I‐1, Figure 1) had a past medical history of DCM and died of progressive congestive heart failure caused by DCM at ages 63 and 69 years, respectively. In Family 1 (Figure 1), all the affected members had echocardiogram‐documented DCM, whereas the unaffected family members had neither history of DCM nor symptoms of DCM, with normal echocardiographic findings. None had structural heart diseases or other diseases contributing to DCM, such as valvular heart disease, coronary heart disease, viral myocarditis, and essential hypertension. The demographic and baseline clinical features of the affected family members alive from Family 1 are summarized in Table 1. Additionally, another cohort of 266 probands inflicted with DCM was clinically investigated in contrast to a total of 418 unrelated healthy subjects without a history of DCM and found that the control individuals (217 male people and 201 female people, with a mean age of 48 years ranging between 37 and 65 years) were matched with the DCM probands (136 male patients and 130 female patients, with an average age of 48 years varying from 36 to 64 years) for ethnicity, age, and sex, as shown in Table S1.
Figure 1. Pedigree and haplotypes for Family 1 affected with dilated cardiomyopathy.

The 4‐generation family with a high incidence of autosomal‐dominant dilated cardiomyopathy was designated as Family 1, with members identified by generations‐numbers (Roman‐Arabic numerals) shown below member symbols. A vertical bar beneath a member denotes the chromosomal region defined by genetic linkage analysis. Polymorphic markers spanning the linkage region on chromosome 15q13.1–q13.3 were displayed to the left of the pedigree, with members' genotypes (represented by numbers) for markers given next to the chromosome bars. The filled bars mean disease haplotype. Key recombination events occurred with marker D15S1019 in member III‐16, and with marker D15S1010 in member III‐3. “+” indicates a carrier of the heterozygous mutation of c.430G>T in the KLF13 gene; an “–” indicates a noncarrier.
Table 1.
Demographic and Phenotypic Characteristics of the Living Pedigree Members Inflicted With Dilated Cardiomyopathy Harboring the Heterozygous KLF13‐E144X Mutation
| Individual (Family 1) | Sex | Age, y | Phenotype | LVEDD, mm | LVESD, mm | LVEF, % | LVFS, % |
|---|---|---|---|---|---|---|---|
| II‐4 | F | 68 | DCM | 69 | 60 | 31 | 15 |
| II‐5 | M | 66 | DCM | 63 | 54 | 33 | 17 |
| II‐9 | M | 61 | DCM, ASD | 77 | 66 | 29 | 14 |
| III‐2 | F | 47 | DCM | 68 | 55 | 38 | 19 |
| III‐3 | M | 45 | DCM | 74 | 63 | 30 | 16 |
| III‐9 | M | 44 | DCM | 56 | 48 | 30 | 14 |
| III‐11 | M | 44 | DCM | 60 | 50 | 39 | 19 |
| III‐16 | F | 40 | DCM | 58 | 46 | 47 | 24 |
| III‐19 | M | 36 | DCM, ASD | 56 | 44 | 44 | 22 |
| IV‐2 | F | 22 | DCM | 54 | 43 | 43 | 29 |
| IV‐6 | M | 21 | DCM | 53 | 41 | 46 | 24 |
ASD indicates atrial septal defect; DCM, dilated cardiomyopathy; F, female; LVEDD, left ventricular end‐diastolic diameter; LVEF, left ventricular ejection fraction; LVESD, left ventricular end‐systolic diameter; LVFS, left ventricular fractional shortening; and M, male.
Mapping of a Novel Genetic Locus for DCM on Chromosome 15q13.1–q13.3
A genome‐wide scan was fulfilled in the 40 living members from Family 1 with a high incidence of DCM (Figure 1) with 386 microsatellite markers with a 10 cM‐resolution. Genetic linkage analysis suggested a significant linkage of DCM to marker D15S165, with a maximum 2‐point LOD score of 5.117510 at recombination fraction (θ)=0.00 (Table 2). The 2‐point LOD scores for D15S165 remained >3 regardless of variations in the penetrance from 60% to 99% and variations in the phenocopy prevalence from 0% to 5%. The pairwise LOD scores indicated no significant linkage to markers in other chromosomal regions (Table 2). To confine the DCM‐linked chromosomal region, 5 additional markers (D15S1002, D15S1019, D15S3976, D15S1010, and D15S144) near the marker D15S165 were genotyped, and based on haplotype analysis (Figure 1), a novel genetic locus for DCM was ultimately mapped to chromosome 15q13.1–q13.3 (GRCh38, chr15:29371221‐32 798 467), a 4.77‐cM (≈3.43 Mbp) interval between markers D15S1019 and D15S1010. There are 99 genes defined within the locus between markers D15S1019 and D15S1010, including 27 protein‐encoding genes, 35 nonencoding RNA genes, and 37 pseudogenes (Table S2).
Table 2.
Two‐Point Logarithm of Odds Scores for the Markers on Chromosome 15q13.1‐q13.3 at Different Recombination Fractions (θ) in Family 1 With Dilated Cardiomyopathy
| Marker | LOD scores at θ | ||||||
|---|---|---|---|---|---|---|---|
| 0.00 | 0.01 | 0.05 | 0.10 | 0.20 | 0.30 | 0.40 | |
| D15S975 | (−∞) | 0.063799 | 1.283779 | 1.619547 | 1.580413 | 1.169251 | 0.545682 |
| D15S1002 | (−∞) | 1.339973 | 2.415505 | 2.594912 | 2.276644 | 1.590878 | 0.691651 |
| D15S1019 | 5.117510 | 5.034579 | 4.694259 | 4.248125 | 3.276644 | 2.180703 | 0.964652 |
| D15S165 | 5.117510 | 5.034579 | 4.694259 | 4.248125 | 3.276644 | 2.180703 | 0.964652 |
| D15S976 | 4.816480 | 4.737913 | 4.415505 | 3.992852 | 3.072524 | 2.034575 | 0.885471 |
| D15S1010 | 3.913390 | 3.847918 | 3.579244 | 3.227035 | 2.460170 | 1.596620 | 0.658374 |
| D15S144 | (−∞) | 2.742278 | 3.136751 | 3.038610 | 2.470470 | 1.667027 | 0.719827 |
| D15S1007 | (−∞) | 2.320679 | 2.733177 | 2.658946 | 2.144605 | 1.408812 | 0.569412 |
θ indicates recombination fraction; and LOD, logarithm of odds.
Identification of DCM‐Causative Mutations in KLF13
WES was carried out for 3 affected members (II‐5, III‐11, and III‐16) and 2 unaffected members (II‐6 and III‐14) from Family 1. Analysis of the candidate genes within the mapping locus from marker D15S1019 to D15S1010, only the mutation chr15: 31619845G>T (GRCh37.p13: NC_000015.9), equivalent to chr15:31327642G>T (GRCh38.p14: NC_000015.10) or NM_015995.4:c.430G>T;p.E144X in the KLF13 gene, coding for an important cardiac transcription factor, was identified and validated to be in cosegregation with DCM in Family 1 by Sanger sequencing analysis. The sequence chromatograms illustrating the heterozygous c.430G>T variation in KLF13 and its WT control sequences are given in Figure 2A. Notably, in the mapped chromosomal region, only the rare variant in the KLF13 gene was confirmed to cosegregate with DCM in the family, and no other rare variants were shared by all affected family members and were absent in the unaffected family members. Additionally, via sequencing analysis of the coding regions and splicing junctions of KLF13 in another cohort of 266 index patients inflicted with DCM, 2 new KLF13 mutations, NM_015995.4:c.580G>T;p.E194X and NM_015995.4:c.595T>C;p.C199R, were discovered in 2 index patients, respectively. The sequence electropherograms showing the heterozygous KLF13 mutations c.580G>T and c.595T>C and their WT control sequences are exhibited in Figure 2B and 2C, respectively. The pedigrees of the 2 index patients carrying the discovered KLF13 mutations from Family 2 and Family 3 are displayed in Figure 3. Genetic analysis of the available members from Family 2 or Family 3 showed that the discovered KLF13 mutation cosegregated with autosomal‐dominant DCM in the whole family, with complete penetrance. The demographic and baseline clinical characteristics of the affected family members alive from Family 2 and Family 3 are summarized in Table 3. Notably, these families had high incidences of miscarriages, of which women members III‐2 and III‐16 in Family 1, member III‐6 in Family 2, and member III‐4 in Family 3 had a history of miscarriage. The 3 heterozygous KLF13 mutations were neither observed in 418 unrelated controls nor released in multiple population genetics databases including the UK Biobank, the Single Nucleotide Polymorphism database, and the Genome Aggregation Database, indicating novel mutations.
Figure 2. New KLF13 mutations responsible for dilated cardiomyopathy.

