

Summary
Background
Emerging evidence shows that α-synuclein seed amplification assays (SAAs) have the potential to differentiate people with Parkinson’s disease from healthy controls. We used the well characterised, multicentre Parkinson’s Progression Markers Initiative (PPMI) cohort to further assess the diagnostic performance of the α-synuclein SAA and to examine whether the assay identifies heterogeneity among patients and enables the early identification of at-risk groups.
Methods
This cross-sectional analysis is based on assessments done at enrolment for PPMI participants (including people with sporadic Parkinson’s disease from LRRK2 and GBA variants, healthy controls, prodromal individuals with either rapid eye movement sleep behaviour disorder (RBD) or hyposmia, and non-manifesting carriers of LRRK2 and GBA variants) from 33 participating academic neurology outpatient practices worldwide (in Austria, Canada, France, Germany, Greece, Israel, Italy, the Netherlands, Norway, Spain, the UK, and the USA). α-synuclein SAA analysis of CSF was performed using previously described methods. We assessed the sensitivity and specificity of the α-synuclein SAA in participants with Parkinson’s disease and healthy controls, including subgroups based on genetic and clinical features. We established the frequency of positive α-synuclein SAA results in prodromal participants (RBD and hyposmia) and non-manifesting carriers of genetic variants associated with Parkinson’s disease, and compared α-synuclein SAA to clinical measures and other biomarkers. We used odds ratio estimates with 95% CIs to measure the association between α-synuclein SAA status and categorical measures, and two-sample 95% CIs from the resampling method to assess differences in medians between α-synuclein SAA positive and negative participants for continuous measures. A linear regression model was used to control for potential confounders such as age and sex.
Findings
This analysis included 1123 participants who were enrolled between July 7, 2010, and July 4, 2019. Of these, 545 had Parkinson’s disease, 163 were healthy controls, 54 were participants with scans without evidence of dopaminergic deficit, 51 were prodromal participants, and 310 were non-manifesting carriers. Sensitivity for Parkinson’s disease was 87·7% (95% CI 84·9–90·5), and specificity for healthy controls was 96·3% (93·4–99·2). The sensitivity of the α-synuclein SAA in sporadic Parkinson’s disease with the typical olfactory deficit was 98·6% (96·4–99·4). The proportion of positive α-synuclein SAA was lower than this figure in subgroups including LRRK2 Parkinson’s disease (67·5% [59·2–75·8]) and participants with sporadic Parkinson’s disease without olfactory deficit (78·3% [69·8–86·7]). Participants with LRRK2 variant and normal olfaction had an even lower α-synuclein SAA positivity rate (34·7% [21·4–48·0]). Among prodromal and at-risk groups, 44 (86%) of 51 of participants with RBD or hyposmia had positive α-synuclein SAA (16 of 18 with hyposmia, and 28 of 33 with RBD). 25 (8%) of 310 non-manifesting carriers (14 of 159 [9%] LRRK2 and 11 of 151 [7%] GBA) were positive.
Interpretation
This study represents the largest analysis so far of the α-synuclein SAA for the biochemical diagnosis of Parkinson’s disease. Our results show that the assay classifies people with Parkinson’s disease with high sensitivity and specificity, provides information about molecular heterogeneity, and detects prodromal individuals before diagnosis. These findings suggest a crucial role for the α-synuclein SAA in therapeutic development, both to identify pathologically defined subgroups of people with Parkinson’s disease and to establish biomarker-defined at-risk cohorts.
Funding
PPMI is funded by the Michael J Fox Foundation for Parkinson’s Research and funding partners, including: Abbvie, AcureX, Aligning Science Across Parkinson’s, Amathus Therapeutics, Avid Radiopharmaceuticals, Bial Biotech, Biohaven, Biogen, BioLegend, Bristol-Myers Squibb, Calico Labs, Celgene, Cerevel, Coave, DaCapo Brainscience, 4D Pharma, Denali, Edmond J Safra Foundation, Eli Lilly, GE Healthcare, Genentech, GlaxoSmithKline, Golub Capital, Insitro, Janssen Neuroscience, Lundbeck, Merck, Meso Scale Discovery, Neurocrine Biosciences, Prevail Therapeutics, Roche, Sanofi Genzyme, Servier, Takeda, Teva, UCB, VanquaBio, Verily, Voyager Therapeutics, and Yumanity.
Introduction
Biomarkers for Parkinson’s disease that reflect underlying pathological features would improve the accuracy of early diagnosis, clarify subtypes, and accelerate clinical trials.1 The pathological hallmark of Parkinson’s disease is the accumulation of misfolded, aggregated α-synuclein in the substantia nigra and other areas of the brain.2,3 α-synuclein aggregates have also been identified in peripheral nervous system tissue.4,5 Misfolded protein amplification techniques, originally developed for the detection of the self-propagating scrapie isoform of the prion protein (PrPSc) in prion diseases,6 have been applied to detect α-synuclein seeds in Parkinson’s disease and other synucleinopathies.7,8 These assays have been reported under the names real-time quaking-induced conversion,9 protein misfolding cyclic amplification,10 and most recently, the consensus name, seed amplification assay (SAA).11,12
Previous studies have shown that α-synuclein SAAs performed on CSF distinguish people with Parkinson’s disease from healthy controls with high sensitivity and specificity.12,13 Preliminary studies have also shown α-synuclein SAA positive results in a high proportion of people from at-risk groups, such as those with isolated rapid eye movement sleep behaviour disorder (RBD).14 One study showed excellent inter-laboratory agreement when samples from the same participants were run on three different assay platforms.12
Although these and other studies have contributed to substantial progress in understanding the potential of the α-synuclein SAA for in vivo molecular characterisation of Parkinson’s disease, large-scale studies confirming and extending these results are needed. In this report, we describe the α-synuclein SAA results for more than 1100 participants in the Parkinson’s Progression Markers Initiative (PPMI) study, including people with Parkinson’s disease with and without associated genetic variants, healthy controls, and people at risk for Parkinson’s disease (either with prodromal features or non-manifesting carriers of genetic variants). The goals of this analysis were to establish assay sensitivity and specificity using a large number of samples, to test the ability of the α-synuclein SAA to detect the early signs of Parkinson’s disease pathophysiological changes in at-risk individuals, and to leverage clinical and biomarker data within the PPMI study cohort to examine clinical and genetic heterogeneity among people with Parkinson’s disease on the basis of α-synuclein SAA status.
Methods
Study design and participants
This study is a cross-sectional analysis using data from the PPMI cohort. PPMI is an international observational study recruiting patients through outpatient neurology practices at academic centres in Austria, Canada, France, Germany, Greece, Israel, Italy, the Netherlands, Norway, Spain, the UK, and the USA, with the goal of identifying clinical and biological markers of disease heterogeneity and progression in Parkinson’s disease. The PPMI study is registered with ClinicalTrials.gov (number NCT01141023). Detailed information about inclusion criteria, informed consent, demographic data, and the study design can be found on the PPMI website. The data were last accessed on Dec 15, 2022.
Participants in this study were included in one of the five PPMI cohorts: participants with Parkinson’s disease, healthy controls, participants with parkinsonism but with scans without evidence of dopamine deficiency (SWEDD), participants who are prodromal (including those with RBD or hyposmia), and non-manifesting carriers of genetic variants associated with Parkinson’s disease; all were recruited between July 7, 2010, and July 4, 2019. The diagnosis for each group was made by site investigators who are movement disorder specialists and confirmed by a central consensus committee review. α-synuclein SAA results were not available to investigators or the consensus committee at the time of diagnosis, and thus were not incorporated into the classification of participants. The study was approved by the institutional review board at each site, and participants provided written informed consent.
Participants with sporadic Parkinson’s disease (non-carriers of LRRK2 or GBA variants) were enrolled if they were within 2 years of diagnosis; had not been administered Parkinson’s disease medications at the time of enrolment; were at Hoehn and Yahr stage 1–2; had abnormal dopamine transporter (DAT)-SPECT; and had two of either: resting tremor, bradykinesia, rigidity (required to have either resting tremor or bradykinesia), or asymmetric resting tremor or asymmetric bradykinesia. Furthermore, participants with Parkinson’s disease who were carriers of either the LRRK2 Gly2019Ser or the GBA Asn409Ser variants were included in this study. The inclusion criteria for genetic Parkinson’s disease were the same as other participants with Parkinson’s disease, except that those with a genetic variant were not required to have an abnormality on DAT-SPECT, had to be within 7 years of diagnosis, and could be receiving treatment for Parkinson’s disease.
