

Abstract
Ferroptosis is an iron-dependent form of cell death driven by oxidation of polyunsaturated (PUFA) phospholipids. Large-scale genetic screens have uncovered a specialized role for PUFA ether phospholipids (ePLs) in promoting ferroptosis. Our understanding of the enzymes involved in PUFA ePL production, however, remains incomplete. Here we show using a combination of pathway mining of genetic dependency maps, AlphaFold-guided structure predictions, and targeted lipidomics that the uncharacterized transmembrane protein TMEM164 – genetic ablation of which has been shown to protect cells from ferroptosis – is a cysteine active-center enzyme that selectively transfers C20:4 acyl chains from phosphatidylcholine to lyso-ePLs to produce PUFA-ePLs. Genetic deletion of TMEM164 across a set of ferroptosis-sensitive cancer cell lines caused selective reductions in C20:4-ePLs with minimal effects on C20:4-diacyl PLs, and this lipid profile produced a variable range of protection from ferroptosis, supportive of an important, but contextualized role for C20:4-ePLs in this form of cell death.
Graphical Abstract

Polyunsaturated phospholipids (PUFA-PLs) serve critical functions in cell biology that include modulation of the structure (e.g., fluidity) of membrane bilayers and as precursors for an array of signaling molecules (e.g., eicosanoids, endocannabinoids)1. PUFA-PLs are susceptible to enzymatic and non-enzymatic oxidation and controlling these oxidative processes is critical for cell homeostasis and viability2. For instance, excess PUFA-PL oxidation can lead to ferroptosis, an iron-dependent form of non-apoptotic cell death3. A key enzyme involved in counteracting excessive PUFA oxidation is glutathione peroxidase 4 (GPX4), which converts lipid hydroperoxides to lipid alcohols, and the genetic or pharmacological disruption of GPX4 promotes ferroptosis in susceptible cell types4,5. Ferroptosis has been implicated in a wide range of human diseases, including cancer and various degenerative disorders3. Accordingly, mapping the biochemical pathways that regulate ferroptosis may not only improve our understanding of the mechanistic underpinnings for this cell death process, but also offer therapeutic targets for enhancing or suppressing ferroptosis sensitivity in specific disease settings.
Recent genome-wide CRISPR–Cas9 suppressor screens have pointed to an important role for PUFA ether-PLs (ePLs) in regulating ferroptosis, specifically identifying genes encoding peroxisomal biogenesis proteins or peroxisomal enzymes involved in ePL biosynthesis as pro-ferroptotic6,7. Mechanistic studies further revealed that cancer cells can convert to a ferroptosis-resistant state in vivo that is associated with reductions in PUFA-ePL content6. Concurrent with this work, independent investigations have begun to showcase the potential for genome-wide mapping of co-essential genes to identify orphan members of biochemical pathways, including the discovery that the previously uncharacterized membrane protein TMEM189 plays a key role in ePL biosynthesis through acting as a plasmanylethanolamine desaturase8. Additional studies support this function9,10, as well as the potential for TMEM189, at least in certain settings, to regulate ferroptotic activity7.
Despite the specialized role emerging for PUFA-ePLs in ferroptosis, it is notable that the pro-ferroptotic ePL enzymes identified so far appear to exhibit generalized substrate scopes that lack acyl chain specificity, which contrasts with other lipid metabolic enzymes implicated in ferroptosis such as ACSL4 and LPCAT3 (or MBOAT5) that display a strong preference for PUFA (C20:4 or arachidonate) substrates11–13. Here, we show using a combination of pathway reconstruction based on genetic dependency maps, structural predictions by AlphaFold, lipidomics, and biochemical and cell biological assays that the uncharacterized transmembrane protein TMEM164 acts as a cysteine-dependent, C20:4-preferring acyltransferase that generates PUFA-ePLs in human cells. Notably, TMEM164 uses C20:4-PC (rather than C20:4-CoA) as an acyl chain donor, providing molecular annotation to an enzymatic activity first described ~30 years ago in the literature14,15. We further show that genetic disruption of TMEM164 leads to substantial and selective reductions in C20:4-ePLs and a range of protection from ferroptosis across different human cancer cell lines. These data, taken together, support emerging models describing a context-dependent role for PUFA-ePLs in ferroptosis and designate TMEM164 as a key enzyme specifically responsible for regulating this class of lipids.
Results
TMEM164 shows co-dependency with ePL and PUFA lipid pathways
We hypothesized that additional genes involved in PUFA-ePL metabolism might show co-dependency relationships with established ePL enzymes in the Cancer Dependency Map8,16. Among such genes, TMEM164, which codes for an uncharacterized integral membrane protein, captured our attention because it not only displayed a co-dependency relationship with ePL enzymes (e.g., AGPS, FAR1, TMEM189), but also with enzymes specifically involved in PUFA (C20:4) lipid metabolism (e.g., ACSL4, LPCAT3) (Fig. 1a). Additionally, we noticed that SELENOI, which functions as the final enzyme in the biosynthesis of phosphatidylethanolamine (PE) and ePE17, displayed prominent co-dependency with TMEM164, potentially pointing to a particular role for this protein in ePE metabolism. GO enrichment analysis of the top 40 co-dependencies for TMEM164 also supported a specific association with ether lipid metabolism (Fig. 1b). Interestingly, TMEM164 further showed a stronger connectivity to ACSL4 and LPCAT3 compared to most of the other established ePL enzymes (e.g., FAR1, AGPS, GNPAT) (Fig. 1c), suggesting that TMEM164 may represent a biochemical link between ePL and PUFA lipid pathways. Also consistent with this hypothesis, recent genome-wide CRISPR–Cas9 suppressor screens have revealed that genetic disruption of TMEM164, like other ePL and PUFA lipid pathway members, impairs ferroptosis6,18,19. We independently verified that TMEM164-null (sgTMEM164) 786-O human renal cell carcinoma cells show greatly reduced sensitivity to ferroptosis induction by the GPX4 inhibitors ML210 and RSL3 (Extended Data Fig. 1).
Figure 1. A role for TMEM164 in the regulation of C20:4 ether lipids.
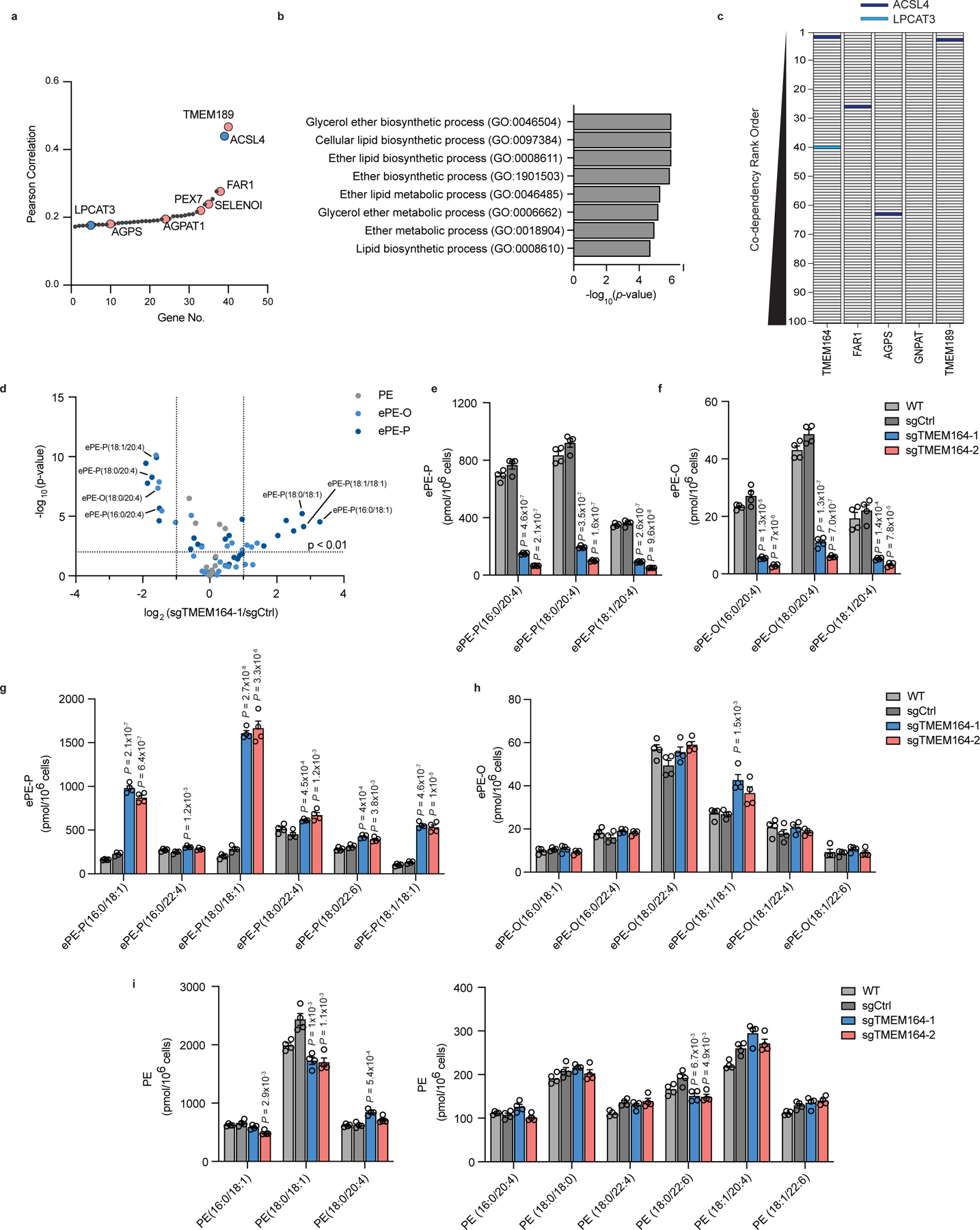
a, Cancer dependency map analysis of TMEM164 showing the genes with the top-40 highest co-dependency scores. Blue and red designate genes involved in C20:4 and ePL metabolism, respectively. b, GO enrichment analysis of the top 40 co-dependency genes with TMEM164. c, Depiction of the location of C20:4 lipid metabolic genes ACSL4 (red) and LPCAT3 (blue) in the relative rank order of the top 100 co-dependencies for TMEM164 versus established ePL-related genes FAR1, AGPS, GNPAT, and TMEM189. Note that LPCAT3 was only found in the top-100 co-dependencies for TMEM164. d, Volcano plot of targeted lipidomic analysis comparing ether PE (ePE) lipids in sgCtrl and sgTMEM164 786-O cells. Data represent mean values from four independent experiments for sgCtrl and sgTMEM164-1 cells. e, f, C20:4 ePE-P (e) and ePE-O (f) lipid measurements in parental (WT), sgCtrl, and sgTMEM164 786-O cells. Two independently generated sgTMEM164 cell populations were analyzed (−1 and −2). g, h, Additional ePE-P (g) and ePE-O (h) lipid measurements in WT, sgCtrl, and sgTMEM164 786-O cells. i, Diacyl PE lipid measurements in WT, sgCtrl, and sgTMEM164 786-O cells. For e-i, data represent mean values ± S.E.M from four independent experiments per group. P-values were derived using a Two-sided Student’s t-test performed relative to sgCtrl cells.
TMEM164 regulates C20:4-ePL production in human cells
We next performed a targeted lipidomic analysis of sgTMEM164 786-O cells using a tandem liquid chromatography mass spectrometry (LC-MS)-based protocol that quantitatively measured ePE lipids, including both acyl chain composition and the presence (plasmenyl, ePE-P) or absence (plasmanyl, ePE-O) of a vinyl ether bond in the sn-1 position (Extended Data Fig. 2). We found that, in comparison to control cells expressing a non-targeted sgRNA (sgCtrl), sgTMEM164 cells displayed major decreases in C20:4 ePEs (Fig. 1d–f and Supplementary Dataset 1) accompanied by substantial elevations in saturated and monounsaturated ePEs (Fig. 1d, g, h and Supplementary Dataset 1). These results pointed to a broad acyl-chain remodeling outcome associated with TMEM164 deletion that is reminiscent of the reshuffling of acyl chains in diacyl PLs that accompanies the genetic or pharmacological disruption of the C20:4-preferring acyltransferases LPCAT3 or MBOAT720–22. One difference in the remodeling profile caused by TMEM164 disruption, however, was the very minor increase in C22:4 ePEs (Fig. 1g, h and Supplementary Dataset 1), which contrasted with the more dramatic elevation in C22:4 PEs found in LPCAT3-null cells21,22. In contrast to the dramatic ePE changes, the diacyl PE content of sgTMEM164 cells was only modestly altered (Fig. 1i and Supplementary Dataset 1). Reductions in C20:4 ether phosphatidylcholine (ePC) lipids were also observed in sgTMEM164 cells, alongside a more substantial elevation in C20:4 diacyl PC lipids (compared to C20:4 diacyl PE lipids) (Extended Data Fig. 3 and Supplementary Dataset 1). Finally, we did not observe changes in free fatty acids (e.g., C20:4, C18:0, C18:1) in sgTMEM164 cells (Extended Data Fig. 4a and Supplementary Dataset 1).
Isotopic tracing experiments provided further evidence that TMEM164 was directly responsible for incorporating C20:4 acyl chains into ePEs. We specifically found that sgTMEM164 786-O cells showed greatly impaired incorporation of deuterated C20:4 [arachidonic acid (AA)-d8] into both ePE-P and ePE-O lipids compared to sgCtrl cells (Fig. 2a, b and Supplementary Dataset 1). In contrast, the genetic disruption of TMEM164 did not substantially affect the incorporation of AA-d8 into diacyl PE pools (Fig. 2c and Supplementary Dataset 1). Interestingly, we observed much greater incorporation of isotopically labeled monounsaturated fatty acid (C18:1, OA-d9) into non-C20:4 ePLs in sgTMEM164 cells (Fig. 2d and Supplementary Dataset 1), mirroring the increased endogenous concentrations of (monoun)saturated ePEs observed in these cells (Fig. 1c, d and Supplementary Dataset 1). This change was likewise restricted to ePLs, as equivalent amounts of monounsaturated diacyl PEs were generated in OA-d9-treated sgCtrl and sgTMEM164 cells (Fig. 2e and Supplementary Dataset 1). Also supportive of a role for TMEM164 in specifically incorporating C20:4 acyl chains into ePEs, we found that sgTMEM164 cells had higher quantities of the (monoun)saturated lyso-ePE (eLPE) co-substrates for this transfer reaction, while lyso-PE (LPE) lipids remained unchanged (Extended Data Fig. 4b, c and Supplementary Dataset 1). This result further offered a potential mechanistic explanation for the elevations in (monoun)saturated ePEs and increased incorporation of OA-d9 into ePEs observed in sgTMEM164 cells, where heightened eLPEs may serve as substrates for alternative acyltransferases that lack C20:4 specificity.
Figure 2. Impaired incorporation of C20:4 (arachidonic acid (AA))-d8 into ePE lipids in TMEM164-deficienct cells.