Sequencing electropherograms showing 2 nonsense mutations and 1 missense mutation in the KLF13 gene as well as their corresponding wild‐type sequences were exhibited. Arrows direct to the heterozygous mutations or homozygous wild‐type sequences. A, Sequencing electropherograms of the affected proband and an unaffected member from Family 1, showing the heterozygous KLF13 mutation c.430G>T and its wild‐type control. B, Sequencing electropherograms of the affected proband and an unaffected member from Family 2, showing the heterozygous KLF13 mutation c.580G>T and its wild‐type control. C, Sequencing electropherograms of the affected proband and an unaffected member from Family 3, showing the heterozygous KLF13 mutation c.595T>C and its wild‐type control.
Figure 3. Pedigrees of Family 2 and Family 3 inflicted with dilated cardiomyopathy.

The 2 families with a high incidence of dilated cardiomyopathy were designated arbitrarily as Family 2 and Family 3, respectively, with members recognized by generations‐numbers. “+” a carrier of the heterozygous KLF13 mutation c.580G>T in Family 2 or c.595T>C in Family 3; “–”, a noncarrier.
Table 3.
Clinical Features and KLF13 Mutations of the Living Family Members Suffering From Dilated Cardiomyopathy
| Family | Individual | Sex | Age, y | Phenotype | LVEDD, mm | LVESD, mm | LVEF, % | LVFS, % | KLF13 mutations |
|---|---|---|---|---|---|---|---|---|---|
| Family 2 | II‐8 | F | 62 | DCM | 77 | 66 | 29 | 14 | c.580G>T |
| III‐3 | M | 66 | DCM | 57 | 47 | 37 | 18 | c.580G>T | |
| III‐6 | M | 61 | DCM, atrial septal defectt | 62 | 53 | 32 | 16 | c.580G>T | |
| III‐10 | F | 47 | DCM | 57 | 46 | 40 | 20 | c.580G>T | |
| IV‐2 | M | 45 | DCM | 56 | 44 | 44 | 22 | c.580G>T | |
| Family 3 | II‐3 | M | 65 | DCM | 63 | 55 | 26 | 13 | c.595T>C |
| III‐4 | F | 41 | DCM | 58 | 45 | 43 | 22 | c.595T>C |
DCM indicates dilated cardiomyopathy; F, female; LVEDD, left ventricular end‐diastolic diameter; LVEF, left ventricular ejection fraction; LVESD, left ventricular end‐systolic diameter; LVFS, left ventricular fractional shortening; and M, male.
Failed Transactivation Function of the Mutant KLF13 Proteins
As shown in Figure 4A and 4C, WT KLF13 transcriptionally activated the ACTC1 or MYH7 promoter in a dose‐dependent pattern. Specifically, 0.025, 0.05, 0.1, 0.2, 0.4, and 0.8 μg of WT KLF13‐pcDNA3.1 transcriptionally activated the ACTC1 promoter in Hela cells by ≈8 (7.5442±2.0915)‐fold, ≈13 (13.3960±0.3900)‐fold, ≈22 (21.8076±0.4779)‐fold, ≈27 (26.6103±0.4405)‐fold, ≈33 (33.4114±0.8934)‐fold, and ≈33 (32.7506±1.6561)‐fold, respectively (Figure 4A); while 0.025, 0.05, 0.1, 0.2, 0.4, and 0.8 μg of WT KLF13‐pcDNA3.1 transcriptionally activated the MYH7 promoter in Hela cells by ≈18 (18.1939±0.8119)‐fold, ≈21 (21.0893±0.4158)‐fold, ≈38 (38.3222±0.1754)‐fold, ≈44 (44.3251±1.4307)‐fold, ≈44 (43.7500±1.5372)‐fold, and ≈44 (44.3071±1.6442)‐fold, respectively (Figure 4C). As shown in Figure 4B, the same amount (0.4 μg) of WT E144X‐, E194X‐, and C199R‐mutant KLF13‐pcDNA3.1 transcriptionally activated the ACTC1 promoter in Hela cells by ≈28 (27.6201±1.1025)‐fold, ≈5 (5.2639±0.6986)‐fold, ≈7 (6.9302±0.5445)‐fold, and ≈9 (8.7595±0.9887)‐fold, respectively (WT versus E144X: t=29.6671, P=0.0001; WT versus E194X: t=29.1432, P=0.0001; WT versus C199R: t=22.0597, P=0.0003); when the same amount (0.2 μg) of WT KLF13‐pcDNA3.1 was cotransfected with empty pcDNA3.1 or E144X‐ or E194X‐ or C199R‐mutant KLF13‐pcDNA3.1, the induced transactivation of the ACTC1 promoter was ≈15 (14.7123±1.0725)‐fold, ≈12 (11.6692±0.8285)‐fold, ≈14 (14.0204±2.1855)‐fold, and ≈14 (14.3422±1.3787)‐fold, respectively (WT versus WT+E144X: t=20.0334, P=0.0004; WT versus WT+E194X: t=9.6231, P=0.0023; WT versus WT+C199R: t=13.0278, P=0.0014). Similarly, as shown in Figure 4D, homozygous WT KLF13 transcriptionally activated the MYH7 promoter by ≈49 (48.5386±2.2430)‐fold whereas homozygous E144X‐, E194X‐, and C199R‐mutant KLF13 activated the MYH7 promoter by ≈14 (13.7403±1.6790)‐fold, ≈13 (13.0757±1.1269)‐fold, and ≈15 (15.2651±2.0540)‐fold, respectively (WT versus E144X: t=21.5118, P=0.0003; WT versus E194X: t=24.4700, P=0.0001; WT versus C199R: t=18.9490, P=0.0005); whereas heterozygous E144X‐, E194X‐, and C199R‐mutant KLF13 activated the MYH7 promoter by ≈20 (20.2243±0.8723)‐fold, ≈22 (21.9228±1.0852)‐fold, and ≈22 (22.0585±1.2527)‐fold, respectively (WT versus WT+E144X: t=20.3774, P=0.0002; WT versus WT+E194X: t=18.5013, P=0.0003; WT versus WT+C199R: t=17.8525, P=0.0004). These results indicate that E144X‐, E194X‐, and C199R‐mutant KLF13 proteins have decreased transcriptional activation in either homozygous or heterozygous status compared with their WT counterparts.
Figure 4. Decreased transcriptional activity of the mutant KLF13 proteins.
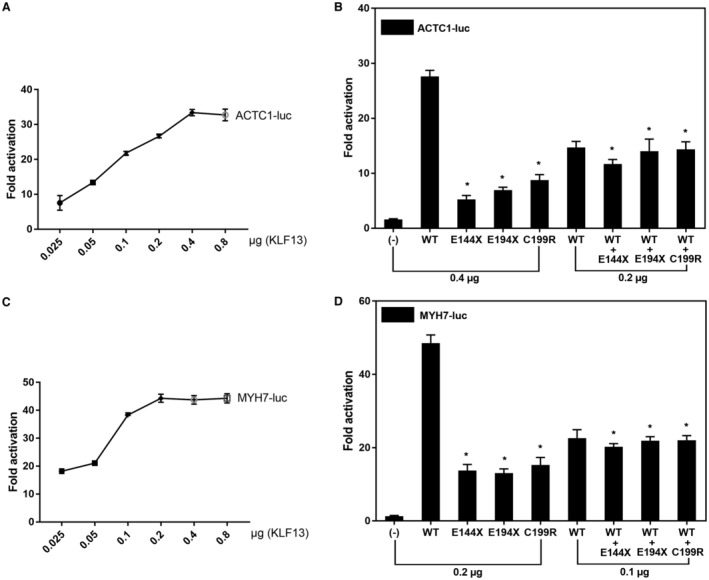
A, In cultivated HeLa cells, WT KLF13 showed dose‐dependent transactivation of the ACTC1 promoter. B, Activation of the ACTC1 promoter‐driven firefly luciferase in Hela cells by WT, E144X, E194X, or C199R mutant, alone or together, showed significantly decreased transactivation by the mutant KLF13 proteins. C, WT KLF13 displayed dose‐dependent transcriptional activation of the MYH7 promoter in cultured Hela cells. D, Activation of the MYH7 promoter‐driven luciferase in Hela cells by WT or E144X, E194X, C199R, separately or in combination, showed significantly reduced transactivation by the mutants. All the experiments were performed in triplicate, with the results shown as mean±SD. Here, each * indicates P<0.005, when compared with the same amounts of WT. WT indicates wild type.
