

Abstract
Introduction:
Acute myeloid leukemia (AML) is the most common and deadly type of leukemia affecting adults. It is typically managed with rounds of non-targeted chemotherapy followed by hematopoietic stem cell transplant but this is only possible in patients who can tolerate these harsh treatments and many are elderly and frail. With the identification of novel tumor-specific cell surface receptors, there is great conviction that targeted antibody therapies will soon become available for these patients.
Areas covered:
In this review, we describe the current landscape of known target receptors for monospecific and bispecific antibody-based therapeutics for AML. Here we characterize each of the receptors and targeted antibody-based therapeutics in development, illustrating the rational design behind each therapeutic compound. We then discuss the bispecific antibodies in development and how they improve immune surveillance of AML. For each therapeutic, we also summarize the available pre-clinical and clinical data, including data from discontinued trials.
Expert opinion:
One antibody-based therapeutic has already been approved for AML treatment, the CD33-targeting antibody-drug conjugate, gemtuzumab ozogamicin. Many more are currently in pre-clinical and clinical studies. These antibody-based therapeutics can perform tumor-specific, elaborate cytotoxic functions and there is growing confidence they will soon lead to personalized, safe AML treatment options that induce durable remissions.
Keywords: Acute myeloid leukemia, antibody, antibody-drug conjugate, bispecific antibody, CD33, CD47, fusion protein, immunotherapy, LILRB4, targeted therapy
1. Introduction
Leukemia is the common name for several malignant disorders that present with clonal expansion of hematopoietic cells in the bone marrow, resulting in increased numbers of cells of the affected lineage in the circulating blood [1]. Leukemia can occur in primitive to mature hematopoietic cells of the myeloid or lymphoid lineage, including mixed cells in some cases [2]. With a current incidence in the United States of 18.1/100,000/year in men and 11/100,000/year in women, leukemia is the 8th and 10th most incident cancer in the US overall for men and women, respectively. It is currently the 7th leading cause of cancer death overall [3].
In recent years, treatment options for leukemia patients have been moving from traditional chemotherapeutic approaches towards targeted and immunotherapeutic strategies, with the identification of tumor-specific antigens [4], the introduction of monoclonal antibodies (mAbs) [5] and decoding of the function of immune checkpoints in preventing adequate antitumor immune response [6].
Advances in immunotherapy include targeting the immune checkpoint Programmed Cell Death Protein 1 and Ligand 1 (PD-1/PD-L1) axis to boost antitumor immunity, but the efficacy of this approach has been limited to a small subset of highly immunogenic tumors [7,8]. The identification of novel immune checkpoints has provided clinicians with the possibility of its therapeutic application in a broader range of cancers, including hematopoietic malignancies such as Acute Myeloid Leukemia (AML) and Acute Lymphocytic Leukemia (ALL).
In ALL, immunotherapies such as blinatumomab are already included in standard-of-care protocols [9] but this approach has been mostly ineffective in AML thus far [10]. Hematopoietic stem cell transplantation (HSCT) remains the most effective treatment for AML, but many AML patients are elderly and thus not suitable for HSCT due to transplant-related risks [11,12]. Elderly patients are eligible for novel hypomethylating agents (HMAs) and venetoclax which may significantly extend life [13,14], but relapsing disease commonly occurs.
AML relapses are facilitated by leukemic stem cells (LSCs), a low-frequency subpopulation of leukemia cells that have self-renewal capacity and inherent capability for drug resistance [15]. Hematopoietic stem cells (HSCs) and LSCs utilize many of the same signaling pathways for their maintenance and survival [16], leading to significant efforts toward the immunophenotypic distinction between them and the development of novel targeted therapies such as antibody drug-conjugates (ADCs), T-cell engaging antibody constructs, adoptive transfer with chimeric antigen receptor (CAR) T cells, and dendritic cell vaccination capable of redirecting the patients’ own immune systems to target leukemic cells specifically. All of these treatment modalities are currently being investigated intensively in clinical trials [10].
In this review, we will summarize the literature on select current and emerging AML-specific receptor targets (Fig. 1). This review will focus primarily on targets for antibody-based immunotherapy under clinical development (Table 1).
Figure 1. Overview of receptor targets for antibody-based therapy in acute myeloid leukemia (AML) patients.
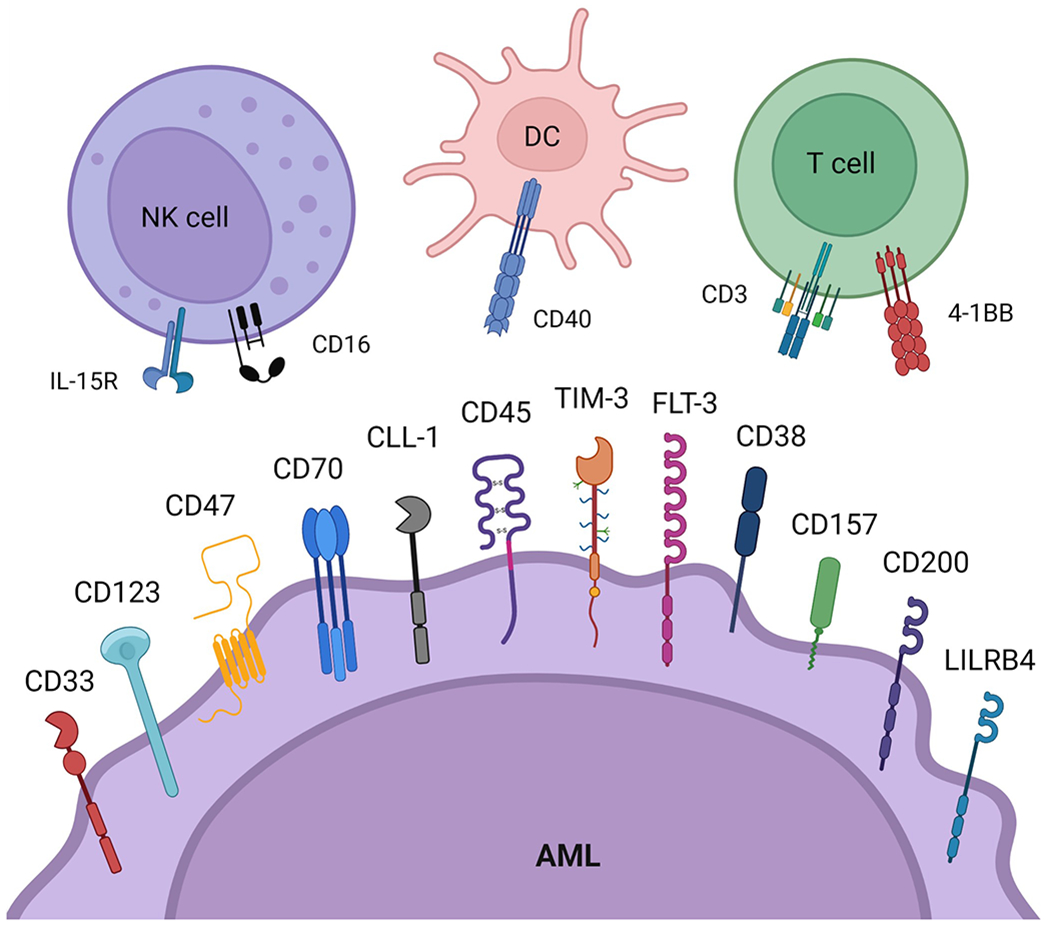
The receptors shown on the surface of the “AML” cell are currently in pre-clinical and clinical studies as targets for antibody-based therapy of AML myeloblasts and leukemic stem cells. The receptors illustrated on the surface of DCs, NK cells and T cells are targets for multispecific antibody-based therapies that directly crosslink these immune cells to AML myeloblasts and leukemic stem cells, leading to potentiation of antigen-presentation and immune effector function against the malignant cells. Created with BioRender.com.
Table 1.
Summary of selected antibody-based therapeutics for AML in active or inactive trials.
| Target | Therapeutic | Sponsor | Therapeutic Class | Development Status | Antibody Format | Natl. Clinical Trial ID(s) |
|---|---|---|---|---|---|---|
| CD33 | Gemtuzumab ozogamicin (GO) | Pfizer | ADC (anti-CD33 mAb X calicheamicin) | In clinical use (marketed product) | Human IgG4 | NCT00927498, NCT00372593 |
| Vadastuximab talirine (SGN-CD33A) | Seagen, Inc. | ADC (anti-CD33 mAb X PBD dimer) | Phase 3 (terminated) | Human IgG1 | NCT01902329, NCT02785900 | |
| Lintuzumab (SGN-33) | Seagen, Inc. | mAb | Phase 2b/3 (terminated) | Human IgG1 | NCT00528333, NCT00006045 | |
| 225Ac-Lintuzumab | Actinium Pharma., Inc. | mAb labeled with α-emitting radionuclide | Phase 1/2 | Human IgG1 conjugated to 225Ac, a radionuclide yielding 4 α-particles | NCT03867682 | |
| BI 836858 | Boehringer Ingelheim Pharma., Inc. | mAb (afucosylated Fc, ADCC-enhanced) | Phase 1/2 | Human IgG1 | NCT01690624, NCT02632721, NCT03013998 | |
| CD123 | Talacotuzumab (CSL362) | Janssen | mAb | Phase 2/3 (terminated) | Human IgG1 | NCT02472145, NCT01632852 |
| Pivekimab sunirine (IMGN632) | ImmunoGen, Inc. / Jazz Pharma., Inc. | ADC (anti-CD123 mAb X IGN) | Phase 1/2 | Human IgG1 | NCT04086264 | |
| Tagraxofusp (SL-401) | Stemline Therapeutics, Inc. | ADC-like fusion protein (IL-3 X diptheria toxin) | Phase 1b/2 | IL-3 fused to truncated diptheria toxin payload | NCT03113643, NCT05442216 | |
| CD47 / SIRPα | Magrolimab (5F9) | Gilead | mAb | Phase 3 | Human IgG4 | NCT02678338, NCT03248479, NCT04435691, NCT04778397, NCT05079230 |
| Evorpacept (ALX148) | ALX Oncology, Inc. | Fusion protein (SIRPα domains X inactive IgG1 Fc) | Phase 1/2 | Decoy SIRPα domains bound to Fc-silenced IgG1 Fc | NCT04755244 | |
| TTI-622 | Pfizer | Fusion protein (SIRPα domains X IgG4 Fc) | Phase 1b | Decoy SIRPα domains bound to IgG4 Fc | NCT03530683 | |
| Anzurstobart (CC-95251) | Celgene / Bristol Myers Squibb | mAb | Phase 1 | Human IgG1 with K322A CDC-silencing mutation | NCT03783403, NCT05168202 | |
| CC-90002 | Celgene | mAb | Phase 1 (terminated) | Human IgG4 with S228P and L235E mutations | NCT02641002 | |
| CD70 | Cusatuzumab (ARGX-110) | Argenx / Janssen | mAb (afucosylated Fc, ADCC-enhanced) | Phase 2 | Human IgG1 | NCT03030612, NCT04150887, NCT04023526 |
| CLL-1 | DCLL9718S | Genentech | ADC (anti-CLL-1 mAb X PBD dimer) | Phase 1 | Human IgG1 | NCT03298516 |
| CD45 | Apamistamab-I-131 (Iomab-B) | Actinium Pharma., Inc. | Radiolabeled mAb (for HSCT pre-conditioning) | Phase 3 | Human IgG1ҡ | NCT02665065 |
| TIM-3 | Sabatolimab (MBG453) | Novartis | mAb | Phase 1b/2 | Human IgG4ҡ with S228P mutation to prevent Fab-arm exchange | NCT03066648, NCT04150029 |
| CD38 | Daratumumab | Genmab / Janssen | mAb | Phase 2 | Human IgG1ҡ | NCT03067571 |
| Isatuximab | ImmunoGen, Inc. / Sanofi | mAb | Phase 2 | Human IgG1 | NCT03860844 | |
| FLT-3 | FLYSYN | SYNIMMUNE GmbH | mAb (ADCC-enhanced) | Phase 1 (completed) | Human IgG1 with S239D-I332E mutations to enhance affinity to FcγRIIIa | NCT02789254 |
| CD157 | MEN1112 / OBT357 | Menarini Group / Oxford BioTherapeutics | mAb (afucosylated Fc, ADCC-enhanced) | Phase 1 (terminated) | Human IgG1 | NCT02353143 |
| CD200 | Samalizumab (ALXN6000) | Alexion Pharma., Inc. | mAb | Phase 1b/2 (terminated) | Human IgG2/4ҡ | NCT03013998 |
| LILRB4 | IO-202 | Immune-Onc Therapeutics, Inc. | mAb | Phase 1 | Human IgG1 | NCT04372433 |
| MK-0482 | Merck | mAb | Phase 1 | Human IgG4 | NCT05038800 | |
| CD33xCD3 | AMG 330 | Amgen | BiTE | Phase 1 (terminated) | Tandem scFv | NCT02520427 |
| AMG 673 | Amgen | BiTE (Half-Life Extended) | Phase 1 (terminated) | Tandem scFv fused to IgG1 Fc domain | NCT03224819 | |
| Vixtimotamab (AMV564) | Amphivena Therapeutics, Inc. | Tandem diabody | Phase 1/2 | Tandem diabody | NCT03144245 | |
| JNJ-67571244 | Janssen | Asymmetric IgG (Duobody) | Phase 1 | Asymmetric IgG4 with αCD33 and αCD3 Fab arms and Fc with S228P-F234A-L235A mutations | NCT03915379 | |
| CD123xCD3 | Vibecotamab (XmAb14045, SQZ622) | Xencor | Fab-scFv-Fc bispecific | Phase 2 | αCD123 Fab-αCD3 scFv fused to XmAb® bispecific Fc domain | NCT02730312, NCT05285813 |
| MGD024 Flotetuzumab / MGD006 |
MacroGenics, Inc. | DART | Phase 1/2 | Diabody with stabilizing disulfide at VH-VL C-terminus, MGD024 fused to IgG1 Fc with L234A-L235A mutations | NCT02152956, NCT05362773 | |
| APVO436 | Aptevo Therapeutics, Inc. | scFv-Fc-scFv bispecific (ADAPTIR) | Phase 1b | Human IgG1 Fc fused to αCD123 scFvs at N-terminus and αCD3 scFvs at C-terminus with L234A-L235A-G237A-K322A mutations | NCT03647800 | |
| CD47xCD40 | SL-172154 | Shattuck Labs, Inc. | Fusion protein (Similar to IgG(H)-scFv2 bispecific) | Phase 1 | Human IgG4 Fc fused to decoy SIRPα domains at hinge and decoy CD40L domains at C-terminus | NCT05275439 |
| CD47×4–1BB | DSP107 | KAHR Medical | Fusion protein (Three SIRPα domains X 4–1BBL domain) |
Phase 1 | Three decoy SIRPα domains fused directly to 4–1BBL domain | NCT04937166 |
| CLL-1xCD3 | Tepoditamab (MCLA-117) | Merus N.V. | Asymmetric IgG (Biclonic) | Phase 1 (terminated) |
Human IgG1 with hetero VH and common VL | NCT03038230 |
| FLT-3xCD3 | AMG 427 | Amgen | BiTE (Half-Life Extended) | Phase 1 | Tandem scFv fused to IgG1 Fc domain | NCT03541369 |
| CLN-049 | Cullinan Oncology, Inc. | IgG(H)-scFv2 bispecific | Phase 1 | Human IgG1 fused to αCD3 scFvs at Fc C-terminus with C226S-C229S-E233P-L234V-L235A-ΔG236-D265G-N297Q-A327Q-A330S mutations | NCT05143996 | |
| CD33xCD16xIL-15R | GTB3650 | GT Biopharma, Inc. | TriKE | Phase 1b | αCD33 scFv and human IL-15 fused to αCD16 heavy chain single-domain antibody (sdAb) | NCT03214666 |
2. Antibody-based therapies
Antibodies, also referred to as immunoglobulins (Ig), are comprised of two heavy and two light chains held together by covalent disulfide bridges and noncovalent bonds and grouped into different isotypes (IgA, IgD, IgE, IgG, and IgM) depending on which type of heavy chain they contain [17,18]. Porter’s research on the Ig structure paved the way for other milestones in antibody-based therapies, such as the introduction of the hybridoma technology in 1975 by Kohler and Milstein [19,20], a major medical breakthrough enabling the generation of therapeutic monoclonal antibodies. Through direct crosslinking of immune effector cells and complement, therapeutic monoclonal antibodies (mAbs) can elicit anti-tumor responses such as antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC). Therapeutic mAbs may also interfere with the anti-inflammatory and oncogenic signaling of receptor targets on the tumor cell surface, leading to host anti-tumor immune responses and decreased tumor progression [21,22].
