

Abstract
Programmed death 1 (PD-1) and its ligands, PD-L1 and PD-L2, deliver inhibitory signals that regulate the balance between T cell activation, tolerance, and immunopathology. Immune responses to foreign and self-antigens require specific and balanced responses to clear pathogens and tumors and yet maintain tolerance. Induction and maintenance of T cell tolerance requires PD-1, and its ligand PD-L1 on nonhematopoietic cells can limit effector T cell responses and protect tissues from immune-mediated tissue damage. The PD-1:PD-L pathway also has been usurped by microorganisms and tumors to attenuate antimicrobial or tumor immunity and facilitate chronic infection and tumor survival. The identification of B7-1 as an additional binding partner for PD-L1, together with the discovery of an inhibitory bidirectional interaction between PD-L1 and B7-1, reveals new ways the B7:CD28 family regulates T cell activation and tolerance. In this review, we discuss current understanding of the immunoregulatory functions of PD-1 and its ligands and their therapeutic potential.
Keywords: costimulation, T cell, autoimmunity, infectious disease, tumor
INTRODUCTION
The concept of T cell costimulation has evolved over time. The two-signal model for T cell activation was proposed by Kevin Lafferty as a model for the activation of naive T cells (1). According to this model, T cells require two signals to become fully activated. The first signal, which gives specificity to the immune response, is provided by the interaction of antigenic peptide−MHC complex with the T cell receptor (TCR). The second, antigen-independent costimulatory signal, is delivered to T cells by antigen-presenting cells (APCs) to promote T cell clonal expansion, cytokine secretion, and effector function. In the absence of the second signal, antigen-specific lymphocytes fail to respond effectively and are functionally inactivated, or anergic, and resistant to subsequent activation by the antigen. The critical inhibitory function of cytotoxic T lymphocyte–associated antigen 4 (CTLA-4, also known as CD152) was revealed by the fatal lymphoproliferative phenotype of Ctla4−/− mice (2, 3). This function demonstrated that T cell pathways could provide negative as well as positive second signals and provided the first indication that negative second signals could regulate T cell tolerance. The discovery of more members of the B7:CD28 family has revealed additional costimulatory pathways that can provide positive and negative second signals to antigen-experienced effector T cells. The functions of these newer pathways have broadened the concept of costimulation.
This review focuses on recent advances in our understanding of one of the newer pathways in the B7:CD28 family, the pathway consisting of the programmed death 1 (PD-1; also known as CD279) receptor and its ligands, PD-L1 (B7-H1; CD274) and PD-L2 (B7-DC; CD273). The PD-1 receptor was discovered in 1992 as a gene upregulated in a T cell hybridoma undergoing cell death (4). The important negative regulatory function of PD-1 was revealed by the autoimmune-prone phenotype of Pdcd1−/− mice in 1999 (5, 6). Since the ligands for PD-1 were identified in 2000 (7, 8) and 2001 (9, 10), there has been steady progress in understanding the functions of PD-1 and its ligands. Here, we first describe the structure and expression of PD-1, PD-L1, and PD-L2. Next, we review recent advances in understanding PD-1 and PD-L signaling. We then summarize recent studies that identify B7-1 (CD80) as a binding partner for PD-L1 and indicate that PD-L1 interactions with B7-1 can lead to bidirectional inhibitory responses in T cells (11). Finally, we discuss our current understanding of the roles played by PD-1 and its ligands in regulating T cell activation and tolerance and consider the therapeutic potential of manipulation of PD-1 and its ligands.
STRUCTURE OF GENES
PD-1 is a 288 amino acid (aa) type I transmembrane protein composed of one immunoglobulin (Ig) superfamily domain, a ~20 aa stalk, a transmembrane domain, and an intracellular domain of approximately 95 residues containing an immunoreceptor tyrosine-based inhibitory motif (ITIM) as well as an immunoreceptor tyrosine-based switch motif (ITSM). PD-1 is encoded by the Pdcd1 gene on chromosome 1 in mice and chromosome 2 in humans. In both species, Pdcd1 consists of 5 exons. Exon 1 encodes a short signal sequence, whereas exon 2 encodes an Ig domain. The stalk and transmembrane domains make up exon 3, and exon 4 codes for a short 12 aa sequence that marks the beginning of the cytoplasmic domain. Exon 5 contains the C-terminal intracellular residues and a long 3′UTR.