Disrupted Synergistic Transactivation Between Mutant KLF13 and GATA4
As shown in Figure 5A, the same amount (0.1 μg) of WT, E144X‐, E194X‐, and C199R‐mutant KLF13‐pcDNA3.1 transactivated the ACTC1 promoter by ≈22 (21.8553±2.1319)‐fold, ≈3 (3.0649±0.2210)‐fold, ≈3 (3.0519±0.0946)‐fold, and ≈3 (2.9397±0.3180)‐fold, respectively (WT versus E144X: t=12.4275, P=0.0006; WT versus E194X: t=12.4710, P=0.0006; WT versus C199R: t=12.4631, P=0.0006); when cotransfected with 0.1 μg of GATA4‐pcDNA3.1, the transcriptional activation effect of each group became ≈43 (43.0359±2.7444)‐fold, ≈6 (6.3061±0.7807)‐fold, ≈7 (6.5421±0.8767)‐fold and ≈6 (5.7587±0.3689)‐fold, respectively (WT+GATA4 versus E144X+GATA4: t=22.2965, P=0.0001; WT+GATA4 versus E194X+GATA4: t=21.9398, P=0.0001; WT+GATA4 versus 199R+GATA4: t=23.3169, P=0.0001). Similarly, as shown in Figure 5B, 0.1 μg of the WT, E144X‐, E194X‐, and C199R‐mutant KLF13‐pcDNA3.1 transcriptionally activated the MYH7 promoter by ≈36 (36.3427±1.3384)‐fold, ≈10 (9.6471±0.3500)‐fold, ≈11 (10.7531±0.7837)‐fold, and ≈11 (10.3900±0.8694)‐fold, respectively (WT versus E144X: t=33.4224, P<0.0001; WT versus E194X: t=28.5768, P=0.0001; WT versus C199R: t=28.1653, P=0.0001); whereas in the presence of GATA4‐pcDNA3.1, the transcriptional activation effect became ≈78 (78.2231±3.7218)‐fold, ≈15 (15.0268±0.6177)‐fold, ≈16 (15.9773±0.8759)‐fold, and ≈16 (16.1415±0.5888)‐fold, respectively (WT+GATA4 versus E144X+GATA4: t=29.0132, P=0.0001; WT+GATA4 versus E194X+GATA4: t=28.1974, P=0.0001; WT+GATA4 versus 199R+GATA4: t=28.5364, P=0.0001). These results suggest that the E144X, E194X, and C199R mutations abolished the synergistic transactivation between KLF13 and GATA4 when compared with their WT counterparts.
Figure 5. Disrupted synergistic transactivation between mutant KLF13 and GATA4.

A, In cultured Hela cells, the transactivation of the ACTC1 promoter by GATA4 in synergy with E144X, E194X or C199R mutant was abolished by each mutation when compared with that by GATA4 and WT KLF13. B, In the presence of GATA4, transactivation of the MYH7 promoter‐driven luciferase in Hela cells by WT, E144X, E194X, or C199R mutant revealed that the mutations ablated the synergistic transcriptional activation between each mutant and GATA4. All the experiments were performed in triplicate, with the results expressed as mean±SD. Here, each * indicates P<0.001, in comparison with its WT counterpart. WT indicates wild type.
Diminished DNA‐Binding Ability of the Mutant KLF13 Proteins
As is shown in Figure 6, nuclear extracts form Hela cells transfected with WT KLF13‐pcDNA3.1 had the ability to specifically bind to biotinylated ACTC1‐ or MYH7‐DNA probes because the binding could be competed by the corresponding excess cold probes. The ability of E144X‐, E194X‐, or C199R‐mutant KLF13 protein to bind to the ACTC1‐DNA probes diminished to an undetectable level (Figure 6A). Similarly, E144X‐, E194X‐, or C199R‐mutant KLF13 protein showed a diminished binding ability to the MYH7‐DNA probes (Figure 6B).
Figure 6. Diminished DNA‐binding ability of the mutant KLF13 proteins.

A, The ability of WT KLF13, E144X, E194X or C199R mutant to bind to the ACTC1‐DNA probe. B, The ability of wild‐type KLF13 (WT), E144X, E194X, or C199R mutant to bind to the MYH7‐DNA probe. Electrophoretic mobility shift assay indicated that WT bound specifically to the ACTC1‐DNA probe (A) or the MYH7‐DNA probe (B), whereas all the mutants showed diminished DNA‐binding affinity to the ACTC1‐DNA (A) or MYH7‐DNA probe (B). WT indicates wild type.
Distinct Nuclear Distribution of the Mutant KLF13 Proteins
As shown in Figure 7, in transfected Hela cells, WT, E194X‐, or C199R‐mutant KLF13 protein was normally localized to the cellular nuclei. However, the E144X‐mutant KLF13 protein was distributed not only in the nuclei but also in the cytoplasm.
Figure 7. Distinct subcellular distribution of the mutant KLF13 proteins.
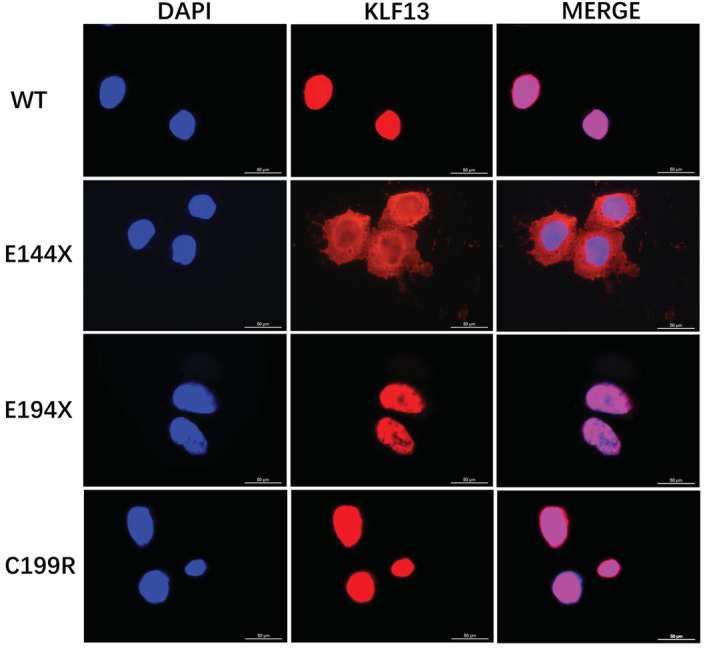
Subcellular localization of WT KLF13, E144X, E194X, and C199R mutant in Hela cells were detected by immunofluorescence. The red color shows the Alexa‐Fluor 594 conjugated secondary antibodies against the anti‐KLF13 antibody and the nuclei were stained with 4′,6‐diamidino‐2‐phenylindole (DAPI; blue). WT, E194X, and C199R mutant were localized exclusively to the nuclei whereas E144X mutant was distributed not only in the nuclei but also in the cytoplasm. Bar=50 μm. WT indicates wild type.
DISCUSSION
In the current investigation, by sequencing analysis of the candidate genes in the mapped region, human chromosome 15q13.1–q13.3, 3 mutations in the KLF13 gene, c.430G>T (p.E144X), c.580G>T (p.E194X), and c.595T>C (p.C199R), were identified to cosegregate with the DCM phenotype in 3 families, respectively, with complete penetrance. Functional assays demonstrated that the 3 mutants had no transactivation on the target genes ACTC1 and MYH7, 2 genes causally linked to DCM. 1 , 2 , 3 , 63 , 64 , 65 Furthermore, the 3 mutations abrogated the synergistic transactivation between KLF13 and GATA4, another gene contributing to DCM. 66 , 67 , 68 Besides, the 3 mutants failed to bind the promoters of ACTC1 and MYH7, and the E144X‐mutant KLF13 showed a defect in cellular nuclear distribution. Hence, it is very likely that genetically defective KLF13 contributes to DCM in these mutation carriers.
In human beings, KLF13 maps on chromosome 15q13.3, coding for a Krüppel‐like transcription factor (KLF) with 288 amino acids, which belongs to a family of transcription factors containing 3 classical zinc finger DNA‐binding domains and binds to GC‐rich DNA sequences as well as related GT and CACCC boxes in the promoters of target genes. 69 In addition to the DNA‐binding domains, which function to bind the promoters of target genes and interact with other transcriptionally cooperative partners, the KLF13 protein possesses 3 other key structural domains, encompassing a transcriptional activation domain, which serves to transactivate the expression of downstream genes, a transcriptional inhibition domain, which works to transcriptionally inhibit the expression of target genes, and a nuclear localization signal responsible for cellular nuclear localization. 70 Previous studies have demonstrated that KLF13 is highly expressed in the hearts of both humans and vertebrates during the whole life from embryos to adults and plays a pivotal role in cardiovascular morphogenesis and postnatal structural remodeling. 71 , 72 , 73 , 74 , 75 , 76 , 77 Recently, multiple experimental investigations together with our present research have substantiated that KLF13 transactivates the expression of multiple downstream genes, including MYH7, ACTC1, CCND1, NPPB, NPPA, and VEGFA, alone or synergistically with GATA4, GATA6, and TBX5. 72 , 73 , 74 Moreover, pathogenetic mutations in its target genes MYH7 and ACTC1 as well as its transcriptionally cooperative partners GATA4, GATA6, and TBX5 have been reported to cause DCM in humans. 63 , 64 , 65 , 66 , 67 , 68 , 78 , 79 , 80 , 81 These findings strongly indicate that KLF13 haploinsufficiency is an alternative molecular mechanism of DCM in a subset of patients.