Antibody-drug conjugates (ADCs) are mAbs tethered to a cytotoxic payload by a chemical linker. Once bound to its target, the ADC-target complex is internalized via receptor-mediated endocytosis and its cytotoxic payload is released intracellularly by means of hydrolyzation of the linker in lysosomes, leading to apoptosis or other means of targeted cell death. ADCs dramatically improve the specificity of cytotoxic chemotherapy delivery, increasing efficacy and reducing systemic exposure and toxicity [23,24,25].
Fc-fusion proteins are antibody-like therapeutics in which the Fc domain of an antibody is covalently linked to other proteins, usually at the flexible hinge region of the Fc. The Fc domain improves the half-life of the therapeutic in human serum and the fused proteins can then function as the antigen-binding regions [26]. With this format, researchers can use any peptide as the antigen-binding region of their therapeutic, such as decoy peptide ligands used to antagonize receptor function.
2.1. Targets for antibody-based therapy of AML
CD33
CD33 is a mostly myeloid-specific receptor and member of the sialic acid binding immunoglobulin-like lectins (Siglecs) that has endocytic properties when bound by bivalent antibodies that result in internalization of the antigen/antibody complex [27]. The cytoplasmic tail contains an immunoreceptor tyrosine-based inhibitory signaling motif (ITIM) and an ITIM-like domain. These domains are phosphorylated by SRC family kinases [28,29,30]. Once phosphorylated, they act as docking sites for tyrosine phosphatases SHP-1 and SHP-2. CD33 is regulated by Suppressor of Cytokine Signaling 3 (SOCS3), which contains an SH2 central domain that competes with SHP-1 and SHP-2 for binding to the ITIM. SOCS3 binding recruits the ECS (Elongin B/C-Cul2/Cul5-SOCS-box protein) E3 ubiquitin ligase complex, which ubiquitinates its target, leading to accelerated proteasomal degradation of CD33, preventing binding of antibodies [31,32].
Though CD33 is expressed on normal myeloid progenitors [33], CD33 is also expressed on more than 90% of leukemic blasts [34] and a portion of LSCs [35], making it an attractive target for antibody-based strategies. Gemtuzumab ozogamicin (GO, Mylotarg™) is a humanized anti-CD33 monoclonal antibody covalently linked to a semisynthetic derivative of calicheamicin, a potent cytotoxic agent [36]. In 2000, the Food and Drug Administration (FDA) granted GO accelerated approval for older patients with relapsed/refractory (R/R) AML who were deemed unsuitable for intensive treatment [37]. However, a phase III trial (SWOG-S0106) evaluating the effects of adding GO to standard induction therapy (daunorubicin and cytarabine) in patients with newly diagnosed (ND) AML was stopped prematurely due to increased mortality (5.5% vs 1.4%) associated with the GO plus chemotherapy arm. Pfizer voluntarily withdrew GO from the US market in 2010 [38,39,40].
In the UK, large clinical studies (MRC AML15/16) implemented lower doses [41,42]. MRC AML15 showed a highly significant benefit of GO for younger patients with “favorable” cytogenetics. In this population, 5-year overall survival (OS) for patients who received GO at induction was 79%, compared to 51% for patients who did not receive GO. Patients with “adverse” cytogenetics were unlikely to benefit [41]. MRC AML16 included older untreated patients and in this population, 3-year cumulative incidence of relapse was significantly lower (68% vs. 76%) and 3-year OS was significantly higher (25% vs. 20%) with just a single dose of GO at induction [42]. Additionally, the phase III ALFA-0701 trial in France reported significantly improved median event-free survival in patients randomized to the GO treatment arm vs. standard-of-care chemotherapy alone (17.3 vs 9.5 months) [43].
In 2017, GO was re-approved by the FDA for ND and R/R adult AML owing to this new data on its clinical safety and efficacy [37]. In 2020, GO was also approved by the FDA for CD33+ AML in pediatric patients following a multicenter study of patients aged 0-29 (AAML0531; NCT00372593). In this study, the addition of GO to traditional chemotherapy conferred a significantly improved 3-year event-free survival (53.1% vs 46.9%) due to reduction in relapse risk [44].
With the success of GO, CD33 has become a very enticing AML target. Other antibody-drug conjugates (ADCs) with a CD33-targeting antibody fused to a cytotoxic payload have shown promise in early-stage clinical studies, but none have cleared the final hurdle of late-stage trials. Vadastuximab talirine (SGN-CD33A), an anti-CD33 mAb linked to a cytotoxic pyrrolobenzodiazepine (PBD) dimer, was granted Orphan Drug designation by the FDA and on the basis of extremely promising phase I data (NCT01902329) [45], a phase III study (CASCADE) was initiated in treatment-naïve, medically-unfit (TNU) AML patients. Disappointingly, the trial was terminated early due to increased rates of infection in the ADC-treated patients (NCT02785900).
Lintuzumab (HuM195, SGN-33), one of the first humanized anti-CD33 mAbs developed, showed pre-clinical efficacy at inducing ADCC and fixing complement [46], but efficacy was minimal in phase IIb (NCT00528333) and phase III (NCT00006045) clinical trials [47]. However, lintuzumab did exhibit an excellent safety profile in these patients and modified versions of the compound such as 225Ac-Lintuzumab with a fused α-particle emitting radionuclide are currently being studied in early-stage trials (NCT03867682).
Another antibody under clinical study is BI 836858, a conventional anti-CD33 IgG1 mAb with an engineered Fc. The Fc was engineered to increase binding to FcγRIIIa on immune effector cells in the tumor microenvironment and this improved binding to FcγRIIIa dramatically increased ADCC in pre-clinical studies [48]. Unfortunately, this Fc-engineered antibody did not show efficacy as monotherapy in a phase I clinical trial in patients with R/R AML (NCT01690624) [49] but HMAs have been shown to enhance the function of BI 836858 in pre-clinical studies by upregulating NK group 2D ligand [48]. Thus, additional clinical studies of the antibody in combination with HMAs have been initiated (NCT02632721, NCT03013998).
CD123
IL-3R is a glycoprotein on cell membranes composed of an α chain (CD123) paired with a ß subunit (CD131) [50]. The CD123 portion binds IL-3 with low affinity and high specificity at its N-terminal domain, while its C-terminal domain is responsible for intracellular signaling [51]. Physiologically, CD123 is highly expressed on pluripotent progenitor cells and signaling through CD123 leads to cellular proliferation and differentiation [52]. Leukemic stem cells and blasts, both at initial AML diagnosis and at relapse, have significantly increased CD123 expression, making CD123 an attractive AML target [53,54].
Talacotuzumab (CSL362) is an IgG1 mAb targeting CD123. This mAb has been shown to induce potent in vitro ADCC against CD123-expressing AML blasts/LSCs and reduction of leukemic cell growth in murine xenograft models of human AML. In addition, the antibody inhibits IL-3 mediated signaling, reducing the proliferation of leukemic progenitor cells [55]. Disappointingly, talacotuzumab in combination with decitabine showed no improvement in efficacy versus decitabine alone for the treatment of AML in a medically-unfit, older population (NCT02472145), resulting in early trial discontinuation [56]. However, in a phase I study assessing single agent talacotuzumab as post-remission therapy in patients with CD123+ AML at high risk of relapse (NCT01632852), 10/20 patients maintained complete remission (CR) at 24 weeks after start of treatment and 3/6 patients with minimal residual disease positive (MRD+) AML converted to MRD− status, supporting the use of anti-CD123 mAbs for maintenance therapy and MRD eradication [57]. Pivekimab sunirine (IMGN632), an ADC of a novel CD123-targeting antibody with a highly potent DNA alkylating payload known as indolinobenzodiazepine pseudodimer (IGN) [58], is now in a phase Ib/II trial in expansion cohorts of MRD+ patients to assess the conversion rate to MRD− disease (NCT04086264) [59].
Another interesting compound, tagraxofusp (SL-401), is an ADC-like fusion protein comprised of human IL-3 fused to a truncated diptheria toxin payload. Tagraxofusp monotherapy has been trialed and FDA-approved for treatment of rare blastic plasmacytoid dendritic cell neoplasm (BPDCN) [60]. It has also been trialed in AML but resistant populations of AML cells developed in response to tagraxofusp monotherapy. This was found to be mediated by DNA methylation and downregulation of diphthamide genes and reversible with azacitidine treatment [61]. BPDCN and some AMLs were also found to be sensitive to Bcl-2 inhibition with venetoclax [62]. A phase Ib combination trial (NCT03113643) of tagraxofusp with venetoclax and/or azacitidine is ongoing and objective responses have been observed in ND AML (complete remission or complete remission with incomplete hematologic recovery (CR/CRi) in 9/14 subjects) but not in R/R AML patients (0/12 subjects) [63]. Consequently, a phase II study (TAGALONG; NCT05442216) of tagraxofusp/azacitidine is planning to recruit ND secondary AML (myelodysplasia progression to AML) patients in 2023.
CD47
CD47 is a transmembrane protein that mainly functions as an anti-phagocytic signal, enabling CD47+ cells such as healthy HSCs to evade phagocytic elimination by macrophages and other phagocytes. Inhibition of phagocytosis occurs when CD47 binds to its cognate receptor Signal Regulatory Protein Alpha (SIRPα) on macrophages leading to tyrosine phosphatase activation and inhibition of myosin accumulation at the phagocytic synapse site. CD47 is found to be constitutively upregulated on myeloid leukemias, and overexpression of CD47 by these malignant cells allows them to evade phagocytosis [64]. Blockade of this CD47/SIRPα interaction has been shown to induce engulfment and elimination of leukemic cells [65,66].
Magrolimab (5F9) is a humanized IgG4 monoclonal antibody with a high affinity for human CD47. In pre-clinical murine models, magrolimab showed potent antitumor activity against AML, non-Hodgkin lymphoma, cutaneous T-cell lymphoma, ALL, and multiple myeloma [67]. Although an initial phase I trial (CAMELLIA) demonstrated issues of decreased hemoglobin, increased transfusion requirements and problems with ABO compatibility testing [68], follow-up phase Ib/II trials (NCT03248479, NCT04435691) tested lower-dose magrolimab in combination with venetoclax and/or azacitidine in TNU AML leading to patient responses, with 30/72 in the TP53-mutant cohort achieving CR/CRi [69]. Based on these encouraging results, phase III (ENHANCE) trials in TP53-mutant (NCT04778397) and TNU (NCT05079230) AML are recruiting patients.
Evorpacept (ALX148) is an antibody-like Fc-fusion protein comprised of SIRPα decoy receptors fused to an IgG1 Fc domain with Fc-silencing mutations [70]. There is an ongoing phase I/II clinical trial in AML patients (ASPEN-05) assessing ALX148 in combination with venetoclax and azacitidine (NCT04755244). Another Fc-fusion protein called TTI-622 composed of SIRPα domains bound to IgG4 Fc is also in clinical development. This fusion protein is a successor to TTI-621, a compound composed of the same CD47-binding SIRPα domains bound to IgG1 Fc. The new version of the therapeutic was designed to reduce adverse events such as anemia and thrombocytopenia by limiting binding to red blood cells and FcγR interactions while still delivering a moderate pro-phagocytic signal through its intact IgG4 Fc [71]. This compound is being tested in a phase I clinical trial in high-risk AML patients (NCT03530683) in combination with venetoclax and/or azacitidine. These combinations cleverly aim to deliver strong pro-phagocytic signals to phagocytes that should synergize well with the phagocytic disinhibition provided by TTI-622 [72].
Anzurstobart (CC-95251), an anti-SIRPα IgG1 monoclonal antibody with complement-silencing mutation K322A in the Fc domain exhibited high affinity to the different SIRPα variants and while it has been Fc-engineered to prevent complement activation, it does promote ADCP of tumor cells in vitro when combined with the tumor-antigen opsonizing antibody rituximab. CC-95251 demonstrated an excellent safety profile with no evidence of blood count depletion in cynomolgus monkeys and two phase I trials in AML have been initiated (NCT03783403, NCT05168202) [73].