Splice variants of PD-1 have been cloned from activated human T cells (12). These transcripts lack exon 2, exon 3, exons 2 and 3, or exons 2 through 4. All these variants, except for the splice variant lacking exon 3 only (PD-1Δex3), are expressed at similar levels as full-length PD-1 in resting peripheral blood mononuclear cells (PBMCs). All variants are significantly induced upon activation of human T cells with anti-CD3 and anti-CD28. The PD-1Δex3 variant encodes an mRNA that lacks the transmembrane domain and resembles soluble CTLA-4, which plays an important role in autoimmunity (13). This variant is enriched in the synovial fluid and sera of patients with rheumatoid arthritis (14).
PD-L1 is a 290 aa type I transmembrane protein encoded by the Cd274 gene on mouse chromosome 19 and human chromosome 9. Cd274 comprises seven exons, the first of which is noncoding and contains the 5′UTR. The next three exons contain the signal sequence, IgV-like domain, and IgC-like domains, respectively. The transmembrane domain and the intracellular domains are contained in the next two exons (exons 5 and 6). The last exon contains intracellular domain residues plus the 3′UTR. The intracellular domain of PD-L1 is short, only about 30 aa, and highly conserved in all reported species. There is no known function for the intracellular tail of PD-L1.
There is one reported splice variant of PD-L1 in humans (15) consisting of a sequence lacking the IgV-like domain encoded in exon 2. This mutant should not be able to bind PD-1, although the function of this splice variant has not yet been reported. No splice variants have been identified for mouse PD-L1.
PD-L2 is a type I transmembrane protein encoded by the Pdcd1lg2 gene adjacent to Cd274 and separated by only 23 kb of intervening genomic DNA in mouse and 42 kb in human. The gene comprises six exons in mouse and seven in human. Exon 1 is non-coding, whereas the second exon contains the signal sequence. The IgV-like domain is composed of exon 3, the IgC-like domain is exon 4, and exon 5 contains a short stalk, transmembrane region, and the beginning of the cytoplasmic domain. In mouse exon 5, there is a stop codon that results in a cytoplasmic domain of only 4 aa. In human, exon 6 and 7 contain an additional coding region resulting in a cytoplasmic domain of 30 aa. The longer form of the cytoplasmic domain is found in human, macaque, chimp, dog, cow, pig, and horse but is lost in mouse and rat. The long form of the cytoplasmic domain has no appreciable signaling motifs but is conserved across diverse species, suggesting that the cytoplasmic tail of PD-L2 may have a functional role.
There are three PD-L2 splice variants identified from activated human PBMCs (9, 16). Analogous to the splice variant described in PD-L1, one form drops out the IgV-like exon and presumably loses the capacity to bind PD-1 (9). A second form (called type II) drops out the IgC-like domain. Another form (called type III) loses the IgC-like domain and the transmembrane residues but preserves the intracellular residues. The type II form would be expected to bind PD-1, as most binding activity resides in the IgV-like domain (17), and the type III form might represent a soluble ligand for PD-1.
STRUCTURE OF PROTEINS
The unbound three-dimensional structure of PD-1 has been obtained by X-ray crystallography and shows that the β-strands of the Ig superfamily fold are well conserved between CTLA-4 and PD-1 (root mean squared deviation of 1.5 Å comparing common α carbons) (18). The CDR3 loop in PD-1 is loosely ordered and does not have conserved amino acids, unlike the binding interface of CTLA-4, which is centered on the MYPPPY motif of the CDR3 loop and is highly ordered owing to the consecutive prolines in this motif. None of the PD-1 CDR3 amino acids was found to be important for binding PD-L by scanning mutagenesis. Biophysical studies have addressed the question of self-association of PD-1. Fluorescence resonance energy transfer analyses of full-length PD-1 expressed in CHO cells and analytical ultracentrifugation on a soluble extracellular PD-1 IgV-like domain showed PD-1 to be monomeric. Whether PD-1 requires dimerization to transduce signals is unclear.