Interestingly, 2 members from Family 1 and 1 member from Family 2 also had an atrial septal defect. Notably, previous investigations have implicated KLF13 loss‐of‐function mutations with distinct types of congenital heart disease, including transposition of the great arteries, double outlet of the right ventricle, tricuspid valve atresia, ventricular septal defect, patent ductus arteriosus, bicuspid aortic valve, and atrial septal defect. 71 , 82 Li and coworkers 71 sequenced KLF13 in a cohort of 309 patients with congenital heart disease and identified 2 heterozygous variants, c.487C>T (P.163 S) and c.467G>A (p.S156N), in 2 patients with tricuspid valve atresia and transposition of the great arteries, respectively. Functional analyses demonstrated that the variant c.467G>A (p.S156N) had significantly increased functionality, including increased protein expression, enhanced transactivation on the BNP promoter, and increased physical and functional interactions with TBX5; whereas the other variant, c.487C>T (p.P163S), inhibited the transcriptional activation of the target gene and the physical and functional interactions with TBX5, with no abnormality in protein expression. 71 Wang and colleagues 82 made a whole‐exosome sequencing analysis in a Chinese family with double‐outlet right ventricle and ventricular septal defect and found a novel heterozygous variation in the KLF13 gene, c.370G>T (p.E124*), which cosegregated with the disease. Biological analysis revealed that the E124*‐mutant KLF13 protein failed to transactivate its cardiac target genes ACTC1 and ANP and disrupted the synergistic transactivation between KLF13 and GATA4, as well as GATA6. 82 Additionally, pathogenetic mutations in its target genes MYH7 and ACTC1 as well as its transcriptionally cooperative partners GATA4, GATA6, and TBX5 have also been reported to cause multiple forms of congenital heart disease in humans. 83 , 84 , 85 Therefore, this study supports the notion that genetically compromised KLF13 predisposes to congenital heart disease. Of note, in clinical practice, we do see some patients with previous atrial septal defect surgery present years later with DCM. Two rare diseases in the same individual seem unlikely so this does make sense and would recommend that this is the subject of future research. Analyzing the databases of adult congenital heart disease would be very interesting.
It has been revealed in experimental animals that genetically compromised KLF13 contributes to heart disease. In Xenopus, ample expression of KLF13 in the heart and vessels was observed during embryogenesis, and knockdown of Klf13 in the embryos gave rise to myocardial trabecular hypoplasia and atrial septal defect, similar to those observed in mice or humans carrying hypomorphic alleles of GATA4. 73 Moreover, defects in growth, proliferation and differentiation of cardiac progenitor cells were observed in Xenopus embryos with downregulated KLF13 levels. 74 Additionally, in Xenopus embryos, KLF13‐depletion led to an almost complete absence of GATA4, GATA5 and TBX5 gene expression, a severely reduced expression level of GATA6, NKX2.5 and CCND1. 73 Expression of ANP as well as αMLC was severely reduced at an early stage in KLF13‐depleted embryos but was upregulated at a late stage, reflecting cardiac dysfunction. 73 The phenotypes of the Xenopus embryos with Klf13 knocked down, small hypoplastic hearts, pointed to an important role of KLF13 in cardiac cell survival, proliferation and differentiation. Consistent with the findings that KLF13 and GATA4 are mutual cofactors, the injection of GATA4 mRNA rescued the cardiac defects of KLF13‐depleted embryos in a dose‐dependent manner. 73 In mice, KLF13 is abundantly expressed at all stages of embryogenesis in the heart, including ventricular trabeculae, atrioventricular cushion, truncus arteriosus, and atrial myocardium. 73 , 86 In mice with homozygous deletion of Klf13, heart enlargement, cardiac vacuolar lesion, and embryonic lethality occurred. 87 Although mice with heterozygous knockout of Klf13 showed no obvious cardiac structural anomaly, compound heterozygous ablation of both Tbx5 and Klf13 remarkably decreased the postnatal viability and increased the penetrance of cardiac septal abnormality resulted from Tbx5 haploinsufficiency. 73 In rat cardiomyocytes, down‐regulating expression of KLF13 suppressed cell viability and promoted cell apoptosis; whereas overexpression of KLF13 increased cell viability and decreased cell apoptosis. 77 In addition, as a major mediator of glucocorticoid receptor signaling key to the regulation of myocardial function, KLF13 protected adult cardiomyocytes from DNA damage and demise. 75 Besides, as a direct target gene of KLF13, CCND1 could suppress DCM caused by TTN insufficiency. 88 Taken collectively, these results suggest that KLF13 dysfunction contributes to congenital heart disease and DCM.
During the past few years, the field is trying to make a better distinction between monogenic causes of DCM and polygenic contributors. The ClinGen consortium curated a long list of DCM‐associated genes and concluded that only a handful of genes were truly monogenic associated with DCM. Other studies came to the same conclusion. 2 , 37 In the current study, the discovery of KLF13 as a new DCM‐associated gene was based on a genome‐wide scan and functional ascertainment and adequately accounted for background population variation. Therefore, KLF13 is likely to be the monogenic cause of DCM, though we could not rule out the possibility that other genes might also make a minor contribution. Presently, the diagnostic gene panels that are used to test patients with DCM are being reduced in the number of genes. Only the true robust DCM‐associated genes are being tested, also to reduce the number of reported variants of uncertain significance. 2 , 37 Hence, it is better to include this gene in diagnostic gene panels for DCM after more immediate evidence is available. The prevalence of KLF13 variants in other cohorts of patients with DCM will be of extreme importance in the validation of KLF13 as a DCM‐associated gene. In addition, in a knock‐in animal model expressing a mutant KLF13, DCM spontaneously occurs.
Notably, 2 patients with peripartum DCM were excluded from the current investigation. Specifically, the 2 patients with peripartum DCM were prospectively excluded from the extended cohort of 268 probands with DCM, and neither was in family 1 or family 2, hence not included in the analysis. However, peripartum cardiomyopathy has been reported as a part of familial DCM, 89 and aggregating evidence has demonstrated the strong genetic basis of familial peripartum cardiomyopathy. 90 , 91 Factually, in roughly 20% of patients with peripartum DCM, screening for cardiomyopathy‐causative genes reveals pathogenic mutations, with TTN truncations most commonly implicated, occurring in approximately 10% of patients with peripartum DCM. 91 , 92 Hence, it is the right time to offer genetic testing to patients with peripartum DCM, 93 and referral for genetic testing should be considered if there is a positive family history of cardiomyopathy or sudden cardiac death. 90
In the current research, we used the traditional positional candidate gene analysis strategy to identify a new disease‐causing gene, as we did previously. 59 , 94 , 95 , 96 Recently, with the rapid advances in massively parallel sequencing approaches, the old approach of Sanger sequencing of a single candidate gene is being replaced by new sequencing technologies, including whole‐exome and whole‐genome sequencing technologies, which are applied more and more in genetic diagnostic routines because of a high ratio of efficiency to cost. 97 Next‐generation sequencing provides the opportunity to fulfill parallel analysis of a large number of genes in an unbiased approach and has been demonstrated to be successful in identifying novel pathogenic mutations responsible for Mendelian diseases. 97 However, though it is slow and costly, traditional Sanger's method has higher fidelity as compared to next‐generation sequencing methods and there are limitations associated with next‐generation sequencing methods themselves, including limited bioinformatical tools, existence of functionally‐undefined genes and failure to analyze noncoding regions for WES. 97 , 98
CONCLUSIONS
In conclusion, this research maps a novel genetic locus of DCM on human chromosome 15q13.1‐q13.3 and identifies KLF13 as a new gene underpinning DCM, which adds new insight into the molecular pathogenesis underlying DCM, implying potential implications for precision medicine of DCM in a subset of patients.
Sources of Funding
This work was supported by the National Natural Science Foundation of China (82070379), the Basic Research Project of Shanghai, China (20JC1418800), and the Natural Science Foundation of Shanghai, China (21ZR1455500).
Disclosures
None.
Supporting information
Table S1–S2
Acknowledgments
The authors cordially thank the study participants including the patients and healthy subjects.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.122.027578
For Sources of Funding and Disclosures, see page 14.
Contributor Information
Ying‐Jia Xu, Email: xuyingjia@5thhospital.com.
Yi‐Qing Yang, Email: yangyiqing@fudan.edu.cn.