SWEDD participants were enrolled with Parkinson’s disease inclusion criteria, with the exception that their initial DAT-SPECT did not show evidence of decreased striatal radio-ligand uptake. Healthy controls were age-matched and sex-matched people without known neurological signs or symptoms. As per the study protocol, healthy controls were recruited at a rate of approximately one control to every two participants with sporadic Parkinson’s disease.
Prodromal participants included individuals without a diagnosis of Parkinson’s disease, but who had prodromal features associated with risk of Parkinson’s disease, including RBD (confirmed by polysomnogram) or otherwise unexplained severe hyposmia (defined as at or less than the 15th percentile using the University of Pennsylvania Smell Identification Test [UPSIT; Sensonics, Philadelphia, PA, USA]) in olfactory performance based on internal population norms.15 Enrolment of participants with RBD and hyposmia was stratified to enrich for cases with abnormal DAT-SPECT. Non-manifesting carriers of either the LRRK2 or GBA variant were included without enrichment for a DAT deficit. For all participants, cohort assignment was done as described in the study protocol and confirmed by the PPMI clinical consensus review committee.
Clinical and pathological assessments
All participants underwent a series of clinical tests described previously.16 Assessments included the Movement Disorders Society Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) parts I (non-motor aspects or experiences of daily living), II (motor aspects or experiences of daily living), and III (motor examination; recorded in the off-state for treated participants), Montreal Cognitive assessment, RBD questionnaire, University of Pennsylvania Smell Identification test (UPSIT), Scales for Outcomes in Parkinson’s Autonomic Dysfunction, and the 15-item Geriatric Depression Scale. Post-mortem assessments were done using previously described methods.2,17
Biomarker assessments
All participants had biosampling done, including blood, CSF, and urine. CSF samples were collected, stored, and shipped according to the PPMI protocol. For this study, samples collected at baseline were analysed by α-synuclein SAA (one sample per participant). In addition to CSF α-synuclein SAA assessment, the other CSF biomarkers assessed included β-amyloid 1–42, total-tau, phosphorylated-tau, and quantitative total α-synuclein. These biomarkers were assessed using methods described previously.18 Plasma neurofilament light chain19 and urine bis(monoacylglycerol) phosphate were also assessed. Finally, DAT-SPECT was performed as previously described.20 A visual interpretation of SPECT images was used to assign patients to the Parkinson’s disease, healthy control, and SWEDD groups. Quantitative analysis using striatal-specific binding ratios (SBRs), corrected for age and sex, were used to compare DAT-SPECT with clinical and biomarker data. A cutoff of less than 65% of age-expected and sex-expected binding in the lower putamen was used to define a quantitatively abnormal DAT-SPECT result.
α-synuclein seed amplification assay
The Amprion α-synuclein SAA developed by Concha-Marambio and colleagues21 has been described previously, following a detailed protocol. Briefly, the 200-μL reaction mixture included 0·3 mg/mL recombinant α-synuclein (Amprion, San Diego, CA, USA), 0·5 mol/L NaCl (Lonza, Basel, Switzerland), 100 mmol/L PIPES-NaOH (pH 6·50; MilliporeSigma, Burlington, MA, USA), and 20% volume per volume CSF. Recombinant α-synuclein was expressed with C-terminal His-tag in Escherichia coli BL21(DE3) and purified using immobilised metal affinity chromatography. All samples were analysed with a single batch of the substrate. One 3/32 inch Si3N4 bead (Tsubaki Nakashima, Osaka, Japan) was added per well using a house-made bead dispenser. Beads were blocked with 1% bovine serum albumin 100 mmol/L PIPES-NaOH (pH 6·50) for 1 h and washed twice with 100 mmol/L PIPES-NaOH (pH 6·50). Samples were run in three technical replicates within 96-well plates, using three FLUOstar Omega readers set to 37°C. Each plate was shaken for 1 min every 29 min, and fluorescence was measured after each cycle for 150 h. The primary outcome of a sample being either α-synuclein SAA positive or negative was determined in the following manner. The highest raw fluorescence from each well was used in a probabilistic algorithm to establish whether each of the three replicates was a positive or negative, and the results of the triplicate were used to establish the assay output for each sample. If all three replicates from a given sample were positive, the sample was deemed positive for α-synuclein SAA. If zero or one replicates were positive, the sample was deemed negative. If two replicates were positive, the sample was deemed inconclusive. A second-level criterion within the algorithm compared the average maximum fluorescence of the three replicates from inconclusive samples, and samples with highly variable or low average maximum fluorescence were deemed negative. The α-synuclein SAA data are available in the PPMI Laboratory of Neuroimaging database (Amprion; project number 155). All α-synuclein SAA analyses were performed by LC-M, CMF, YM, PAU, and HN, masked to participant demographic features and diagnosis. Blinding was protected by shipping samples randomly assigned by cohort using unique specimen identifications.
Statistical analysis
This study was performed using all PPMI participants with available baseline samples at the time of performance of the assay. SAS version 9.4 software was used for all statistical analyses and figures. Sensitivity and specificity with 95% Wald CIs were calculated for Parkinson’s disease, healthy controls, and SWEDD groups, and subgroups based on sex and olfactory performance. For cohorts with one group having sample sizes fewer than 40, Wilson’s method was used to calculate the 95% CIs for sensitivity and specificity. Descriptive statistics at baseline, including median (IQR) for continuous measures because of the skewness of the data and frequency (percentage) for categorical measures, were calculated by cohort, subgroup, and α-synuclein SAA status for demographics, MDS-UPDRS scores, CSF biomarkers, UPSIT percentile, and DAT-SPECT SBR. For groups with fewer than three total samples, IQRs were not provided for continuous outcomes. Similarly, for categorical outcomes, percentages for groups with total samples fewer than ten were not provided. Separately for each Parkinson’s disease subgroup, the two-sample 95% CI from the resampling method using ten thousand replicates was used to assess differences in medians between α-synuclein SAA positive and α-synuclein-SAA negative participants for age, disease duration, MDS-UPDRS scores, expected DAT-SPECT SBR, mean striatum SBR, serum neurofilament light chain, and total di-18:1 bis(monoacylglycerol) phosphate. Moreover, to measure the association between α-synuclein SAA status and sex, Hoehn and Yahr stage, race, hyposmia status, and CSF biomarkers, odds ratio (OR) estimates with 95% Wald CIs for relevant categorical outcomes were reported. To account for skewness and the upper limit of detection, each CSF biomarker was dichotomised. Because of the strong relationships for both sex and age with a positive α-synuclein SAA result within the LRRK2 Parkinson’s disease cohort, we implemented a modelling strategy, adjusting for age and sex, for any positive univariate associations observed within that cohort. For skewed continuous variables, a log transform was applied to address the violation of model assumptions. Statistical analyses were not provided for groups with a low sample size (eg GBA Parkinson’s disease with negative α-synuclein SAA).
Role of the funding source
Research officers (MF, SH, LO, TS) at the funding institution were involved in the study design, data interpretation, and writing of the report. The funders were not involved in data collection or data analysis.
Results
This analysis included 1123 participants who were enrolled between July 7, 2010, and July 4, 2019: 545 individuals with Parkinson’s disease (sporadic, n=373; LRRK2 Gly2019Ser variant, n=123; GBA Asn409Ser variant, n=49), 163 healthy controls, 54 SWEDD participants, 51 prodromal partici pants (hyposmia, n=18; RBD, n=33), and 310 non-manifesting carriers (LRRK2, n=159; GBA, n=151). 19 people (sporadic Parkinson’s disease, n=4; LRRK2 Parkinson’s disease, n=3; healthy controls, n=3; SWEDD, n=3; prodromal, n=1; and non-manifesting carriers, n=5) had inconclusive α-synuclein SAA results and were excluded from the analysis. Sensitivity analyses conducted under the conservative assumption that the assay was always incorrect in these cases showed similar results to those obtained excluding inconclusive results (data not shown). Prodromal participants were older and more likely to be male than any other group. Non-manifesting carriers were more likely to be female than any other group. Other demographic features are shown in table 1.