Molecular models of PD-L1 and PD-L2 have been generated on the basis of the crystal structures of B7-1 and B7-2 to guide site-directed alanine scanning mutagenesis (17). The binding interfaces of both PD-L1 and PD-L2 to PD-1 are on their IgV-like domains. Interestingly, certain mutants of PD-L1 and PD-L2 that were unable to bind PD-1 could stimulate T cell proliferation in conjunction with anti-CD3. These non-PD-1 binding mutants were also able to stimulate Pdcd1−/− T cells, providing evidence for another receptor for PD-L1 and PD-L2.
EXPRESSION OF PD-1 AND ITS LIGANDS
PD-1 can be expressed on T cells, B cells, natural killer T cells, activated monocytes, and dendritic cells (DCs) (Figure 1). PD-1 is not expressed on resting T cells but is inducibly expressed after activation (19). Although PD-1 cell surface protein expression can be detected within 24 h of stimulation, functional effects of PD-1 ligation are observed within a few hours following T cell activation (20). Ligation of TCR or BCR can upregulate PD-1 on lymphocytes, and the level of mRNA transcription does not strictly correlate with protein production (21). In normal human reactive lymphoid tissue, PD-1 is expressed on germinal center–associated T cells (133). PD-1 compartmentalization in intracellular stores has been described in a regulatory T cell population (22, 22a). PD-1 is inducibly expressed on APCs on myeloid CD11c+ DCs and monocytes in humans (23), but its function on these cells is not clear. There are no data to support a function for PD-1 in the absence of antigen receptor signaling.
Figure 1.
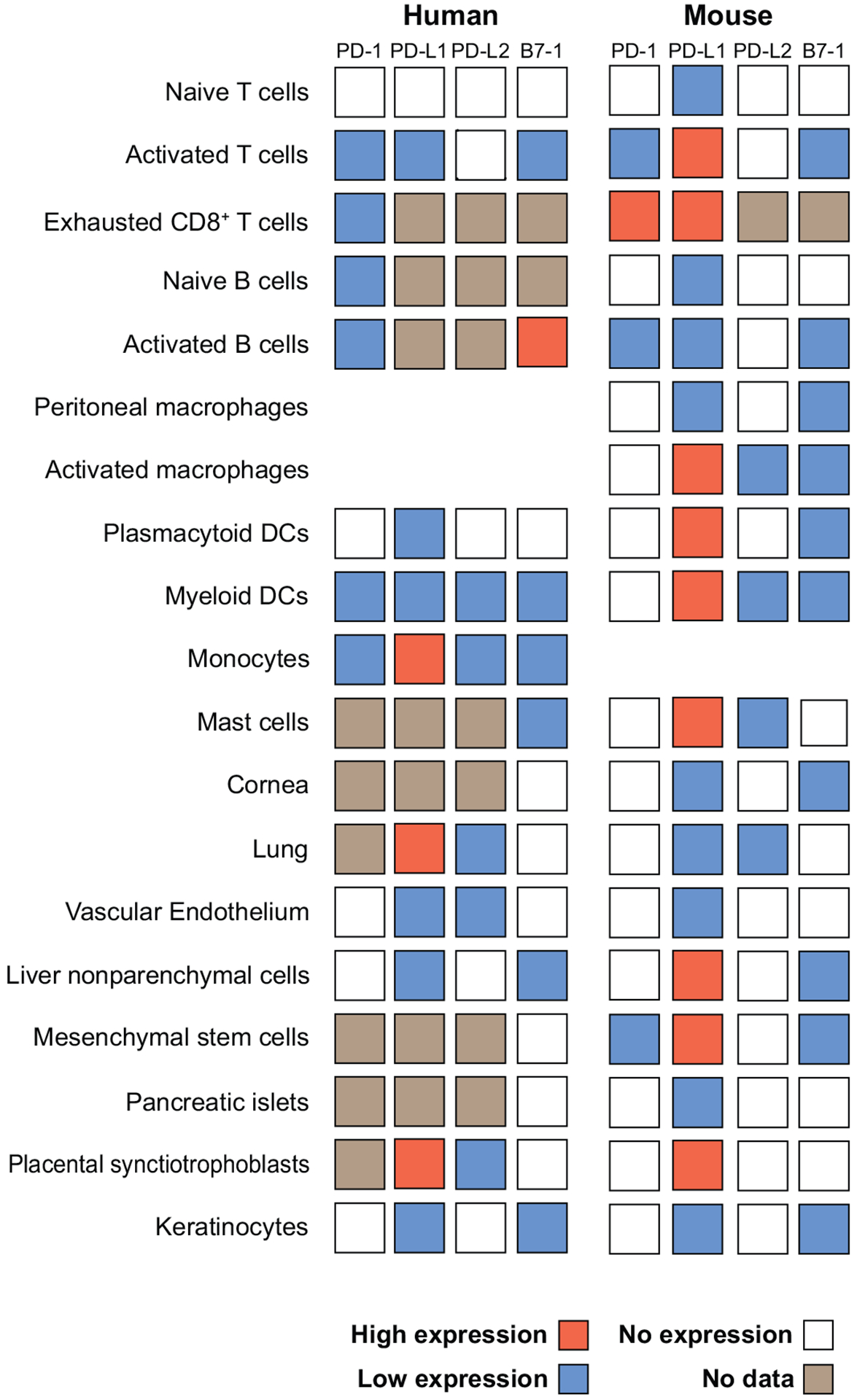
Comparison of expression of PD-1, PD-L1, PD-L2, and B7-1 on human and mouse cells. PD-1, PD-L1, PD-L2, and B7-1 are expressed on a wide range of human (7–9, 20, 23, 37, 55, 89, 143, 146–149) and mouse cells (4, 9, 24, 31, 85, 87, 113, 143, 147, 150–153).