References
- 1. Jordan E, Peterson L, Ai T, Asatryan B, Bronicki L, Brown E, Celeghin R, Edwards M, Fan J, Ingles J, et al. Evidence‐based assessment of genes in dilated cardiomyopathy. Circulation. 2021;144:7–19. doi: 10.1161/CIRCULATIONAHA.120.053033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mazzarotto F, Tayal U, Buchan RJ, Midwinter W, Wilk A, Whiffin N, Govind R, Mazaika E, de Marvao A, Dawes TJW, et al. Reevaluating the genetic contribution of monogenic dilated cardiomyopathy. Circulation. 2020;141:387–398. doi: 10.1161/CIRCULATIONAHA.119.037661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen SN, Mestroni L, Taylor MRG. Genetics of dilated cardiomyopathy. Curr Opin Cardiol. 2021;36:288–294. doi: 10.1097/HCO.0000000000000845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jain A, Norton N, Bruno KA, Cooper LT Jr, Atwal PS, Fairweather D. Sex differences, genetic and environmental influences on dilated cardiomyopathy. J Clin Med. 2021;10:2289. doi: 10.3390/jcm10112289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Giri P, Mukhopadhyay A, Gupta M, Mohapatra B. Dilated cardiomyopathy: a new insight into the rare but common cause of heart failure. Heart Fail Rev. 2022;27:431–454. doi: 10.1007/s10741-021-10125-6 [DOI] [PubMed] [Google Scholar]
- 6. Nuzzi V, Cannatà A, Manca P, Castrichini M, Barbati G, Aleksova A, Fabris E, Zecchin M, Merlo M, Boriani G, et al. Atrial fibrillation in dilated cardiomyopathy: outcome prediction from an observational registry. Int J Cardiol. 2021;323:140–147. doi: 10.1016/j.ijcard.2020.08.062 [DOI] [PubMed] [Google Scholar]
- 7. Piers SR, Androulakis AF, Yim KS, van Rein N, Venlet J, Kapel GF, Siebelink HM, Lamb HJ, Cannegieter SC, Man SC, et al. Nonsustained ventricular tachycardia is independently associated with sustained ventricular arrhythmias in nonischemic dilated cardiomyopathy. Circ Arrhythm Electrophysiol. 2022;15:e009979. doi: 10.1161/CIRCEP.121.009979 [DOI] [PubMed] [Google Scholar]
- 8. Rossano JW, Kantor PF, Shaddy RE, Shi L, Wilkinson JD, Jefferies JL, Czachor JD, Razoky H, Wirtz HS, Depre C, et al. Elevated heart rate and survival in children with dilated cardiomyopathy: a multicenter study from the pediatric cardiomyopathy registry. J Am Heart Assoc. 2020;9:e015916. doi: 10.1161/JAHA.119.015916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stolfo D, Albani S, Savarese G, Barbati G, Ramani F, Gigli M, Biondi F, Dal Ferro M, Zecchin M, Merlo M, et al. Risk of sudden cardiac death in New York Heart Association class I patients with dilated cardiomyopathy: a competing risk analysis. Int J Cardiol. 2020;307:75–81. doi: 10.1016/j.ijcard.2020.02.025 [DOI] [PubMed] [Google Scholar]
- 10. Tabish AM, Azzimato V, Alexiadis A, Buyandelger B, Knöll R. Genetic epidemiology of titin‐truncating variants in the etiology of dilated cardiomyopathy. Biophys Rev. 2017;9:207–223. doi: 10.1007/s12551-017-0265-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Giudicessi JR, Shrivastava S, Ackerman MJ, Pereira NL. Clinical impact of secondary risk factors in TTN‐mediated dilated cardiomyopathy. Circ Genom Precis Med. 2021;14:e003240. doi: 10.1161/CIRCGEN.120.003240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Enzan N, Matsushima S, Ide T, Kaku H, Tohyama T, Funakoshi K, Higo T, Tsutsui H. Beta‐blocker use is associated with prevention of left ventricular remodeling in recovered dilated cardiomyopathy. J Am Heart Assoc. 2021;10:e019240. doi: 10.1161/JAHA.120.019240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Domae K, Miyagawa S, Yoshikawa Y, Fukushima S, Hata H, Saito S, Kainuma S, Kashiyama N, Iseoka H, Ito E, et al. Clinical outcomes of autologous stem cell‐patch implantation for patients with heart failure with nonischemic dilated cardiomyopathy. J Am Heart Assoc. 2021;10:e008649. doi: 10.1161/JAHA.117.008649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Raafs AG, Boscutti A, Henkens MTHM, van den Broek WWA, Verdonschot JAJ, Weerts J, Stolfo D, Nuzzi V, Manca P, Hazebroek MR, et al. Global longitudinal strain is incremental to left ventricular ejection fraction for the prediction of outcome in optimally treated dilated cardiomyopathy patients. J Am Heart Assoc. 2022;11:e024505. doi: 10.1161/JAHA.121.024505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marrow BA, Cook SA, Prasad SK, McCann GP. Emerging techniques for risk stratification in nonischemic dilated cardiomyopathy: JACC review topic of the week. J Am Coll Cardiol. 2020;75:1196–1207. doi: 10.1016/j.jacc.2019.12.058 [DOI] [PubMed] [Google Scholar]
- 16. Wada K, Misaka T, Yokokawa T, Kimishima Y, Kaneshiro T, Oikawa M, Yoshihisa A, Takeishi Y. Blood‐based epigenetic markers of FKBP5 gene methylation in patients with dilated cardiomyopathy. J Am Heart Assoc. 2021;10:e021101. doi: 10.1161/JAHA.121.021101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sveinbjornsson G, Olafsdottir EF, Thorolfsdottir RB, Davidsson OB, Helgadottir A, Jonasdottir A, Jonasdottir A, Bjornsson E, Jensson BO, Arnadottir GA, et al. Variants in NKX2‐5 and FLNC cause dilated cardiomyopathy and sudden cardiac death. Circ Genom Precis Med. 2018;11:e002151. doi: 10.1161/CIRCGEN.117.002151 [DOI] [PubMed] [Google Scholar]
- 18. Akhtar MM, Lorenzini M, Cicerchia M, Ochoa JP, Hey TM, Sabater Molina M, Restrepo‐Cordoba MA, Dal Ferro M, Stolfo D, Johnson R, et al. Clinical phenotypes and prognosis of dilated cardiomyopathy caused by truncating variants in the TTN gene. Circ Heart Fail. 2020;13:e006832. doi: 10.1161/CIRCHEARTFAILURE.119.006832 [DOI] [PubMed] [Google Scholar]
- 19. Hey TM, Rasmussen TB, Madsen T, Aagaard MM, Harbo M, Mølgaard H, Nielsen SK, Haas J, Meder B, Møller JE, et al. Clinical and genetic investigations of 109 index patients with dilated cardiomyopathy and 445 of their relatives. Circ Heart Fail. 2020;13:e006701. doi: 10.1161/CIRCHEARTFAILURE.119.006701 [DOI] [PubMed] [Google Scholar]
- 20. Verdonschot JAJ, Hazebroek MR, Krapels IPC, Henkens MTHM, Raafs A, Wang P, Merken JJ, Claes GRF, Vanhoutte EK, van den Wijngaard A, et al. Implications of genetic testing in dilated cardiomyopathy. Circ Genom Precis Med. 2020;13:476–487. doi: 10.1161/CIRCGEN.120.003031 [DOI] [PubMed] [Google Scholar]
- 21. Kato K, Ohno S, Sonoda K, Fukuyama M, Makiyama T, Ozawa T, Horie M. LMNA missense mutation causes nonsense‐mediated mRNA decay and severe dilated cardiomyopathy. Circ Genom Precis Med. 2020;13:435–443. doi: 10.1161/CIRCGEN.119.002853 [DOI] [PubMed] [Google Scholar]
- 22. Nielsen SK, Hansen F, Schrøder HD, Wibrand F, Gustafsson F, Mogensen J. Recessive inheritance of a rare variant in the nuclear mitochondrial gene for AARS2 in late‐onset dilated cardiomyopathy. Circ Genom Precis Med. 2020;13:560–562. doi: 10.1161/CIRCGEN.120.003086 [DOI] [PubMed] [Google Scholar]
- 23. Levitas A, Muhammad E, Zhang Y, Perea Gil I, Serrano R, Diaz N, Arafat M, Gavidia AA, Kapiloff MS, Mercola M, et al. A novel recessive mutation in SPEG causes early onset dilated cardiomyopathy. PLoS Genet. 2020;16:e1009000. doi: 10.1371/journal.pgen.1009000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Quiat D, Witkowski L, Zouk H, Daly KP, Roberts AE. Retrospective analysis of clinical genetic testing in pediatric primary dilated cardiomyopathy: testing outcomes and the effects of variant reclassification. J Am Heart Assoc. 2020;9:e016195. doi: 10.1161/JAHA.120.016195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Qiao Q, Zhao CM, Yang CX, Gu JN, Guo YH, Zhang M, Li RG, Qiu XB, Xu YJ, Yang YQ. Detection and functional characterization of a novel MEF2A variation responsible for familial dilated cardiomyopathy. Clin Chem Lab Med. 2020;59:955–963. doi: 10.1515/cclm-2020-1318 [DOI] [PubMed] [Google Scholar]