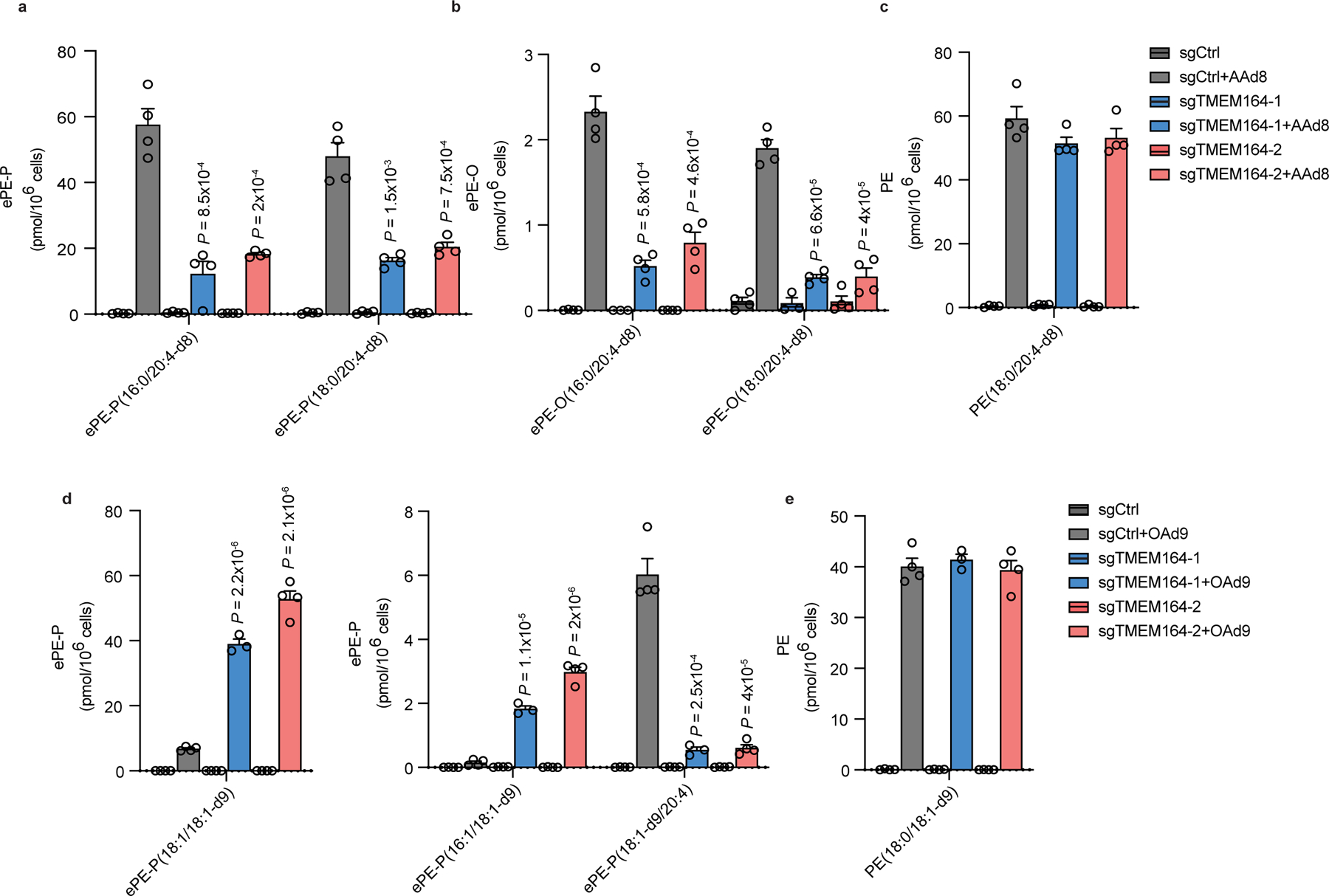
a-c, Measurement of AA-d8 incorporation into C20:4 (a) ePE-P, (b) ePE-O and (c) diacyl PE lipid species in sgCtrl and sgTMEM164 786-O cells. d, e, Measurement of oleic acid (OA; C18:1)-d9 incorporation into C18:1 (d) ePE-P and (e) ester PE lipid species in sgCtrl and sgTMEM164 786-O cells. For a-e, cells were treated with 25 µM of AA-d8 or OA-d9 for 4 h prior to lipid measurements. Data represent mean values ± S.E.M from three-four independent experiments per group. P-values were derived using a Two-sided Student’s t-test performed relative to sgCtrl cells.
Our lipidomic data, taken together, pointed to a specific function for TMEM164 in regulating C20:4 ePLs that was reminiscent of the catalytic activities of well-established acyltransferases from the MBOAT family (LPCAT3, MBOAT7) involved in the formation of C20:4 diacyl phospholipids. However, TMEM164 shared no discernible sequence motif or predicted domain homology with the MBOAT acyltransferases. We therefore considered whether TMEM164 might be part of a distinct transmembrane protein/enzyme family.
Computational evidence that TMEM164 is an acyltransferase
In parallel with our experimental characterization of TMEM164 in ePL metabolism, we were engaged in a separate line of inquiry involving integrated deep homology searches and AlphaFold2 modeling23 of the AIG1/ADTRP family of animal and fungal multi-pass transmembrane (TM) proteins (PFAM domain family PF04750) that we recently discovered to hydrolyze the FAHFA (fatty acid esters of hydroxylated fatty acids) class of lipids through a likely Thr/His catalytic dyad24. Iterative PsiBLAST25 and HhPRED26 searches detected two distant orphan clusters at 10–15% identity: i) YwaF proteins in bacteria and archaea (PF09529), and ii) TMEM164 proteins in animals (PF14808). The derivation of structurally accurate AlphaFold2 models23, either from proteome model databases27 or built with ColabFold28, allowed for a broader structural comparison with DALI29 unhindered by low to negligible sequence similarity that, in conjunction with reciprocal homology searches, linked the AIG1/ADTRP, YwaF, and TMEM164 families to additional bacterial/archaeal (YpjA) and protozoa (Hypo(thetical)) proteins (Fig. 3a and Extended Data Fig. 5a–c).
Figure 3. Integrated homology mapping and AlphaFold2 analysis identifies a predicted shared structure between TMEM164 and the AIG1/ADTRP lipid hydrolases.
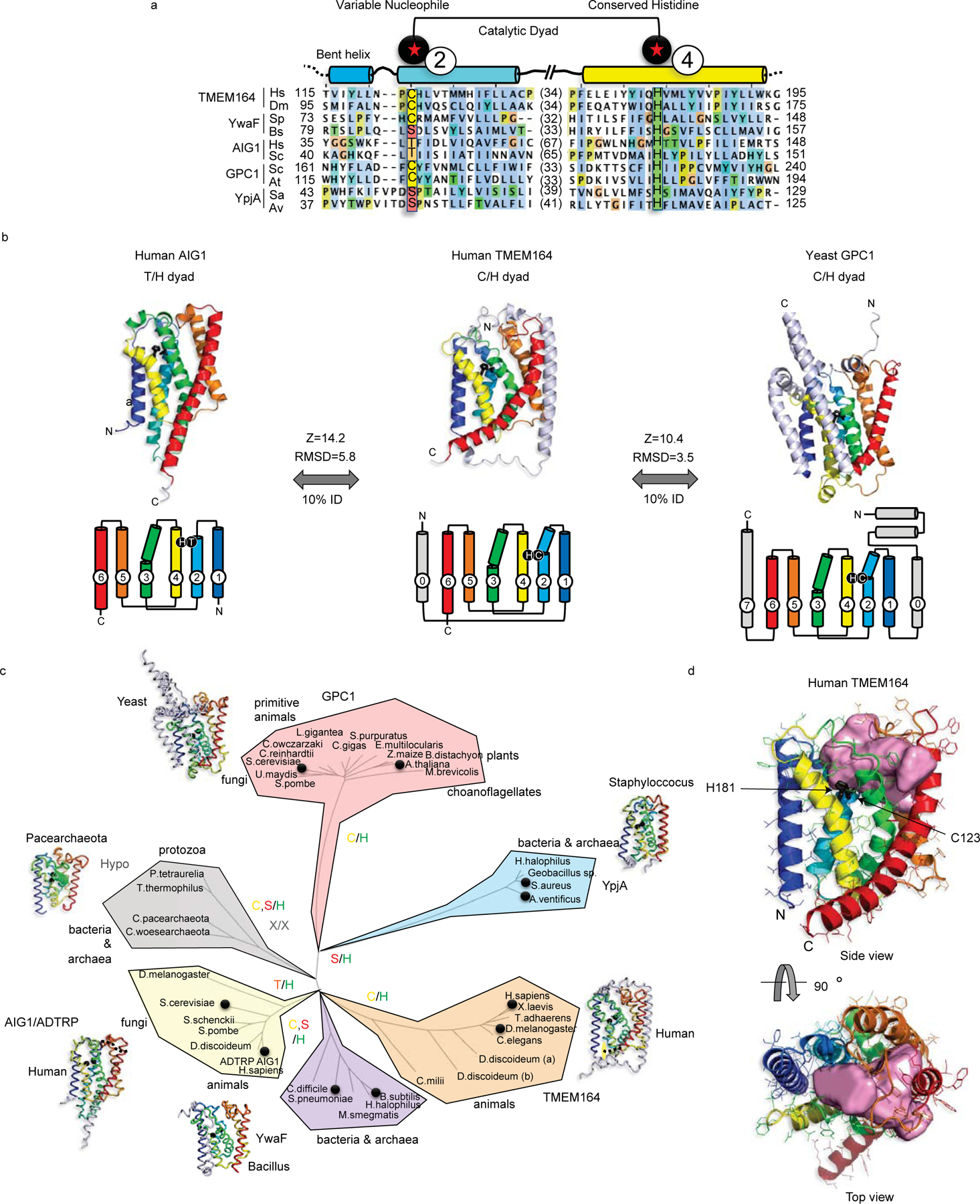
a, DALI superposition of the AlphaFold2-predicted structures of human (H.sapiens; Hs) and D. melanogaster (Dm) TMEM164 reveals a catalytic dyad of C123 and H181 in the human chain (respectively on TM helices 2 and 4 of the conserved 6TM fold) that aligns with the Thr/His dyad of AIG1-class FAHFA hydrolases (from H.sapiens and S. cerevisiae (Sc)), and defines a superfamily of nucleophile-variable/His-invariant enzymes that share a conserved membrane topology with otherwise sparse sequence identity. Prokaryotic YwaF and YpjA enzymes respectively include S.pneumoniae (Sp) and B.subtilis (Bs), and S.aureus (Sa) and A.ventificus (Av) proteins, while GPC1 enzymes are drawn from S.cerevisiae and A.thaliana (At). See Extended Data Fig. 5 for a more complete sequence alignment. b, Fold and topology comparison of human TMEM164 with human AIG1 and yeast GPC1 enzymes highlighting a 6TM core fold (with helices 1–6 color-ramped to match the topology diagrams; additional helices in grey) with active site Cys or Thr nucleophiles in blue TM2, placed opposite to the invariant His in yellow TM4 (sidechains in black stick format). Z-scores and RMSD values (in Å) for comparison of TMEM164 and AIG1 or GPC1 AlphaFold2 models are provided. c, Evolutionary tree of the AIG1/TMEM164/GPC1/YwaF/YpjA (or ATGY) superfamily drawn by DALI-derived fold similarity with variable nucleophilic residues noted in each branch. Black circles mark sequences present in the abridged alignment in a. d, CavityPlus analysis of the human TMEM164 6TM core showing an internal cavity (with predicted portals to the lipid bilayer) that abuts the C123/H181 catalytic dyad (in black stick form). Figures drawn with Jalview (http://jalview.org) and PyMOL (http://pymol.org).
The AlphaFold2 model showed the preservation of a six transmembrane (6TM) helical core across all of the families (Fig. 3b, c and Extended Data 5d). Interestingly, however, the equivalent residues to the Thr/His catalytic dyad in the AIG1/ADTRP family were substituted with a conserved Cys/His pair in TMEM164 proteins, while YwaF and YpiA proteins variably displayed Ser, Thr, or Cys nucleophilic residues alongside the invariant His (Fig. 3a, b and Extended Data Fig. 5a, d). The loose cluster of hypothetical proteins (in bacteria, archaea and protozoa) showed divergent or missing active site residues (Fig. 3c and Extended Data Fig. 5b, c). The conserved 6TM core of human TMEM164, trained against the UniProt-enlarged AlphaFold2 model databases with the fast structure search engines of RUPEE and FoldSeek30,31, collected one additional fold family at less than 10% sequence identity (that had eluded HhPRED queries), centered in fungi, plants, and primitive animals (PF10998), and headlined by the yeast glycerophosphocholine acyltransferase GPC1 (or GPCAT)32 (Fig. 3a–c and Extended Data Fig. 5a–d). Superposition of distant TMEM164 and GPC1 enzymes revealed the shared presence of the predicted Cys/His catalytic dyad in the 6TM core (Fig. 3a–c).
The AlphaFold2-predicted structure of human TMEM164 analyzed by CASTp and CavityPlus33,34 further revealed an internal cavity with predicted portals to the lipid bilayer and the cytosol abutting the C123/H181 putative catalytic dyad (Fig. 3d). CavPHARMER34 produced a pharmacophore scaffold within the TMEM164 internal cavity that was compatible with accommodating the C20:4 ePE product of a predicted TMEM164-catalyzed reaction (Extended Data Fig. 6).
These structural modeling studies drawing connectivity to other enzymes with experimentally defined roles in lipid metabolism (AIG1 and GPC1 branches), combined with our lipidomic data, pointed to a function for TMEM164 as a cysteine-dependent integral membrane acyltransferase that specifically generates C20:4 ePLs in human cells.
TMEM164 uses C20:4 PC as an acyl chain donor
In considering candidate substrates for the putative acyltransferase activity of TMEM164, we noted a body literature dating back to the early 1980s describing a transmembrane enzyme activity that transferred the sn-2 C20:4 acyl chain from PC lipids to lyso-ePL to form C20:4 ePL14,15. While this CoA-independent acyltransferase activity (Fig. 4a) has been detected in multiple cell and tissue types14,15, it has remained, to date, molecularly uncharacterized. We found that membrane lysates from sgCtrl 786-O cells formed C20:4 ePE when incubated with deuterated C20:4 PC (and to a lesser extent deuterated C20:4 PE) and lyso-ePE, while this activity was ablated in sgTMEM164 786-O cells (Fig. 4b). No such PC/PE-dependent acyltransferase activity was observed in sgCtrl membrane lysates incubated with saturated or monounsaturated PC or PE lipids (Fig. 4c), consistent with previous studies showing stringent preference for the C20:4 acyl chain14,15. Expression of WT-TMEM164, but not a C123A-TMEM164 mutant, restored the C20:4 PC-dependent acyltransferase activity of membrane lysates from sgTMEM164 cells (Fig. 4d and Extended Data Fig. 7a). We also found that the C20:4 PC-dependent acyltransferase activity of sgCtrl membrane lysates was blocked by the cysteine-directed alkylating agent N-ethylmaleimide (Extended Data Fig. 7b), as has been described previously for this activity in other cell types14,15. Finally, returning to the AlphaFold2 model of the TMEM164 structure, we noted that the internal cavity predicted to bind lipid substrate was sufficient in size to accommodate the C20:4 PC substrate and was also proximal to hydrophobic tunnels extending out to the lipid bilayer of the membrane, which may represent entrance and exit routes for substrates and products of the acyltransferase reaction (Extended Data Fig. 6). These results, taken together, indicate that TMEM164 functions as a cysteine-dependent acyltransferase generating C20:4 ePEs from C20:4 PC and lyso-ePE substrates.
Figure 4. TMEM164 acts as a C20:4-PL-dependent acyltransferase that generates C20:4-ePE lipids.