Notably, an unconjugated anti-CD47 IgG4 monoclonal antibody with Fc-silencing mutations called CC-90002 was recently tested in a phase I trial in R/R AML and MDS patients (NCT02641002). The antibody exhibited cytotoxicity in pre-clinical models without inducing hemagglutination which commonly occurs with bivalent anti-CD47 mAbs but this trial was discontinued due to a perceived lack of efficacy. However, further research has shown that the efficacy of the antibody in pre-clinical models was dependent on macrophage effector function and thus requires residual FcγR function to engage effector macrophages [74]. Further engineering of this antibody’s Fc-FcγR binding interactions may be required to improve its efficacy.
CD70
CD27 is a tightly regulated co-stimulatory molecule, activated through its unique ligand CD70, enabling activation of innate and adaptive immunity [75]. CD70 is a transmembrane protein comprised of an extracellular domain, a stalk region and a transmembrane domain from the C-terminal TNF homology domain. CD27 activates signaling pathways resulting in the activation of transcription factors of the NFκB family and MAP kinases. By activation of these pathways, CD27 enhances cellular proliferation and induces anti-apoptotic signaling [76]. Riether et al. demonstrated that both AML blasts and progenitors consistently express CD70 and its cognate receptor CD27 and that CD27/CD70 signaling propagates disease [77].
Cusatuzumab (ARGX-110) is an ADCC-enhanced anti-CD70 mAb that significantly reduced soluble CD27 levels in treated LSCs, suggesting that the CD27/CD70 interaction is efficiently blocked. Custatuzumab was initially tested in combination with azacitidine in AML as HMAs like azacitidine are thought to increase CD70 expression in leukemia stem/progenitor cells (LSPCs) by demethylation of the CD70 promoter and downregulation of miR-29b levels, resulting in the upregulation of CD70 transcription factors. Xenotransplantation experiments were performed to analyze the synergistic potential of this combination and showed significantly reduced LSCs compared to monotherapy with no effect on peripheral blood lymphocytes [78]. Based on these results, a small phase I/II combination trial (NCT03030612) was initiated. Encouraging results from this study led to a larger follow-up Ib study (ELEVATE; NCT04150887) which demonstrated impressive responses. CR/CRi was achieved in 30/44 patients [79]. A phase II trial (CULMINATE; NCT04023526) is now ongoing.
CLL-1
C-type lectin-like receptors play a pivotal role in the fight against infection and maintain homeostasis and self-tolerance by recognizing damage-associated and pathogen-associated—molecular patterns [80,81]. CLL-1 (also known as CLEC12A or MICL) is a type II transmembrane glycoprotein, consisting of a single extracellular carbohydrate recognition domain with 6 N-glycosylation sites, a transmembrane region, and an intracellular N-terminus with two motifs of I/VxYxxL and YxxM. The I/VxYxxL domain is an ITIM that negatively regulates immune cell activation by recruitment of inhibitory phosphatases [82,83,84]. Several studies have demonstrated that CLL-1 is preferentially expressed on leukemic progenitor cells compared to normal progenitors [54,85,86] and therapeutics targeting it are now under development.
Zheng et al. developed a novel anti-CLL1 ADC with a self-destructive disulfide linker conjugated to an engineered cysteine residue at K149C on the human IgG light chain and this anti-CLL-1-ds-PBD was highly effective at depleting tumor cells in cynomolgus monkeys with no target-independent toxicities [87]. The ADC was licensed to Genentech as DCLL9718S and trialed in untreated AML patients (NCT03298516) but disappointingly, the study was stopped during dose escalation due to hepatic toxicity. Researchers still remain optimistic that CLL-1 is a viable target for AML therapeutics, with multiple CLL-1-targeting autologous cell therapies in early-stage clinical trials for R/R AML, such as KITE-222 (NCT04789408) and CLL-1.CAR-T (NCT04219163).
CD45
CD45 (also known as leukocyte common antigen or LCA) is a tyrosine phosphatase expressed on all hematopoietic cells with the exception of mature erythrocytes and platelets. As CD45 is expressed on a spectrum of primitive to mature leukocytes, antagonism may not be a safe therapeutic strategy for AML, but researchers have shown that CD45-specific cytotoxic agents can provide safe and effective preconditioning of patient bone marrow prior to HSCT [88].
Apamistamab-I-131 (Iomab-B), an innovative antibody-based therapeutic targeting CD45, aims to utilize this mechanism to improve the toxicity profile of HSCT-preconditioning and thus lower the fitness requirements for patients to receive an often lifesaving transplant. Iomab-B is a radiolabeled IgG1ҡ antibody that targets the CD45 receptor, delivering cytotoxic ß particles to CD45+ cells. Iomab-B is currently under development as a conditioning agent for use prior to allogeneic HSCT for patients 55 and older with active R/R AML and was granted Orphan Drug designation for this indication by the EMA and FDA [89]. A phase III trial (SIERRA; NCT02665065) is currently evaluating the efficacy of Iomab-B in combination with a Reduced Intensity Conditioning (RIC) regimen compared to a conventional care regimen prior to HSCT in 150 older patients with active R/R AML. Incredibly, preliminary data has shown that of the patients receiving the full therapeutic dose of Iomab-B and RIC prior to HSCT, 100% have been successfully engrafted, compared to successful engraftment of 18% of patients who received a physician’s choice of salvage therapy in the control study arm. Additionally, 38 patients who did not respond to the conventional care regimen have crossed over to the study’s treatment arm and all 38 have been successfully engrafted as well. [89].
TIM-3
Anti-T-cell immunoglobulin and mucin-domain containing molecule-3 (TIM-3) is a receptor primarily known for promoting immune tolerance that is expressed by exhausted T cells in the peripheral blood and bone marrow in many physiologic settings [90,91], including AML [92]. TIM-3 is also found on NK cells and on myeloid cells, including malignant AML cells, in which there is an autocrine stimulatory loop that works through the interaction between TIM-3 and Galectin-9 ligand, leading to phosphorylation of ERK and protein kinase B (PKB, also known as Akt). This process results in the induction of the β-catenin pathway and NFκB activation, leading to clonal selection of LSCs and tumor progression. Crucially, blocking TIM-3/Galectin-9 interaction with antagonistic mAbs has been shown to prevent LSC renewal and rescue NK cell-mediated cytotoxicity [93,94,95].
Sabatolimab (MBG453) is a humanized Fc-modified anti-TIM-3 IgG4ҡ (S228P) antibody being developed for the treatment of AML and high-risk MDS. The antibody blocks binding of Galectin-9 to TIM-3, preventing leukemic stem-cell renewal and inducing apoptosis. Sabatolimab also facilitates ADCP of AML cells through Fc-FcγRIa engagement but does not experience Fab-arm exchange with endogenous IgG4 antibodies due to its S228P mutation at the Fc hinge region [96]. A phase Ib study of sabatolimab/HMA (NCT03066648) demonstrated moderate clinical efficacy with CR/CRi achieved in 11/34 evaluable patients [97] and a phase II study of this combination is now ongoing in TNU AML patients (STIMULUS-AML1; NCT04150029) [98].
CD38
CD38 is a transmembrane glycoprotein expressed on hematopoietic cells that regulates cytokine release, cellular adhesion and migration toward sites of inflammation. CD38 is expressed by cells of the lymphoid and myeloid lineage as well as red blood cells and platelets. However, the highest expression of CD38 is found on plasma cells, making CD38 an ideal target for targeted immunotherapy in plasma cell malignancies such as multiple myeloma (MM). AML blasts also have notably high CD38 expression and pre-clinical studies have shown that anti-CD38 mAbs can induce significant ADCP and prevent AML cell trafficking in a xenograft model [99].
Isatuxumab and daratumumab are CD38-targeting monoclonal IgG1 antibodies that have been FDA-approved for MM. Both antibodies function through immune effector cell mechanisms but isatuximab can induce apoptosis of CD38+ cells directly while daratumumab requires engagement of effector immune cells [100]. Both have entered phase II trials for AML, with isatuximab being trialed in combination with chemotherapy in pediatric patients (NCT03860844) and daratumumab being trialed in adults (NCT03067571).
FLT-3
FLT-3 (FMS-like tyrosine kinase 3 or CD135) is an appealing target in AML because it is a proto-oncogene found on the cell surface of malignant leukemic blasts in most AML patients with limited expression on normal cells [101]. Recent FDA approvals of FLT-3-targeting kinase inhibitors midostaurin and gilteritinib have demonstrated the efficacy of targeting FLT-3 in AML. FLT-3 is found to be mutated in nearly a third of patients, and development of the second-generation gilteritinib has improved specificity for FLT-3 internal tandem duplication and tyrosine kinase domain mutations which has improved responses in FLT-3-mutant R/R AML patients [102,103].
Antibody therapies targeting FLT-3 uniquely bind the FLT-3 extracellular domain which has low genotypic variance, allowing for greater applicability of these therapeutics to all AML patients. FLYSYN is an ADCC-enhanced FLT3-targeting IgG1 antibody that mediated markedly enhanced cellular cytotoxicity against FLT3+ cell lines as well as AML patient blasts in pre-clinical studies [104]. This antibody was tested in a phase I trial in MRD+ AML patients (NCT02789254) and showed moderate preliminary efficacy with ≥1 log MRD reduction in 11/31 patients [105] but no follow-up clinical study has been initiated, as the sponsor is developing more potent FLT-3-targeting modalities.
CD157
CD157 is a glycosylphosphatidylinositol (GPI)-anchored glycoprotein found on the surface of granulocytes, monocytes and myeloid precursors. This cell surface receptor binds extracellular matrix (ECM) proteins like fibronectin with high affinity in a complex with β1 and β2-integrins, allowing for intracellular signal transduction through FAK that promotes cell adhesion and migration [106]. Adhesion to fibronectin is known to protect AML blasts from chemotherapy-induced cytotoxicity, an effect known as cell adhesion-mediated drug resistance, and an ex vivo study by Yakymiv et al. showed that this fibronectin-mediated protective effect against cytosine arabinoside (AraC) treatment proved to be stronger in CD157+ than in CD157− primary AML cells, suggesting that CD157 has a role in facilitating leukemia cell interactions with ECM components and modulates the sensitivity of AML cells to chemotherapy. [107]. A CD157-targeting mAb (MEN1112/OBT357) has demonstrated potent ADCC activity in ex vivo primary AML and autologous PBMC co-culture studies regardless of FcγRIIIa polymorphism, due to its afucosylated IgG1 format [108] and studies in non-human primates demonstrated low immunogenicity and toxicity [109]. However, in a clinical study (ARMY-1, NCT02353143), unpredicatable liver toxicities and limited clinical responses led to trial discontinuation. Targeting CD157 in AML may still hold potential, but will require appropriate clinical contextualization.
CD200
CD200 is an immunoglobulin (Ig)-like immune checkpoint receptor with two extracellular Ig-like domains that bind its cognate receptor CD200R1, expressed on some hematopoietic cell precursors to save them from autoimmune destruction [110]. CD200 is also consistently overexpressed on AML LSCs, providing these malignant cells with immune privilege [111]. Disappointingly, an early-phase clinical trial of the anti-CD200 IgG2/4ҡ Samalizumab in R/R AML (NCT03013998) was discontinued, but this was somewhat unsurprising as this antibody only showed pre-clinical efficacy in lymphoma models [112]. Research by Herbrich et al. in AML xenografts demonstrated that CD200+ AML progressed more rapidly and had diminished T cell responses relative to CD200− AML. And importantly, treatment with a novel anti-CD200 IgG1 mAb led to AML destruction in both PBMC-humanized immunocompetent and azacitidine/venetoclax-resistant immunodeficient mouse models, indicating that anti-CD200 therapy may be clinically effective in the post-HSCT setting and in treatment-resistant MRD+ AML [113].
2.2. LILRB family of receptor targets
Leukocyte immunoglobulin (Ig)-like receptors (LILR) are a family of 11 immunoregulatory transmembrane glycoproteins consisting of multiple Ig-like extracellular domains, a transmembrane region, and an intracellular cytoplasmic tail [114]. LILRs were first reported in 1997 [115,116], and have since been referred to as LIRs, immunoglobulin-like transcripts (ILTs), and CD85. LILRs include six immune-activating LILRs (LILRA1-6) and five immune-inhibiting LILRs (LILRB1-5) [117]. Although LILRs are expressed only in primates, there are two murine relatives: PirB and gp49B1 [118]. With the exception of LILRA3, which exists in a soluble protein form, functional LILRAs 1, 2, and 4-6 contain either two or four Ig-like extracellular domains, as well as short intracellular cytoplasmic tails that are associated with immunoreceptor tyrosine-based activation motifs (ITAMs) with a “classical” amino acid pattern of YxxI/Lx(6-12)YxxI/L [119,120]. Charged arginines and lysines on the ITAMs recruit Syk/ZAP70 family kinases, which activate downstream immune signals and increase secretion of inflammatory cytokines [121]. In contrast, inhibitory LILRBs have long intracellular cytoplasmic tails containing up to four immunoreceptor tyrosine-based inhibitory motifs (ITIMs) each with a canonical pattern of six amino acids: S/I/V/LxYxxI/V/L [122]. Binding of specific ligands to an LILRB’s Ig-like extracellular domain leads to phosphorylation of intracellular ITIM tyrosine residues, which then recruit inhibitory phosphatases SHP-1, SHP-2, and SHIP1. These inhibitory phosphatases suppress the downstream tyrosine phosphorylation and Ca2+ mobilization necessary for cells to generate an inflammatory response [123,124]. Differences in each LILRB’s structure and patterns of expression across different cell lineages as well as their interactions with different ligands alter their effects on immune suppression.
LILRBs are expressed predominantly on hematopoietic cells, although specific LILRB family members are also present on neurons, osteoclasts, endothelial cells, and stromal cells [125]. These receptors mediate anti-inflammatory signals which help cells to avoid autoimmune attack by monocytes, macrophages, neutrophils, and lymphoid cells [126,127]. However, in patients with AML, expression of LILRBs on AML cells and myeloid-derived suppressor cells (MDSCs) can alter the tumor microenvironment (TME) by suppressing the function of nearby inflammatory cells [128]. Studies have shown that LILRBs are both anti-inflammatory and oncogenic, supporting tumors through immune suppression in the TME and promoting migration of tumors to distant sites [129]. As the LILRB family of receptors can act as both immune checkpoints and tumor-supporting factors, they are of great therapeutic interest in hematologic and solid tumors.