The two PD-1 ligands differ in their expression patterns. PD-L1 is constitutively expressed on mouse T and B cells, DCs, macrophages, mesenchymal stem cells, and bone marrow–derived mast cells (24). PD-L1 expression is also found on a wide range of nonhematopoietic cells (see Figure 1) and is upregulated on a number of cell types after activation. Both type I and type II interferons (IFNs) upregulate PD-L1 (25, 26). Analyses of the human PD-L1 promoter demonstrate that both constitutive and inducible PD-L1 expression are dependent on two IFN regulatory factor-1 (IRF-1) binding sites that are between 200 and 320 bp upstream of the transcriptional start site (27). These IRF-1 binding sites are also found in mouse, although their importance has not been directly tested. Several studies have examined which signaling pathways are required for PD-L1 expression by using pharmacological inhibitors. PD-L1 expression in cell lines is decreased when MyD88, TRAF6, and MEK are inhibited (28). JAK2 has also been implicated in PD-L1 induction (27, 28). Loss or inhibition of phosphatase and tensin homolog (PTEN), a cellular phosphatase that modifies phosphatidylinositol 3-kinase (PI3K) and Akt signaling, increases post-transcriptional PD-L1 expression in cancers (29).
PD-L2 expression is much more restricted than PD-L1 expression. PD-L2 is inducibly expressed on DCs, macrophages, and bone marrow–derived mast cells. PD-L2 is also expressed on 50% to 70% of resting peritoneal B1 cells, but not on conventional B2 B cells (30). PD-L2 expression on B1 cells tracks with a restricted VH usage that is skewed toward VH11/VH12. PD-L2+ B1 cells bind phosphatidylcholine and may be important for innate immune responses against bacterial antigens. Less is known about transcriptional regulation of PD-L2. Its induction by IFN-γ is partially dependent on NF-κB (31). PD-L2 can also be induced on monocytes and macrophages by GM-CSF, IL-4, and IFN-γ (24, 32).