Table 1:
Demographics and clinical features
| Parkinson’s disease (N=545) | Healthy control (N=163) | SWEDD (N=54) | Prodromal (N=51) | Non-manifesting carriers (N=310) | |
|---|---|---|---|---|---|
|
| |||||
| Age, years | 63·4 (56·3-69·1) | 62·6 (55·0-69·2) | 63·3 (52·4-68·5) | 67·7 (65·9-73·2) | 61·7 (56·6-66·8) |
| Sex | |||||
| Male sex | 337 (62%) | 107 (66%) | 32 (59%) | 40 (78%) | 134 (43%) |
| Female sex | 208 (38%) | 56 (34%) | 22 (41%) | 11 (22%) | 176 (57%) |
| Disease duration, years since diagnosis | 0·6 (0·3-1·8) | NA | 0·3 (0·2-0·9) | NA | NA |
| Hoehn and Yahr stage, 3-5 | 10 (2%) | 0 | 1 (2%) | 0 | 0 |
| Race | |||||
| White | 508 (93%) | 151 (93%) | 51 (94%) | 48 (94%) | 299 (96%) |
| All other races | 36 (7%) | 11 (7%) | 3 (6%) | 2 (4%) | 6 (2%) |
| American Indian or Alaska Native | 1 (<1%) | 0 | 0 | 0 | 0 |
| Asian (including Native Hawaiian OR other Pacific Islander) | 9 (2%) | 0 | 1 (2%) | 0 | 0 |
| Black | 6 (1%) | 8 (5%) | 1 (2%) | 1 (2%) | 0 |
| Multiracial | 17 (3%) | 2 (1%) | 0 | 0 | 4 (1%) |
| Not reported | 4 (1%) | 2 (1%) | 1 (2%) | 2 (4%) | 7 (2%) |
| Hispanic or Latino ethnicity | 31 (6%) | 3 (2%) | 2 (4%) | 18 (35%) | 15 (5%) |
| Hyposmia, ≤15th percentile | 390 (72%) | 14 (9%) | 10 (19%) | 44 (86%) | 40 (13%) |
| Missing hyposmia data | 9 (2%) | 0 | 0 | 1 (2%) | 1 (<1%) |
| Abnormal dopamine transporter lowest putamen specific binding ratio | 0·30 (0·24-0·38) | 0·95 (0·81-1·17) | 0·95 (0·81-1·13) | 0·59 (0·45-0·71) | 1·04 (0·88-1·20) |
| Positive α-synuclein seed amplification assay | 478 (88%) | 6 (4%) | 5 (9%) | 44 (86%) | 25 (8%) |
| LRRK2 variant | 123 (23%) | 0 | 0 | NA | 159 (51%) |
| GBA variant | 49 (9%) | 0 | 0 | NA | 151 (49%) |
Data shown as n (%), median (IQR), or mean (SD). Statistical analyses comparing cohorts were not performed. NA=not applicable. NMC=non-manifesting carriers. SWEDD=participants with scans without evidence of dopaminergic deficit.
The sensitivity of α-synuclein SAA for detecting all Parkinson’s disease cases, combining sporadic and genetic cases, was 87·7% (95% CI 84·9–90·5; table 2). α-synuclein SAA was positive slightly more often for SWEDD participants than for healthy controls (9% vs 4%). This result is in keeping with the literature showing that a small number of individuals with borderline DAT-SPECT imaging results have progressive parkinsonism.22
Table 2:
Sensitivity of CSF α-synuclein seed amplification assay for Parkinson’s disease, and specificity for healthy controls and SWEDD
| N | Specificity (95% CI) | Sensitivity (95% CI) | |
|---|---|---|---|
|
| |||
| Healthy controls | 163 | 96·3% (93·4-99·2) | NA |
| SWEDD | 54 | 90·7% (83·0-98·5) | NA |
| All Parkinson’s disease cases | 545 | NA | 87·7% (84·9-90·5) |
| Hyposmic | 390 | NA | 97·2% (95·5-98·8) |
| Normosmic | 146 | NA | 63·0% (55·2-70·8) |
| Sporadic Parkinson’s disease | 373 | NA | 93·3% (90·8-95·8) |
| LRRK2 mutation Parkinson’s disease | 123 | NA | 67·5% (59·2-75·8) |
| GBA mutation Parkinson’s disease | 49 | NA | 95·9% (90·4-100·0) |
| LRRK2 mutation Parkinson’s disease | |||
| Male participants | 65 | NA | 78·5% (68·5-88·5) |
| Female participants | 58 | NA | 55·2% (42·4-68·0) |
| Hyposmic | 69 | NA | 89·9% (82·7-97·0) |
| Normosmic | 49 | NA | 34·7% (21·4-48·0) |
| Normosmic and female participants | 24 | NA | 12·5% (4·3-31·0) |
NA=not applicable. SWEDD=participants with scans without evidence of dopaminergic deficit.
The proportion of participants with positive α-synuclein SAA varied across subgroups based on genetic and clinical features. Among genetic Parkinson’s disease subgroups, the proportion of participants with positive α-synuclein SAA results was highest for GBA Parkinson’s disease (95·9%; 95% CI 90·4–100·0), followed by sporadic Parkinson’s disease (93·3%; 90·8–95·8), and lowest for LRRK2 Parkinson’s disease (67·5%; 59·2–75·8; table 2). Among clinical features, hyposmia was the most robust predictor of a positive result. Among all participants with Parkinson’s disease with hyposmia, the sensitivity of α-synuclein SAA was 97·2% (95·5–98·8) compared with 63·0% (55·2–70·8) among all participants with Parkinson’s disease without olfactory dysfunction. Combining genetic and clinical features, the sensitivity of α-synuclein SAA in sporadic Parkinson’s disease with a typical olfactory deficit was 98·6% (96·4–99·4), and in participants with sporadic Parkinson’s disease without olfactory deficit it was 78·3% (69·8–86·7). By contrast, the sensitivity for LRRK2 variant carriers with hyposmia was 89·9% (82·7–97·0) compared with 34·7% (21·4–48·0) for LRRK2 carriers with normal olfaction (figure 1, table 2). Additionally, for LRRK2 participants, the likelihood of a positive α-synuclein SAA result was lower for female participants (55·2%; 42·4–68·0) than male participants (78·5%; 68·5–88·5). Three of 24 female normosmic LRRK2 carriers had a positive a-synuclein SAA result (12·5%; 4·3–31·0). α-synuclein SAA results did not differ between male participants and female participants with sporadic Parkinson’s disease or GBA Parkinson’s disease (table 3). Other clinical features were significantly associated with α-synuclein SAA status, but the magnitude of these associations was modest and only observed in the LRRK2 subgroup. Consistent with the aforementioned results, LRRK2 variant carriers with negative α-synuclein SAA results were more likely to be female (65% vs 35%; OR 2·9; 95% CI 1·3 to 6·5), and were also older (median age 69·1 vs 62·0; difference 7·1; 4·9 to 9·2) than those with positive results (table 3). The LRRK2 Parkinson’s disease group with negative α-synuclein SAA results had slightly less self-reported impairment (MDS-UPDRS II; median difference −3; 95% CI −5·6 to −0·4) and slightly less severe motor features by examination (MDS-UPDRS III; −7; −12 to −2) than the α-synuclein SAA positive LRRK2 Parkinson’s disease group. There were no significant associations between autonomic function, cognitive test results, depression scores, or RBD scores and α-synuclein SAA status for any groups (data not shown). Biomarker differences were also generally modest and were observed only in LRRK2 carriers (appendix p 1). LRRK2 variant carriers with negative α-synuclein SAA had higher (less severe) DAT lowest putamen SBR (median difference 0·12; 0·05 to 0·19). In univariate analysis, the LRRK2 Parkinson’s disease group with negative α-synuclein SAA had higher serum neurofilament light chain than the SAA positive group (median difference 7·7; 1·3 to 14·1). However, this association was not present after adjustment for age and sex (difference in log serum neurofilament light chain −0·19; −0·40 to 0·01). There were no apparent associations with other biomarkers (appendix p 1).