LILRB4
The LILRB target of greatest interest in AML currently is LILRB4 (also known as CD85K, ILT3, LIR5 and HM18) [129], an inhibitory type I transmembrane protein in the LILRB family of receptors expressed primarily on monocytic cells (monocytes, macrophages, DCs) [130] as well as plasmablasts [131] and Tregs [132]. LILRB4 can also be found on in vitro differentiated progenitor mast cells, endothelial cells, and osteoclasts [125,130,133].
Unlike other LILRBs, LILRB4 has only two extracellular Ig-like domains (D1 and D2), a transmembrane domain, and three intracellular ITIMs. The structure of D2 is similar to the D4 domain of other LILRBs; however, it contains unique 310 helical turns that reduce D2’s contact with D1. The resulting D1-D2 interface folds into an obtuse angle that is both conformationally and electrostatically incapable of binding to MHC I – unlike the Ig-like domains of LILRB1 and LILRB2 [134]. Certain surface regions of D1 and the D1-D2 hinge region enable LILRB4 to bind with novel non-MHC class I ligands such as Apolipoprotein E (ApoE) [129] and fibronectin [135].
Physiologically, LILRB4’s role is to negatively regulate monocyte and T cell activity to protect cells from autoimmunity. On monocytes and macrophages, LILRB4 crosslinking to FcγRIII leads to recruitment of SHP-1 and inhibition of FcγRIII-mediated downstream signaling and Ca2+ mobilization [123] while LILRB4 crosslinking to FcγRI leads to recruitment of non-SHP-1 phosphatases that downregulate production of TNFα [136] and suppress FcγRI-dependent immune functions [137]. On other cell types, LILRB4 is capable of inhibiting T cell activation. For example, LILRB4 on DCs promotes conversion of alloreactive CD4+CD45RO+CD25+ T cells to Tregs which proceed to further tolerize DCs [138]. A 2009 study in humanized mice suggested that LILRB4 also suppresses the release of pro-inflammatory cytokines IL-1 alpha, IL-6, and type I IFN, as well as CXCL10 and CXCL11 which aid in mobilization of effector T cells [139].
Monocytic AML subtypes (M4/M5-AML) hijack this anti-inflammatory physiology by overexpressing the LILRB4 receptor [129]. Intracellularly, tyrosines at residues 412 and 442 of the distal two ITIM domains are phosphorylated when LILRB4 binds Apolipoprotein E (ApoE) ligand. This recruits SHP-2, leading to nuclear trafficking of NFκB and induction of downstream effectors including arginase 1 (ARG1) and urokinase receptor (uPAR) which inhibit T cell proliferation and promote tissue infiltration respectively [140]. Additionally, fibronectin in the stromal TME has been shown to serve as a ligand for LILRB4 on myeloid cells in the TME that supports myeloid cell phenotypic differentiation into tumor-associated suppressive cells [141] (Fig. 2). Researchers have demonstrated that blocking LILRB4 with antagonistic antibodies disinhibits T cell activation and prohibits differentiation of myeloid cells into tumor-associated suppressive cells while also impeding LILRB4+ tumor cell migration [129,141,142]. Furthermore, as LILRB4 is significantly overexpressed on monocytic AML cells, cytotoxic antibody-based therapeutics targeting LILRB4 such as CAR-T cells and ADCs have been developed and these have shown great efficacy against monocytic AML in the pre-clinical setting [143,144] (Fig. 3).
Figure 2. LILRB4 signaling in the acute myeloid leukemia (AML) immune and stromal tumor microenvironment (TME).

LILRB4 on AML cells binds Apolipoprotein E (ApoE) and this recruits SHP-2 leading to upregulation of NFκB and induction of downstream effectors including arginase 1 (ARG1) and urokinase receptor (uPAR) which inhibit T cell proliferation and promote tissue infiltration respectively. Additionally, fibronectin serves as a ligand for LILRB4 on myeloid cells in the stromal TME supporting phenotypic differentiation into tumor-associated suppressive cells such as tolerogenic dendritic cells that downregulate inflammatory signaling leading to failure of leukocyte adhesion and local T cell suppression. Created with BioRender.com.
Figure 3. Mechanisms of antibody-based therapeutics targeting LILRB4 in acute myeloid leukemia (AML).

Unconjugated antagonistic antibodies against LILRB4 prevent binding of its ApoE and fibronectin ligands, leading to disinhibition of T cell proliferation and suppression of anti-inflammatory myeloid cell differentiation while simultaneously prohibiting migration of LILRB4+ tumor cells to distant sites. Humanized anti-LILRB4 mAbs in an IgG1 format with intact, functional Fc can also engage effector NK cells and macrophages in antibody dependent cell-mediated cytotoxicity and phagocytosis respectively. Furthermore, as LILRB4 is significantly overexpressed on monocytic AML cells, cytotoxic antibody-derived therapeutics targeting LILRB4 such as anti-LILRB4 chimeric antigen receptor (CAR) T cells and anti-LILRB4 antibody-drug conjugates (ADCs) have been developed that have shown great efficacy against monocytic AML subtypes. Created with BioRender.com.
LILRB4 immunoregulatory signaling is a critical and tumor-specific feature of AML and the TME. Thus, targeting of LILRB4 with antibody-based immunotherapy is an area of great clinical interest. There are currently three anti-LILRB4 mAbs in ongoing clinical trials and two of these (IO-202 and MK-0482) are being tested in AML patients. The first anti-LILRB4 antibody approved for clinical use in AML was IO-202, an IgG1 mAb currently under phase I evaluation in hematologic malignancy (NCT04372433) [145]. IO-202 was granted Orphan Drug designation for treatment of AML and Fast Track designation for the treatment of R/R AML by the FDA. Clinical efficacy data has not yet been publicly released, but pre-clinical studies of IO-202 have demonstrated that the antibody is efficacious in hLILRB4-transgenic and xenograft mouse AML models and well-tolerated in cynomolgus monkeys [145]. MK-0482, an anti-LILRB4 IgG4 antibody, is also under phase I clinical evaluation in patients with R/R AML (NCT05038800). Of note, two autologous cell therapies, an anti-LILRB4 CAR-T and an anti-LILRB4 synthetic T cell antigen receptor T cell (STAR-T) have entered phase I trials in patients with R/R myelomonocytic (M4) or monocytic (M5) AML (NCT04803929) and R/R AML (NCT05518357), respectively.
Pre-clinical studies have shown that other receptors in the LILRB family may also prove useful as AML targets but due to their more complex signaling patterns in the AML immune TME or relatively uncharacterized biology, antibody therapeutics targeting these receptors have not made it to the clinical stage for AML. However, AML xenograft studies of antibody therapeutics independently targeting LILRB1 [146] and LILRB3 [147] have importantly demonstrated that blockade of LILRB signaling increased AML cell death and effector function of NK cells/CTLs as well as suppression of anti-inflammatory MDSC and Treg infiltration into the AML immune TME.
3. Bispecific antibody-based therapies
The term bispecific antibody (bsAb) describes a large family of therapeutic molecules with two antigen recognition sites. The bsAbs used to target leukemias are typically immune cell engagers (ICEs) in which one antigen recognition site targets a leukemia-specific antigen (LSA) while the other targets an immune cell antigen (commonly CD3 on T cells), allowing for synchronous targeting and facilitating of host immunity in an MHC-independent manner [148]. Bispecific antibodies were first generated in the 1960s when antigen-binding fragments from two different polyclonal sera were re-associated into bispecific F(ab’)2 molecules. With the development of hybridoma technology in 1975 and antibody engineering in the last three decades, it is now possible to generate antibodies of more than one defined specificity with relative ease [149]. Generally, these are divided into two major classes: those lacking an Fc region (Non-IgG-Like) and those bearing an Fc region (IgG-Like) [150,151] (Fig. 4). De Gast et al. developed the first bsAb used in hematological malignancies, a bispecific T cell engager (BiTE) targeting CD3xCD19, without significant clinical response in non-Hodgkin lymphoma but with toxicities limited to grade II fever and chills following infusion [152]. Over twelve years later, the CD3xCD19 bsAb blinatumomab was approved by the FDA for second-line use in B-cell precursor ALL [153].
Figure 4. Bispecific antibody-based immune cell engager (ICE) formats commonly used to target acute myeloid leukemia (AML).

Bispecific antibody-based therapeutic formats commonly used to target AML can be broadly classified into Non-IgG-Like ICEs lacking an IgG Fc and IgG-Like ICEs containing a Fc domain. The Non-IgG-Like BiTE is made up of a scFv targeting a leukemia-specific antigen (LSA) linked to a scFv targeting a T cell antigen, typically CD3. A Non-IgG-Like DART bispecific agent consists of variable domains of two antigen binding specificities linked to two independent polypeptide chains connected noncovalently. There is also an additional covalent linker in the form of a disulfide bridge, enhancing stability and promoting efficient crosslinking of AML and T cells. The TandAb is a Non-IgG-Like bispecific ICE with improved avidity through its bivalent binding of both the LSA and T cell antigen. The IgG-Like ICE class has superior stability via its Fc domain which can bind neonatal Fc receptors in vascular endothelial cells, preventing lysosomal degradation of these bsAbs. This class contains the heterodimeric Asymmetric IgGs which appear similar to conventional homodimeric IgG antibodies but have two structurally different Fab arms which bind to LSAs and T cell antigens respectively. The IgG-Like BiTE (Half Life Extended) is simply a BiTE fused to an IgG Fc domain to improve serum half-life of the compound. The IgG-Like Fab-scFv-Fc is a heterodimeric antibody-like protein with an LSA-targeting Fab arm and T cell antigen-targeting scFv fused to a Fc domain. And the IgG-Like scFv2-Fc-scFv2 and IgG(H)-scFv2 bispecifics each leverage bivalent formats to increase avidity to their target antigens. The former incorporates two LSA-targeting scFvs fused to the N-terminus and two T cell antigen-targeting scFvs fused to the C-terminus of an IgG Fc domain, while the latter is a canonical IgG antibody fused to two T cell antigen-targeting scFvs at the Fc C-terminus. Created with BioRender.com.
3.1. Targets for bispecific antibody-based therapy of AML
CD33xCD3
AMG 330 and AMG 673, a CD33xCD3 BiTE and half-life extended (HLE) BiTE respectively, were developed to recruit T cells to recognize and kill CD33+ human AML [154]. Aigner et al. demonstrated that T-cells from AML patient samples were efficiently engaged by AMG 330 and that primary malignant cells were sensitive to BiTE-mediated killing by allogeneic T-cells [155]. AMG 330 was tested in a phase I trial in adult R/R AML patients (NCT02520427), where it was given as a continuous IV infusion because of its short half-life (<2 h). In an update presented at ASCO 2020, AMG 330 provided early evidence of acceptable safety profile, drug tolerability and anti-leukemic activity [156,157,158]. AMG 673 fused scFvs with binding specificities for CD33 and CD3 to the N-terminus of an IgG Fc region to increase the BiTE’s terminal half-life from a few hours to a few days. Preliminary results from a phase I trial (NCT03224819) in R/R AML patients showed evidence of blast reduction [159] but trials of both AMG 330 and 673 have been discontinued due to sponsor prioritization decisions.
Vixtimotamab (AMV564) is a CD33xCD3 tandem diabody with two scFvs for each target. In this bivalent format, AMV564 has high avidity and a size exceeding the renal clearance threshold. Preliminary data from a first-in-human trial in R/R AML (NCT03144245) demonstrated limited toxicity and evidence of T cell activation and bone marrow blast reduction in most patients [160].
JNJ-67571244 is a CD33xCD3 asymmetric IgG4 mAb with Fc-silencing mutations generated using controlled Fab-arm exchange (Genmab DuoBody® technology) [161]. The anti-CD33 Fab arm of JNJ-67571244 uniquely binds the genotypically-conserved membrane-proximal IgC2 domain rather than the genotypically-variant membrane-distal IgV domain, allowing for consistent binding across CD33 genotypes. This bsAb was specifically cytotoxic to CD33+ AML blasts in primary samples and well-tolerated in cynomolgus monkeys, paving the way for a phase I clinical trial (NCT03915379) of the compound as monotherapy in patients with R/R AML or high-risk MDS [162]. The initial trial has been completed but data is not yet publicly available.
CD123xCD3
Vibecotamab (XmAb14045, SQZ622) is a CD123xCD3 bsAb that has a full-length Fc-engineered IgG, allowing for intermittent (weekly) administration. A phase I study in R/R AML (NCT02730312) demonstrated few clinical responses [163] but biomarker data from this study has led to initiation of a phase II trial (NCT05285813) aiming to eradicate MRD positivity in a cohort of AML patients in morphologic remission.
Another emerging bispecific technology is the dual-affinity retargeting (DART) agent, consisting of two scFvs covalently linked to enhance stability. DART crosslinking of leukemic cells to effector T cells has been demonstrated to assist cytolytic synapse formation and T cell killing [164]. Flotetuzumab (MGD006) is a DART with two independent polypeptides fusing the heavy-chain variable domain of one antibody to the light-chain variable domain of the other that has shown activity in AML [165]. In a phase I/II study of MGD006 (NCT02152956), CR/CRi was achieved in 6/30 AML patients with primary induction failure or relapse [166]. Although this compound showed preliminary efficacy in early-stage trials, there were concerns of cytokine release and a next-generation “DART-Fc” currently known as MGD024 has been developed to avoid this issue and improve circulating half-life. This DART-Fc has a mutation in the CD3-targeting scFv to limit CD3ε binding and a fused IgG1 Fc to increase the compound’s half-life in circulation [167]. MGD024 will be tested in an upcoming phase I trial (NCT05362773).