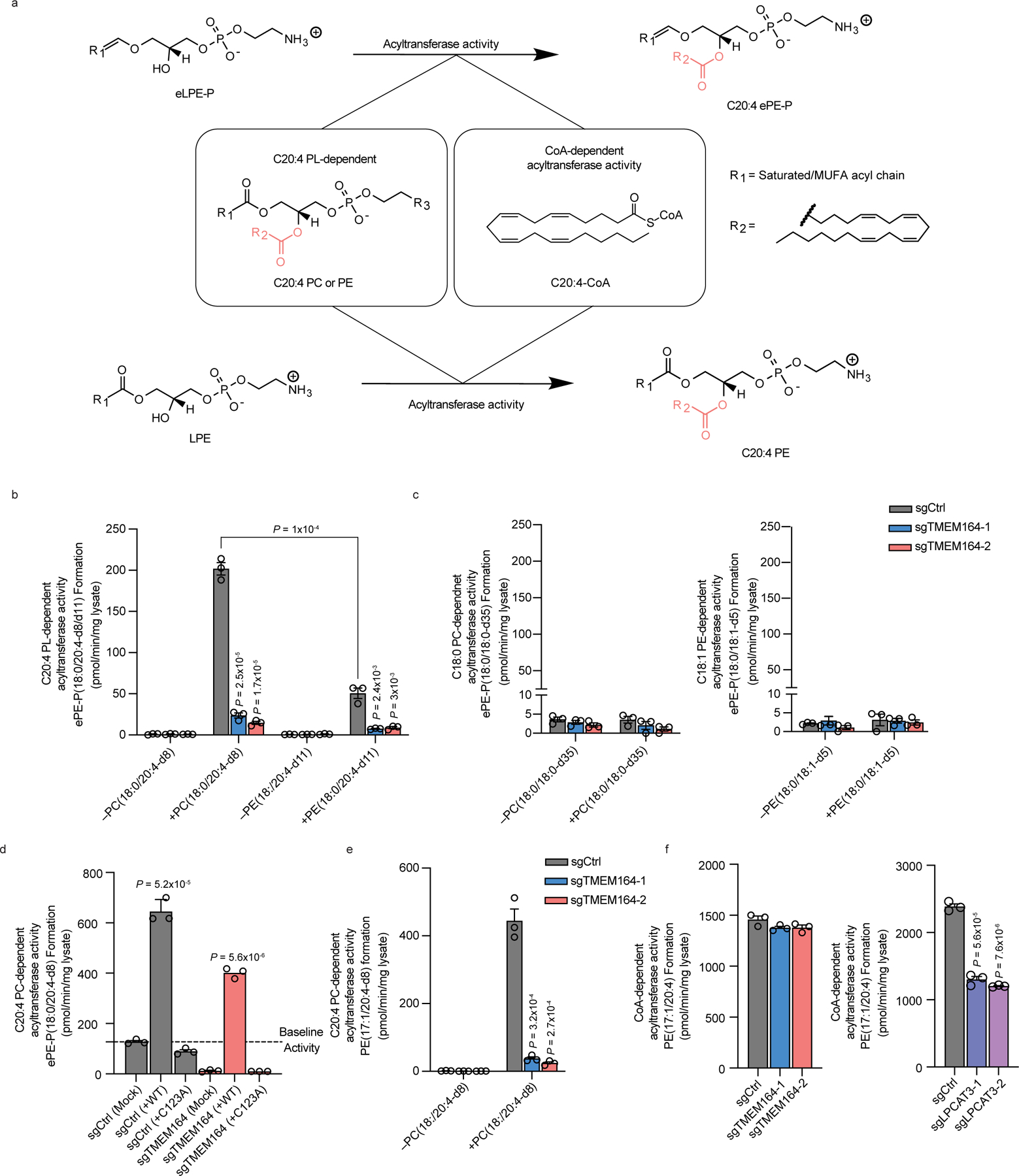
a, Diagram of CoA-dependent or C20:4-phospholipid (PL)-dependent acyltransferase reactions responsible for generating C20:4 (e)PE lipids. b, Measurement of the formation of ePE-P(C18:0/C20:4-d8/d11) from diacyl PC(C18:0/C20:4-d8) or PE(C18:0/C20:4-d11) and lyso-ePE-P(C18:0) as donor and acceptor substrates, respectively, assayed with membrane lysates from sgCtrl and sgTMEM164 786-O cells. c, Measurement of the formation of ePE-P(C18:0/C18:0-d35/d5) from diacyl PC(C18:0/C18:0-d35) or PE(C18:0/C18:1-d5) and lyso-ePE-P(C18:0) as donor and acceptor substrates, respectively, assayed with membrane lysates from sgCtrl and sgTMEM164 786-O cells. d, Measurement of the formation of ePE-P(C18:0/C20:4-d8) from diacyl PC(C18:0/C20:4-d8) and lyso-ePE-P(C18:0) from membrane lysates of sgCtrl and sgTMEM164-2 786-O cells engineered to recombinantly express FLAG-tagged WT-TMEM164 or a C123A-TMEM164 mutant. The cell lysates containing WT-TMEM164 were diluted 1:1 with mock lysate to normalize expression to the C123A-TMEM164 mutant (see Extended Data Fig. 7a). e, Measurement of the formation of diacyl PE(17:1/20:4-d8) from diacyl PC(C18:0/C20:4-d8) and LPE(C17:1) as donor and acceptor substrates, respectively, assayed with membrane lysates from sgCtrl and sgTMEM164 786-O cells. f, Measurement of the formation of diacyl PE(C17:1/C20:4) using C20:4-CoA and LPE(C17:1) as donor and acceptor substrates, respectively, assayed with membrane lysates from sgCtrl, sgTMEM164, and sgLPCAT3 786-O cells. For b-f, membrane lysates (1 µg/µL) were treated with 50 µM each of donor and acceptor substrates for 1 h at 37° C prior to analysis. Data represent mean values ± S.E.M from three independent experiments per group. P-values were derived using a Two-sided Student’s t-test performed relative to sgCtrl cells unless otherwise indicated.
While our findings supported that TMEM164 represents the historically described C20:4 PC-dependent acyltransferase activity responsible for C20:4 ePL formation, we wondered whether this protein might also utilize C20:4 CoA as a substrate. This question initially proved challenging to address, as neither isotopically labeled C20:4 CoA nor nonnatural acyl chain (e.g., C17 acyl chain) variants of lyso-ePLs are commercially available. We observed, however, that sgCtrl, but not sgTMEM164 786-O cell membrane lysates, also supported the formation of C20:4 diacyl PE using deuterated C20:4 PC and C17:1 lyso-PE as acyl chain donor and acceptor lipids, respectively (Fig. 4e). This discovery allowed for subsequent testing of CoA-dependent acyltransferase activity for TMEM164 by incubating sgCtrl and sgTMEM164 membrane lysates with C20:4 CoA and C17:1 lyso-PE and measuring production of the unnatural lipid C20:4/C17:1 PE. Using this assay, we found that sgCtrl and sgTMEM164 lysates showed equivalent C20:4 CoA-dependent acyltransferase activity, which was instead substantially reduced in membrane lysates from sgLPCAT3 cells (Fig. 4f). On the other hand, sgLPCAT3 membrane lysates exhibited unaltered C20:4 PC-dependent acyltransferase activity using both C17:1 lyso-PE and C18:0 lyso-ePE-P as acceptor substrates (Extended Data Fig. 7c, d), indicating that LPCAT3 and TMEM164 serve as exclusively C20:4 CoA- and C20:4 PC-dependent acyltransferases, respectively.
C123 is required for TMEM164 acyltransferase activity
We recombinantly expressed WT-TMEM164 and several C123X point mutants in HEK293T cells and found that none of the C123 mutants exhibited substantial acyltransferase activity (Fig. 5a, b). Several of the C123X mutants expressed at ~50% lower levels than WT-TMEM164 (e.g., C123A, C123S, and C123V), and the C123R mutant was not detectable (Fig. 5a). The C123Y mutant, however, expressed at similar levels to WT-TMEM164 (Fig. 5a), which was a noteworthy finding because base editing methods using a cytidine deaminase convert cysteine residues to tyrosine35. We therefore used cytosine base editing to create a TMEM164-C123Y knock-in population of 786-O cells (Extended Data Fig. 8a) and compared their properties to 786-O cells treated with a sgRNA targeting a non-human (GFP) control gene (sgCtrl). We found that the TMEM164-C123Y cells displayed substantial reductions in C20:4 PC-dependent acyltransferase activity (Fig. 5c) and C20:4 ePE lipids (Fig. 5d, Extended Data Fig. 8b and Supplementary Dataset 1) with concomitant elevations in saturated/monounsaturated ePE lipids (Fig. 5e) and unchanged diacyl PE lipids (Extended Data Fig. 8c and Supplementary Dataset 1). Finally, the TMEM164-C123Y knock-in 786-O cells also displayed reduced sensitivity to GPX4 inhibitor-induced ferroptosis (Fig. 5f and Extended Data Fig. 8d). Considering that the biochemical and cellular changes observed in the TMEM164-C123Y knock-in 786-O cells generally matched those found in TMEM164-null (sgTMEM164) 786-O cells (see Extended Data Fig. 1c, d and Fig. 1d–i), these results indicate that the contributions of TMEM164 to ePL metabolism and ferroptosis depend on the C123-mediated acyltransferase activity of this protein.
Figure 5. Characterization of the enzymatic activity and ferroptosis contributions of TMEM164.
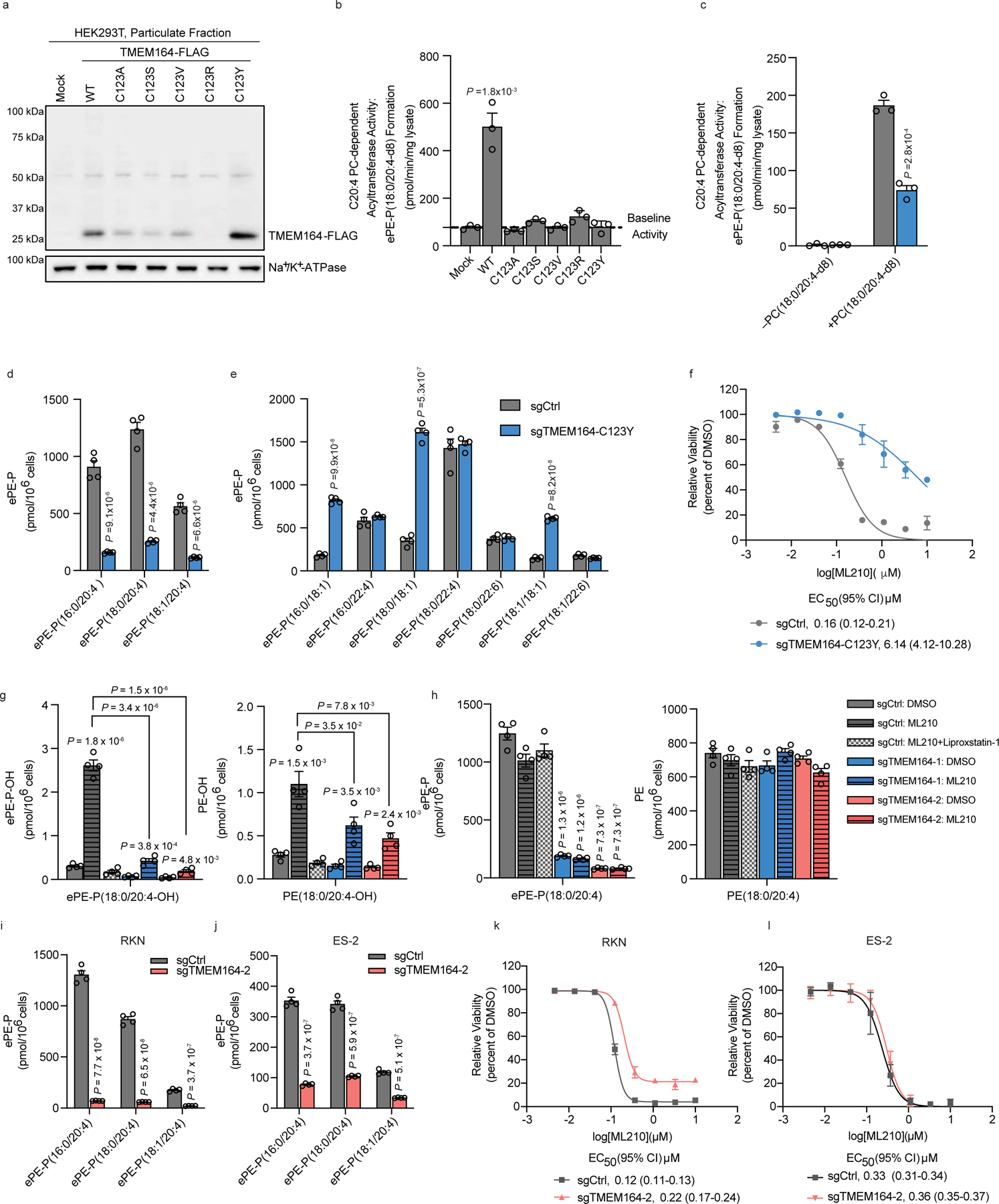
a, Western blot of membrane lysates from HEK293T cells expressing C-terminally FLAG-tagged WT-TMEM164 or C123X-TMEM164 mutants. b, c, Measurement of the formation of ePE-P(C18:0/C20:4-d8) from diacyl PC(C18:0/C20:4-d8) (50 µM) and lyso-e-PE-P(C18:0) (50 µM) as substrates assayed with membrane lysates (1 µg/uL, 1 h incubation at 37° C) from (b) HEK293T cells expressing the indicated TMEM164 proteins or (c) sgCtrl and sgTMEM164-C123Y based-edited 786-O cell populations. d, e, Measurement of C20:4 ePE-P (d) and additional ePE-P (e) lipids in sgCtrl and sgTMEM164-C123Y base-edited 786-O cell populations. f, Viability of sgCtrl and sgTMEM164-C123Y base-edited 786-O cell populations treated with the indicated concentrations of ML210 measured at 24 h post-treatment. g, h, Oxidized (g) and non-oxidized (h) PE(C18:0/C20:4) and ePE-P(C18:0/2C0:4) lipid measurements in sgCtrl and sgTMEM164 786-O cells treated with ML210 for 2 h. sgCtrl: ML210 + Liproxstatin-1 group was pre-treated with the ferroptosis-blocking compound Liproxstatin-1 (0.5 µM) for 2 h prior to ML210 treatment. i, j, C20:4 ePE lipid measurements in sgCtrl and sgTMEM164 RKN (i) and ES-2 (j) cell lines. k,l, Viability of sgCtrl and sgTMEM164 RKN (k) and ES-2 (l) treated with indicated concentration of ML210 measured at 24 h post-treatment. For b and c data represent mean values ± S.E.M from three independent experiments per group. For d-e and g-j, data represent mean values ± S.E.M from four independent experiments per group. *p< 0.01, ** p < 0.001 (Two-sided Student’s t-test performed relative to sgCtrl cells). For f, k and l, data represent mean values ± S.E.M. from two independent experiments. For b,c, d-e and g-j P-values were derived using a Two-sided Student’s t-test performed relative to sgCtrl cells.
Impact of TMEM164 loss on oxidized ePEs and ferroptosis
The primary lipid mediators of ferroptosis are generally accepted to represent oxidized variants of PUFA phospholipids3. Consistent with the ferroptosis resistance displayed by sgTMEM164 786-O cells, we found that ML210-induced formation of oxidized ePE-P(C18:0/C20:4-OH) was substantially suppressed in these cells compared to sgCtrl cells (Fig. 5g and Supplementary Dataset 1). Interestingly, sgTMEM164 786-O cells also showed significant attenuation of ML210-induced oxidized diacyl PE(C18:0/C20:4-OH) (Fig. 5g and Supplementary Dataset 1) despite displaying unchanged diacyl PE(C18:0/C20:4) (Fig. 5h and Supplementary Dataset 1). This result may indicate that a portion of the oxidized diacyl PE(C18:0/C20:4-OH) was generated by bulk elevations in the free radical content of ML210-treated cells36, which in turn may be lowered by the substantial suppression of oxidized ePE-P(C18:0/C20:4-OH) in sgTMEM164 786-O cells (Fig. 5g and Supplementary Dataset 1).
Recent studies have indicated that PUFA ether lipids contribute to ferroptosis in a context-dependent manner and, more generally, suggest that this cell death process may lack unified regulatory pathways37. To explore this concept further, we evaluated the effects of genetically disrupting TMEM164 in two additional ferroptosis-sensitive cell lines – the leiomyosarcoma cell line RKN and the ovarian clear cell carcinoma cell line ES-2. In both cell lines, loss of TMEM164 resulted in robust decreases in C20:4 ePE lipids (Fig. 5i, j and Supplementary Dataset 1) with minimal changes C20:4 diacyl PEs (Extended Data Fig. 9a, b), generally mirroring the changes observed in sgTMEM164 786-O cells (Fig. 1e). Interestingly, however, each TMEM164-deficient cell line showed a unique set of additional ePE alterations, with sgTMEM164 RKN cells exhibiting modest and variable changes in (monoun)saturated ePEs compared to sgTMEM164 786-O or sgTMEM164 ES-2 cells, and sgTMEM164 ES-2 cells displaying substantial elevations in most of the quantified non-C20:4 ePEs, including several alternative PUFA ePEs (e.g., C22:4 and C22:6 ePEs) (Extended Data Fig. 9c, d and Supplementary Dataset 1). These distinct lipid profiles for sgTMEM164 RKN cells and ES-2 cells correlated with partial and negligible protection from ML210-induced ferroptosis, respectively (Fig. 5k, l). While we do not yet fully understand which specific lipid changes caused by disruption of TMEM164 contribute to ferroptosis sensitivity, it is possible that reductions in C20:4 ePLs combined with predominant elevations in (monoun)saturated ePLs confer maximal protection (as observed in 786-O cells), while concomitant increases in alternative PUFA ePLs may create an alternative source for pro-ferroptotic lipids that counteract the anti-ferroptotic effects of decreases in C20:4 ePLs (as observed in ES-2 cells).