Figure 1:
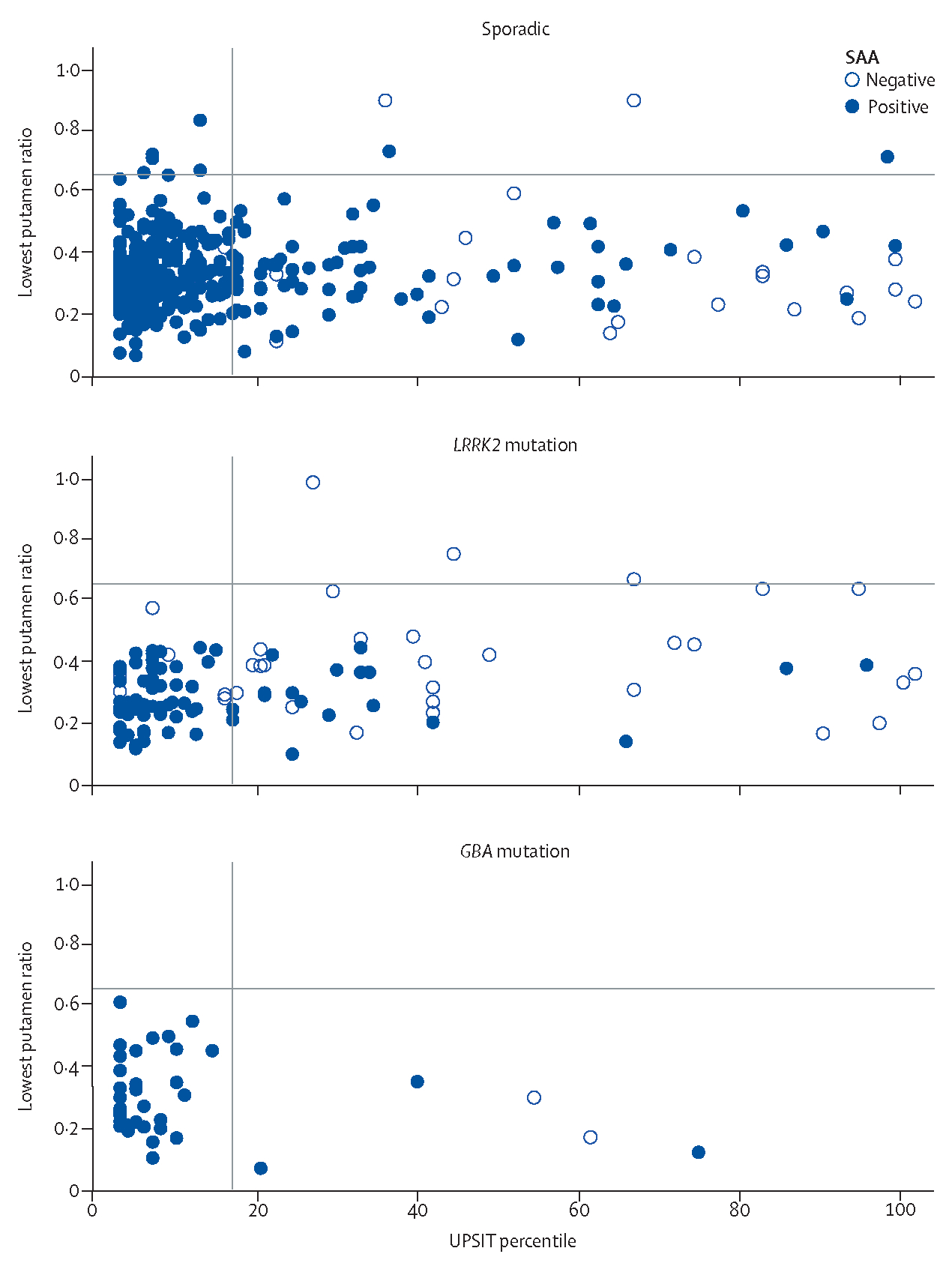
Association between dopamine transporter binding, olfaction, and α-synuclein SAA results among participants with manifest Parkinson’s disease The figure shows the relationship between α-synuclein SAA status and dopamine transporter imaging measured by the percent of age-expected and sex-expected lowest putamen specific binding ratio, a measure of dopamine transporter loss in the most sensitive striatal region16 and the University of Pennsylvania Smell Identification Test age and sex percentile of normal.15 The horizontal line represents the dopamine transporter-SPECT lowest putamen specific binding ratio of less than 65% (individuals less than 65% are in the Parkinson’s disease range), and the vertical line represents the age-adjusted and sex-adjusted University of Pennsylvania Smell Identification Test percentile of 15% or less cutoff (individuals less than 15% have hyposmia). SAA=seed amplification assay.
Table 3:
Clinical characteristics by α-synuclein SAA status for sporadic Parkinson’s disease, LRRK2 mutation Parkinson’s disease, GBA mutation Parkinson’s disease, and healthy controls
| Sporadic Parkinson’s disease | LRRK2 mutation Parkinson’s disease | GBA mutation Parkinson’s disease | Healthy controls | |||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|||||
| SAA positive (N=348) | SAA negative (N=25) | SAA positive (N=83) | SAA negative (N=40) | SAA positive (N=47) | SAA negative (N=2) | SAA positive (N=6) | SAA negative (N=157) | |
|
| ||||||||
| Age, years* | 62·6 (55·3 to 68·9) | 66·4 (58·4 to 71·1) | 62·0 (55·0 to 66·5) | 69·1 (66·0 to 72·4) | 62·8 (56·3 to 69·5) | 67·8 (NA) | 64·1 (60·1 to 65·2) | 62·0 (54·8 to 69·2) |
| Male sex† | 227 (65%) | 16 (64%) | 51 (61%) | 14 (35%) | 27 (57%) | 2 (NA) | 5 (NA) | 102 (65%) |
| Female sex | 121 (35%) | 9 (36%) | 32 (39%) | 26 (65%) | 20 (43%) | 0 (NA) | 1 (NA) | 55 (35%) |
| Disease duration, years since diagnosis | 0·4 (0·2 to 0·7) | 0·4 (0·2 to 0·6) | 2·4 (1·3 to 4·7) | 2·4 (1·2 to 4·4) | 3·1 (1·4 to 5·3) | 1·0 (NA) | NA | NA |
| White | 320 (92%) | 24 (96%) | 78 (94%) | 39 (98%) | 45 (96%) | 2 (NA) | 6 (NA) | 145 (92%) |
| MDS-UPDRS I | 5 (3 to 7) | 6 (4 to 12) | 7 (4 to 11) | 7 (2 to 12) | 7 (5 to 11) | 4 (NA) | 4 (1 to 5) | 2 (1 to 4) |
| MDS-UPDRS II‡ | 5 (3 to 8) | 8 (4 to 12) | 7 (4 to 10) | 4 (2 to 8) | 8 (5 to 12) | 8 (NA) | 0 (0 to 1) | 0 (0 to 0) |
| MDS-UPDRS III§ | 20 (15 to 26) | 22 (17 to 27) | 24 (15 to 31) | 17 (12 to 26) | 30 (20 to 39) | 25 (NA) | 0 (0 to 3) | 0 (0 to 2) |
| Total MDS-UPDRS | 31 (23 to 41) | 37 (25 to 48) | 37 (27 to 55) | 32 (21 to 42) | 43 (34 to 57) | 44 (NA) | 6 (1 to 7) | 3 (1 to 7) |
| Hyposmia, ≤15th percentile¶ | 274 (79%) | 4 (16%) | 62 (75%) | 7 (18%) | 43 (91%) | 0 (NA) | 3 (NA) | 11 (7%) |
| Missing hyposmia data | 2 (1%) | 1 (4%) | 4 (5%) | 1 (3%) | 1 (2%) | 0 | 0 | 0 |
| Abnormal dopamine transporter lowest putamen specific binding ratio‖ | 0·31 (0·25 to 0·38) | 0·32 (0·23 to 0·38) | 0·26 (0·22 to 0·37) | 0·38 (0·29 to 0·47) | 0·28 (0·21 to 0·37) | 0·23 (NA) | 1·01 (0·87 to 1·27) | 0·95 (0·81 to 1·17) |
Data shown as median (IQR) or n (%). Difference in medians with 95% CIs were used for within group comparisons of α-synuclein SAA status for age, disease duration, University of Pennsylvania Smell Identification test percentile, and age-percent and sex-percent expected specific binding ratio. Odds ratio with 95% CIs were used for within group comparisons of α-synuclein SAA status for sex, Hoehn and Yahr stage, race, and hyposmia status. MDS-UPDRS=Movement Disorders Society Unified Parkinson’s Disease Rating Scale. NA=not applicable. OR=odds ratio. SAA=seed amplification assay.