APVO436 is a bispecific targeting CD123xCD3 which has been uniquely engineered with ADAPTIR® technology to engage CD123 with scFv domains linked to the N-termini and engage T cells with CD3-specific scFv domains linked to the C-termini of the Fc domain. The Fc-domain has also been mutated to reduce engagement of Fc gamma receptors but maintains FcRn-binding capabilities. This scFv2-Fc-scFv2 structure is notable for its bivalent avidity for both target antigens and its reduced cytokine release relative to other bispecific antibody formats. AML xenograft studies of APVO436 showed dose-dependent anti-leukemic activity and prolonged survival with minimal cytokine release [168]. A phase Ib trial in R/R AML (NCT03647800) is ongoing and the early data has critically shown no dose-limiting toxicities [169].
CD47xCD40
SL-172154 is a fusion protein consisting of two human SIRPα domains and two human CD40L domains fused to a human IgG4 Fc. In pre-clinical studies, the SIRPα domains of the fusion protein were found to antagonize the CD47 checkpoint on tumor cells, leading to phagocytosis. Additionally, the CD40L domains potently stimulated CD40 on antigen-presenting cells, potentiating antigen release and cross-presentation to CD8+ T cells [170]. SL-172154 is now under phase I clinical study in high-risk AML and MDS patients (NCT05275439).
CD47×4-1BB
DSP107 (KAHR Medical) is a fusion protein consisting of three human SIRPα domains fused to a single human 4-1BBL domain. This ICE aims to exploit CD47 checkpoint inhibition while simultaneously engaging 4-1BB on T cells. 4-1BB is an attractive target for this purpose as it is considered a surrogate marker for the tumor-reactive T cell subset of Tumor-Infiltrating Lymphocytes (TILs) in the TME but it is not expressed on resting T cells in the peripheral blood like CD3 is. Agonistic 4-1BB-targeting antibodies like urelumab have not been successful clinically thus far, as these antibodies cannot fulfill the signaling requirements of 4-1BB without FcγR-crosslinking which can be overly cytotoxic. The DSP107 fusion protein aims to overcome this issue as it is immobilized by trimeric binding to CD47 on tumor cells, allowing for stable delivery of the 4-1BBL ligand to its 4-1BB receptor on TILs in the TME. DSP107 was found to potently and specifically stimulate TILs in AML xenograft studies [171] and a phase I clinical trial of the compound is now recruiting patients (NCT04937166).
CLL-1xCD3
Tepoditamab (MCLA-117) is a modified full-length human bispecific IgG1 antibody targeting CLL-1xCD3 which has proven capable of inducing targeted cytotoxicity against primary AML cells in pre-clinical studies [172]. Tepoditamab was shown to efficiently redirect T cells to kill tumor cells while sparing normal HSCs [173]. Preliminary data from an ongoing phase I trial (NCT03038230) showed that no maximum tolerated dose was reached, but unfortunately only 1/58 patients achieved morphologic leukemia-free status [174]. Researchers initially aimed to improve clinical activity of the compound through pharmacometric modeling, optimizing T cell activation and selecting patients based on CLL-1 levels, but it now appears that tepoditamab will not be developed further.
ABL602, a bsAb in pre-clinical development, also targets CLL-1xCD3 but has a unique asymmetric 2+1 structure which exhibits stronger bivalent binding to CLL-1 antigen than tepoditamab. ABL602 also exhibits greater tumor-specific killing of AML and lower levels of cytokine release than tepoditamab in vitro [175], indicating it may have an improved toxicity and efficacy profile in patients.
FLT-3xCD3
AMG 427 is a FLT-3xCD3 HLE BiTE composed of anti-FLT-3 and anti-CD3 scFvs bound in tandem with an IgG Fc fused to the CD3-targeting domain. In ex vivo studies, AMG 427 induced high cytotoxicity of primary AML blasts with increased FLT-3 expression. This was enhanced by the presence of an anti-PD-1 antibody [176]. A phase I trial of AMG 427 in R/R AML is enrolling patients (NCT03541369).
CLN-049 is a FLT-3xCD3 IgG(H)-scFv2 bispecific consisting of a bivalent anti-FLT-3 IgG1 with Fc-silencing mutations fused to anti-CD3 scFv domains at the C-termini of the heavy chains. In pre-clinical studies of AML cell lines, even cell lines with low FLT-3 expression were efficiently lysed upon treatment with sub-nanomolar concentrations of CLN-049 when co-cultured with heterologous PBMCs, while FLT-3-expressing hematopoietic progenitor cells and dendritic cells were not found to be sensitive to CLN-049 killing. However, target expression level was associated with increased cytokine release, so CLN-049 may have the highest therapeutic index in patients with FLT-3low AML blasts [177]. A phase I trial (NCT05143996) of CLN-049 in R/R AML is currently recruiting patients.
4. Conclusion
Acute myeloid leukemia (AML) remains the most common and deadliest form of leukemia, with about 20,000 new cases and 11,500 deaths attributed to AML in the United States in 2022 [178]. In recent years, the elucidation of tumor-specific AML and immune microenvironment receptors has prompted generation of novel antibody-based therapeutics targeting these receptors. Many of these mAbs, ADCs, bsAbs, and even trispecific [179] antibody-based therapeutics have reached the clinical testing stage and results from these trials will soon aid in the development of personalized therapeutic options that will greatly improve prognoses for some populations of AML patients, such as those deemed ineligible for HSCT. Targeted therapies offer these patients the realistic possibility of achieving strong remissions and overcoming issues of treatment resistance and deadly relapsing disease in the years to come.
5. Expert opinion
While treatment options have improved for patients with acute myeloid leukemia (AML) over the past decade, outcomes remain unacceptably poor. Standard of care induction therapy remains the intensive non-targeted chemotherapy combination of cytarabine and an anthracycline derivative infused over 7 days and 3 days respectively (“7+3”). Consolidative therapy with higher doses of cytarabine, and/or hematopoietic stem cell transplant (HSCT) is then utilized to improve the durability of remissions and increase the incidence of cure, however many patients cannot tolerate the intensity of these therapies, and minimal residual disease may still persist leading to subsequent relapse.
For older patients not appropriate for standard intensive chemotherapy, progress in recent years includes the development and FDA approval of the Bcl-2 inhibitor venetoclax in combination with hypomethylating agents (HMAs). With this combination, median overall survival now exceeds 12 months, which is improved from previous expectations, but still unsatisfactory.
Targeted therapeutics offer the promise of robust efficacy against AML blasts and leukemic stem cell (LSC) progenitors, with limited toxicity, by executing precision targeting of tumor-specific and tumor microenvironment (TME)-specific antigens. Bone marrow biopsy and peripheral blood analysis at diagnosis (and at relapse) can provide key information including cytogenetic and molecular profiling of the AML blasts and progenitors, as well as the unique characteristics of each patient’s TME. This information can allow clinicians to select targeted therapeutic options that will improve long-term prognoses for the populations of AML patients at highest risk, such as the large population of older AML patients currently unable to receive HSCTs due to fitness requirements and those with adverse cytogenetics.
In the future, there is great optimism that antibody-based targeted immunotherapies will significantly improve survival in even the highest-risk AML patient populations.
Future AML research will continue to be focused on the promising areas of tumor- and TME-specific targeted therapies such as antibody-based therapeutics and adoptive cell therapies, as these areas have seen great progress in recent years, for example, the re-approval of gemtuzumab ozogamicin and ongoing clinical trials investigating of targeted antibody-based agents (e.g., magrolimab and IO-202). Importantly, early detection and prevention are other areas of great interest in AML research that stand to greatly improve patient outcomes in the coming years. Specifically, there has been recent progress in the understanding of cytogenetic profiles that predispose individuals to therapy-related AML (t-AML) such as those with unstable genetic variants susceptible to carcinogenesis induced by radiation or alkylating agents and in the identification of prognostic biomarkers known to lead myelodysplastic syndrome (MDS) to evolve into AML. Further research in these areas will help clinicians prevent t-AML in vulnerable patients and find ways to stop MDS in its tracks before it progresses to the especially lethal AML.
From our perspective, we remain confident that the future of AML therapy will be both more effective and more individualized. Targeted induction therapies for AML will be tailored to each patient’s cytogenetic and molecular profile, leading to decreased toxicity and deeper and more durable remissions. While most eligible higher risk AML patients will still likely require HSCT to offer the best long-term efficacy, we are hopeful that longitudinal clinical studies of newer immunotherapies will confirm the role of targeted immunotherapies to improve and enhance the ability to maintain durable remissions in all patients.
Article highlights.
Acute myeloid leukemia (AML) is the most common and deadly adult leukemia.
AML is managed with toxic non-targeted chemotherapy and hematopoietic stem cell transplant in patients who can tolerate it.
Older patients can be treated with more benign hypomethylating agents (HMAs) and venetoclax but most patients experience lethal relapsing disease.
Target receptors (e.g. CD33, CD123, CD47, LILRB4) on the AML cell surface have been identified in recent years and antibody-based therapeutics specifically targeting these receptors have shown promise in pre-clinical and clinical studies.
Antibody-based therapeutics have been designed to specifically agonize or antagonize cell signaling pathways downstream of these receptors. They can also be engineered to deliver cytotoxic payloads as antibody-drug conjugates and connect humoral immunity to cell-based cytotoxic immune functions through engineering of the antibody Fc region.
Additionally, bispecific antibody-based therapeutics have been developed to specifically bind effector immune cells such as T cells and NK cells and direct them to malignant cells via concurrent specific binding of tumor-specific antigens.
Continued research into novel AML-specific targets and their fundamental biological functions will aid development of rationally designed antibody-based therapeutics that will provide safer and more effective treatment options for all AML patients.
Acknowledgements
We also thank Drs. Charlene Liao and Paul Woodard for their careful and critical editing of the manuscript.
Funding
We would like to acknowledge the National Center for Advancing Translational Sciences (TL1TR003169) and the National Cancer Institute (1R01CA248736 and 1R01CA263079) of the National Institutes of Health, the Cancer Prevention and Research Institute of Texas (RP180435, RP150551, RP190561, and RP220032) and Immune-Onc Therapeutics, Inc. (Sponsored Research Grant #111077) for their financial support.
Declaration of interest
N Zhang., Z An and CC Zhang had several patent applications licensed to Immune-Onc Therapeutics, Inc. Z An is a Scientific Advisory Board (SAB) member with Immune-Onc Therapeutics, Inc. NG Daver has received research funding from BMS, Pfizer, Immunogen, NovImmune, Genentech, Abbvie, Astellas, Daiichi-Sankyo, Hanmi, Roche and Forty-Seven, and serves as a consultant/advisor to Pfizer, BMS, Amgen, Gilead, Forty-Seven, Genentech, Novartis, Jazz, Immunogen, Astellas, Abbvie, Genentech, Trillium, Syndax and Kite. CD DiNardo has received research funding (to institution) from Abbvie, Astex, BMS, Cleave, Foghorn, Immune-Onc Therapeutics, Inc., Loxo and Servier, serves as a consultant/advisor to Abbvie, BMS, Genmab, Gilead, GSK, Jazz, Kura, Novartis, Servier and Takeda, and is on the scientific advisory board of Notable Labs.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Abbreviations:
- ACT
adoptive cellular therapy
- ADC
antibody drug conjugate
- ADCC
antibody dependent cell-mediated cytotoxicity
- ADCP
antibody dependent cellular phagocytosis
- AML
acute myeloid leukemia
- ALL
acute lymphocytic leukemia
- ApoE
apolipoprotein E
- AZA
azacitidine
- Bcl-2
B-cell lymphoma 2
- BiTE
bispecific T-cell engager
- BPDCN
blastic plasmacytoid dendritic cell neoplasm
- bsAb
bispecific antibody
- CAR-T
chimeric antigen receptor T cells
- CDC
complement dependent cytotoxicity
- CLL-1
C-type lectin-like molecule 1
- CR
complete remission
- CRi
complete remission with incomplete hematologic recovery
- CyTOF
cytometry by time of flight
- DART
dual-affinity re-targeting protein
- DC
dendritic cell
- F(ab)
antigen-binding fragment
- F(ab’)2
bivalent antigen-binding fragment
- Fc
fragment crystallizable
- FcγRs
Fc gamma receptors
- FcRn
neonatal Fc receptor
- FLT-3
fms like tyrosine kinase 3
- GO
gemtuzumab ozogamicin
- HLE
half-life extended
- IgG
immunoglobulin G
- IGN
indolinobenzodiazepine pseudodimer
- ITAM
immunoreceptor tyrosine-based activation motif
- ITIM
immunoreceptor tyrosine-based inhibitory signaling motif
- HMAs
hypomethylating agents
- HSC
hematopoietic stem cell
- HSCT
hematopoietic stem cell transplant
- LILR
leukocyte immunoglobulin-like receptor
- LSC
leukemic stem cell
- M4 AML
myelomonocytic AML
- M5 AML
monocytic AML
- mAb
monoclonal antibody
- MDS
myelodysplastic syndrome
- MDSCs
myeloid-derived suppressor cells
- MM
multiple myeloma
- MRD
minimal residual disease
- NFκB
nuclear factor kappa-light-chain-enhancer of activated B cells
- OS
overall survival
- PBD
pyrrolobenzodiazepine dimer
- PBMC
peripheral blood mononuclear cell
- PD-L1
programmed death-ligand 1
- PD-1
programmed cell death protein 1
- PR
partial response
- R/R
relapsed/refractory
- scFv
single-chain variable fragment
- SHIP-1
Src homology region 2 domain-containing inositol polyphosphate 5-phosphatase 1
- SHP-1
Src homology region 2 domain-containing phosphatase-1
- SHP-2
Src homology region 2 domain-containing phosphatase-2
- Siglecs
sialic acid binding immunoglobulin-like lectins
- SIRPα
Signal Regulatory Protein Alpha
- TAM
tumor-associated macrophage
- TIM-3
Anti-T-cell immunoglobulin and mucin-domain containing molecule-3
- TME
tumor microenvironment
- Treg
regulatory T cell
- TriKE
trispecific killer engager
- TNU
treatment-naïve, medically-unfit
- VH
variable heavy chain
- VL
variable light chain
Footnotes
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Juliusson G, Hough R. Leukemia. Progress in Tumor Research 43, 87–100 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Mastelaro de Rezende M, Ferreira AT, Paredes-Gamero EJ. Leukemia stem cell immunophenotyping tool for diagnostic, prognosis, and therapeutics. Journal of Cellular Physiology 235(6), 4989–98 (2020). [DOI] [PubMed] [Google Scholar]
- 3.American Cancer Society Cancer Statistics Center. http://cancerstatisticscenter.cancer.org. Accessed October 10, 2022.