- 26. Almomani R, Herkert JC, Posafalvi A, Post JG, Boven LG, van der Zwaag PA, Willems PHGM, van Veen‐Hof IH, Verhagen JMA, Wessels MW, et al. Homozygous damaging SOD2 variant causes lethal neonatal dilated cardiomyopathy. J Med Genet. 2020;57:23–30. doi: 10.1136/jmedgenet-2019-106330 [DOI] [PubMed] [Google Scholar]
- 27. Robinson HK, Zaklyazminskaya E, Povolotskaya I, Surikova Y, Mallin L, Armstrong C, Mabin D, Benke PJ, Chrisant MR, McDonald M, et al. Biallelic variants in PPP1R13L cause paediatric dilated cardiomyopathy. Clin Genet. 2020;98:331–340. doi: 10.1111/cge.13812 [DOI] [PubMed] [Google Scholar]
- 28. Ganapathi M, Argyriou L, Martínez‐Azorín F, Morlot S, Yigit G, Lee TM, Auber B, von Gise A, Petrey DS, Thiele H, et al. Bi‐allelic missense disease‐causing variants in RPL3L associate neonatal dilated cardiomyopathy with muscle‐specific ribosome biogenesis. Hum Genet. 2020;139:1443–1454. doi: 10.1007/s00439-020-02188-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Al‐Yacoub N, Colak D, Mahmoud SA, Hammonds M, Muhammed K, Al‐Harazi O, Assiri AM, Al‐Buraiki J, Al‐Habeeb W, Poizat C. Mutation in FBXO32 causes dilated cardiomyopathy through up‐regulation of ER‐stress mediated apoptosis. Commun Biol. 2021;4:884. doi: 10.1038/s42003-021-02391-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Landim‐Vieira M, Johnston JR, Ji W, Mis EK, Tijerino J, Spencer‐Manzon M, Jeffries L, Hall EK, Panisello‐Manterola D, Khokha MK, et al. Familial dilated cardiomyopathy associated with a novel combination of compound heterozygous TNNC1 variants. Front Physiol. 2020;10:1612. doi: 10.3389/fphys.2019.01612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Di RM, Yang CX, Zhao CM, Yuan F, Qiao Q, Gu JN, Li XM, Xu YJ, Yang YQ. Identification and functional characterization of KLF5 as a novel disease gene responsible for familial dilated cardiomyopathy. Eur J Med Genet. 2020;63:103827. doi: 10.1016/j.ejmg.2019.103827 [DOI] [PubMed] [Google Scholar]
- 32. Miura A, Kondo H, Yamamoto T, Okumura Y, Nishio H. Sudden unexpected death of infantile dilated cardiomyopathy with JPH2 and PKD1 gene variants. Int Heart J. 2020;61:1079–1083. doi: 10.1536/ihj.20-155 [DOI] [PubMed] [Google Scholar]
- 33. Garnier S, Harakalova M, Weiss S, Mokry M, Regitz‐Zagrosek V, Hengstenberg C, Cappola TP, Isnard R, Arbustini E, Cook SA, et al. Genome‐wide association analysis in dilated cardiomyopathy reveals two new players in systolic heart failure on chromosomes 3p25.1 and 22q11.23. Eur Heart J. 2021;42:2000–2011. doi: 10.1093/eurheartj/ehab030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McAfee Q, Chen CY, Yang Y, Caporizzo MA, Morley M, Babu A, Jeong S, Brandimarto J, Bedi KC Jr, Flam E, et al. Truncated titin proteins in dilated cardiomyopathy. Sci Transl Med. 2021;13:eabd7287. doi: 10.1126/scitranslmed.abd7287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Restrepo‐Cordoba MA, Wahbi K, Florian AR, Jiménez‐Jáimez J, Politano L, Arad M, Climent‐Paya V, Garcia‐Alvarez A, Hansen RB, Larrañaga‐Moreira JM, et al. Prevalence and clinical outcomes of dystrophin‐associated dilated cardiomyopathy without severe skeletal myopathy. Eur J Heart Fail. 2021;23:1276–1286. doi: 10.1002/ejhf.2250 [DOI] [PubMed] [Google Scholar]
- 36. Enomoto H, Mittal N, Inomata T, Arimura T, Izumi T, Kimura A, Fukuda K, Makino S. Dilated cardiomyopathy‐linked heat shock protein family D member 1 mutations cause up‐regulation of reactive oxygen species and autophagy through mitochondrial dysfunction. Cardiovasc Res. 2021;117:1118–1131. doi: 10.1093/cvr/cvaa158 [DOI] [PubMed] [Google Scholar]
- 37. Stroeks SLVM, Hellebrekers DMEI, Claes GRF, Tayal U, Krapels IPC, Vanhoutte EK, van den Wijngaard A, Henkens MTHM, Ware JS, Heymans SRB, et al. Clinical impact of re‐evaluating genes and variants implicated in dilated cardiomyopathy. Genet Med. 2021;23:2186–2193. doi: 10.1038/s41436-021-01255-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Almannai M, Luo S, Faqeih E, Almutairi F, Li Q, Agrawal PB. Homozygous SPEG mutation is associated with isolated dilated cardiomyopathy. Circ Genom Precis Med. 2021;14:e003310. doi: 10.1161/CIRCGEN.120.003310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ware SM, Wilkinson JD, Tariq M, Schubert JA, Sridhar A, Colan SD, Shi L, Canter CE, Hsu DT, Webber SA, et al. Genetic causes of cardiomyopathy in children: first results from the pediatric cardiomyopathy genes study. J Am Heart Assoc. 2021;10:e017731. doi: 10.1161/JAHA.121.020840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sedaghat‐Hamedani F, Rebs S, El‐Battrawy I, Chasan S, Krause T, Haas J, Zhong R, Liao Z, Xu Q, Zhou X, et al. Identification of SCN5a p.C335R variant in a large family with dilated cardiomyopathy and conduction disease. Int J Mol Sci. 2021;22:12990. doi: 10.3390/ijms222312990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fischer B, Dittmann S, Brodehl A, Unger A, Stallmeyer B, Paul M, Seebohm G, Kayser A, Peischard S, Linke WA, et al. Functional characterization of novel alpha‐helical rod domain desmin (DES) pathogenic variants associated with dilated cardiomyopathy, atrioventricular block and a risk for sudden cardiac death. Int J Cardiol. 2021;329:167–174. doi: 10.1016/j.ijcard.2020.12.050 [DOI] [PubMed] [Google Scholar]
- 42. Li M, Xia S, Xu L, Tan H, Yang J, Wu Z, He X, Li L. Genetic analysis using targeted next‐generation sequencing of sporadic Chinese patients with idiopathic dilated cardiomyopathy. J Transl Med. 2021;19:189. doi: 10.1186/s12967-021-02832-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gaertner A, Bloebaum J, Brodehl A, Klauke B, Sielemann K, Kassner A, Fox H, Morshuis M, Tiesmeier J, Schulz U, et al. The combined human genotype of truncating TTN and RBM20 mutations is associated with severe and early onset of dilated cardiomyopathy. Genes (Basel). 2021;12:883. doi: 10.3390/genes12060883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rico Y, Ramis MF, Massot M, Torres‐Juan L, Pons J, Fortuny E, Ripoll‐Vera T, González R, Peral V, Rossello X, et al. Familial dilated cardiomyopathy and sudden cardiac arrest: new association with a SCN5A mutation. Genes (Basel). 2021;12:1889. doi: 10.3390/genes12121889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Heliö K, Mäyränpää MI, Saarinen I, Ahonen S, Junnila H, Tommiska J, Weckström S, Holmström M, Toivonen M, Nikus K, et al. GRINL1A complex transcription unit containing GCOM1, MYZAP, and POLR2M genes associates with fully penetrant recessive dilated cardiomyopathy. Front Genet. 2021;12:786705. doi: 10.3389/fgene.2021.786705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Robles‐Mezcua A, Rodríguez‐Miranda L, Morcillo‐Hidalgo L, Jiménez‐Navarro M, García‐Pinilla JM. Phenotype and progression among patients with dilated cardiomyopathy and RBM20 mutations. Eur J Med Genet. 2021;64:104278. doi: 10.1016/j.ejmg.2021.104278 [DOI] [PubMed] [Google Scholar]