SAA negative LRRK2 Parkinson’s disease were older than their SAA positive counterparts (OR 7·0 [95% CI 4·9 to 9·2]).
SAA negative LRRK2 Parkinson’s disease were more often male than their SAA positive counterparts (OR 2·9 [95% CI 1·3 to 6·5]).
The difference in median MDS-UPDRS II between SAA negative LRRK2 Parkinson’s disease and their SAA positive counterparts was −3 (95% CI −5·6 to −0·4).
The difference in median MDS-UPDRS III between SAA negative LRRK2 Parkinson’s disease and their SAA positive counterparts was −7 (95% CI −12 to −2).
Hyposmia was more common in α-synuclein SAA positive sporadic Parkinson’s disease than in α-synuclein SAA negative sporadic Parkinson’s disease (OR 19, 95% CI 6·3-57·4), and the same was true for LRRK2 Parkinson’s disease (OR 16·7 [95% CI 6·3 to 44·3]).
The difference in median specific binding ratio between SAA negative LRRK2 Parkinson’s disease and their SAA positive counterparts was 0·12 (95% CI 0·05 to 0·19).
44 (86%) of 51 participants with RBD and hyposmia had positive α-synuclein SAA results (appendix p 2). For the 18 participants who were prodromal and recruited based on smell loss, 16 had positive α-synuclein SAA results (appendix p 2), and 28 of 33 RBD cases had positive α-synuclein SAA results. The positive RBD cases also tended to have abnormal olfaction. All but one participant of these 27 who were α-synuclein SAA positive were at or less than the 15th percentile of the UPSIT score, where as four of five α-synuclein SAA negative individuals were normosmic. There were no other clinical features that were associated with a positive α-synuclein SAA result in prodromal participants. In the prodromal (RBD or hyposmia) groups, 29·6% of the participants who were α-synuclein SAA positive had DAT-SPECT results more than the cutoff of greater than or equal to 65% of age-expected and sex-expected binding uptake associated with prodromal Parkinson’s disease,23 supportive of a model in which a positive α-synuclein SAA result could precede abnormal DAT imaging (figure 2). A smaller number of RBD cases (three of 33) had positive DAT imaging but negative α-synuclein SAA results (figure 2). Of 310 non-manifesting carriers, 25 (8%) had positive α-synuclein SAA results, of whom 9% (14 of 159) were LRRK2 and 7% (11 of 151) were GBA non-manifesting carriers (figure 2).
Figure 2: Association between dopamine transporter binding, olfaction, and α-synuclein SAA results among prodromal and non-manifesting carriers of either LRRK2 or GBA variants.

The figure shows the relationship between α-synuclein SAA status and dopamine transporter imaging measured by the percent of age-expected and sex-expected lowest putamen specific binding ratio, a measure of dopamine transporter loss in the most sensitive striatal region16 and the University of Pennsylvania Smell Identification Test age and sex percentile of normal.15 The horizontal line represents the dopamine transporter-SPECT lowest putamen specific binding ratio of less than 65% (individuals less than 65% are in the Parkinson’s disease range), and the vertical line represents the age-adjusted and sex-adjusted University of Pennsylvania Smell Identification Test percentile of 15% or less cutoff (individuals less than 15% have hyposmia).
Autopsy data were available for 15 participants, all with a clinical diagnosis of Parkinson’s disease. 14 participants had typical pathological findings, including Lewy bodies and Lewy neurites, and were α-synuclein SAA positive. The one α-synuclein SAA negative case was a normosmic participant with Parkinson’s disease who also carried the LRRK2 Gly2019Ser variant. At autopsy, this patient had nigral cell loss and depigmentation, but no Lewy bodies or neurites (data not shown).
Discussion
This study uses the comprehensive clinical and biomarker PPMI dataset to show that α-synuclein SAA provides information on Parkinson’s disease genetic and clinical heterogeneity and identifies at-risk individuals, possibly at an early stage of degeneration. We showed that α-synuclein-SAA is highly accurate in differentiating Parkinson’s disease from healthy controls, but observed variability among genetic subgroups, in particular among participants with LRRK2 Parkinson’s disease. The proportion of participants with positive α-synuclein SAA results was highest among patients with sporadic Parkinson’s disease with a typical olfactory deficit. By contrast, the proportion of α-synuclein SAA positive results was lower in those with normal olfaction and in LRRK2 variant carriers. In groups defined by genetic variant carrier status, we found the highest proportion of α-synuclein SAA positive results in people with GBA Parkinson’s disease, followed closely by sporadic Parkinson’s disease, and a substantially lower proportion in LRRK2 Parkinson’s disease. Preserved olfaction was associated with negative α-synuclein SAA results across genetic subgroups, and this association was most striking in participants who were both normosmic and LRRK2 variant carriers. This finding was particularly true among female participants; only 12·5% of female normosmic LRRK2 carriers (three of 24) had a positive α-synuclein SAA result.
In cases that came to post-mortem examination, all participants with positive α-synuclein SAA results had typical Lewy pathology, whereas the one case with negative α-synuclein SAA result (an LRRK2 carrier with preserved olfaction) had no Lewy pathology. These clinical and pathological data suggest that there might be different pathologies in α-synuclein SAA positive and negative LRRK2 Parkinson’s disease, despite a similar clinical phenotype. Our results showing a lower frequency of α-synuclein SAA positivity in participants with Parkinson’s disease carrying a LRRK2 variant is consistent with a previous study of 15 participants with LRRK2 Gly2019Ser Parkinson’s disease and 16 non-manifesting carriers.24 Notably, the proportion of α-synuclein SAA negative cases that we found (approximately a third) closely mirrors the frequency of LRRK2 Parkinson’s disease cases without Lewy pathology at autopsy reported in the literature.25,26 These consistent results indicate that α-synuclein SAA is a marker of pathology characterised by α-synuclein aggregates rather than a clinical Parkinson’s disease phenotype, and enables ante-mortem differentiation of cases with atypical pathological findings.
Along with preserved olfaction, people with Parkinson’s disease with a LRRK2 variant and a negative α-synuclein SAA result were older, more likely to be female, and had slightly less motor impairment than LRRK2 carriers who had a positive α-synuclein SAA. These results are consistent with a modest clinical–pathological correlation within LRRK2 Parkinson’s disease, that can be stratified by α-synuclein SAA result. Furthermore, the higher number of female participants among LRRK2 carriers who are α-synuclein SAA negative might partly account for the observation that the expected male-to-female predominance seen in sporadic Parkinson’s disease is not present in LRRK2 carriers.27
Regarding prodromal and at-risk individuals, the results of this study confirm that most participants with RBD with an abnormal polysomnogram and DAT deficit are α-synuclein SAA positive. We extended our assessment to individuals at risk for Parkinson’s disease on the basis of impaired olfaction who also had a DAT deficit, and found that they are equally likely to be α-synuclein SAA positive. Of the five (of 33) RBD cases who were α-synuclein SAA negative, three had abnormal DAT imaging and four also had normal olfactory function, possibly explained by the observation that some people with RBD progress to multiple system atrophy.28 This disorder is associated with less olfactory impairment than Parkinson’s disease.29 Previous studies evaluating the use of α-synuclein SAA in multiple system atrophy have produced varying results, depending on the assay condition, and multiple system atrophy has been shown to have distinct α-synuclein SAA aggregation properties relative to Parkinson’s disease.10,30 For this study, we used an assay that targeted the detection of Parkinson’s disease aggregates, and which has a lower sensitivity to detect glial cytoplasmic inclusions in multiple system atrophy.