- 4.Gubin MM, Artyomov MN, Mardis ER, et al. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest. 125(9), 3413–21 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Newsome BW, Ernstoff MS. The clinical pharmacology of therapeutic monoclonal antibodies in the treatment of malignancy; have the magic bullets arrived? Br. J. Clin. Pharmacol 66, 6–19 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharma P, Allison JP. The future of immune checkpoint therapy. Science 348, 56–61 (2015). [DOI] [PubMed] [Google Scholar]; • Great summary of human T-cell based responses to immune checkpoint blockade
- 7.Bagchi S, Yuan R, Engleman EG. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annual Review of Pathology 16, 223–49 (2021). [DOI] [PubMed] [Google Scholar]
- 8.Iwai Y, Ishida M, Tanaka Y, et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. PNAS 99(19), 12293–7 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barsan V, Ramakrishna S, Davis KL. Immunotherapy for the Treatment of Acute Lymphoblastic Leukemia. Curr. Oncol. Rep 22, 11–19 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Lichtenegger FS, Krupka C, Köhnke T, et al. Immunotherapy for acute myeloid leukemia. Seminars in Hematology 52(3), 207–14 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Aoki J, Kanamori H, Tanaka M, et al. Impact of age on outcomes of allogeneic hematopoietic stem cell transplantation with reduced intensity conditioning in elderly patients with acute myeloid leukemia. Am. J. Hematol 91(3), 302–7 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Takami A Hematopoietic stem cell transplantation for acute myeloid leukemia. International Journal of Hematology 107(5), 513–8 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Lessi F, Laurino M, Papayannidis C, et al. Hypomethylating Agents (HMAs) as Salvage Therapy in Relapsed or Refractory AML: An Italian Multicentric Retrospective Study. Biomedicines 9(8), 972 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. NEJM 383(7), 617–29 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat. Med 15, 1010–2 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Gurska LM, Ames K, Gritsman K. Signaling Pathways in Leukemic Stem Cells. Advances in Experimental Medicine and Biology 1143, 1–39 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lipman NS, Jackson LR, Trudel LJ, et al. Monoclonal Versus Polyclonal Antibodies: Distinguishing Characteristics, Applications, and Information Resources. ILAR J. 46(3), 258–68 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Buss NAPS, Henderson SJ, McFarlane M, et al. Monoclonal antibody therapeutics: History and future. Current Opinion in Pharmacology 12(5), 615–22 (2012). [DOI] [PubMed] [Google Scholar]; • Useful history of mAb therapeutics and their applications
- 19.Porter RR. Separation and Isolation of Fractions of Rabbit Gamma-Globulin Containing the Antibody and Antigenic Combining Sites. Nat. Cell Biol 182, 670–1 (1958). [DOI] [PubMed] [Google Scholar]
- 20.Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256(5517), 495–7 (1975). [DOI] [PubMed] [Google Scholar]
- 21.Redman JM, Hill EM, AlDeghaither D, et al. Mechanisms of action of therapeutic antibodies for cancer. Molecular Immunology 67(2), 28–45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams BA, Law A, Hunyadkurti J, et al. Antibody therapies for acute myeloid leukemia: Unconjugated, toxin-conjugated, radio-conjugated and multivalent formats. Journal of Clinical Medicine 8(8), 1261 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Good overview of the commonly used formats of antibody-based therapy for AML
- 23.Gorovits B, Krinos-Fiorotti C. Proposed mechanism of off-target toxicity for antibody-drug conjugates driven by mannose receptor uptake. Cancer Immunol. Immunother 62, 217–23 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalim M, Chen J, Wang S, et al. Intracellular trafficking of new anticancer therapeutics: antibody-drug conjugates. Drug Des. Devel. Ther 11, 2265–76 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walter RB. Brief overview of antibody-drug conjugate therapy for acute leukemia. Expert Opin. Biol. Ther 21(7), 795–9 (2021). [DOI] [PubMed] [Google Scholar]; • Concise review of the ADCs that have been tested in acute leukemia patients
- 26.Czajkowsky DM, Hu J, Shao Z, et al. Fc-fusion proteins: new developments and future perspectives. EMBO Mol. Med 4(10), 1015–28 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pelosi E, Castelli G, Testa U. Targeting LSCs through membrane antigens selectively or preferentially expressed on these cells. Blood Cells Mol. Dis 55(4), 336–46 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Taylor VC, Buckley CD, Douglas M, et al. The myeloid-specific sialic acid-binding receptor, CD33, associates with the protein-tyrosine phosphatases, SHP-1 and SHP-2. J. Biol. Chem 274, 11505–12 (1999). [DOI] [PubMed] [Google Scholar]
- 29.Ulyanova T, Blasioli J, Woodford-Thomas TA, et al. The sialoadhesin CD33 is a myeloid-specific inhibitory receptor. Eur. J. Immunol 29, 3440–9 (1999). [DOI] [PubMed] [Google Scholar]
- 30.Paul SP, Taylor LS, Stansbury EK, et al. Myeloid specific human CD33 is an inhibitory receptor with differential ITIM function in recruiting the phosphatases SHP-1 and SHP-2. Blood 96, 483–90 (2000). [PubMed] [Google Scholar]
- 31.Orr SJ, Morgan NM, Elliott J, et al. CD33 responses are blocked by SOCS3 through accelerated proteasomal-mediated turnover. Blood 109, 1061–8 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nature Reviews Immunology 7(4), 255–66 (2007). [DOI] [PubMed] [Google Scholar]; • Nice review of the Siglec family, with particular focus on CD33-related receptors
- 33.Pearce D, Taussig D, Bonnet D. Implications of the Expression of Myeloid Markers on Normal and Leukemic Stem Cells. Cell Cycle 5(3), 271–3 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Griffin JD, Linch D, Sabbath K, et al. A monoclonal antibody reactive with normal and leukemic human myeloid progenitor cells. Leuk. Res 8(4), 521–34 (1984). [DOI] [PubMed] [Google Scholar]
- 35.Vercauteren S, Zapf R, Sutherland H. Primitive AML progenitors from most CD34(+) patients lack CD33 expression but progenitors from many CD34(−) AML patients express CD33. Cytotherapy 9(2), 194–204 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Selby C, Yacko LR, Glode AE. Gemtuzumab Ozogamicin: Back Again. J. Adv. Pract. Oncol 10(1), 68–82 (2019). [PMC free article] [PubMed] [Google Scholar]
- 37.Baron J, Wang ES. Gemtuzumab ozogamicin for the treatment of acute myeloid leukemia. Expert Review of Clinical Pharmacology 11(6), 549–59 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bross PF, Beitz J, Chen G, et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res 7(6), 1490–6 (2001). [PubMed] [Google Scholar]
- 39.Petersdorf SH, Kopecky KJ, Slovak M, et al. A phase 3 study of gemtuzumab ozogamicin during induction and post consolidation therapy in younger patients with acute myeloid leukemia. Blood 121, 4854–60 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gottardi M, Sperotto A, di Rorà AGL, et al. Gemtuzumab ozogamicin in acute myeloid leukemia: Past, present and future. Minerva Medica 111(5), 395–410 (2020). [DOI] [PubMed] [Google Scholar]; • Comprehensive history of and lessons learned from the approval process of gemtuzumab ozogamicin (GO), the only approved antibody-based therapy for AML
- 41.Burnett AK, Hills RK, Milligan D, et al. Identification of Patients with Acute Myeloblastic Leukemia Who Benefit from the Addition of Gemtuzumab Ozogamicin: Results of the MRC AML15 Trial. Journal of Clinical Oncology 29(4), 369–377 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Burnett AK, Russell NH, Hills RK, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy improves survival in older patients with acute myeloid leukemia. Journal of Clinical Oncology 30(32), 3924–31 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Lambert J, Pautas C, Terré C, et al. Gemtuzumab ozogamicin for de novo acute myeloid leukemia: Final efficacy and safety updates from the open-label, phase III ALFA-0701 trial. Haematologica 104(1), 113–9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gamis AS, Alonzo TA, Meshinchi S, et al. Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: results from the randomized phase III Children’s Oncology Group trial AAML0531. J. Clin. Oncol 32(27), 3021–32 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stein EM, Walter RB, Erba HP, et al. A Phase 1 Trial of Vadastuximab Talirine as Monotherapy in Patients with CD33-positive Acute Myeloid Leukemia. Blood 131, 387–96 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sutherland MK, Yu C, Lewis TS, et al. Anti-Leukemic Activity of Lintuzumab (Sgn-33) in Preclinical Models of Acute Myeloid Leukemia. MAbs 1, 481–90 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feldman EJ, Brandwein J, Stone R, et al. Phase III Randomized Multicenter Study of a Humanized Anti-CD33 Monoclonal Antibody, Lintuzumab, in Combination with Chemotherapy, Versus Chemotherapy Alone in Patients with Refractory or First-Relapsed Acute Myeloid Leukemia. J. Clin. Oncol 23, 4110–6 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Vasu S, He S, Cheney C. Decitabine enhances anti-CD33 monoclonal antibody BI 836858-mediated natural killer ADCC against AML blasts. Blood 127(23), 2879–89 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vasu S, Altman JK, Uy GL, et al. A phase I study of the fully human, fragment crystallizable-engineered, anti-CD-33 monoclonal antibody BI 836858 in patients with previously-treated acute myeloid leukemia. Haematologica 107(3), 770–3 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu K, Zhu M, Huang Y, et al. CD123 and its potential clinical application in leukemias. Life Sciences 122, 59–64 (2015). [DOI] [PubMed] [Google Scholar]; • Good review of CD123 in leukemia, a major target for antibody-based AML therapy
- 51.Barry SC, Korpelainen E, Sun Q, et al. Roles of the N and C terminal domains of the interleukin-3 receptor α chain in receptor function. Blood 89(3), 842–52 (1997). [PubMed] [Google Scholar]
- 52.Testa U, Pelosi E, Frankel A. CD 123 is a membrane biomarker and a therapeutic target in hematologic malignancies. Biomarker Research 2(1), 4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Muñoz L, Nomdedeu JF, López O, et al. Interleukin-3 receptor alpha chain (CD123) is widely expressed in hematologic malignancies. Haematologica 86, 1261–9 (2001). [PubMed] [Google Scholar]
- 54.Haubner S, Perna F, Köhnke T, et al. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia 33(1), 64–74 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Busfield SJ, Biondo M, Wong M, et al. Targeting of acute myeloid leukemia in vitro and in vivo with an anti-CD123 mAb engineered for optimal ADCC. Leukemia 28, 2213–21 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Montesinos P, Roboz GJ, Bulabois CE, et al. Safety and efficacy of talacotuzumab plus decitabine or decitabine alone in patients with acute myeloid leukemia not eligible for chemotherapy: results from a multicenter, randomized, phase 2/3 study. Leukemia 35(1), 62–74 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith BD, Roboz GJ, Walter RB, et al. First-in Man, Phase 1 Study of CSL362 (Anti-IL3Rα / Anti-CD123 Monoclonal Antibody) in Patients with CD123+ Acute Myeloid Leukemia (AML) in CR at High Risk for Early Relapse. Blood 124(21), 120 (2014). [Google Scholar]
- 58.Kovtun Y, Jones GE, Adams S, et al. A CD123-targeting antibody-drug conjugate, IMGN632, designed to eradicate AML while sparing normal bone marrow cells. Blood Advances 2(8), 848–58 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Daver NG, Sweet KL, Montesinos P, et al. A Phase 1b/2 Study of IMGN632, a CD123-Targeting Antibody-Drug Conjugate (ADC), As Monotherapy or in Combination with Venetoclax and/or Azacitidine for Patients with CD123-Positive Acute Myeloid Leukemia. Blood 136(Supplement 1), 50–1 (2020).32430504 [Google Scholar]
- 60.Pemmaraju N, Lane AA, Sweet KL, et al. Tagraxofusp in blastic plasmacytoid dendritic-cell neoplasm. NEJM 380(17), 1628–37 (2019). [DOI] [PubMed] [Google Scholar]
- 61.Togami K, Pastika T, Stephansky J, et al. DNA methyltransferase inhibition overcomes diphthamide pathway deficiencies underlying CD123-targeted treatment resistance. J. Clin. Invest 129(11), 5005–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Montero J, Stephansky J, Cai T, et al. Blastic Plasmacytoid Dendritic Cell Neoplasm Is Dependent on BCL2 and Sensitive to Venetoclax. Cancer Discov. 7(2), 156–64 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lane AA, Stein AS, Garcia JS, et al. Safety and Efficacy of Combining Tagraxofusp (SL-401) with Azacitidine or Azacitidine and Venetoclax in a Phase 1b Study for CD123 Positive AML, MDS, or BPDCN. Blood 138(Supplement 1), 2346 (2021). [Google Scholar]
- 64.Jaiswal S, Jamieson CH, Pang WW, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 138(2), 271–85 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Key discovery of CD47/SIRPα pathway as AML evasion mechanism
- 65.Chao MP, Weissman IL, Majeti R. The CD47-SIRPalpha pathway in cancer immune evasion and potential therapeutic implications. Curr. Opin. Immunol 24, 225–32 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chao MP, Takimoto CH, Feng DD, et al. Therapeutic Targeting of the Macrophage Immune Checkpoint CD47 in Myeloid Malignancies. Frontiers in Oncology 9, 1380 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Current overview of AML therapeutics targeting CD47/SIRPα, with particular focus on magrolimab which has been granted Fast Track designation by the FDA