Discussion
The molecular characterization of orphan enzyme activities has benefited from several recent advances, including – i) complete genome sequences of diverse organisms, which enables deep homology mapping of large protein families38,39; ii) protein structure prediction programs, such as AlphaFold, which facilitate the discovery of shared folds for very distally related proteins23; and iii) large-scale gene essentiality screens across many hundreds of cell lines, which can assign genes to a common biochemical pathway based on correlated dependency profiles8,16. Here, we have made use of each of these approaches to determine that TMEM164, a previously uncharacterized integral membrane protein implicated in the regulation of ferroptosis7,18,19, acts as an acyltransferase that transfers C20:4 acyl chains from PC to lyso-ePLs to generate C20:4-ePLs (Fig. 6) – a biochemical reaction first described nearly thirty years ago in the literature14,15, but for which the responsible enzyme has remained unknown. We provide evidence that a conserved cysteine (C123) is required for the acyltransferase activity of TMEM164. This cysteine, along with a conserved histidine (H181) also align based on sequence and structural homology predictions with the putative Thr/His catalytic dyad of the AIG1/ADTRP family of lipid hydrolases. Based on this information, we posit that C123 serves as a catalytic nucleophile of TMEM164, first reacting with C20:4 PC to form an acyl-enzyme intermediate, followed by transfer of this C20:4 acyl group to lyso-ePLs to form ePLs (Extended Data Fig. 10). Such an acyltransferase mechanism involving acyl-donating PC substrates and presumed protein-bound acyl-enzyme intermediates has precedent in other lipid metabolic pathways, including those that form cholesterol esters40 and N-acyl phosphatidylethanolamines41. Studies to date with the distantly related enzyme GPC1 indicate that it uses acyl-CoA32, suggesting that members of the AIG1/TMEM164/GPC1/YwaF/YpjA (or ATGY) enzyme family may use diverse acyl-donating substrates.
Figure 6. TMEM164 bridges C20:4 diacyl PL and ePL biosynthesis.
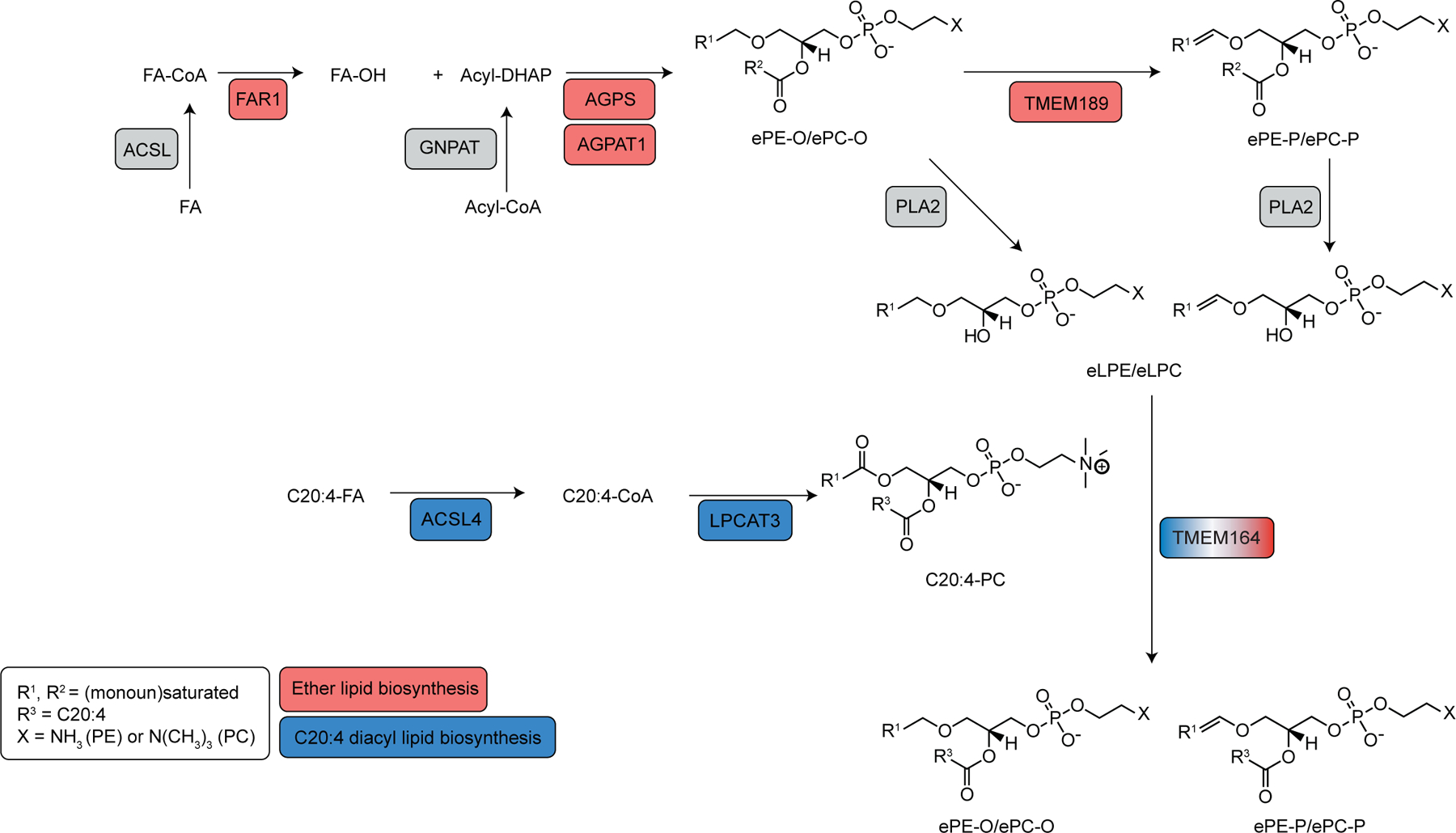
Metabolic pathway diagram describing how TMEM164 links C20:4-diacyl PL and ePE lipids. C20:4 and ePL enzymes showing co-dependency with TMEM164 are highlighted in blue and red, respectively. FA, fatty acid.
The geometry of the minimal catalytic dyad predicted for the TMEM164 and AIG1/ADTRP enzymes resembles part of the Cys/His/Asn and Ser/His/Asp catalytic triads in papain cysteine proteases and trypsin proteases, respectively. The trypsin superfamily further encompasses viral enzymes evolved to use Cys/His/Asp triads, and, likewise, human trypsins can function with Cys or Thr nucleophiles42–44, attesting to the plasticity of nucleophile catalysis in a conserved fold setting. These hydrolytic triads can be further whittled to a nucleophile/His dyad––accompanied by an ‘oxyanion hole’ of H-bond donors––grafted into non-protease scaffolds, suggesting a minimal origin for functional esterase active sites45. The nucleophilic promiscuity of the ATGY enzyme family appears to represent a particularly provocative example of evolutionary (re)invention of a catalytic engine married to an integral membrane fold suited to different steps in lipid metabolism. While the structural model generated by AlphaFold2 confidently assigned TMEM164 to the ATGY family, experimentally determined structures of this protein may be needed to determine additional important mechanistic details, including, for instance, how this enzyme specifically uses C20:4 PC as a substrate over related lipids such as C20:4 CoA and PCs bearing alternative sn-2 acyl chains.
The remarkable specificity displayed by TMEM164 for transferring C20:4 acyl chains suggests that this enzyme has a specialized role in regulating PUFA ePL content, which is reminiscent of the roles that enzymes like LPCAT3 and MBOAT7 play in C20:4 phospholipid metabolism20–22. In each case, when the C20:4 acyltransferase is genetically disrupted, the decreases in direct C20:4 (e)PL products is accompanied by secondary changes in other (e)PLs. The increases in eLPE species observed in TMEM164-deficient cells point to a model where this co-substrate accumulates in the absence of TMEM164 and can then be re-acylated by other as-of-yet unidentified non-C20:4-preferring acyltransferases to remodel the ePL lipid network. One peculiar feature of the TMEM164-disrupted lipid profile was the substantial elevations in C20:4 PCs, which we interpret to indicate that TMEM164 not only regulates basal concentrations of its enzymatic products (C20:4 ePLs), but also the acyl-chain donor substrate C20:4 PC.
Our finding that genetic deletion of TMEM164 offered varying degrees of protection against ferroptosis across different cancer cell lines, despite a consistent suppression of C20:4 ePE, is consistent with other emerging data that point to a contextualized role ether lipid pathways in regulating ferroptosis37. While further studies are needed to understand the specific biochemical states of cells that result in a pro-ferroptotic function for PUFA ePLs, our broader lipidomic data of sgTMEM164 cells may point to the variable expression of alternative acyltransferases as one contributory factor, as these enzymes have the potential to reshuffle the overall acyl chain composition of ePLs in a manner that may enhance or detract from the presumed ferroptosis-suppressing effect of lowering C20:4 ePEs. Similarly, AGPS deletion has been found, in certain cell contexts, to universally decrease ePEs, but also promote elevations in PUFA diacyl-PLs, resulting in negligible protection against ferroptosis37. We did not observe substantial elevations in PUFA diacyl-PLs following disruption of TMEM164 in any of the three cell lines examined herein (Fig. 1i and Extended Data Fig. 9), indicating that other secondary lipid changes may contribute to the variable ferroptosis protection associated with TMEM164 loss. In the case of 786-O cells, which showed the maximal reduction in ferroptosis sensitivity following TMEM164 deletion, we also cannot rule out a potentially protective antioxidative contribution from the vinyl ether bond of elevated non-PUFA ePEs46.
Projecting forward, the discovery that TMEM164 is a specialized acyltransferase responsible for regulating C20:4 ePL content in human cells raises several important questions. For instance, we found that TMEM164 also accepted ester lyso-PLs as substrates, but diacyl PLs were generally unperturbed in TMEM164-disrupted cells. The diacyl C20:4 PL content was instead regulated by LPCAT3. Does this reflect distinct subcellular localizations for LPCAT3 and TMEM164 that restrict their respective activities to specific lipid substrate classes in the cell? Our efforts to date to analyze TMEM164 with commercially available antibodies have been largely unsuccessful, as we have only been able to detect a weak signal for recombinantly expressed TMEM164 in HEK293T cell membrane lysate and have not yet been able to visualize endogenous TMEM164 in cell lines like 786-O. Future efforts to generate higher quality antibodies capable of detecting endogenous TMEM164 are therefore warranted. The presence of a conserved cysteine required for TMEM164 activity also suggests the potential to create covalent inhibitors of the enzyme, which may have value as chemical probes to further study C20:4 ether lipid contributions to ferroptosis and other biological processes. Searches of our legacy chemical proteomic datasets have revealed sparse coverage of TMEM16447,48 including the tryptic peptide containing C123, which may reflect low abundance of this integral membrane protein or limited reactivity with the chemical probes evaluated so far. We also emphasize the importance of further elucidating the biochemical features of human cells that predict ferroptosis sensitivity to TMEM164, and ePL metabolic pathways more generally, as these features may serve as biomarkers for promoting context-dependent ferroptosis in diseases like cancer. When contemplating potential functions for TMEM164 and C20:4 ePLs beyond ferroptosis, we call attention to the provocative Cancer Dependency Map profiles shared by TMEM164, TMEM189, and ACSL4, where subsets of hematological cancer cell lines show impaired growth when the genes encoding these proteins are deleted (https://depmap.org/). Previous studies have also demonstrated that certain cancer cells express heightened ePL content49,50. Future lipidomic analyses of TMEM164-dependent cancer cells may accordingly be illuminating. In summary, our work, combined with recent functional assignments of TMEM189 as a plasmanylethanolamine desaturase8–10, has furnished a near-complete metabolic pathway for producing C20:4-ePLs, a lipid class with important contributions to cell biological processes such as ferroptosis.
Methods
Materials
All chemicals were obtained from Sigma Aldrich unless indicated otherwise. All lipids were purchased from Avanti Polar lipids or Cayman Chemical Company. RSL3 and ML210 were purchased from SelleckChem.
Cell lines
All cell lines tested negative for mycoplasma contamination. Cells were maintained at 37°C with 5% CO2, washed with Dulbecco’s PBS (DPBS, Corning, 21-031-CV), and detached with trypsin-EDTA 0.05% (Gibco) for cell sub-culturing or cell-based assays. HEK293T (ATCC, CRL-3216) cells were cultured in DMEM (Corning, 15-013-CV) supplemented with 10% (v/v) fetal bovine serum (FBS, Omega Scientific), penicillin (100 U/mL), streptomycin (100 µg/mL) and L-glutamine (2 mM). 786-O (ATCC, CRL-1932), RKN (JCRB, 0176), and ES-2 (ATCC, CRL-1978) cells were cultured in RPMI (Corning, 15-040-CV) supplemented with 10% (v/v) FBS, penicillin (100 U/mL), streptomycin (100 µg/mL) and L-glutamine (2 mM).
C20:4 PL-dependent acyltransferase LC-MS based substrate assay
HEK293T, 786-O-Cas9 sgCtrl or sgTMEM164 cell lysates were lysed in assay buffer A [30 mM Tris-HCl (pH 7.4), 5 mM EDTA, 120 mM NaCl] and complete protease inhibitor, EDTA-free (Roche) using a probe sonicator (Branson Sonifier model 250) with 8 pulses (30% duty cycle, output setting = 4) and membrane fraction was obtained by centrifugation at 100,000 x g for 45 min at 4°C. Membrane lysates were prepared by sonicating cell pellets in assay buffer A and total protein concentrations were determined using the Bio-Rad DC protein assay kit. Membrane lysates were diluted to 1 mg/mL using assay buffer A. 2 mg/mL stocks of both the lipid acyl acceptor [e.g., lyso-ePE-P(18:0); 4.29 mM] and lipid acyl donor [e.g., PC(18:0/20:4-d8); 2.47 mM] were prepared by either drying down commercial stocks under N2 stream or weighed out from dry stocks then resuspended in appropriate amount of assay buffer A. The lipid substrate cocktail was then prepared by diluting the lipid stock solutions to 66 µM in a final volume of 190 µL per reaction using assay buffer A (i.e., 1.25 × 10−8 moles of each lipid). Assay was performed by combining 60 µL of diluted membrane lysates with 190 µL of lipid substrate cocktail for a final volume of 250 µL and incubated at 37°C with shaking (200 rpm) for 60 min. The reaction was quenched by the addition of 450 µL 2:1 CHCl3/MeOH (v/v) with 100 pmol PE(17:0/20:4) as an internal standard. The mixture was vortexed and centrifuged at 2,000 x g for 10 min at 4°C to separate the aqueous and organic phases.
The organic phase was extracted and dried down under N2 stream. Lipids were suspended in 100 µL 2:1 CHCl3/MeOH and stored at −80°C until analyzed. The organic phase was analyzed by LC/MS–based multiple reaction monitoring (MRM) (Agilent Technologies 6460 or 6470 Triple Quad). MS analysis was performed using negative mode ESI with the following parameters: drying gas temperature, 350°C; drying gas flow, 9 l/min; nebulizer pressure, 45 Ψ; sheath gas temperature, 375°C; sheath gas flow, 12 l/min; fragmentor voltage, 100 V; and capillary voltage, 3.5 kV. The separation of the analyte was achieved using a 50 mm × 4.6 mm 5 μm Gemini C18 column (Phenomenex) coupled to a guard column (Gemini: C18: 4 × 3 mm). The LC solvents were as follows: solvent A, H2O/MeOH (95:5, v/v) with 0.1% NH4OH (v/v); and solvent B, iPrOH/MeOH/H2O (60:35:5, v/v/v) with 0.1% NH4OH (v/v). The LC gradient following after injection: 20% B at 0.1 mL/min for 5 min; then increase to 85% B at 0.4 mL/min for 15 min; increase to 100% B at 0.5 mL/min for 5 min, run at 100% B at 0.5 mL/min for 6 min; then go back to 20% B and equilibrate at 0.5 mL/min for 3 min. ePE-P(18:0/20:4-d8) (758.5 m/z → 311.3 m/z), ePE-P(18:0/20:4-d11) (761.5 m/z → 314.3 m/z), ePE-P(18:0/18:0-d35) (765.5 m/z → 318.3 m/z), ePE-P(18:0/18:1-d5) (733.5 m/z → 286.3 m/z), PE(17:1/20:4-d8) (758.5 m/z → 311.3 m/z), and PE(17:0/20:4) (internal standard, 752.5 m/z → 303.3 m/z) were measured.