We observed several participants with prodromal RBD and hyposmia with positive α-synuclein SAA results but DAT imaging in the normal range. By contrast, no prodromal hyposmia participants had negative α-synuclein SAA results and positive DAT imaging. Only participants with RBD potentially explained by multiple system atrophy had negative α-synuclein SAA and DAT binding consistent with Parkinson’s disease. Approximately 7–10% of non-manifesting carriers of either GBA or LRRK2 Parkinson’s disease-related variants had positive α-synuclein SAA results. This figure is similar to the lifetime penetrance of Parkinson’s disease among GBA Asn409Ser carriers,31 but substantially lower than estimates of Parkinson’s disease penetrance among LRRK2 Gly2019Ser carriers.32,33 Taken together, these findings are consistent with a temporal pattern of biomarker abnormalities whereby there might be a long-term period in which abnormalities in α-synuclein SAA are present before changes appear in physiological markers, such as DAT-SPECT results, and that this pattern might be more variable in LRRK2 carriers than GBA carriers. Notably, among the GBA non-manifesting carriers, those few individuals who are α-synuclein SAA positive are also more likely to have olfactory dysfunction, even in the absence of a DAT deficit. Our results provide biomarker support for a long-term prodromal period. This concept has been proposed in the literature, but did not have supporting evidence from nigral neuropathological findings or dopamine imaging studies.34 Subsequent studies in larger prodromal cohorts are needed to confirm this hypothesis.
The results of our study have immediate implications for clinical trial design. For LRRK2-targeted therapies, stratification based on α-synuclein SAA results might be necessary to assess therapeutic benefits. For GBA-targeted therapies, it would be reasonable to exclude α-synuclein SAA negative participants, since GBA Parkinson’s disease is overwhelmingly α-synuclein SAA positive,35 and, thus, a negative result would raise questions about a Parkinson’s disease diagnosis. α-synuclein SAA could also be combined with other biomarkers, such as markers of Alzheimer’s pathology, to classify individuals with mixed pathology.30 Similarly, for α-synuclein targeted therapies, the possibility that α-synuclein-SAA negative participants will respond differently to treatment should be considered in patient selection and sample size estimates. For planned clinical trials targeting at-risk populations, α-synuclein SAA results should be considered for identifying the earliest stages of synucleinopathy and those individuals likely to progress to a clinical disorder with typical Lewy pathology.
Our study has several strengths in its methods. First, we used samples from a large and well characterised cohort consisting of several clinically relevant subgroups. Participants were comprehensively evaluated with a series of clinical evaluations of motor and non-motor features and biomarkers, including DAT imaging and fluid biomarkers, permitting the comparison of α-synuclein SAA results against these measures. The number of participants with Parkinson’s disease and concurrent matched controls and the inclusion of prodromal cohorts in the PPMI cohort allowed for subgroup analyses and the extension of previous results to new populations. Second, α-synuclein SAA analysis was performed in a masked manner using a robust, validated assay platform. Third, the participants were diagnosed by expert neurologists on the basis of standardised criteria within the PPMI study.
The limitations of this study should be acknowledged. First, there was no a priori hypothesis or sample size estimation. Rather, we took advantage of the large and readily available PPMI cohort. On the basis of the diagnostic performance of the assay in previous studies,7 we expect a high power. We feel that this is a reasonable expectation given that this is the largest cohort that has been interrogated with α-synuclein SAA. Second, non-parametric methods were used to compare α-synuclein SAA status in participants with Parkinson’s disease for some outcomes because of the skewness of the data and the low number of samples in some groups. Additional samples would enable the use of more powerful methods. Third, the analyses presented in this report are all cross-sectional. Since PPMI has clinical and biological samples collected over time, a report on longitudinal data, including motor and non-motor features, will be an important topic for future analysis and might identify other clinical correlates. Fourth, the number of prodromal (hyposmia and RBD) cases with normal DAT imaging was small. This gap prevents us from drawing definitive conclusions about the temporal ordering of α-synuclein SAA and DAT imaging abnormalities. Fifth, there are several unanswered questions regarding the genetic forms of Parkinson’s disease. This cohort consisted of only Gly2019Ser LRRK2 carriers and Asn409Ser GBA carriers. α-synuclein SAA results in patients with other LRRK2 or GBA variants, as well as variants in other Parkinson’s disease-associated genes such as PRKN and PINK1, could not be assessed. Again, longitudinal studies and studies of additional at-risk PPMI participants, who are currently being recruited, might address these issues.
In summary, this study extends our understanding of the usefulness of α-synuclein SAA for in vivo molecular assessment of Parkinson’s disease. We show in a large, deeply phenotyped cohort that α-synuclein SAA is highly accurate in typical Parkinson’s disease, but results vary depending on the presence of the LRRK2 Gly2019Ser variant, as well as clinical features, particularly hyposmia. Within LRRK2 variant carriers, there are also differences in age, sex, and motor performance that are associated with α-synuclein SAA status. Another key finding is that prodromal and non-manifesting carriers, especially carriers of the GBA Asn409Ser variant, have evidence of abnormal α-synuclein aggregation before other detectable clinical or biomarker changes, including alterations in DAT imaging. One implication of this result is that the prodromal period in Parkinson’s disease might be longer than had been projected previously and might start before changes in dopaminergic integrity, at least in some individuals. Taken together, these findings have immediate implications for clinical trial design, both to identify pathologically defined subgroups of people with Parkinson’s disease and to establish biomarker-defined at-risk cohorts. Longitudinal research is needed to investigate the prognostic value of α-synuclein SAA and whether changes in quantitative measures of α-synuclein aggregation indicate progressive pathology over time.
Supplementary Material
Research in context.
Evidence before this study
We searched PubMed with the search terms: “Parkinson’s disease (PD)”, “prodromal”, “Non-manifest carriers”, “GBA”, “LRRK2”, “real-time quaking-induced conversion (RT-QuIC)”, “protein misfolding cyclic amplification (PMCA)”, AND “seed amplification assay (SAA)” for articles published in English on or before Oct 25, 2022, in any field. This is a large and rapidly growing field of literature, and many studies were identified, including case-series of people with Parkinson’s disease with and without genetic variants, individuals with isolated rapid eye movement sleep behaviour disorder (RBD), and four studies of non-manifesting carriers of genetic variants associated with Parkinson’s disease. Although some studies addressed variation in α-synuclein seed amplification results among groups based on genetic carrier status, and others have investigated results in at-risk groups, substantial knowledge gaps still exist in confirming the results of these studies in large, well characterised cohorts and in showing associations with clinical features and other biomarkers.
Added value of this study
To our knowledge, this is the largest report of comparative data from a cohort of participants with Parkinson’s disease, healthy controls, individuals with clinical syndromes prodromal to Parkinson’s disease (hyposmia and RBD), and non-manifesting carriers of LRRK2 Gly2019Ser and GBA Asn409Ser mutations. The strengths of our data include a large sample size, robust clinical dataset, a high percentage of dopamine transformer scans completed, and the ability to compare non-manifesting carriers to similarly aged healthy controls, which allows for intergroup comparisons and subgroup analysis. The key novel findings in this study include: a marked variability in rates of positive α-synuclein SAA results, particularly among LRRK2 variant carriers depending on olfactory performance and sex; and α-synuclein SAA positivity in prodromal and non-manifesting carriers without dopaminergic imaging abnormalities in a high number of participants, whereas the converse (ie, dopaminergic imaging abnormalities in the absence of a positive α-synuclein SAA result) is less common, indicating that α-synuclein SAA might be an early indicator of synucleinopathy. We also confirmed the high diagnostic accuracy of α-synuclein SAA for sporadic Parkinson’s disease compared with healthy controls, and that α-synuclein SAA is negative in most non-manifesting carriers, suggesting that the presence of synuclein aggregates in CSF is not a lifelong trait but rather acquired at some point close to disease onset.