- 67.Liu J, Wang L, Zhao F, et al. Pre-clinical development of a humanized anti-CD47 antibody with anti-cancer therapeutic potential. PLoS One 10, e0137345 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vyas P, Knapper S, Kelly R, et al. Initial phase 1 results of the first-in-class anti-CD47 antibody HU5F9-G4 in relapsed/refractory acute myeloid leukemia patients. Eur. Hematol. Assoc. Meeting 23, Abstract PF232 (2018). [Google Scholar]
- 69.Daver NG, Vyas P, Kambhampati S, et al. S132: TOLERABILITY AND EFFICACY OF THE FIRST-IN-CLASS ANTI-CD47 ANTIBODY MAGROLIMAB COMBINED WITH AZACITIDINE IN FRONTLINE PATIENTS WITH TP53-MUTATED ACUTE MYELOID LEUKEMIA: PHASE 1B RESULTS. Hemasphere 6(Suppl.), 33–4 (2022). [Google Scholar]
- 70.Oronsky B, Carter C, Reid T, et al. Just eat it: A review of CD47 and SIRP-α antagonism. Seminars in Oncology 47(2), 117–24 (2020). [DOI] [PubMed] [Google Scholar]
- 71.Petrova PS, Viller NN, Wong M, et al. TTI-621 (SIRPalphaFc): A CD47-Blocking Innate Immune Checkpoint Inhibitor with Broad Antitumor Activity and Minimal Erythrocyte Binding. Clin. Cancer Res 23, 1068–79 (2017). [DOI] [PubMed] [Google Scholar]
- 72.Daver NG, Maris M, Ramchandren R, et al. PB1820: CD47-BLOCKER TTI-622 COMBINED WITH AZACITIDINE IN PATIENTS WITH TP53-MUTATED ACUTE MYELOID LEUKEMIA (AML) AND WITH AZACITIDINE + VENETOCLAX IN ELDERLY OR UNFIT PATIENTS WITH TP53-WILDTYPE AML. Hemasphere 6(Suppl.), 1700–1 (2022). [Google Scholar]
- 73.Chan H, Trout C, Mikolon D, et al. Discovery and Preclinical Characterization of CC-95251, an Anti-SIRPα Antibody That Enhances Macrophage-Mediated Phagocytosis of Non-Hodgkin Lymphoma (NHL) Cells When Combined with Rituximab. Blood 138, 2271 (2021). [Google Scholar]
- 74.Narla RK, Modi H, Bauer D, et al. Modulation of CD47-SIRPα innate immune checkpoint axis with Fc-function detuned anti-CD47 therapeutic antibody. Cancer Immunol. Immunother 71, 473–89 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hintzen RQ, Lens SM, Beckmann MP, et al. Characterization of the human CD27 ligand, a novel member of the TNF gene family. J. Immunol 152(4), 1762–73 (1994). [PubMed] [Google Scholar]
- 76.Wajant H Therapeutic targeting of CD70 and CD27. Expert Opinion on Therapeutic Targets 20(8), 959–73 (2016). [DOI] [PubMed] [Google Scholar]; • Concise review of the CD27/CD70 receptor/ligand and strategies for targeting it
- 77.Riether C, Schürch CM, Bührer ED, et al. CD70/CD27 signaling promotes blast stemness and is a viable therapeutic target in acute myeloid leukemia. Journal of Experimental Medicine 214(2), 359–80 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Riether C, Pabst T, Höpner S, et al. Targeting CD70 with cusatuzumab eliminates acute myeloid leukemia stem cells in patients treated with hypomethylating agents. Nature Medicine 26(9), 1459–67 (2020). [DOI] [PubMed] [Google Scholar]
- 79.Roboz GJ, Pabst T, Aribi A, et al. Safety and Efficacy of Cusatuzumab in Combination with Venetoclax and Azacitidine (CVA) in Patients with Previously Untreated Acute Myeloid Leukemia (AML) Who Are Not Eligible for Intensive Chemotherapy; An Open-Label, Multicenter, Phase 1b Study. Blood 138(Supplement 1), 369 (2021). [Google Scholar]
- 80.Chiffoleau E C-type lectin-like receptors as emerging orchestrators of sterile inflammation represent potential therapeutic targets. Front. Immunol 9, 227 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Neumann K, Castineiras-Vilarino M, Hockendorf U, et al. Clec12a is an inhibitory receptor for uric acid crystals that regulates inflammation in response to cell death. Immunity 40, 389–99 (2014). [DOI] [PubMed] [Google Scholar]
- 82.Bakker AB, van den Oudenrijn S, Bakker AQ, et al. C-type lectin-like molecule-1: a novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res. 64, 8443–50 (2004). [DOI] [PubMed] [Google Scholar]
- 83.Marshall AS, Willment JA, Lin HH, et al. Identification and characterization of a novel human myeloid inhibitory C-type lectin-like receptor (MICL) that is predominantly expressed on granulocytes and monocytes. J. Biol. Chem 279, 14792–802 (2004). [DOI] [PubMed] [Google Scholar]
- 84.Darwish NH, Sudha T, Godugu K, et al. Acute myeloid leukemia stem cell markers in prognosis and targeted therapy: potential impact of BMI-1, TIM-3 and CLL-1. Oncotarget 7, 57811–20 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen CH, Floyd H, Olson NE, et al. Dendritic-cell-associated C-type lectin 2 (DCAL-2) alters dendritic-cell maturation and cytokine production. Blood 107, 1459–67 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bill M, van Kooten Niekerk PB, Woll PS, et al. Mapping the CLEC12A expression on myeloid progenitors in normal bone marrow; implications for understanding CLEC 12A-related cancer stem cell biology. J. Cell Mol. Med 22, 2311–8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Study of CLL-1 expression patterns on myeloid progenitors which has important implications for CLL-1 as a biomarker and therapeutic target for AML
- 87.Zheng B, Yu SF, Rosario G, et al. An anti–CLL-1 antibody–drug conjugate for the treatment of acute myeloid leukemia. Clin. Cancer Res 25(4), 1358–68 (2019). [DOI] [PubMed] [Google Scholar]
- 88.Matthews DC, Appelbaum FR, Eary JF, et al. Phase I study of (131)I-anti-CD45 antibody plus cyclophosphamide and total body irradiation for advanced acute leukemia and myelodysplastic syndrome. Blood 94(4), 1237–47 (1999). [PubMed] [Google Scholar]
- 89.Gyurkocza B, Nath R, Seropian SE, et al. High Rates of Transplantation in the Phase III Sierra Trial Utilizing Anti-CD45 (Iodine) 131I-Apamistamab (Iomab-B) Conditioning with Successful Engraftment and Tolerability in Relapsed/Refractory (R/R) Acute Myeloid Leukemia (AML) Patients after Lack of Response to Conventional Care and Targeted Therapies. Tandem Meetings of ASTCT and CIBMTR 22, Abstract 40 (2022). [Google Scholar]
- 90.Monney L, Sabatos C, Gaglia J, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 415, 536–41 (2002). [DOI] [PubMed] [Google Scholar]
- 91.Sabatos C, Chakravarti S, Cha E, et al. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat. Immunol 4, 1102–10 (2003). [DOI] [PubMed] [Google Scholar]
- 92.Tan J, Huang S, Huang J, et al. Increasing Tim-3+CD244+, Tim-3+CD57+, and Tim-3+PD-1+ T Cells in Patients with Acute Myeloid Leukemia. Asia Pac. J. Clin. Oncol 16(3), 137–41 (2020). [DOI] [PubMed] [Google Scholar]
- 93.Kikushige Y, Miyamoto T, Yuda J, et al. A TIM-3/Galectin-9 Autocrine Stimulatory Loop Drives Self-Renewal of Human Myeloid Leukemia Stem Cells and Leukemia Progression. Cell Stem Cell 17, 341–52 (2016). [DOI] [PubMed] [Google Scholar]
- 94.Goncalves Silva I, Ruegg L, Gibbs BF, et al. The Immune Receptor Tim-3 Acts as a Trafficker in a Tim-3/Galectin-9 Autocrine Loop in Human Myeloid Leukemia Cells. Oncoimmunology 5(7), e1195535 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Goncalves Silva I, Yasinska IM, Sakhnevych SS, et al. The Tim-3-Galectin-9 Secretory Pathway is Involved in the Immune Escape of Human Acute Myeloid Leukemia Cells. EBioMedicine 22, 44–57 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schwartz S, Patel N, Longmire T, et al. Characterization of sabatolimab, a novel immunotherapy with immuno-myeloid activity directed against TIM-3 receptor. Immunother. Adv 2(1), ltac019 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Brunner AM, Esteve J, Porkka K, et al. Efficacy and safety of sabatolimab (MBG453) in combination with hypomethylating agents (HMAs) in patients (Pts) with very high/high-risk myelodysplastic syndrome (vHR/HR-MDS) and acute myeloid leukemia (AML): final analysis from a phase Ib study. Blood 138(Supplement 1), 244 (2021). [Google Scholar]
- 98.Zeidan AM, Westermann J, Kovacsovics T, et al. AML-484 First Results of a Phase II Study (STIMULUS-AML1) Investigating Sabatolimab + Azacitidine + Venetoclax in Patients with Newly Diagnosed Acute Myeloid Leukemia (ND AML). Clin. Lymphoma Myeloma Leuk 22(Supplement 2), S255 (2022). [Google Scholar]
- 99.Farber M, Chen Y, Arnold L, et al. Targeting CD38 in acute myeloid leukemia interferes with leukemia trafficking and induces phagocytosis. Sci. Rep 11, 22062 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhu C, Song Z, Wang A, et al. Isatuximab Acts Through Fc-Dependent, Independent, and Direct Pathways to Kill Multiple Myeloma Cells. Front. Immunol 11, 1771 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rosnet O, Bühring HJ, Marchetto S, et al. Human FLT3/FLK2 receptor tyrosine kinase is expressed at the surface of normal and malignant hematopoietic cells. Leukemia 10(2), 238–48 (1996). [PubMed] [Google Scholar]
- 102.Daver NG, Schlenk RF, Russell NH, et al. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia 33, 299–312 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Perl AE, Martinelli G, Cortes JE, et al. Gilteritinib or chemotherapy for relapsed or refractory-mutated AML. NEJM 381, 1728–40 (2019). [DOI] [PubMed] [Google Scholar]
- 104.Hofmann M, Große-Hovest L, Nübling T, et al. Generation, selection and preclinical characterization of an Fc-optimized FLT3 antibody for the treatment of myeloid leukemia. Leukemia 26, 1228–37 (2012). [DOI] [PubMed] [Google Scholar]
- 105.Heitmann JS, Dörfel D, Kayser S, et al. First-in-human phase I dose escalation and expansion study evaluating the Fc optimized FLT3 antibody Flysyn in acute myeloid leukemia patients with minimal residual disease. Blood 136, 8–9 (2020).32614959 [Google Scholar]
- 106.Morone S, Augeri S, Cuccioloni M, et al. Binding of CD157 protein to fibronectin regulates cell adhesion and spreading. J. Biol. Chem 289, 15588–601 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yakymiv Y, Augeri S, Bracci C, et al. CD157 signaling promotes survival of acute myeloid leukemia cells and modulates sensitivity to cytarabine through regulation of anti-apoptotic Mcl-1. Sci. Rep 11, 21230 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Krupka C, Lichtenegger FS, Kohnke T, et al. Targeting CD157 in AML using a novel, Fc-engineered antibody construct. Oncotarget 8, 35707–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yakymiv Y, Augeri S, Fissolo G, et al. CD157: From Myeloid Cell Differentiation Marker to Therapeutic Target in Acute Myeloid Leukemia. Cells 8(12), 1580 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gorczynski RM. CD200:CD200R-Mediated Regulation of Immunity. ISRN Immunology 2012, 682168 (2012). [Google Scholar]
- 111.Ho JM, Dobson SM, Voisin V, et al. CD200 expression marks leukemia stem cells in human AML. Blood Adv. 4, 5402–13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kretz-Rommel A, Qin F, Dakappagari N, et al. Blockade of CD200 in the Presence or Absence of Antibody Effector Function: Implications for Anti-CD200 Therapy. J. Immunol 180(2), 699–705 (2008). [DOI] [PubMed] [Google Scholar]
- 113.Herbrich S, Baran N, Cai T, et al. Overexpression of CD200 is a stem cell-specific mechanism of immune evasion in AML. Journal for ImmunoTherapy of Cancer 9, e002968 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu J, Wu Q, Shi J, et al. LILRB4, from the immune system to the disease target. American Journal of Translational Research 12(7), 3149–66 (2020). [PMC free article] [PubMed] [Google Scholar]
- 115.Colonna M, Navarro F, Bellón T, et al. A Common Inhibitory Receptor for Major Histocompatibility Complex Class I Molecules on Human Lymphoid and Myelomonocytic Cells. J. Exp. Med 186(11), 1809–18 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Identification of LILR family of receptors
- 116.Borges L, Hsu ML, Fanger N, et al. A family of human lymphoid and myeloid Ig-like receptors, some of which bind to MHC class I molecules. J. Immunol 159(11), 5192–6 (1997). [PubMed] [Google Scholar]
- 117.Abdallah F, Coindre S, Gardet M, et al. Leukocyte Immunoglobulin-Like Receptors in Regulating the Immune Response in Infectious Diseases: A Window of Opportunity to Pathogen Persistence and a Sound Target in Therapeutics. Front. Immunol 12, 717998 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Relevant overview of physiologic roles of LILRs in pathogen response
- 118.Kang X, Kim J, Deng M, et al. Inhibitory leukocyte immunoglobulin-like receptors: Immune checkpoint proteins and tumor sustaining factors. Cell Cycle 15(1), 25–40 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Concise and cancer-specific summary of LILR biology
- 119.Shaw AS, Gauen LK, Zhu Y. Interactions of TCR tyrosine based activation motifs with tyrosine kinases. Seminars in Immunology 7, 13–20 (1995). [DOI] [PubMed] [Google Scholar]
- 120.Isakov N Immunoreceptor tyrosine-based activation motif (ITAM), a unique module linking antigen and Fc receptors to their signaling cascades. J. Leukoc. Biol 61, 6–16 (1997). [DOI] [PubMed] [Google Scholar]
- 121.van der Touw W, Chen HM, Pan PY, et al. LILRB receptor-mediated regulation of myeloid cell maturation and function. Cancer Immunol. Immunother 66(8), 1079–87 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Daeron M, Latour S, Malbec O, et al. The same tyrosine-based inhibition motif, in the intracytoplasmic domain of Fc gamma RIIB, regulates negatively BCR-, TCR-, and FcR-dependent cell activation. Immunity 3, 635–46 (1995). [DOI] [PubMed] [Google Scholar]