Coenzyme A-dependent acyltransferase LC-MS based substrate assay
HEK293T cell lysates over-expressing hTMEM164-FLAG or hLPCAT3-FLAG cell lysates were lysed in assay buffer B [10 mM Tris-HCl (pH 7.4), 1 mM EDTA, 150 mM NaCl] and complete protease inhibitor, EDTA-free (Roche) using a probe sonicator (Branson Sonifier model 250) with 15 pulses (30% duty cycle, output setting = 3) and membrane fraction was obtained by centrifugation at 100,000 x g for 45 min at 4°C. Membrane lysates were prepared by sonicating cell pellets in assay buffer B and total protein concentrations were determined using the Bio-Rad DC protein assay kit. Membrane lysates were diluted to 0.01 mg/mL using assay buffer B. 2 mg/mL stocks of both the lipid acyl acceptor [e.g., lyso-PE(17:1); 4.29 mM) and lipid acyl donor (e.g., C20:4-CoA; 1.81 mM) were prepared by either drying down commercial stocks under N2 stream or weighed out from dry stocks then resuspended in appropriate amount of assay buffer B. The lipid substrate cocktail was then prepared by diluting the lipid stock solutions to 66 µM in a final volume of 190 µL per reaction using assay buffer B (i.e., 1.25 × 10−8 moles of each lipid). Assay was performed by combining 60 µL of diluted membrane lysates with 190 µL of lipid substrate cocktail for a final volume of 250 µL and incubated at 25°C with shaking (200 rpm) for 10 min. The reaction was quenched by the addition of 450 µL 2:1 CHCl3/MeOH (v/v) with 100 pmol PE(17:0/20:4) as an internal standard. The mixture was vortexed and centrifuged at 2,000 x g for 10 min at 4°C to separate the aqueous and organic phases.
The organic phase was analyzed by LC/MS–based multiple reaction monitoring (MRM) (Agilent Technologies 6460 or 6470 Triple Quad). MS analysis was performed using ESI with the following parameters: drying gas temperature, 350°C; drying gas flow, 9 l/min; nebulizer pressure, 45 Ψ; sheath gas temperature, 375°C; sheath gas flow, 12 l/min; fragmentor voltage, 100 V; and capillary voltage, 3.5 kV. The separation of the analyte was achieved using a 50 mm × 4.6 mm 5 μm Gemini C18 column (Phenomenex) coupled to a guard column (Gemini: C18: 4 × 3 mm). The LC solvents were as follows: solvent A, H2O/MeOH (95:5, v/v) with 0.1% NH4OH (v/v); and solvent B, iPrOH/MeOH/H2O (60:35:5, v/v/v) with 0.1% NH4OH (v/v). The LC gradient following after injection: 20% B at 0.8 mL/min for 6 min; then increase to 100% B at 1 mL/min for 2 min; decrease to 20% B at 1 mL/min for 1 min; then equilibrate at 20% B at 0.8 mL/min for 0.5 min. PE(17:1/20:4) (750.5 m/z → 303.3 m/z) and PE(17:0/20:4) (internal standard, 752.5 m/z → 303.3 m/z) were measured.
Metabolomic analysis for cell samples
Cells were washed twice with cold DPBS, and the total cell metabolome was extracted in 4 mL 2:1:1 CHCl3/MeOH/DPBS (v/v/v) solution containing the internal standard mix of 100 pmol PC(12:0/12:0) and 100 pmol PE(12:0/12:0). The mixture was vortexed vigorously and centrifuged at 2,000 x g for 5 min at 4°C. The bottom organic phase was collected, and the remaining aqueous phase was acidified with 100 µL formic acid and re-extracted by the addition of 2 mL CHCl3. Both organic extracts were pooled, dried down under N2 stream, and reconstituted in 150 µL 2:1 CHCl3/MeOH (v/v) for LC/MS analysis.
Metabolites analyzed in this study were quantified using LC/MS–based multiple reaction monitoring (MRM) methods (Agilent Technologies 6460 or 6470 Triple Quad). MS analysis was performed using ESI with the following parameters: drying gas temperature, 350°C; drying gas flow, 9 L/min; nebulizer pressure, 45 Ψ; sheath gas temperature, 375°C; sheath gas flow, 12 l/min; fragmentor voltage, 100 V; and capillary voltage, 3.5 kV. The MRM transitions for the targeted LC/MS analysis are presented in the compiled lipidomics data spreadsheet. The separation of metabolites was achieved using a 50 mm × 4.6 mm 5 μm Gemini C18 column (Phenomenex) coupled to a guard column (Gemini: C18: 4 × 3 mm). For negative mode analysis, H2O:MeOH (95:5, v/v) with 0.1% NH4OH (v/v) and iPrOH:MeOH:H2O (60:35:5, v/v) with 0.1% NH4OH (v/v) were used as solvent A and B, respectively. For positive mode analysis of (e)PC, 20 mM ammonium acetate in H2O and 20 mM ammonium acetate in MeOH were used as buffer A and B, respectively. The LC gradient for negative mode analysis was the following after injection: 20% B at 0.1 mL/min for 5 min; then increase to 85% B at 0.4 mL/min for 15 min; increase to 100% B at 0.5 mL/min for 5 min, run at 100% B at 0.5 mL/min for 2 min; then go back to 20% B and equilibrate at 0.5 mL/min for 5 min. The LC gradient for positive mode analysis of (e)PC was the following after injection: start from 75% B and increase to 99% B at 0.35 mL/min for 22 min; run at 99% B for 17 min; then go back to 75% B and equilibrate for 3 min. The LC gradient for positive mode analysis of eLPE-P was the following after injection: start from 15% B for 2 min; increase to 30% B for 0.5 min; increase to 48% B for 8.5 min; increase to 82% B for 0.5 min; increase to 99% B for 0.5 min; then decrease back to 15% B for the final 4 min. Lipid species were quantified by measuring areas under the curve in comparison to the corresponding internal standards and then normalizing to the cell numbers.
The LC-MS method for eLPE-P and LPE analysis was adopted from a previous publication51. MS analysis was performed in positive mode using ESI with the following parameters: drying gas temperature, 325°C; drying gas flow, 8 L/min; nebulizer pressure, 25 Ψ; sheath gas temperature, 350°C; sheath gas flow, 11 l/min; fragmentor voltage, 150 V; and capillary voltage, 3.5 kV. Solvent A was ACN:H2O (60:40, v/v) with 0.1% formic acid and 10mM ammonium formate, and solvent B was iPrOH:ACN (90:10, v/v). The LC gradient was the following after injection: start from 15% B for 2 min; increase to 30% B for 0.5 min; increase to 48% B for 8.5 min; increase to 82% B for 0.5 min; increase to 99% B for 0.5 min; then decrease back to 15% B for the final 4 min.
Isobaric ePE-O and ePE-P lipids were separated by ~1.5 min retention time difference, with ePE-O eluting before ePE-P as previously reported10,52. Treatment of lipid extracts with 10% (v/v) of formic acid or HCl (3N) in a pilot experiment was used to confirm the difference in retention time between ePE-O and ePE-P species (Extended Data Fig. 3). ePC-O and ePC-P species were not distinguished in this study, as has been described in previous protocols6.
In situ metabolic labeling assay
786-O-Cas9 sgCtrl and sgTMEM164-2 cells were plated at 500,000 cells in a 6 cm2 dish the day prior to the assay and then treated with DMSO, C20:4-d8 FFA or C18:1-d9 FFA (Cayman) (25 µM) for 4 h. Following treatment, cells were collected, washed twice with ice-cold PBS and pellets were stored at −80°C for further analysis. Metabolomics analysis was performed in negative mode as described above. ePE-P(16:0/20:4-d8) (730.5 m/z → 311.3 m/z), ePE-P(18:0/20:4-d8) (758.5 m/z → 311.3 m/z), ePE-P(16:1/18:1-d9) (707.5 m/z → 290.3 m/z), ePE-P(18:1/18:1-d9) (735.5 m/z → 290.3 m/z), ePE-P(18:1-d9/20:4) (757.5 m/z → 303.3 m/z), ePE-O(16:0/20:4-d8) (732.5 m/z → 311.3 m/z), ePE-O(18:0/20:4-d8) (760.5 m/z → 311.3 m/z), PE-P(18:0/20:4-d8) (774.5 m/z → 311.3 m/z), PE(18:0/18:1-d9) (753.5 m/z → 290.3 m/z), PE(12:0/12:0) (internal standard, 578.3 m/z → 199.2 m/z) were measured.
Metabolomic analysis of oxidized PE and ePE lipids
786-O-Cas9 sgCtrl and sgTMEM164-2 cells were plated at 500,000 cells in a 6 cm2 dish the day prior to the assay. The next day sgCtrl cells were pretreated with Liproxstatin-1 (Cayman) (500 nM) for 2 h, and subsequently sgCtrl, sgTMEM164-1, and sgTMEM164-2 cells were treated with either DMSO or ML210 (2 µM) for 2 h. Following treatment, cells were washed twice with cold DPBS, and resuspended in 1 mL DPBS with 50 µM DTPA. The total cell metabolome was extracted in 4 mL 2:1:1 CHCl3/MeOH/DPBS (v/v/v) solution containing 100 pmol PE(17:0/20:4) as an internal standard. The mixture was vortexed vigorously and centrifuged at 2,000 x g for 5 min at 4°C. The bottom organic phase was collected and dried down under N2 stream and reconstituted in 60 µL 2:1 CHCl3/MeOH (v/v) for LC/MS analysis.
The organic phase was analyzed by LC/MS–based multiple reaction monitoring (MRM) (Agilent Technologies 6460 or 6470 Triple Quad). MS analysis was performed using ESI with the following parameters: drying gas temperature, 350°C; drying gas flow, 9 l/min; nebulizer pressure, 50 Ψ; sheath gas temperature, 375°C; sheath gas flow, 12 l/min; fragmentor voltage, 100 V; and capillary voltage, 3.5 kV. The separation of the analyte was achieved using a 50 mm × 4.6 mm 5 μm Gemini C18 column (Phenomenex) coupled to a guard column (Gemini: C18: 4 × 3 mm). The LC solvents were as follows: solvent A, H2O/MeOH (95:5, v/v) with 0.1% NH4OH (v/v); and solvent B, iPrOH/MeOH/H2O (60:35:5, v/v/v) with 0.1% NH4OH (v/v). The LC gradient following after injection: 20% B at 0.1 mL/min for 5 min; then increase to 85% B at 0.4 mL/min for 15 min; increase to 100% B at 0.5 mL/min for 5 min, run at 100% B at 0.5 mL/min for 6 min; then go back to 20% B and equilibrate at 0.5 mL/min for 2 min. ePE-P(18:0/20:4-OH) (766.5 m/z → 319.2 m/z), ePE-P(18:0/20:4) (750.5 m/z → 303.3 m/z), PE(18:0/20:4-OH) (782.5 m/z → 319.2 m/z), PE(18:0/20:4) (766.5 m/z → 303.3 m/z), PE(17:0/20:4) (internal standard, 752.5 m/z → 303.3 m/z) were measured.
Generation of CRISPR-mediated TMEM164- and LPCAT3-null cells
sgTMEM164 and sgLPCAT3 786-O-Cas9 cells were generated as described previously3,4. Additionally, sgTMEM164 cohorts in cancer cell lines followed similar procedures. Briefly, 786-O, ES-2, and RKN cells were transduced with pLX-311-Cas9 vector (Addgene 96924), which results in constitutive expression of SpCas9 and contains the blasticidin S-resistance gene. Lentiviral sgRNAs targeting human TMEM164 and LPCAT3 along with a control sgRNA were cloned into the pXPR_BRD050 constitutive sgRNA expression vector. The sequences for the sgRNAs are described below (sequences are 5’ to 3’).
sgCTRL-fwd:caccgGCGAGGTATTCGGCTCCGCG
sgCTRL-rev:aaacCGCGGAGCCGAATACCTCG
sgTMEM164-1-fwd: caccgCATCCTGAGGCAGACGAAGG
sgTMEM164-1-rev: aaacCCTTCGTCTGCCTCAGGATGC
sgTMEM164-2-fwd:caccGCCTGTGCCTGACCTTCGGGG
sgTMEM164-2-rev:aaacCCCCGAAGGTCAGGCACAGGc
sgLPCAT3-1-fwd: caccGAGACTCAGGCGCTTGAGAGC
sgLPCAT3-1-rev: aaacGCTCTCAAGCGCCTGAGTCTC
sgLPCAT3-2-fwd: caccGACTGAAGCACAATACACAGC
sgLPCAT3-2-rev: aaacGCTGTGTATTGTGCTTCAGTC
sgRNA-encoding plasmids were co-transfected with dr8.9 envelope and VSV-G packaging plasmids into 500,000 HEK293T cells in 6 cm2 dishes using the Fugene6 transfection reagent (Promega). Virus-containing supernatants were collected 48 h after transfection and used to infect target 786-O-Cas9 cells in the presence of 10 mg/mL polybrene (Santa Cruz). The cells were allowed to recover for 48 h. Puromycin (2 µg/mL) and Blasticidin (10 µg/mL) was then added to the cells, and the cells were expanded. Genetic alterations in sgLPCAT3 cells were previously reported21, while genetic alterations in sgTMEM164 cells were confirmed by next-generation sequencing analysis as described below.
Generation of sgTMEM164-C123Y base-edited 786-O cells
Plasmid containing the cytidine deaminase conjugated to dCas9 was a gift from the laboratory of David Liu. sgRNA targeting C123 on TMEM164 as well as a control sgRNA targeting GFP was cloned into the vector. The sequences are described below (sequences are 5’ to 3’).
sgCTRL-fwd: caccgGCGAGGTATTCGGCTCCGCG
sgCTRL-rev: aaacCGCGGAGCCGAATACCTCG
sgTMEM164-C123Y-fwd: caccgGTGACAGGGGTTGAGCAGGT
sgTMEM164-C123Y-rev: aaacACCTGCTCAACCCCTGTCACc
sgRNA-encoding plasmids were co-transfected with dr8.9 envelope and VSV-G packaging plasmids into 500,000 HEK293T cells in 6 cm2 dishes using the Fugene6 transfection reagent (Promega). Virus-containing supernatants were collected 48 h after transfection and used to infect target 786-O cells in the presence of 10 mg/mL polybrene (Santa Cruz). The cells were allowed to recover for 48 h. Puromycin (2 µg/mL) was then added to the cells, and the cells were expanded. Genetic alterations in sgTMEM164-C123Y cells were confirmed by next-generation sequencing analysis as described below.
Targeted genomic sequencing and data analysis
Genomic sites of interested were amplified and indexed using a two-step PCR method as previously described53. Briefly, cells were lysed using 10 mM Tris (pH 7.5), 0.5% Tween 20, 0.02% SDS plus 20 µg/ml freshly added proteinase K and were incubated at 55°C for 2 h before heat inactivation at 90°C for 30 min. Primers containing homology to targeted region and Illumina adapters were used for PCR1 (sequences below, 5’ to 3’). Specifically, in each reaction, 5 µl of genomic DNA extract, 12.5 µl Phusion PCR master mix, 1.25 µl of 10 µM forward and 1.25 µl of 10 µM reverse primers were added to a total volume of 25 µl. The PCR1 reactions were carried out as follows: 98°C for 3 min, then 30 cycles of (20 s of 98°C, 25 s of 60°C, 25 s of 72°C), followed by final extension at 72°C for 2 min. PCR1 products were cleaned using Ampure beads (Beckman) according to manufacturer’s instructions and were eluted in 20 µl of water. For each PCR2 reaction, 5 µl of PCR1 product, 12.5 µl Phusion PCR master mix, 1.25 µl of 10 µM forward and 1.25 µl of 10 µM reverse index primers were added to a total volume of 25 µl. The PCR2 reactions were carried out as follows: 98°C for 3 min, then 12 cycles of (20 s of 98°C, 20 s of 60°C, 25 s of 72°C), followed by final extension at 72°C for 2 min. The PCR2 products were then pooled and cleaned using Ampure beads. The library was quantified using PicoGreen dsDNA assay kits (ThermoFisher) and sequenced on an Illumina Miniseq instrument with 30% PhiX spike-in. Samples reads were demultiplexed based on combinatorial dual indexes. The genome editing quantification was performed using CRISPResso2 (https://crispresso.pinellolab.partners.org)54.