- 47. Hirayama‐Yamada K, Inagaki N, Hayashi T, Kimura A. A novel titin truncation variant linked to familial dilated cardiomyopathy found in a Japanese family and its functional analysis in genome‐edited model cells. Int Heart J. 2021;62:359–366. doi: 10.1536/ihj.20-664 [DOI] [PubMed] [Google Scholar]
- 48. Hakui H, Kioka H, Miyashita Y, Nishimura S, Matsuoka K, Kato H, Tsukamoto O, Kuramoto Y, Takuwa A, Takahashi Y, et al. Loss‐of‐function mutations in the co‐chaperone protein BAG5 cause dilated cardiomyopathy requiring heart transplantation. Sci Transl Med. 2022;14:eabf3274. doi: 10.1126/scitranslmed.abf3274 [DOI] [PubMed] [Google Scholar]
- 49. Xiao L, Wu D, Sun Y, Hu D, Dai J, Chen Y, Wang D. Whole‐exome sequencing reveals genetic risks of early‐onset sporadic dilated cardiomyopathy in the Chinese Han population. Sci China Life Sci. 2022;65:770–780. doi: 10.1007/s11427-020-1951-4 [DOI] [PubMed] [Google Scholar]
- 50. Vikhorev PG, Vikhoreva NN, Yeung W, Li A, Lal S, Dos Remedios CG, Blair CA, Guglin M, Campbell KS, Yacoub MH, et al. Titin‐truncating mutations associated with dilated cardiomyopathy alter length‐dependent activation and its modulation via phosphorylation. Cardiovasc Res. 2022;118:241–253. doi: 10.1093/cvr/cvaa316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cannatà A, Merlo M, Dal Ferro M, Barbati G, Manca P, Paldino A, Graw S, Gigli M, Stolfo D, Johnson R, et al. Association of titin variations with late‐onset dilated cardiomyopathy. JAMA Cardiol. 2022;7:371–377. doi: 10.1001/jamacardio.2021.5890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Khan RS, Pahl E, Dellefave‐Castillo L, Rychlik K, Ing A, Yap KL, Brew C, Johnston JR, McNally EM, Webster G. Genotype and cardiac outcomes in pediatric dilated cardiomyopathy. J Am Heart Assoc. 2022;11:e022854. doi: 10.1161/JAHA.121.022854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yuen M, Worgan L, Iwanski J, Pappas CT, Joshi H, Churko JM, Arbuckle S, Kirk EP, Zhu Y, Roscioli T, et al. Neonatal‐lethal dilated cardiomyopathy due to a homozygous LMOD2 donor splice‐site variant. Eur J Hum Genet. 2022;30:450–457. doi: 10.1038/s41431-022-01043-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Malakootian M, Bagheri Moghaddam M, Kalayinia S, Farrashi M, Maleki M, Sadeghipour P, Amin A. Dilated cardiomyopathy caused by a pathogenic nucleotide variant in RBM20 in an Iranian family. BMC Med Genomics. 2022;15:106. doi: 10.1186/s12920-022-01262-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang Y, Han B, Fan Y, Yi Y, Lv J, Wang J, Yang X, Jiang D, Zhao L, Zhang J, et al. Next‐generation sequencing reveals novel genetic variants for dilated cardiomyopathy in pediatric Chinese patients. Pediatr Cardiol. 2022;43:110–120. doi: 10.1007/s00246-021-02698-8 [DOI] [PubMed] [Google Scholar]
- 56. Hershberger RE, Cowan J, Jordan E, Kinnamon DD. The complex and diverse genetic architecture of dilated cardiomyopathy. Circ Res. 2021;128:1514–1532. doi: 10.1161/CIRCRESAHA.121.318157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ramchand J, Wallis M, Macciocca I, Lynch E, Farouque O, Martyn M, Phelan D, Chong B, Lockwood S, Weintraub R, et al. Prospective evaluation of the utility of whole exome sequencing in dilated cardiomyopathy. J Am Heart Assoc. 2020;9:e013346. doi: 10.1161/JAHA.119.013346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ellinor PT, Sasse‐Klaassen S, Probst S, Gerull B, Shin JT, Toeppel A, Heuser A, Michely B, Yoerger DM, Song BS, et al. A novel locus for dilated cardiomyopathy, diffuse myocardial fibrosis, and sudden death on chromosome 10q25‐26. J Am Coll Cardiol. 2006;48:106–111. doi: 10.1016/j.jacc.2006.01.079 [DOI] [PubMed] [Google Scholar]
- 59. Guo XJ, Qiu XB, Wang J, Guo YH, Yang CX, Li L, Gao RF, Ke ZP, Di RM, Sun YM, et al. PRRX1 loss‐of‐function mutations underlying familial atrial fibrillation. J Am Heart Assoc. 2021;10:e023517. doi: 10.1161/JAHA.121.023517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chahal CAA, Tester DJ, Fayyaz AU, Jaliparthy K, Khan NA, Lu D, Khan M, Sahoo A, Rajendran A, Knight JA, et al. Confirmation of cause of death via comprehensive autopsy and whole exome molecular sequencing in people with epilepsy and sudden unexpected death. J Am Heart Assoc. 2021;10:e021170. doi: 10.1161/JAHA.121.021170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shinya Y, Hiraide T, Momoi M, Goto S, Suzuki H, Katsumata Y, Kurebayashi Y, Endo J, Sano M, Fukuda K, et al. TNFRSF13B c.226G>A (p.Gly76Ser) as a novel causative mutation for pulmonary arterial hypertension. J Am Heart Assoc. 2021;10:e019245. doi: 10.1161/JAHA.120.019245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yaoita N, Satoh K, Satoh T, Shimizu T, Saito S, Sugimura K, Tatebe S, Yamamoto S, Aoki T, Kikuchi N, et al. Identification of the novel variants in patients with chronic thromboembolic pulmonary hypertension. J Am Heart Assoc. 2020;9:e015902. doi: 10.1161/JAHA.120.015902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Frank D, Yusuf Rangrez A, Friedrich C, Dittmann S, Stallmeyer B, Yadav P, Bernt A, Schulze‐Bahr E, Borlepawar A, Zimmermann WH, et al. Cardiac α‐actin (ACTC1) gene mutation causes atrial‐septal defects associated with late‐onset dilated cardiomyopathy. Circ Genom Precis Med. 2019;12:e002491. doi: 10.1161/CIRCGEN.119.002491 [DOI] [PubMed] [Google Scholar]
- 64. Hershberger RE, Parks SB, Kushner JD, Li D, Ludwigsen S, Jakobs P, Nauman D, Burgess D, Partain J, Litt M. Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin Transl Sci. 2008;1:21–26. doi: 10.1111/j.1752-8062.2008.00017.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Petropoulou E, Soltani M, Firoozabadi AD, Namayandeh SM, Crockford J, Maroofian R, Jamshidi Y. Digenic inheritance of mutations in the cardiac troponin (TNNT2) and cardiac beta myosin heavy chain (MYH7) as the cause of severe dilated cardiomyopathy. Eur J Med Genet. 2017;60:485–488. doi: 10.1016/j.ejmg.2017.06.008 [DOI] [PubMed] [Google Scholar]
- 66. Li RG, Li L, Qiu XB, Yuan F, Xu L, Li X, Xu YJ, Jiang WF, Jiang JQ, Liu X, et al. GATA4 loss‐of‐function mutation underlies familial dilated cardiomyopathy. Biochem Biophys Res Commun. 2013;439:591–596. doi: 10.1016/j.bbrc.2013.09.023 [DOI] [PubMed] [Google Scholar]
- 67. Zhao L, Xu JH, Xu WJ, Yu H, Wang Q, Zheng HZ, Jiang WF, Jiang JF, Yang YQ. A novel GATA4 loss‐of‐function mutation responsible for familial dilated cardiomyopathy. Int J Mol Med. 2014;33:654–660. doi: 10.3892/ijmm.2013.1600 [DOI] [PubMed] [Google Scholar]
- 68. Li J, Liu WD, Yang ZL, Yuan F, Xu L, Li RG, Yang YQ. Prevalence and spectrum of GATA4 mutations associated with sporadic dilated cardiomyopathy. Gene. 2014;548:174–181. doi: 10.1016/j.gene.2014.07.022 [DOI] [PubMed] [Google Scholar]
- 69. Scohy S, Gabant P, Van Reeth T, Hertveldt V, Drèze PL, Van Vooren P, Rivière M, Szpirer J, Szpirer C. Identification of KLF13 and KLF14 (SP6), novel members of the SP/XKLF transcription factor family. Genomics. 2000;70:93–101. doi: 10.1006/geno.2000.6362 [DOI] [PubMed] [Google Scholar]
- 70. Song A, Patel A, Thamatrakoln K, Liu C, Feng D, Clayberger C, Krensky AM. Functional domains and DNA‐binding sequences of RFLAT‐1/KLF13, a Krüppel‐like transcription factor of activated T lymphocytes. J Biol Chem. 2002;277:30055–30065. doi: 10.1074/jbc.M204278200 [DOI] [PubMed] [Google Scholar]