- 123.Cella M, Döhring C, Samaridis J, et al. A novel inhibitory receptor (ILT3) expressed on monocytes, macrophages, and dendritic cells involved in antigen processing. J. Exp. Med 185(10), 1743–51 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Initial discovery of the myeloid immune checkpoint LILRB4/ILT3
- 124.Katz HR. Inhibition of inflammatory responses by leukocyte Ig-like receptors. Adv. Immunol 91, 251–72 (2006). [DOI] [PubMed] [Google Scholar]
- 125.Kim-Schulze S, Seki T, Vlad G, et al. Regulation of ILT3 gene expression by processing of precursor transcripts in human endothelial cells. Am. J. Transplant 6(1), 76–82 (2006). [DOI] [PubMed] [Google Scholar]
- 126.Barkal AA, Weiskopf K, Kao KS, et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat. Immunol 19(1), 76–84 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Deng M, Chen H, Liu X, et al. Leukocyte immunoglobulin-like receptor subfamily B: therapeutic targets in cancer. Antibody Therapeutics 4(1), 16–33 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Comprehensive and cancer-specific summary of LILRB therapeutic targets
- 128.Fleming V, Hu X, Weber R, et al. Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front. Immunol 9, 398 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Deng M, Gui X, Kim J, et al. LILRB4 signalling in leukaemia cells mediates T cell suppression and tumour infiltration. Nature 562(7728), 605–9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Critical connection discovered between LILRB4 signaling and AML
- 130.Tedla N, Lee CW, Borges L, et al. Differential expression of leukocyte immunoglobulin-like receptors on cord-blood-derived human mast cell progenitors and mature mast cells. J. Leukoc. Biol 83, 334–43 (2008). [DOI] [PubMed] [Google Scholar]
- 131.Inui M, Hirota S, Hirano K, et al. Human CD43+ B cells are closely related not only to memory B cells phenotypically but also to plasmablasts developmentally in healthy individuals. Int. Immunol 27, 345–55 (2015). [DOI] [PubMed] [Google Scholar]
- 132.Ulges A, Klein M, Reuter S, et al. Protein kinase CK2 enables regulatory T cells to suppress excessive TH2 responses in vivo. Nat. Immunol 16, 267–75 (2015). [DOI] [PubMed] [Google Scholar]
- 133.Mori Y, Tsuji S, Inui M, et al. Inhibitory immunoglobulin-like receptors LILRB and PIR-B negatively regulate osteoclast development. J. Immunol 181(7), 4742–51 (2008). [DOI] [PubMed] [Google Scholar]
- 134.Cheng H, Mohammed F, Nam G, et al. Crystal structure of leukocyte Ig-like receptor LILRB4 (ILT3/LIR-5/CD85k): a myeloid inhibitory receptor involved in immune tolerance. The Journal of Biological Chemistry 286(20), 18013–25 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Su MT, Inui M, Wong YL, et al. Blockade of checkpoint ILT3/LILRB4/gp49B binding to fibronectin ameliorates autoimmune disease in BXSB/Yaa mice. Int. Immunol 33, 447–58 (2021). [DOI] [PubMed] [Google Scholar]
- 136.Lu HK, Rentero C, Raftery MJ, et al. Leukocyte Ig-like receptor B4 (LILRB4) is a potent inhibitor of FcgammaRI-mediated monocyte activation via dephosphorylation of multiple kinases. J. Biol. Chem 284, 34839–48 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study has crucial therapeutic implications for antibodies targeting LILRB4
- 137.Park M, Raftery MJ, Thomas PS, et al. Leukocyte immunoglobulin-like receptor B4 regulates key signalling molecules involved in FcgammaRI-mediated clathrin-dependent endocytosis and phagocytosis. Sci. Rep 6, 35085 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Manavalan JS, Kim-Schulze S, Scotto L, et al. Alloantigen specific CD8+CD28− FOXP3+ T suppressor cells induce ILT3+ ILT4+ tolerogenic endothelial cells, inhibiting alloreactivity. Int. Immunol 16, 1055–68 (2004). [DOI] [PubMed] [Google Scholar]
- 139.Chang CC, Liu Z, Vlad G, et al. Ig-like transcript 3 regulates expression of proinflammatory cytokines and migration of activated T cells. J. Immunol 182(9), 5208–16 (2009). [DOI] [PubMed] [Google Scholar]
- 140.Li Z, Deng M, Huang F, et al. LILRB4 ITIMs mediate the T cell suppression and infiltration of acute myeloid leukemia cells. Cellular & Molecular Immunology 17(3), 272–82 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Paavola KJ, Roda JM, Lin VY, et al. The fibronectin-ILT3 interaction functions as a stromal checkpoint that suppresses myeloid cells. Cancer Immunol. Res 9, 1283–97 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Essential study for understanding of LILRB4 as a stromal checkpoint
- 142.Gui X, Deng M, Song H, et al. Disrupting LILRB4/APOE Interaction by an Efficacious Humanized Antibody Reverses T-cell Suppression and Blocks AML Development. Cancer Immunol. Res 7(8), 1244–57 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Overview of the screening process and functional assays used to discover anti-LILRB4 blocking antibodies
- 143.John S, Chen H, Deng M, et al. A novel anti-LILRB4 CAR-T cell for the treatment of monocytic AML. Mol. Ther 26, 2487–95 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Anami Y, Deng M, Gui X, et al. LILRB4-targeting antibody-drug conjugates for the treatment of acute myeloid leukemia. Mol. Cancer Ther 19, 2330–9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.DiNardo CD, Pollyea DA, Konopleva M, et al. A First-in-human (FIH) phase 1 study of the anti-LILRB4 antibody IO-202 in relapsed/refractory (R/R) myelomonocytic and monocytic acute myeloid leukemia (AML) and R/R chronic myelomonocytic leukemia (CMML). Blood 136(Supplement 1), 19–20 (2020). [Google Scholar]
- 146.Godal R, Bachanova V, Gleason M, et al. Natural killer cell killing of acute myelogenous leukemia and acute lymphoblastic leukemia blasts by killer cell immunoglobulin-like receptor-negative natural killer cells after NKG2A and LIR-1 blockade. Biol. Blood Marrow Transplant 16, 612–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wu G, Xu Y, Schultz RD, et al. LILRB3 supports acute myeloid leukemia development and regulates T-cell antitumor immune responses through the TRAF2-cFLIP-NF-κB signaling axis. Nat. Cancer 2(11), 1170–84 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kontermann RE, Brinkmann U. Bispecific antibodies. Drug Discovery Today 20(7), 838–47 (2015). [DOI] [PubMed] [Google Scholar]
- 149.Brinkmann U, Kontermann RE. The making of bispecific antibodies. mAbs 9(2), 182–212 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Krishnamurthy A, Jimeno A. Bispecific antibodies for cancer therapy: A review. Pharmacology and Therapeutics 185, 122–34 (2018). [DOI] [PubMed] [Google Scholar]
- 151.Labrijn AF, Janmaat ML, Reichert JM, et al. Bispecific antibodies: a mechanistic review of the pipeline. Nat. Rev. Drug Discov 18, 585–608 (2019). [DOI] [PubMed] [Google Scholar]; •• Extremely comprehensive overview of bispecific antibody formats
- 152.De Gast GC, Van Houten AA, Haagen IA, et al. Clinical experience with CD3 x CD19 bispecific antibodies in patients with B cell malignancies. J. Hematother 4(5), 433–7 (1995). [DOI] [PubMed] [Google Scholar]
- 153.Jen EY, Xu Q, Schetter A, et al. FDA Approval: Blinatumomab for Patients with B-cell Precursor Acute Lymphoblastic Leukemia in Morphologic Remission with Minimal Residual Disease. Clin. Cancer Res 25(2), 473–7 (2019). [DOI] [PubMed] [Google Scholar]
- 154.Laszlo GS, Gudgeon CJ, Harrington KH, et al. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood 123(4), 554–61 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Aigner M, Feulner J, Schaffer S, et al. T lymphocytes can be effectively recruited for ex vivo and in vivo lysis of AML blasts by a novel CD33/CD3-bispecific BiTE antibody construct. Leukemia 27(5), 1107–15 (2013). [DOI] [PubMed] [Google Scholar]
- 156.Daver NG, Alotaibi AS, Bücklein V, et al. T-cell-based immunotherapy of acute myeloid leukemia: current concepts and future developments. Leukemia 35(7), 1843–63 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Excellent and current review of T-cell modifying therapies for AML
- 157.Ravandi F, Walter RB, Subklewe M, et al. Updated Results from Phase I Dose-Escalation Study of AMG 330, a Bispecific T-Cell Engager Molecule, in Patients with Relapsed/Refractory Acute Myeloid Leukemia (R/R AML). J. Clin. Oncol 38, 7508 (2020). [Google Scholar]
- 158.Clark MC, Stein A. CD33 directed bispecific antibodies in acute myeloid leukemia. Best Practice and Research: Clinical Haematology 33(4), 101224 (2020). [DOI] [PubMed] [Google Scholar]
- 159.Subklewe M, Stein A, Walter RB, et al. Preliminary Results from a Phase 1 First-in-Human Study of AMG 673, a Novel Half-Life Extended (HLE) Anti-CD33/CD3 BiTE (Bispecific T-Cell Engager) in Patients with Relapsed/Refractory (R/R) Acute Myeloid Leukemia (AML). Blood 134, 833 (2019). [Google Scholar]
- 160.Westervelt P, Roboz GJ, Cortes JE, et al. Phase 1 first-in-human Trial of AMV564, a bivalent bispecific (2×2) CD33/CD3 T-cell engager, in patients with relapsed/refractory acute myeloid leukemia (AML). Blood 132(Supplement 1), 1455 (2018).30111608 [Google Scholar]
- 161.Guy DG, Uy GL. Bispecific Antibodies for the Treatment of Acute Myeloid Leukemia. Current Hematologic Malignancy Reports 13(6), 417–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Nair-Gupta P, Diem M, Reeves D, et al. A novel C2 domain binding CD33xCD3 bispecific antibody with potent T-cell redirection activity against acute myeloid leukemia. Blood Adv. 4(5), 906–19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ravandi F, Bashey A, Stock W, et al. Complete Responses in Relapsed/Refractory Acute Myeloid Leukemia (AML) Patients on a Weekly Dosing Schedule of Vibecotamab (XMAB14045), a CD123 x CD3 T Cell-Engaging Bispecific Antibody; Initial Results of a Phase 1 Study. Blood 136(Supplement 1), 4–5 (2020).32614961 [Google Scholar]
- 164.Moore PA, Zhang W, Rainey GJ, et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood 117, 4542–51 (2011). [DOI] [PubMed] [Google Scholar]
- 165.Isidori A, Cerchione C, Daver NG, et al. Immunotherapy in Acute Myeloid Leukemia: Where We Stand. Frontiers in Oncology 11, 656218 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Uy GL, Aldoss I, Foster MC, et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood 137(6), 751–62 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Alderson RF, Huang L, Zhang X, et al. Combinatorial Anti-Tumor Activity in Animal Models of a Novel CD123 x CD3 Bispecific Dart® Molecule (MGD024) with Cytarabine, Venetoclax or Azacitidine Supports Combination Therapy in Acute Myeloid Leukemia. Blood 138(Supplement 1), 1165 (2021). [Google Scholar]
- 168.Comeau MR, Miller RE, Bader R, et al. APVO436, a bispecific anti-CD123 x anti-CD3 ADAPTIR™ molecule for redirected T-cell cytotoxicity, induces potent T-cell activation, proliferation and cytotoxicity with limited cytokine release. Cancer Res. 78(13_Supplement), Abstract 1786 (2018). [Google Scholar]
- 169.Watts J, Lin TL, Mims A, et al. Post-hoc Analysis of Pharmacodynamics and Single-Agent Activity of CD3xCD123 Bispecific Antibody APVO436 in Relapsed/Refractory AML and MDS Resistant to HMA or Venetoclax Plus HMA. Front. Oncol 11, 806243 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.de Silva S, Fromm G, Shuptrine CW, et al. CD40 Enhances Type I Interferon Responses Downstream of CD47 Blockade, Bridging Innate and Adaptive Immunity. Cancer Immunol. Res 8(2), 230–45 (2010). [DOI] [PubMed] [Google Scholar]
- 171.Cendrowicz E, Jacob L, Greenwald S, et al. DSP107 combines inhibition of CD47/SIRPα axis with activation of 4–1BB to trigger anticancer immunity. J. Exp. Clin. Cancer Res 41(1), 97 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Leong SR, Sukumaran S, Hristopoulos M, et al. An anti-CD3/anti-CLL-1 bispecific antibody for the treatment of acute myeloid leukemia. Blood 129(5), 609–18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.van Loo PF, Hangalapura BN, Thordardottir S, et al. MCLA-117, a CLEC12AxCD3 bispecific antibody targeting a leukemic stem cell antigen, induces T cell-mediated AML blast lysis. Expert Opinion on Biological Therapy 19(7), 721–33 (2019). [DOI] [PubMed] [Google Scholar]
- 174.Mascarenhas J, Huls G, Venditti A, et al. Update from the ongoing phase I multinational study of MCLA-117, a bispecific CLEC12A x CD3 T-cell engager, in patients (pts) with acute myelogenous leukemia (AML). Eur. Hematol. Assoc. Meeting 25, Abstract EP538 (2020). [Google Scholar]
- 175.Lim Y, Lee E, Lee S, et al. A Novel Asymmetrical Anti-CLL-1×CD3 Bispecific Antibody, ABL602, Induces Potent CLL1-Specific Antitumor Activity with Minimized Sensitization of Pro-Inflammatory Cytokines. Blood 138(Supplement 1), 2234 (2021). [Google Scholar]
- 176.Brauchle B, Goldstein RL, Karbowski CM, et al. Characterization of a novel FLT3 BiTE molecule for the treatment of acute myeloid leukemia. Mol. Cancer Ther 19, 1875–88 (2020). [DOI] [PubMed] [Google Scholar]
- 177.Mehta NK, Pfluegler M, Meetze K, et al. A novel IgG-based FLT3xCD3 bispecific antibody for the treatment of AML and B-ALL. Journal for ImmunoTherapy of Cancer 10, e003882 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Surveillance, Epidemiology, and End Results (SEER) SEER*Stat Database: Acute Myeloid Leukemia — Cancer Stat Facts. U. S. National Institutes of Health, National Cancer Institute. https://seer.cancer.gov. Accessed November 29, 2022. [Google Scholar]
- 179.Vallera DA, Felices M, McElmurry R, et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clinical Cancer Research 22(14), 3440–50 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]