Forward Sequencing Primer: ACACGACGCTCTTCCGATCTGGAGAGCCTGAGCAAGAATC
Reverse Sequencing Primer: CTTGGCACCCGAGAATTCCAGGATGAGGTGCTGTACACTATG
Western blot analysis
Cell pellets were lysed in DPBS and complete protease inhibitor, EDTA-free using a probe sonicator (Branson Sonifier model 250) with 15 pulses (30% duty cycle, output setting = 4). Proteins were resolved by SDS–PAGE, transferred to 0.45 μM nitrocellulose membranes (Thermo Fisher) which were blocked with 5% milk in TBST buffer [20 mM Tris-HCl (pH 7.6), 150 mM NaCl with 0.1% tween 20]. Primary antibodies were used at the following concentrations: 1:2,000 anti-Na+/K+-ATPase (Cell Signaling, no. 3010), 1:2,000 anti-FLAG-HRP conjugated (Abcam, ab2493), 1:2,000 anti-TMEM164 polyclonal (Thermo Scientific, PA5-58540). Blots were incubated with primary antibodies in 5% milk in TBST at 4°C overnight. Following another TBST wash (5 times, 5 min), membranes were incubated with 1:5,000 anti-rabbit IgG, HRP conjugated secondary antibody in 5% milk in TBST (Cell Signaling, no. 7074S) at room temperature for 1 h. Membranes were washed with TBST (5 times, 5 min) and developed with ECL western blotting detection reagent kit (Thermo Scientific) and detected with Bio-Rad ChemiDoc MP imaging system.
Recombinant expression of TMEM164 wild type and C123A mutant in sgCtrl and sgTMEM164-2 786-O-Cas9 cells
Full length C-terminally FLAG-tagged TMEM164 and C123A mutant were obtained by ordering a gBlock gene fragment from Integrated DNA Technologies with a silent point mutation at the PAM site for sgTMEM164-2 and attB1/attB2 sites flanking the gene. Gene fragments were cloned into the gateway compatible pLV416 vector by standard gateway cloning. TMEM164-encoding plasmids were co-transfected with dr8.9 envelope and VSV-G packaging plasmids into 4 million HEK293T cells in 10 cm2 dishes using the Fugene6 transfection reagent (Promega). Virus-containing supernatants were collected 48 h after transfection and used to infect sgCtrl or sgTMEM164-2 786-O-Cas9 cells in the presence of 10 mg/mL polybrene (Santa Cruz). The cells were allowed to recover for 48 h. Geneticin (Thermo Fisher) (0.8 mg/mL) was then added to the cells, and the cells were expanded.
Recombinant expression of TMEM164 wild type and active site cavity mutants by transfection of HEK293T cells
TMEM164 mutants were obtained by ordering gBlock gene fragments from Integrated DNA Technologies with attB1/attB2 sites flanking the gene. Gene fragments were cloned into the gateway compatible pLV416 vector using standard gateway cloning techniques. HEK293T cells were transiently transfected with TMEM164 WT or mutants using 1 mg/mL PEI (pH 7.4) and cells were collected for analysis 48 h later.
Ferroptosis assay
786-O, RKN, and ES-2 cells were plated in white, clear-bottom 96-well plates (Greiner) at 5,000 cells per well the day prior to the assay. On the day of the assay, DMSO or a serial dilution of RSL3 or ML210 (3-fold dilution starting from 10 µM, 8 concentrations) were added to the wells and allowed to incubate for 24 h. At 24 h, the plates were removed from the incubator, 50 µL of CellTiter-GLO (CTG) (Promega) was added to each well and allowed to incubate for 30 min at room temperature. Luminescence was measured on a Clariostar plate reader (BMG Labtech).
Computational and structural analyses.
The initial discovery of a greater superfamily beyond AIG1/ADTRP––previously identified as novel FAHFA hydrolases in lipid metabolism––was done with sensitive sequence search algorithms, iterative PsiBLAST and HHPRED, using servers located in the BLAST suite at the NCBI (http://blast.ncbi.nlm.nih.gov) and as part of the bioinformatics Toolkit at the MPI-Tübingen (http://toolkit.tuebingen.mpg.de/tools/hhpred), respectively. In particular, HHPRED considers PsiPRED-generated secondary structure predictions in addition to the iteratively-generated PSSM sequence profiles, and these enhanced search tools can be aimed at diverse proteome-level collections of sequences, as well as various structure and domain databases, to detect distant matches. In this manner with HHPRED, the human AIG1 sequence locates AIG1/ADTRP transmembrane enzyme homologs in D.melanogaster and S.cerevisiae (consistent with the PFAM PF04750 family cluster; PFAM databases are maintained at http://pfam.xfam.org), and also finds more distant, partial matches with prokaryotic YwaF members (like B.subtilis and C.difficile, members of PF09529 family) that show alignment of predicted TM helices and overlap of potential catalytic dyads––with an invariant, C-terminal His, and a variable (Thr, Cys or Ser), more N-terminal, nucleophile. Iterative PsiBLAST runs that reach into sequence families absent from HHPRED, further link the YwaF proteins to members of the Hypothetical clan (principally in archaea, also featuring variable nucleophile catalytic dyads) and the more distant YpjA clan (PF7187, principally Ser/His catalytic dyads). These latter enzymes were used to query eukaryotic proteomes with HHPRED and locate the orphanTMEM164 sequence in humans (as well as D.melanogaster and C.elegans), building a distant homology link to PFAM family PF14808 that exclusively use a Cys/His catalytic dyad.
The structural implications of these weak sequence similarity HHPRED and PsiBLAST matches was tested with the best 2nd generation structure prediction program, TrRosetta, using the server at Nankai University (http://yanglab.nankai.edu.cn/trRosetta), that generated 3D structure models of AIG1/ADTRP, YwaF and YpjA, Hypothetical and TMEM164 proteins, and asserted that they had a common 6TM core fold (Fig. 4a,b, Extended Data Fig. 5a,b) decorated by N- and C-terminal extensions of variable length. The Thr/His catalytic dyad in human AIG1 was distributed across TM2 and TM4, respectively, while the Cys/His dyad in human TMEM164 likewise mapped to structurally equivalent locations in TM2 and TM4. The advent of the new deep-learning-based AlphaFold2 algorithm (http://www.deepmind.com/research/highlighted-research/alphafold), allowed much more precise modeling of representative members of the 5 linked families of sequences with the versatile and fast ColabFold implementation (http://github.com/sokrypton/ColabFold), that reasserted even more accurately, the presence of a common 6TM core fold bearing the enzymatic machinery of this emerging membrane protein superfamily (Fig. 4c, Extended Data Fig. 5e). In many cases now, AlphaFold2 models of superfamily members can be directly retrieved from the AlphaFold database maintained at the EBI (https://alphafold.ebi.ac.uk), that is now incorporated into most UniProt files (https://www.uniprot.org).
Fast searches of the extensive AlphaFold model databases––organized by proteomes––using Rupee and FoldSeek servers (respectively at http://ayoubresearch.com and https://search.foldseek.com) validated the homology links of the 5 sequence families by showing significant fold relationships. In addition, these fold-level screens discovered an additional integral membrane enzyme family headlighted by S.cerevisiae and A.thaliana GPC1 enzymes (members of PF10998 family) at very low, ~10% sequence identity, further sharing the same Cys/His catalytic dyad of the human TMEM164 chain, that had eluded detection by the HHPRED searches. AlphaFold2 models of GPC1 enzymes revealed the same core 6TM fold with catalytic residues in TM2 and TM4, with the largest N- and C-terminal extensions (Fig. 4b, Extended Data Fig. 5d).
The low level of sequence identity between the now six branches of novel enzymes in the AIG and ADTRP/TMEM164/GPC1/YwaF and YpjA (or ATGY) superfamily was insufficient to gauge their evolutionary relationship, so the slow but rigorous DALI algorithm (http://ekhidna2.biocenter.helsinki.fi) was used to perform an all-against-all matching of the AlphaFold2-generated models. The average linkage clustering of DALI Z-scores (Extended Data Fig. 5c), a measure of structural similarity, was used to generate dendrograms as conventional linear trees (Extended Data Fig. 5b) or hyperbolic trees (Fig. 4c, made with the HyperTree program from http://kinase.com/tools/HyperTree.html) of the ATGY superfamily, showing clear separation of the six branches, with their particular and divergent catalytic dyad preferences.
Closer structural analysis of the TMEM164 fold was performed with CavityPlus (http://www.pkumdl.cn/cavityplus), CASTp (http://sts.bioe.uic.edu), and CAVER (http://loschmidt.chemi.muni.cz/caverweb) programs, to delineate the size and shape of the internal cavity in the 6TM helical core (Fig. 4d, Extended Data Fig. 6e) as well as the tunnels that link cavity to the lipid bilayer, and allow passage of substrates and products (Extended Data Fig. 6f–g). CavityPlus also links to CavPharmer that additionally builds a pharmacophore ‘skeleton’ (by analysis of residue constellations surrounding the cavity) that fits within the cavity and abuts the catalytic dyad and is highly suggestive of a diacyl chain phospholipid ligand (which could be a substrate or product).
EC50 calculation
For the acyltransferase assays, acyltransferase activities were determined as a ratio of the area under the peak of the expected product over the area under the peak of either PE(17:0/20:4) (internal standard, 752.5 m/z → 303.3 m/z) or PE(12:0/12:0) (internal standard, 578.3 m/z → 199.2 m/z). Data were then normalized by dividing the acyltransferase activity by the amount of membrane proteome used and the length of the assay. For the ferroptosis assay, relative cell viability was determined by dividing the CTG value from RSL3/ML210 treated wells by the CTG value from the average of the DMSO treated wells and multiplying by 100. EC50 values were determined by plotting a log(inhibitor) vs. normalized response, and the dose-response curves were generated using the Prism software (GraphPad Software, Inc.).
Statistics
Statistical analyses were performed using GraphPad Prism (GraphPad Software, Inc.). All data are shown as mean values ± SEM or SD and two-sided Student’s t-test was used to perform statistical analyses. A p-value of < 0.01 was considered statistically significant for this study.
Extended Data
Extended Data Fig. 1. Characterization of sgTMEM164 786-O cells.
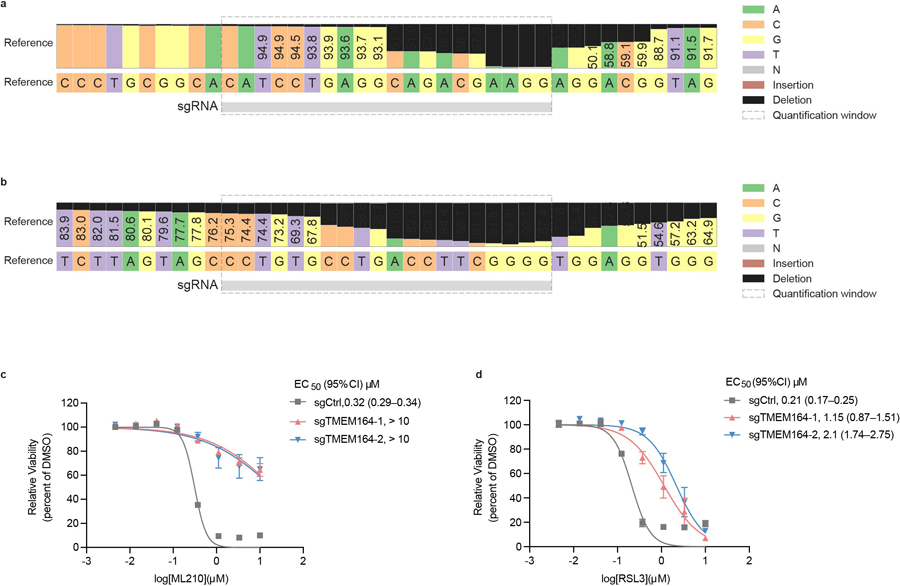
a, b, Confirmation of genetic alterations in 786-O cells at sgRNA sites targeted with (a) sgTMEM164-1 and (b) sgTMEM164-2 by next-generation sequencing analysis. c, d, Genetic deletion of TMEM164 in 786-O cells results in reduced sensitivity to ferroptosis induced by the GPX4 inhibitors ML210 (c) and RSL3 (d) measured at 24 h post-treatment with GPX4 inhibitors. Data represent mean values ± S.E.M. from two independent experiments.
Extended Data Fig. 2. Targeted lipidomic (LC-MS/MS) methods for measuring ePLs.

a, b, Detection of endogenous ePE-P(C18:0/C20:4) in 786-O cells by LC-MS/MS (a) and comparison to commercial ePE-P(C18:0/C20:4) standard (b). Left traces show extracted ion chromatogram of a feature corresponding to ePE-P(C18:0/C20:4) (750.5 → 303.3). Right traces show MS2 spectra for ePE-P(C18:0/C20:4). c,d, Targeted LC-MS/MS analysis of isobaric ePE-O and ePE-P species performed as described in the Methods section revealing that they are separated by approximately 1.5 min difference in retention time. Treatment of lipid extracts from 786-O cells with 10% (v/v) formic acid (c) or HCl (3N) (d) for 30 min at 37°C reveals ePE-P elutes after ePE-O as evidenced by a significant reduction in the ePE-P peak following HCl treatment. AUC, area under the curve.
Extended Data Fig. 3. Alterations in ePC and diacyl PC production in TMEM164-deficient cells.
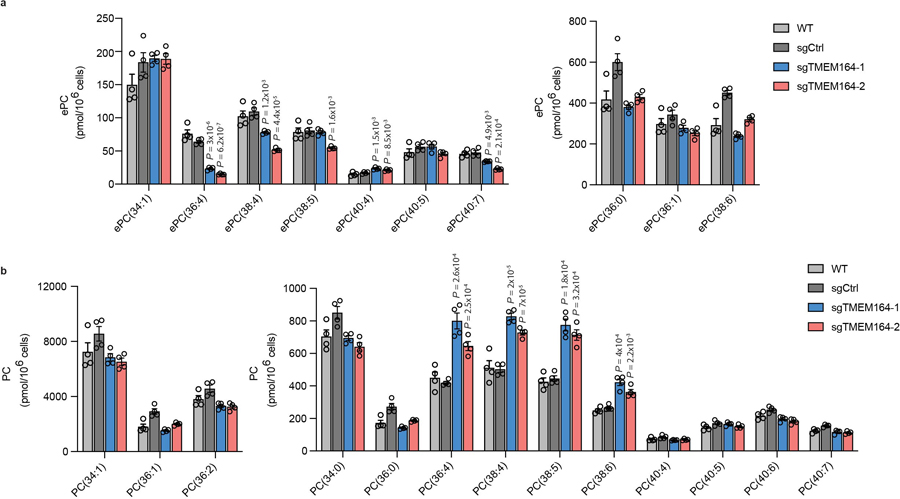
a, b ePC (a) and PC (b) lipid measurements in sgCtrl and sgTMEM164 786-O cells. For a, due to technical limitations we did not distinguish between ePC-O and ePC-P lipids. Data represent mean values ± S.E.M from four independent experiments per group. P-values were derived using a Two-sided Student’s t-test performed relative to sgCtrl cells; only shown for lipids where sgCtrl and parental (WT) data did not differ.
Extended Data Fig. 4. Measurement of free fatty acids (FFAs), eLPE, and LPE in TMEM164-deficient cells.