Implications of all the available evidence
Our results show that the α-synuclein SAA identifies people with Parkinson’s disease with high sensitivity and specificity, provides information about molecular heterogeneity, and detects prodromal individuals before diagnosis. These findings suggest a crucial role for α-synuclein SAA in therapeutic development, both to identify pathologically defined subgroups of people with Parkinson’s disease and to establish biomarker-defined at-risk cohorts.
Acknowledgments
PPMI is funded by the Michael J Fox Foundation for Parkinson’s Research and funding partners, including: Abbvie, AcureX, Aligning Science Across Parkinson’s, Amathus Therapeutics, Avid Radiopharmaceuticals, Bial Biotech, Biohaven, Biogen, BioLegend, Bristol-Myers Squibb, Calico Labs, Celgene, Cerevel, Coave, DaCapo Brainscience, 4D Pharma, Denali, Edmond J Safra Foundation, Eli Lilly, GE Healthcare, Genentech, GlaxoSmithKline, Golub Capital, Insitro, Janssen Neuroscience, Lundbeck, Merck, Meso Scale Discovery, Neurocrine Biosciences, Prevail Therapeutics, Roche, Sanofi Genzyme, Servier, Takeda, Teva, UCB, VanquaBio, Verily, Voyager Therapeutics, and Yumanity.
Footnotes
Declaration of interests
AS declares consultancy for Merck, Parkinson Study Group; and honoraria from Bial. LC-M declares employment for Amprion; stock ownership (employee stock options) for Amprion; and patent or patent applications numbers US 20210277076A1, US 20210311077A1, US 20190353669A1, and US 20210223268A1. CMF declares employment for Amprion; stock ownership (employee stock options) for Amprion; and patent or patent applications numbers US 20210277076A1 and US 20210311077A1. YM declares employment for Amprion; stock ownership (employee stock options) for Amprion; and patent or patent applications number US 20210277076A1. PAU declares employment for Amprion; and stock ownership (employee stock options) for Amprion. HN declares employment for Amprion; and stock ownership (employee stock options) for Amprion. RNA declares consultancies for Janssen, Sanofi, Ono Therapuetics, Avrobio, Takeda, Gain Therapuetics, Merck, GlaxoSmithKline, and Caraway. LMC declares honoraria (royalties) from Elsevier and Wolters Kluwel. TF declares travel grants from the Michael J Fox Foundation. KMe declares consultancies for the Michael J Fox Foundation, AcuRx, Caraway, Cerebral Therapeutics, NRG Therapeutics (scientific advisory board), Nurabio, Retromer Therapeutics (director on the board, part-time chief scientific officer), Schrodinger, Sinopia Biosciences (scientific advisory board), and Vanqua Biosciences (scientific advisory board); stock ownership for Cognition Therapeutics, Eli Lilly (retiree stock holder), Envisagenics, and Retromer Therapeutics; honoraria for the University of Utah; patents or patent applications for Retromer Therapeutics (planned patent); and travel grants from the University of Utah. BM declares consultancies from Roche, Biogen, and the Michael J Fox Foundation; honoraria for Abbvie; and travel grants for Abbvie. KLP declares consultancies for Curasen; was on a scientific advisory board for Curasen and Amprion; honoraria from invited scientific presentations to universities and professional societies not exceeding $5000 per year from California Congress of Clinical Neurology, California Neurological Society, and Johns Hopkins University; and patents or patent applications numbers 17/314,979 and 63/377,293. JS declares consultancies from Invicro, Biogen, and Abbvie; and stock ownership from RealmIDX, MNI Holdings, and LikeMinds. TSi declares consultancies for 4D Pharma, Acadia, AcureX, AskBio, Amneal, Blue Rock Therapeutics, Caraway, Critical Path for Parkinson’s Consortium, Denali, Michael J Fox Foundation, Neuroderm, Roche, Sanofi, Sinopia, Sunovion, Takeda, UCB, Vanqua Bio, and Voyager. CMT declares consultancies for CNS Ratings, Australian Parkinson’s Mission, Biogen, Evidera, Cadent (data safety monitoring board), Adamas (steering committee), Biogen (via the Parkinson Study Group steering committee), Kyowa Kirin (advisory board), Lundbeck (advisory board), Jazz/Cavion (steering committee), and Acorda (advisory board). DW declares receiving salary support from Michael J Fox Foundation for serving on an Executive Steering Committee for the PPMI. AV declares consultancies for CoA Therapeutics, XW Pharma, and Biogen. MF declares employment for the Michael J Fox Foundation. LMAO declares employment for the Michael J Fox Foundation. SJH declares employment for the Michael J Fox Foundation. TSh declares employment for the Michael J Fox Foundation. KMa declares consultancies for Invicro, MJFF, Roche, Calico, Coave, Neuron23, Orbimed, Neuroderm, Sanofi, Takeda, Merck, Lilly, Inhibikase, and Neuramedy. CS declares employment for Amprion; stock ownership for Amprion; honoraria (will receive royalties for the sale of seed amplification assay [SAA]) from Amprion; and patents or patent applications, awarded and amplified in conjunction with Amprion for the SAA assay.
For the full list of the Parkinson’s Progression Markers Initiative, see the appendix
Contributor Information
Andrew Siderowf, Department of Neurology, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA.
Luis Concha-Marambio, Research and Development Unit, Amprion, San Diego, CA, USA.
David-Erick Lafontant, Department of Biostatistics, College of Public Health, University of Iowa, Iowa City, IA, USA.
Carly M Farris, Research and Development Unit, Amprion, San Diego, CA, USA.
Yihua Ma, Research and Development Unit, Amprion, San Diego, CA, USA.
Paula A Urenia, Research and Development Unit, Amprion, San Diego, CA, USA.
Hieu Nguyen, Research and Development Unit, Amprion, San Diego, CA, USA.
Roy N Alcalay, Department of Neurology, Tel Aviv Sourasky Medical Center, Tel Aviv, Israel; Department of Neurology, Columbia University Irving Medical Center, New York, NY, USA.
Lana M Chahine, Department of Neurology, University of Pittsburgh, Pittsburgh, PA, USA.
Tatiana Foroud, Department of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis, IN, USA.
Douglas Galasko, Department of Neurology, University of California, San Diego, CA, USA.
Karl Kieburtz, University of Rochester Medical Center, University of Rochester, Rochester, NY, USA.
Kalpana Merchant, Department of Neurology, Northwestern University Feinberg School of Medicine, Chicago, IL, USA.
Brit Mollenhauer, Department of Neurology, University Medical Center Göttingen, Göttingen, Germany; Paracelsus-Elena Klinik, Kassel, and German Center for Neurodegenerative Diseases, Göttingen, Germany.
Kathleen L Poston, Department of Neurology and Neurological Sciences, Stanford University School of Medicine, Stanford, CA, USA.
John Seibyl, Institute for Neurodegenerative Disorders, New Haven, CT, USA.
Tanya Simuni, Department of Neurology, Northwestern University Feinberg School of Medicine, Chicago, IL, USA.
Caroline M Tanner, Department of Neurology, Weill Institute for Neurosciences, University of California San Francisco, San Francisco, CA, USA; Parkinson’s Disease Research, Education and Clinical Center, San Francisco Veterans Affairs Medical Center, San Francisco, CA, USA.
Daniel Weintraub, Department of Psychiatry, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA; Parkinson’s Disease Research, Education and Clinical Center, Philadelphia Veterans Affairs Medical Center, Philadelphia, PA, USA.
Aleksandar Videnovic, Department of Neurology, Neurological Clinical Research Institute, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA.
Seung Ho Choi, Department of Biostatistics, College of Public Health, University of Iowa, Iowa City, IA, USA.
Ryan Kurth, Department of Biostatistics, College of Public Health, University of Iowa, Iowa City, IA, USA.
Chelsea Caspell-Garcia, Department of Biostatistics, College of Public Health, University of Iowa, Iowa City, IA, USA.
Christopher S Coffey, Department of Biostatistics, College of Public Health, University of Iowa, Iowa City, IA, USA.