- 71. Li W, Li B, Li T, Zhang E, Wang Q, Chen S, Sun K. Identification and analysis of KLF13 variants in patients with congenital heart disease. BMC Med Genet. 2020;21:78. doi: 10.1186/s12881-020-01009-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Darwich R, Li W, Yamak A, Komati H, Andelfinger G, Sun K, Nemer M. KLF13 is a genetic modifier of the Holt‐Oram syndrome gene TBX5. Hum Mol Genet. 2017;26:942–954. doi: 10.1093/hmg/ddx009 [DOI] [PubMed] [Google Scholar]
- 73. Lavallée G, Andelfinger G, Nadeau M, Lefebvre C, Nemer G, Horb ME, Nemer M. The Kruppel‐like transcription factor KLF13 is a novel regulator of heart development. EMBO J. 2006;25:5201–5213. doi: 10.1038/sj.emboj.7601379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Nemer M, Horb ME. The KLF family of transcriptional regulators in cardiomyocyte proliferation and differentiation. Cell Cycle. 2007;6:117–121. doi: 10.4161/cc.6.2.3718 [DOI] [PubMed] [Google Scholar]
- 75. Cruz‐Topete D, He B, Xu X, Cidlowski JA. Krüppel‐like factor 13 is a major mediator of glucocorticoid receptor signaling in cardiomyocytes and protects these cells from DNA damage and death. J Biol Chem. 2016;291:19374–19386. doi: 10.1074/jbc.M116.725903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bayoumi AS, Park KM, Wang Y, Teoh JP, Aonuma T, Tang Y, Su H, Weintraub NL, Kim IM. A carvedilol‐responsive microRNA, miR‐125b‐5p protects the heart from acute myocardial infarction by repressing pro‐apoptotic bak1 and klf13 in cardiomyocytes. J Mol Cell Cardiol. 2018;114:72–82. doi: 10.1016/j.yjmcc.2017.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gu M, Wang J, Wang Y, Xu Y, Zhang Y, Wu W, Liao S. MiR‐147b inhibits cell viability and promotes apoptosis of rat H9c2 cardiomyocytes via down‐regulating KLF13 expression. Acta Biochim Biophys Sin (Shanghai). 2018;50:288–297. doi: 10.1093/abbs/gmx144 [DOI] [PubMed] [Google Scholar]
- 78. Xu L, Zhao L, Yuan F, Jiang WF, Liu H, Li RG, Xu YJ, Zhang M, Fang WY, Qu XK, et al. GATA6 loss‐of‐function mutations contribute to familial dilated cardiomyopathy. Int J Mol Med. 2014;34:1315–1322. doi: 10.3892/ijmm.2014.1896 [DOI] [PubMed] [Google Scholar]
- 79. Zhang XL, Qiu XB, Yuan F, Wang J, Zhao CM, Li RG, Xu L, Xu YJ, Shi HY, Hou XM, et al. TBX5 loss‐of‐function mutation contributes to familial dilated cardiomyopathy. Biochem Biophys Res Commun. 2015;459:166–171. doi: 10.1016/j.bbrc.2015.02.094 [DOI] [PubMed] [Google Scholar]
- 80. Zhou W, Zhao L, Jiang JQ, Jiang WF, Yang YQ, Qiu XB. A novel TBX5 loss‐of‐function mutation associated with sporadic dilated cardiomyopathy. Int J Mol Med. 2015;36:282–288. doi: 10.3892/ijmm.2015.2206 [DOI] [PubMed] [Google Scholar]
- 81. Patterson J, Coats C, McGowan R. Familial dilated cardiomyopathy associated with pathogenic TBX5 variants: expanding the cardiac phenotype associated with Holt‐Oram syndrome. Am J Med Genet A. 2020;182:1725–1734. doi: 10.1002/ajmg.a.61635 [DOI] [PubMed] [Google Scholar]
- 82. Wang SS, Wang TM, Qiao XH, Huang RT, Xue S, Dong BB, Xu YJ, Liu XY, Yang YQ. KLF13 loss‐of‐function variation contributes to familial congenital heart defects. Eur Rev Med Pharmacol Sci. 2020;24:11273–11285. doi: 10.26355/eurrev_202011_23617 [DOI] [PubMed] [Google Scholar]
- 83. van der Linde IHM, Hiemstra YL, Bökenkamp R, van Mil AM, Breuning MH, Ruivenkamp C, Ten Broeke SW, Veldkamp RF, van Waning JI, van Slegtenhorst MA, et al. A Dutch MYH7 founder mutation, p.(Asn1918Lys), is associated with early onset cardiomyopathy and congenital heart defects. Neth Heart J. 2017;25:675–681. doi: 10.1007/s12471-017-1037-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Augière C, Mégy S, El Malti R, Boland A, El Zein L, Verrier B, Mégarbané A, Deleuze JF, Bouvagnet P. A novel alpha cardiac actin (ACTC1) mutation mapping to a domain in close contact with myosin heavy chain leads to a variety of congenital heart defects, arrhythmia and possibly midline defects. PLoS One. 2015;10:e0127903. doi: 10.1371/journal.pone.0127903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yasuhara J, Garg V. Genetics of congenital heart disease: a narrative review of recent advances and clinical implications. Transl Pediatr. 2021;10:2366–2386. doi: 10.21037/tp-21-297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Martin KM, Metcalfe JC, Kemp PR. Expression of Klf9 and Klf13 in mouse development. Mech Dev. 2001;103:149–151. doi: 10.1016/s0925-4773(01)00343-4 [DOI] [PubMed] [Google Scholar]
- 87. Gordon AR, Outram SV, Keramatipour M, Goddard CA, Colledge WH, Metcalfe JC, Hager‐Theodorides AL, Crompton T, Kemp PR. Splenomegaly and modified erythropoiesis in KLF13−/− mice. J Biol Chem. 2008;283:11897–11904. doi: 10.1074/jbc.M709569200 [DOI] [PubMed] [Google Scholar]
- 88. Liao D, Chen W, Tan CY, Wong JX, Chan PS, Tan LW, Foo R, Jiang J. Upregulation of Yy1 suppresses dilated cardiomyopathy caused by Ttn insufficiency. Sci Rep. 2019;9:16330. doi: 10.1038/s41598-019-52796-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. van Spaendonck‐Zwarts KY, van Tintelen JP, van Veldhuisen DJ, van der Werf R, Jongbloed JD, Paulus WJ, Dooijes D, van den Berg MP. Peripartum cardiomyopathy as a part of familial dilated cardiomyopathy. Circulation. 2010;121:2169–2175. doi: 10.1161/CIRCULATIONAHA.109.929646 [DOI] [PubMed] [Google Scholar]
- 90. Sliwa K, Bauersachs J, Arany Z, Spracklen TF, Hilfiker‐Kleiner D. Peripartum cardiomyopathy: from genetics to management. Eur Heart J. 2021;42:3094–3102. doi: 10.1093/eurheartj/ehab458 [DOI] [PubMed] [Google Scholar]
- 91. Spracklen TF, Chakafana G, Schwartz PJ, Kotta MC, Shaboodien G, Ntusi NAB, Sliwa K. Genetics of peripartum cardiomyopathy: current knowledge, future directions and clinical implications. Genes (Basel). 2021;12:103. doi: 10.3390/genes12010103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Goli R, Li J, Brandimarto J, Levine LD, Riis V, McAfee Q, DePalma S, Haghighi A, Seidman JG, Seidman CE, et al. Genetic and phenotypic landscape of peripartum cardiomyopathy. Circulation. 2021;143:1852–1862. doi: 10.1161/CIRCULATIONAHA.120.052395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Arany Z. It is time to offer genetic testing to women with peripartum cardiomyopathy. Circulation. 2022;146:4–5. doi: 10.1161/CIRCULATIONAHA.122.059177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang Y, Xu WY, Jin HW, Sun H, Su XY, Zhuang QN, et al. KCNQ1 gain‐of‐function mutation in familial atrial fibrillation. Science. 2003;299:251–254. doi: 10.1126/science.1077771 [DOI] [PubMed] [Google Scholar]
- 95. Yang Y, Yang Y, Liang B, Liu J, Li J, Grunnet M, Olesen SP, Rasmussen HB, Ellinor PT, Gao L, et al. Identification of a Kir3.4 mutation in congenital long QT syndrome. Am J Hum Genet. 2010;86:872–880. doi: 10.1016/j.ajhg.2010.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Li RG, Xu YJ, Ye WG, Li YJ, Chen H, Qiu XB, Yang YQ, Bai D. Connexin45 (GJC1) loss‐of‐function mutation contributes to familial atrial fibrillation and conduction disease. Heart Rhythm. 2021;18:684–693. doi: 10.1016/j.hrthm.2020.12.033 [DOI] [PubMed] [Google Scholar]
- 97. Kalayinia S, Goodarzynejad H, Maleki M, Mahdieh N. Next generation sequencing applications for cardiovascular disease. Ann Med. 2018;50:91–109. doi: 10.1080/07853890.2017.1392595 [DOI] [PubMed] [Google Scholar]
- 98. Al‐Murshedi F, Meftah D, Scott P. Underdiagnoses resulting from variant misinterpretation: time for systematic reanalysis of whole exome data? Eur J Med Genet. 2019;62:39–43. doi: 10.1016/j.ejmg.2018.04.016 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1–S2