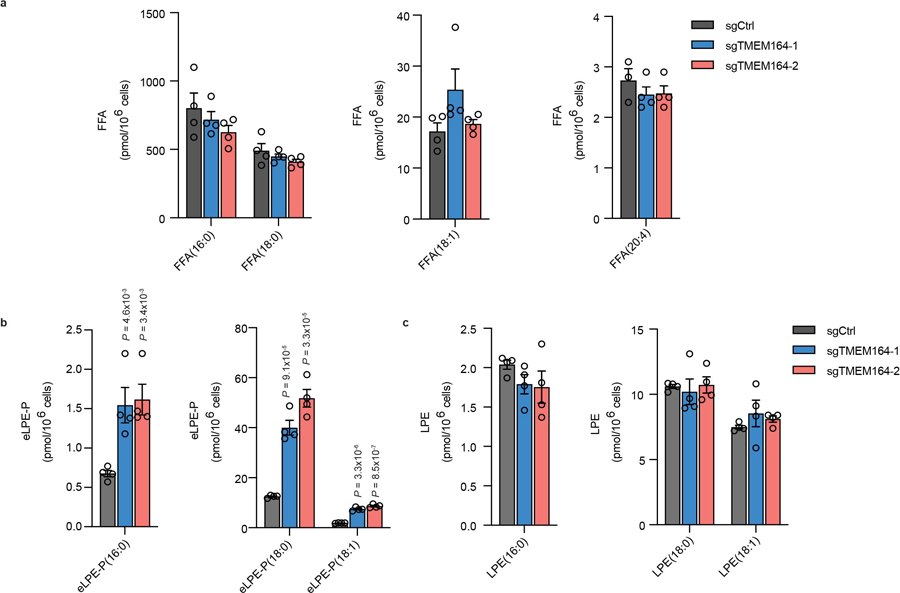
FFA (a), eLPE (b) and LPE (c) lipid measurements in sgCtrl and sgTMEM164 786-O cells. Data represent mean values ± S.E.M from four independent experiments per group. P-values were derived using a Two-sided Student’s t-test performed relative to sgCtrl cells.
Extended Data Fig. 5. Integrated homology mapping and AlphaFold2 analysis identifies a predicted shared structure between TMEM164 and the AIG1/ADTRP lipid hydrolases.
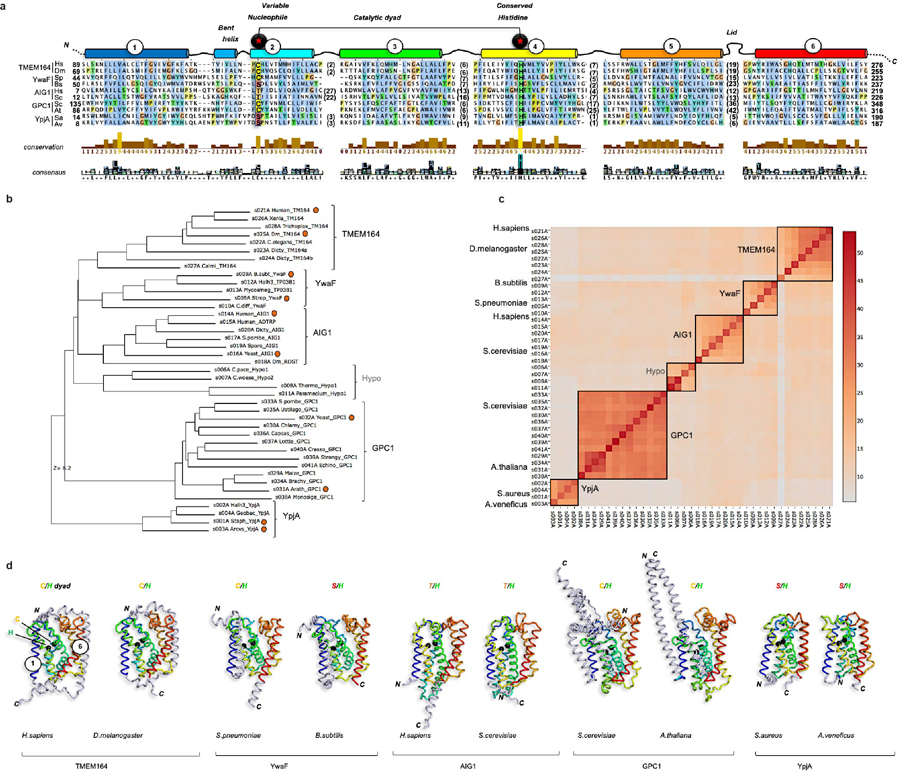
a, Structure-based sequence alignment by DALI of AlphaFold2 models (http://alphafold.ebi.ac.uk) of the H.sapiens (UniProt Q5U3C3) and D.melanogaster (UniProt Q7JRB2) TMEM164 proteins, with representative proteins from the AIG1/ADTRP family (H.sapiens (Q9NVV5); S.cerevisiae (P38842)), prokaryotic YwaF (S.pneumoniae (Q8CYG1); B.subtilis (P25149)) and YpjA proteins (S.aureus (Q2FYH4); A.venificus (F2KNL0)), and GPC1 enzymes (S.cerevisiae (P48236); A.thaliana (Q9FJB4). In some cases, models were built with ColabFold implementation of AlphaFold2 (http://github.com/sokrypton/ColabFold). The alignment was displayed and analyzed with Jalview (http://jalview.org), showing the six blocks of conserved structure corresponding to TM helices 1–6. Length and sequence-variable gaps in the alignment corresponding to loops in the superposed folds were abbreviated by residue stretches in parentheses. Positions of the variable nucleophile and invariant His residues (Cys123 and His181 in human TMEM164, respectively) are boxed and labeled. b, DALI-derived superpositions provide a measure of structural relatedness for the six main branches of the enzyme superfamily displayed in the phylogenetic hypertree in Fig. 4c (http://kinase.com/tools/HyperTree.html), with chains aligned in panel a noted by red circles. Z-score of 8.2 is provided for the most distant branch (YpjA). c, Matrix of DALI Z-scores displaying the clusters of more closely related AlphaFold2-derived structures from all-against-all superpositions performed with the DALI server (http://ekhidna2.biocenter.helsinki.fi), leading to reconstitution of the six branches of the enzyme superfamily. Five of these clusters have sequence alignments that can be retrieved from the PFAM database (http://pfam.xfam.org): TMEM164 (PF14808), AIG1/ADTRP (PF04750), GPC1 (PF10998), YwaF (PF09529) and YpjA (PF7187). d, Tube representations of aligned AlphaFold2 models showing positions of the catalytic dyad with black Ca spheres and preservation of the core 6TM fold (color-ramped from blue TM1 to red TM6) with N-or C-terminal extensions drawn in grey. Figures composed with PyMOL (http://pymol.org).
Extended Data Fig. 6. AlphaFold2 analysis of TMEM164 active site cavity.
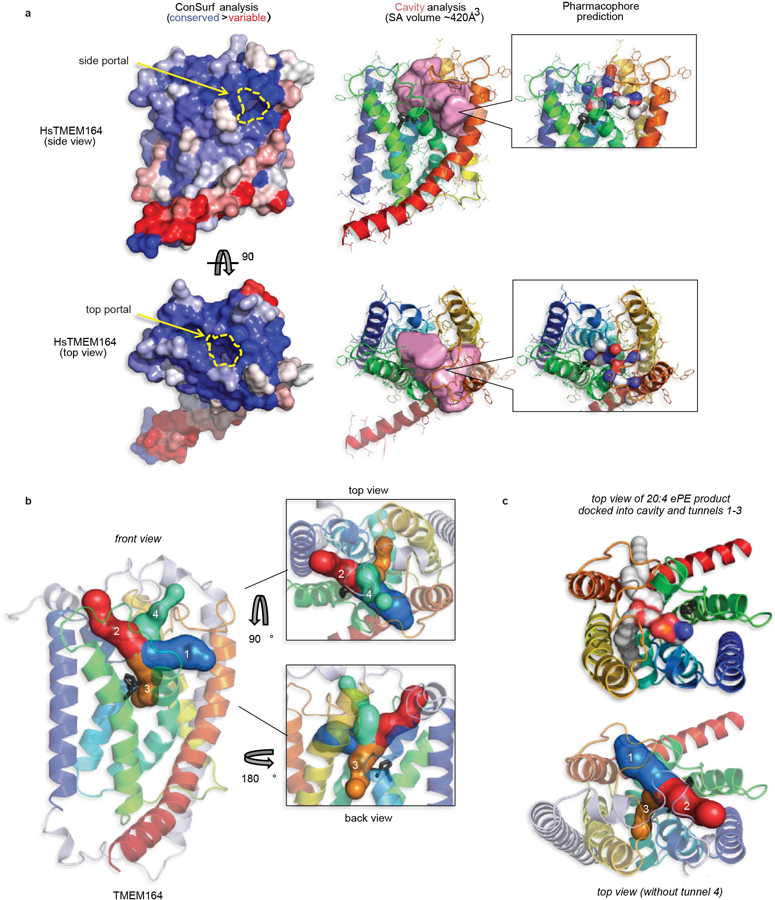
a, The human TMEM164 structural model was processed by the ConSurf server (http://consurf.tau.ac.il) to map the sequence conservation profile of its underlying family to the 3D fold surface color-ramped from blue (conserved) to red (variable) patches. For side and top views, the ConSurf surfaces are aligned to the ribbon model of TMEM164 embedded with the CavityPlus-calculated volume in pink (server at http://pkumdl.cn), showing the positions of predicted portals to the lipid bilayer. In addition, the CavPharmer tool at CavityPlus was used to predict a pharmacophore skeleton for a prospective ligand (which could represent the lipid substrates or product of the enzymatic reaction) for TMEM164, showing a shape and chemical nature in agreement with a predicted function as a C20:4-preferring lyso-ePL acyltransferase. b, The program CAVER (https://loschmidt.chemi.muni.cz/caverweb) was used to visualize the tunnels leading from the central TMEM164 cavity that abuts the Cys123/His181 predicted catalytic dyad (shown in black stick format), to the portals that open to the lipid bilayer (tunnels 1 and 3, respectively in blue and orange, with lengths 12.8 Å and 16.7 Å) and to the outer leaflet of the membrane (tunnels 2 and 4, respectively in red and green, both with lengths of 15.1 Å). c, A top view of the six-transmembrane core of TMEM164 with docked C20:4 ePE, with acyl chains occupying tunnels 1 and 3, and head group cresting into the tunnel 2 space. Tunnels 1 and 3 may represent the principal entrance and exit routes for substrates and product, that place the headgroup in proximity of the active site.
Extended Data Fig. 7. Characterization of C20:4 PC-dependent acyltransferase activity of TMEM164.
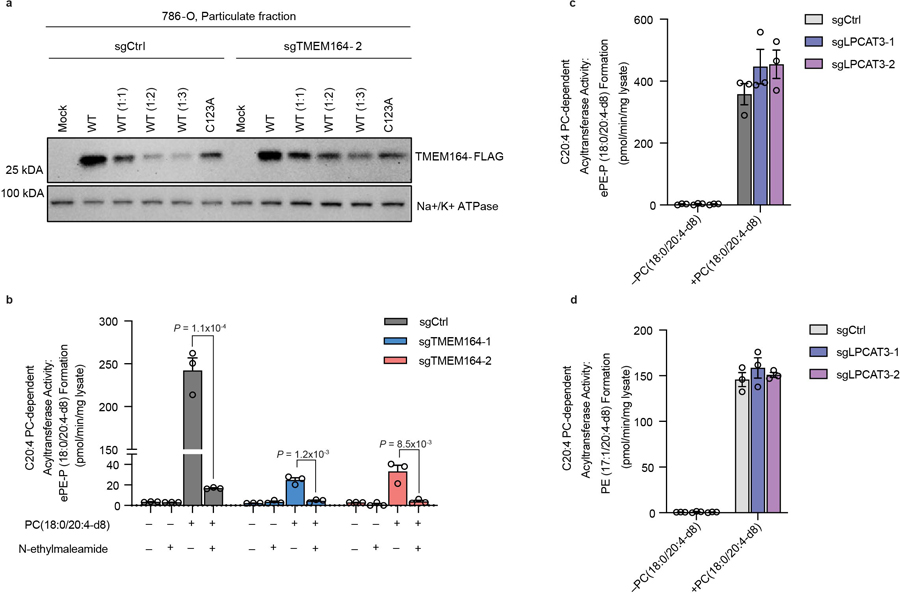
a, Western blot of membrane lysates from sgCtrl and sgTMEM164-2 786-O cells recombinantly expressing C-terminally FLAG-tagged WT-TMEM164 or a C123A-TMEM164 mutant. WT-TMEM164 lysates were serially diluted with mock lysate to identify a dilution (1:1) that matched the expression of C123A-TMEM164. Data are from a single experiment representative of two independent experiments. b, Membrane lysates from sgCtrl or sgTMEM164 786-O cells were treated with N-ethylmaleimide (NEM; 200 µM, 60 min) prior to exposure to diacyl PC(C18:0/C20:4-d8) and lyso-ePE-P(C18:0) substrates and measurement of ePE-P(C18:0/C20:4-d8) product. c, d, Measurement of (c) ePE-P(C18:0/C20:4-d8) and (d) diacyl PE(C17:1/C20:4-d8) formation in membrane lysates from sgCtrl and sgLPCAT3 786-O cells using diacyl PC(C18:0/C20:4-d8) and ePE-P(C18:0) or LPE(C17:1) as donor and acceptor substrate, respectively. For b-d, membrane lysates (1 µg/µL) were treated with 50 µM each of donor and acceptor substrates for 1 h at 37° C prior to analysis. For b-d data represent mean values ± S.E.M from three independent experiments per group. P-values were derived using a Two-sided Student’s t-test performed relative to sgCtrl cells.
Extended Data Fig. 8. Characterization of sgTMEM164-C123Y base-edited cells.
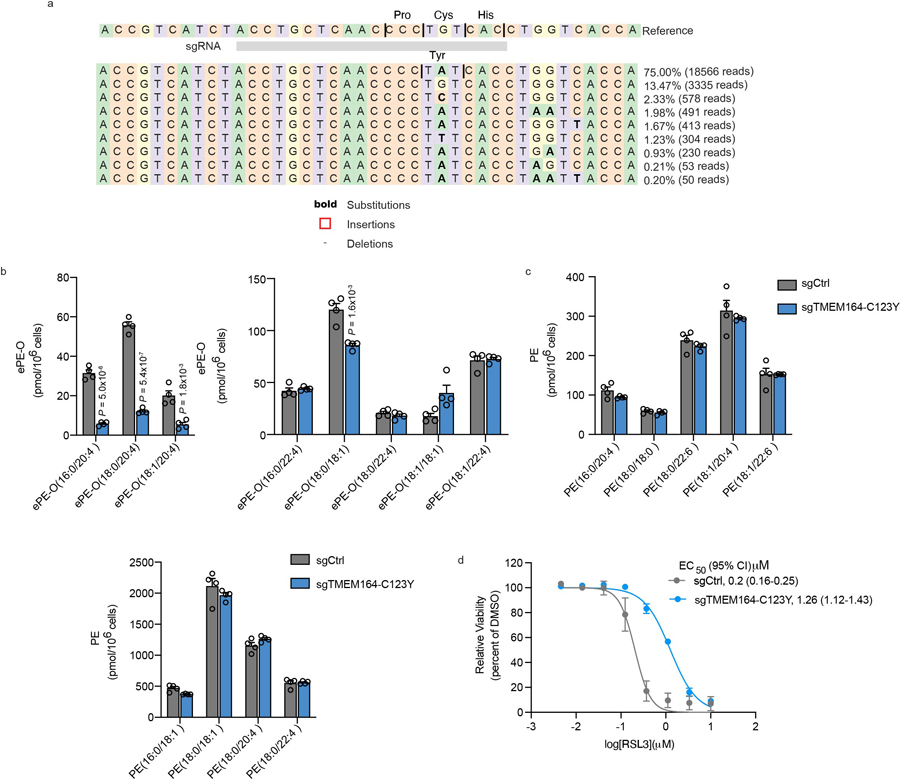
a, Confirmation of base-editing of TMEM164 genomic DNA in 786-O cells at the sgRNA target site for sgTMEM164-C123Y base-edited cell population by next-generation sequencing analysis. b, Measurement of C20:4 (left) and additional (right) ePE-O lipids in sgCtrl and sgTMEM164-C123Y base-edited cell populations. c, Measurement of diacyl PE lipids in sgCtrl and sgTMEM164-C123Y base-edited 786-O cell populations. d, Cell viability of sgCtrl and sgTMEM164-C123Y base-edited cell populations treated with the indicated concentrations of the GPX4 inhibitor RSL3 measured at 24 h post-treatment. For b and c data represent mean values ± S.E.M from four independent experiments per group. For d, data represent mean values ± S.E.M. from two independent experiments. P-values were derived using a Two-sided Student’s t-test performed relative to sgCtrl cells.