Mark Frasier, The Michael J Fox Foundation for Parkinson’s Research, New York, NY, USA.
Luis M A Oliveira, The Michael J Fox Foundation for Parkinson’s Research, New York, NY, USA.
Samantha J Hutten, The Michael J Fox Foundation for Parkinson’s Research, New York, NY, USA.
Todd Sherer, The Michael J Fox Foundation for Parkinson’s Research, New York, NY, USA.
Kenneth Marek, Institute for Neurodegenerative Disorders, New Haven, CT, USA.
Claudio Soto, Research and Development Unit, Amprion, San Diego, CA, USA; Department of Neurology, University of Texas McGovern Medical School at Houston, TX, USA.
Data sharing
Data used in the preparation of this article were obtained from the Parkinson’s Progression Markers Initiative (PPMI) database (https://www.ppmi-info.org/access-data-specimens/download-data). For up-to-date information on the study, visit https://www.ppmi-info.org. All data used in this study, as well as a data dictionary, are free and publicly available at the PPMI website. Additional, related documents including the study protocol and assay methods are also available. Data access can be requested on the website. There are no restrictions on who can request access.
References
- 1.Parnetti L, Gaetani L, Eusebi P, et al. CSF and blood biomarkers for Parkinson’s disease. Lancet Neurol 2019; 18: 573–86. [DOI] [PubMed] [Google Scholar]
- 2.Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24: 197–211. [DOI] [PubMed] [Google Scholar]
- 3.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature 1997; 388: 839–40. [DOI] [PubMed] [Google Scholar]
- 4.Beach TG, Adler CH, Sue LI, et al. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol 2010; 119: 689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donadio V, Incensi A, Leta V, et al. Skin nerve α-synuclein deposits: a biomarker for idiopathic Parkinson disease. Neurology 2014; 82: 1362–69. [DOI] [PubMed] [Google Scholar]
- 6.Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001; 411: 810–13. [DOI] [PubMed] [Google Scholar]
- 7.Shahnawaz M, Tokuda T, Waragai M, et al. Development of a biochemical diagnosis of Parkinson disease by detection of α-synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol 2017; 74: 163–72. [DOI] [PubMed] [Google Scholar]
- 8.Fairfoul G, McGuire LI, Pal S, et al. Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann Clin Transl Neurol 2016; 3: 812–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Groveman BR, Orrù CD, Hughson AG, et al. Rapid and ultra-sensitive quantitation of disease-associated α-synuclein seeds in brain and cerebrospinal fluid by αSyn RT-QuIC. Acta Neuropathol Commun 2018; 6: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shahnawaz M, Mukherjee A, Pritzkow S, et al. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020; 578: 273–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Concha-Marambio L, Farris CM, Holguin B, et al. Seed amplification assay to diagnose early Parkinson’s and predict dopaminergic deficit progression. Mov Disord 2021; 36: 2444–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Russo MJ, Orru CD, Concha-Marambio L, et al. High diagnostic performance of independent alpha-synuclein seed amplification assays for detection of early Parkinson’s disease. Acta Neuropathol Commun 2021; 9: 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rossi M, Candelise N, Baiardi S, et al. Ultrasensitive RT-QuIC assay with high sensitivity and specificity for Lewy body-associated synucleinopathies. Acta Neuropathol 2020; 140: 49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iranzo A, Fairfoul G, Ayudhaya ACN, et al. Detection of α-synuclein in CSF by RT-QuIC in patients with isolated rapid-eye-movement sleep behaviour disorder: a longitudinal observational study. Lancet Neurol 2021; 20: 203–12. [DOI] [PubMed] [Google Scholar]
- 15.Brumm MC, Pierz KA, Lafontant D-E, et al. Updated percentiles for the University of Pennsylvania smell identification test in adults 50 years of age and older. Neurology 2023; published online Feb 27. 10.1212/WNL.0000000000207077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marek K, Chowdhury S, Siderowf A, et al. The Parkinson’s progression markers initiative (PPMI)—establishing a PD biomarker cohort. Ann Clin Transl Neurol 2018; 5: 1460–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 2012; 123: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Irwin DJ, Fedler J, Coffey CS, et al. Evolution of Alzheimer’s disease cerebrospinal fluid biomarkers in early Parkinson’s disease. Ann Neurol 2020; 88: 574–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mollenhauer B, Dakna M, Kruse N, et al. Validation of serum neurofilament light chain as a biomarker of Parkinson’s disease progression. Mov Disord 2020; 35: 1999–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simuni T, Uribe L, Cho HR, et al. Clinical and dopamine transporter imaging characteristics of non-manifest LRRK2 and GBA mutation carriers in the Parkinson’s Progression Markers Initiative (PPMI): a cross-sectional study. Lancet Neurol 2020; 19: 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Concha-Marambio L, Pritzkow S, Shahnawaz M, Farris CM, Soto C. Seed Amplification Assay (SAA) for detection of pathologic αSynuclein aggregates in CSF as biomarker for Parkinson’s disease and related synucleinopathies. Nat Protoc 2023; published online Jan 18. 10.1038/s41596-022-00787-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marek K, Seibyl J, Eberly S, et al. Longitudinal follow-up of SWEDD subjects in the PRECEPT Study. Neurology 2014; 82: 1791–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jennings D, Siderowf A, Stern M, et al. Conversion to Parkinson disease in the PARS hyposmic and dopamine transporter–deficit prodromal cohort. JAMA Neurol 2017; 74: 933–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garrido A, Fairfoul G, Tolosa ES, Martí MJ, Green A. α-synuclein RT-QuIC in cerebrospinal fluid of LRRK2-linked Parkinson’s disease. Ann Clin Transl Neurol 2019; 6: 1024–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalia LV, Lang AE, Hazrati LN, et al. Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol 2015; 72: 100–05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneider SA, Alcalay RN. Neuropathology of genetic synucleinopathies with parkinsonism: review of the literature. Mov Disord 2017; 32: 1504–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen W, Yan X, Lv H, Liu Y, He Z, Luo X. Gender differences in prevalence of LRRK2-associated Parkinson disease: a meta-analysis of observational studies. Neurosci Lett 2020; 715: 134609. [DOI] [PubMed] [Google Scholar]
- 28.Postuma RB, Iranzo A, Hogl B, et al. Risk factors for neurodegeneration in idiopathic rapid eye movement sleep behavior disorder: a multicenter study. Ann Neurol 2015; 77: 830–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Müller A, Müngersdorf M, Reichmann H, Strehle G, Hummel T. Olfactory function in Parkinsonian syndromes. J Clin Neurosci 2002; 9: 521–24. [DOI] [PubMed] [Google Scholar]
- 30.Bellomo G, De Luca CMG, Paoletti FP, Gaetani L, Moda F, Parnetti L. α-synuclein seed amplification assays for diagnosing synucleinopathies: the way forward. Neurology 2022; 99: 195–205. [DOI] [PubMed] [Google Scholar]
- 31.Rana HQ, Balwani M, Bier L, Alcalay RN. Age-specific Parkinson disease risk in GBA mutation carriers: information for genetic counseling. Genet Med 2013; 15: 146–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Healy DG, Falchi M, O’Sullivan SS, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol 2008; 7: 583–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marder K, Wang Y, Alcalay RN, et al. Age-specific penetrance of LRRK2 G2019S in the Michael J. Fox Ashkenazi Jewish LRRK2 Consortium. Neurology 2015; 85: 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Savica R, Rocca WA, Ahlskog JE. When does Parkinson disease start? Arch Neurol 2010; 67: 798–801. [DOI] [PubMed] [Google Scholar]
- 35.Brockmann K, Quadalti C, Lerche S, et al. Association between CSF alpha-synuclein seeding activity and genetic status in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol Commun 2021; 9: 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data used in the preparation of this article were obtained from the Parkinson’s Progression Markers Initiative (PPMI) database (https://www.ppmi-info.org/access-data-specimens/download-data). For up-to-date information on the study, visit https://www.ppmi-info.org. All data used in this study, as well as a data dictionary, are free and publicly available at the PPMI website. Additional, related documents including the study protocol and assay methods are also available. Data access can be requested on the website. There are no restrictions on who can request access.