Extended Data Fig. 9. C20:4 PE and non-C20:4 ePE-P measurements in TMEM164-deficient RKN and ES-2 cells.
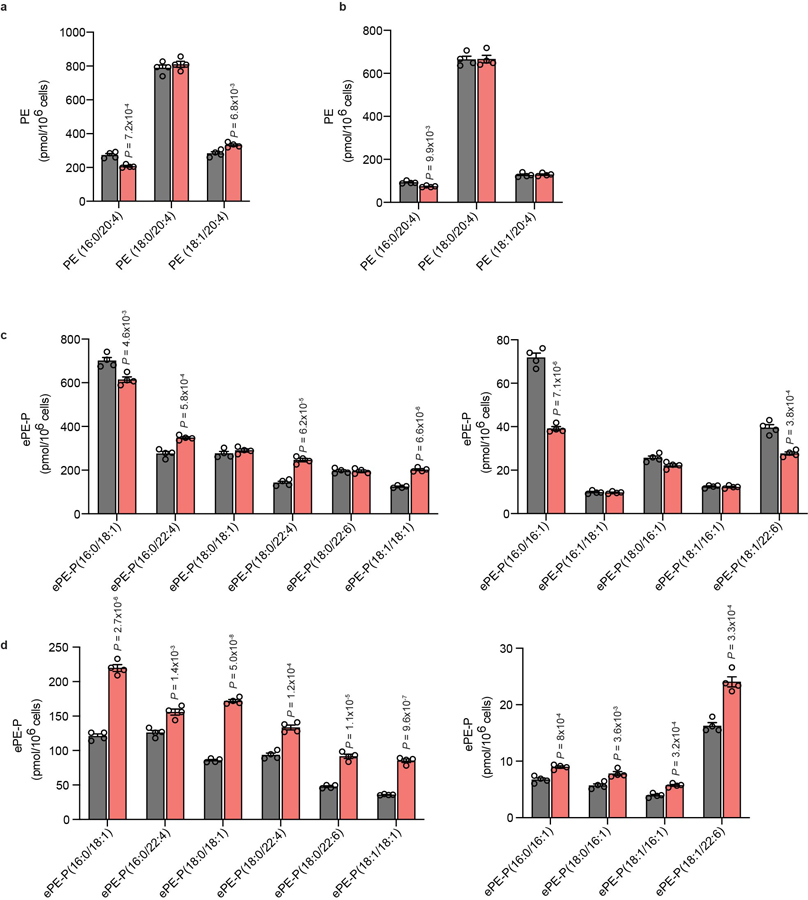
a, b, Measurement of C20:4 PE lipids in sgCtrl and sgTMEM164-2 RKN (a) and ES-2 (b) cell lines. c, d, Measurement of non-C20:4 ePE-P lipids in sgCtrl and sgTMEM164-2 RKN (c) and ES-2 (d) cell lines. Data represent mean values ± S.E.M from four independent experiments per group. P-values were derived using a Two-sided Student’s t-test performed relative to sgCtrl cells.
Extended Data Fig. 10. TMEM164 catalytic mechanism of action.
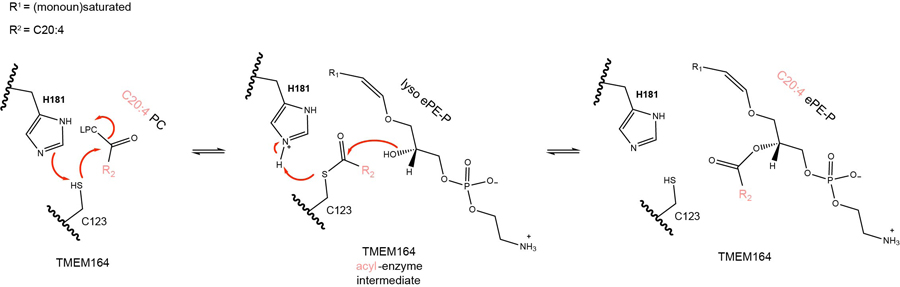
Proposed catalytic mechanism for C20:4 PL-dependent acyltransferase activity of TMEM164. LPC, lysophosphatidylcholine.
Supplementary Material
Acknowledgements.
This work was supported by the NIH (CA231991), The Damon Runyon Cancer Research Foundation (H.L.), and Lundbeck. We are grateful to J. Blankman, H. Reardon, and M. Niphakis for helpful discussions throughout the performance of this research.
Footnotes
Competing Interests
The authors declare no competing financial interests.
Data Availability
Raw sequencing data validating genomic alterations in 786-O base-edited and knockout cell lines are deposited at the Sequence Read Archive (accession number PRJNA902005). Data supporting the findings of this work are available within the paper and its Supplementary Information. The datasets, constructs, and cell models generated and analyzed during the current study are available from the corresponding author upon request.
References
- 1.van Meer G, Voelker DR & Feigenson GW Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9, 112–124 (2008). https://doi.org: 10.1038/nrm2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang WS et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 113, E4966–4975 (2016). https://doi.org: 10.1073/pnas.1603244113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stockwell BR et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171, 273–285 (2017). https://doi.org: 10.1016/j.cell.2017.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang WS et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 (2014). https://doi.org: 10.1016/j.cell.2013.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eaton JK et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat Chem Biol 16, 497–506 (2020). https://doi.org: 10.1038/s41589-020-0501-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zou Y et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 585, 603–608 (2020). https://doi.org: 10.1038/s41586-020-2732-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cui W, Liu D, Gu W & Chu B Peroxisome-driven ether-linked phospholipids biosynthesis is essential for ferroptosis. Cell Death Differ 28, 2536–2551 (2021). https://doi.org: 10.1038/s41418-021-00769-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wainberg M et al. A genome-wide atlas of co-essential modules assigns function to uncharacterized genes. Nat Genet 53, 638–649 (2021). https://doi.org: 10.1038/s41588-021-00840-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gallego-Garcia A et al. A bacterial light response reveals an orphan desaturase for human plasmalogen synthesis. Science 366, 128–132 (2019). https://doi.org: 10.1126/science.aay1436 [DOI] [PubMed] [Google Scholar]
- 10.Werner ER et al. The TMEM189 gene encodes plasmanylethanolamine desaturase which introduces the characteristic vinyl ether double bond into plasmalogens. Proc Natl Acad Sci U S A 117, 7792–7798 (2020). https://doi.org: 10.1073/pnas.1917461117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doll S et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13, 91–98 (2017). https://doi.org: 10.1038/nchembio.2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dixon SJ et al. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem Biol 10, 1604–1609 (2015). https://doi.org: 10.1021/acschembio.5b00245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun WY et al. Phospholipase iPLA2beta averts ferroptosis by eliminating a redox lipid death signal. Nat Chem Biol 17, 465–476 (2021). https://doi.org: 10.1038/s41589-020-00734-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kramer RM & Deykin D Arachidonoyl transacylase in human platelets. Coenzyme A-independent transfer of arachidonate from phosphatidylcholine to lysoplasmenylethanolamine. J Biol Chem 258, 13806–13811 (1983). [PubMed] [Google Scholar]
- 15.Sugiura T, Masuzawa Y, Nakagawa Y & Waku K Transacylation of lyso platelet-activating factor and other lysophospholipids by macrophage microsomes. Distinct donor and acceptor selectivities. J Biol Chem 262, 1199–1205 (1987). [PubMed] [Google Scholar]
- 16.Tsherniak A et al. Defining a Cancer Dependency Map. Cell 170, 564–576 e516 (2017). https://doi.org: 10.1016/j.cell.2017.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horibata Y & Hirabayashi Y Identification and characterization of human ethanolaminephosphotransferase1. J Lipid Res 48, 503–508 (2007). https://doi.org: 10.1194/jlr.C600019-JLR200 [DOI] [PubMed] [Google Scholar]
- 18.Zou Y et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun 10, 1617 (2019). https://doi.org: 10.1038/s41467-019-09277-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zou Y et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol 16, 302–309 (2020). https://doi.org: 10.1038/s41589-020-0472-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee HC et al. LPIAT1 regulates arachidonic acid content in phosphatidylinositol and is required for cortical lamination in mice. Mol Biol Cell 23, 4689–4700 (2012). https://doi.org: 10.1091/mbc.E12-09-0673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reed A et al. LPCAT3 Inhibitors Remodel the Polyunsaturated Phospholipid Content of Human Cells and Protect from Ferroptosis. ACS Chem Biol 17, 1607–1618 (2022). https://doi.org: 10.1021/acschembio.2c00317 [DOI] [PubMed] [Google Scholar]
- 22.Bourgeois T et al. Deletion of lysophosphatidylcholine acyltransferase 3 in myeloid cells worsens hepatic steatosis after a high-fat diet. J Lipid Res 62, 100013 (2021). https://doi.org: 10.1194/jlr.RA120000737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). https://doi.org: 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parsons WH et al. AIG1 and ADTRP are atypical integral membrane hydrolases that degrade bioactive FAHFAs. Nat Chem Biol 12, 367–372 (2016). https://doi.org: 10.1038/nchembio.2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Altschul SF et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–3402 (1997). https://doi.org: 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gabler F et al. Protein Sequence Analysis Using the MPI Bioinformatics Toolkit. Curr Protoc Bioinformatics 72, e108 (2020). https://doi.org: 10.1002/cpbi.108 [DOI] [PubMed] [Google Scholar]
- 27.Varadi M et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res 50, D439–D444 (2022). https://doi.org: 10.1093/nar/gkab1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mirdita M et al. ColabFold: making protein folding accessible to all. Nat Methods (2022). https://doi.org: 10.1038/s41592-022-01488-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holm L Dali server: structural unification of protein families. Nucleic Acids Res (2022). https://doi.org: 10.1093/nar/gkac387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Kempen M et al. FoldSeek: fast and accurate protein structure search. bioRxiv (2022). https://doi.org: 10.1101/2022.02.07.479398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ayoub R & Lee Y RUPEE: A fast and accurate purely geometric protein structure search. PLoS One 14, e0213712 (2019). https://doi.org: 10.1371/journal.pone.0213712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Glab B et al. Cloning of Glycerophosphocholine Acyltransferase (GPCAT) from Fungi and Plants: A NOVEL ENZYME IN PHOSPHATIDYLCHOLINE SYNTHESIS. J Biol Chem 291, 25066–25076 (2016). https://doi.org: 10.1074/jbc.M116.743062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tian W, Chen C, Lei X, Zhao J & Liang J CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res 46, W363–W367 (2018). https://doi.org: 10.1093/nar/gky473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu Y et al. CavityPlus: a web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction. Nucleic Acids Res 46, W374–W379 (2018). https://doi.org: 10.1093/nar/gky380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Komor AC, Kim YB, Packer MS, Zuris JA & Liu DR Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016). https://doi.org: 10.1038/nature17946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yan HF et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther 6, 49 (2021). https://doi.org: 10.1038/s41392-020-00428-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Magtanong L et al. Context-dependent regulation of ferroptosis sensitivity. Cell Chem Biol (2022). https://doi.org: 10.1016/j.chembiol.2022.06.004 [DOI] [PubMed] [Google Scholar]
- 38.Zallot R, Oberg N & Gerlt JA The EFI Web Resource for Genomic Enzymology Tools: Leveraging Protein, Genome, and Metagenome Databases to Discover Novel Enzymes and Metabolic Pathways. Biochemistry 58, 4169–4182 (2019). https://doi.org: 10.1021/acs.biochem.9b00735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown SD & Babbitt PC New insights about enzyme evolution from large scale studies of sequence and structure relationships. J Biol Chem 289, 30221–30228 (2014). https://doi.org: 10.1074/jbc.R114.569350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jonas A Lecithin cholesterol acyltransferase. Biochim Biophys Acta 1529, 245–256 (2000). https://doi.org: 10.1016/s1388-1981(00)00153-0 [DOI] [PubMed] [Google Scholar]
- 41.Ogura Y, Parsons WH, Kamat SS & Cravatt BF A calcium-dependent acyltransferase that produces N-acyl phosphatidylethanolamines. Nat Chem Biol 12, 669–671 (2016). https://doi.org: 10.1038/nchembio.2127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bazan JF & Fletterick RJ Viral cysteine proteases are homologous to the trypsin-like family of serine proteases: structural and functional implications. Proc Natl Acad Sci U S A 85, 7872–7876 (1988). https://doi.org: 10.1073/pnas.85.21.7872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Higaki JN, Evnin LB & Craik CS Introduction of a cysteine protease active site into trypsin. Biochemistry 28, 9256–9263 (1989). https://doi.org: 10.1021/bi00450a004 [DOI] [PubMed] [Google Scholar]
- 44.Baird TT Jr., Wright WD & Craik CS Conversion of trypsin to a functional threonine protease. Protein Sci 15, 1229–1238 (2006). https://doi.org: 10.1110/ps.062179006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Richter F et al. Computational design of catalytic dyads and oxyanion holes for ester hydrolysis. J Am Chem Soc 134, 16197–16206 (2012). https://doi.org: 10.1021/ja3037367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zoeller RA et al. Plasmalogens as endogenous antioxidants: somatic cell mutants reveal the importance of the vinyl ether. Biochem J 338 ( Pt 3), 769–776 (1999). [PMC free article] [PubMed] [Google Scholar]
- 47.Vinogradova EV et al. An Activity-Guided Map of Electrophile-Cysteine Interactions in Primary Human T Cells. Cell 182, 1009–1026 e1029 (2020). https://doi.org: 10.1016/j.cell.2020.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bar-Peled L et al. Chemical Proteomics Identifies Druggable Vulnerabilities in a Genetically Defined Cancer. Cell 171, 696–709 e623 (2017). https://doi.org: 10.1016/j.cell.2017.08.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Snyder F & Wood R Alkyl and alk-1-enyl ethers of glycerol in lipids from normal and neoplastic human tissues. Cancer Res 29, 251–257 (1969). [PubMed] [Google Scholar]
- 50.Albert DH & Anderson CE Ether-linked glycerolipids in human brain tumors. Lipids 12, 188–192 (1977). https://doi.org: 10.1007/BF02533292 [DOI] [PubMed] [Google Scholar]
Methods-only References
- 51.Cajka T, Smilowitz JT & Fiehn O Validating Quantitative Untargeted Lipidomics Across Nine Liquid Chromatography-High-Resolution Mass Spectrometry Platforms. Anal Chem 89, 12360–12368 (2017). https://doi.org: 10.1021/acs.analchem.7b03404 [DOI] [PubMed] [Google Scholar]
- 52.Koch J et al. Unequivocal Mapping of Molecular Ether Lipid Species by LC-MS/MS in Plasmalogen-Deficient Mice. Anal Chem 92, 11268–11276 (2020). https://doi.org: 10.1021/acs.analchem.0c01933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang TP, Newby GA & Liu DR Precision genome editing using cytosine and adenine base editors in mammalian cells. Nat Protoc 16, 1089–1128 (2021). https://doi.org: 10.1038/s41596-020-00450-9 [DOI] [PubMed] [Google Scholar]
- 54.Clement K et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat Biotechnol 37, 224–226 (2019). https://doi.org: 10.1038/s41587-019-0032-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing data validating genomic alterations in 786-O base-edited and knockout cell lines are deposited at the Sequence Read Archive (accession number PRJNA902005). Data supporting the findings of this work are available within the paper and its Supplementary Information. The datasets, constructs, and cell models generated and analyzed during the current study are available from the corresponding author upon request.