Signaling Through PD-1
Signaling through costimulatory receptors primarily functions to modify antigen receptor signaling. PD-1 typically has greater effects on cytokine production than on cellular proliferation, with significant effects on IFN-γ, TNF-α, and IL-2 production. PD-1-mediated inhibitory signals depend on the strength of the TCR signal, with greater inhibition delivered at low levels of TCR stimulation. This reduction can be overcome by costimulation through CD28 (8) or IL-2 (33). PD-1 may exert its effects on cell differentiation and survival directly by inhibiting early activation events that are positively regulated by CD28 or indirectly through IL-2 (33). Both CD28 and IL-2 promote cell expansion and survival through effects on anti-apoptotic, cell cycle, and cytokine genes. IL-2 withdrawal can lead to cell death, another process in which PD-1 has been implicated. There is strong evidence that PD-1 ligation inhibits the induction of the cell survival factor Bcl-xL (20). PD-1 inhibits the expression of transcription factors associated with effector cell function, including GATA-3, Tbet, and Eomes (34). Further studies are required to determine whether PD-1-mediated inhibition is related to its ability to counteract cell survival signals and effector differentiation mediated through CD28, IL-2, Bcl-xL, or a combination of these factors.
PD-1 is phosphorylated on its two intracellular tyrosines upon ligand engagement, and then binds phosphatases that downregulate antigen receptor signaling through direct dephosphorylation of signaling intermediates. Two phosphatases, SH2-domain containing tyrosine phosphatase 1 (SHP-1) and SHP-2, can bind to the ITIM and ITSM motifs of PD-1 (35, 36). PD-1 inhibitory function is lost when the ITSM alone is mutated, demonstrating that this tyrosine plays the primary functional role of PD-1 inhibition (20, 36). The association between SHP-1 and PD-1 appears to be weaker than the interaction of PD-1 with SHP-2. Together, these studies suggest that PD-1 functions by recruiting SHP-2, and possibly SHP-1, to the antigen receptor signaling complex (35).
While the binding of SHP-2 to PD-1 is significantly enhanced by PD-1 ligation (20), proximity of PD-1 to the antigen receptor appears to be important for inhibition by PD-1. PD-1 ligation inhibits antigen receptor signaling only in cis and not in trans, indicating that PD-1 ligation must occur close to the site of antigen receptor engagement (37). CTLA-4 moves from an intracellular store to the immunological synapse between an APC and lymphocyte after antigen recognition, depending on the strength of signal (38). In contrast, PD-1 redistributes from uniform cell surface expression to the synapse during T cell–APC interactions (22a). PD-1 could exert its inhibitory effects by bringing SHP-2 into the synapse during antigen receptor signaling, and cross-linking of PD-1 and CD3 increases the amount of SHP-2, but not SHP-1, associated with PD-1 (39).
PD-1 ligation inhibits PI3K activity and downstream activation of Akt. In contrast, CTLA-4 inhibits Akt activation but does not alter PI3K activity, indicating that these coinhibitory receptors function through distinct mechanisms. PD-1 ligation inhibits phosphorylation of CD3ζ, ZAP70, and PKCθ (40). Other costimulatory receptors, such as CD150, bind SHP-2 through interactions of their ITIM/ITSM domains with the adaptor protein SH2 domain–containing molecule 1A (SH2D1A, also known as SAP) (41). Unlike the CD150 family of cell surface receptors, the ITIM/ITSM motifs of PD-1 do not bind SH2D1A (20). Therefore, PD-1 inhibition likely functions solely through direct interaction with SHP-2, or possibly SHP-1, to directly inhibit early events in the antigen receptor signaling cascades (Figure 2). PD-1 interaction with SHP-1 and SHP-2 is further supported by a peptide immunoprecipitation study that investigated binding partners for the PD-1 ITIM and ITSM motifs using mass spectroscopy. The PD-1 ITIM/ITSM motifs also associate with Lck and Csk (35). These findings suggest that Csk and/or Lck may mediate the phosphorylation of PD-1 in T cells, similar to Lyn phosphorylation of PD-1 in a B cell line (36).
Figure 2.
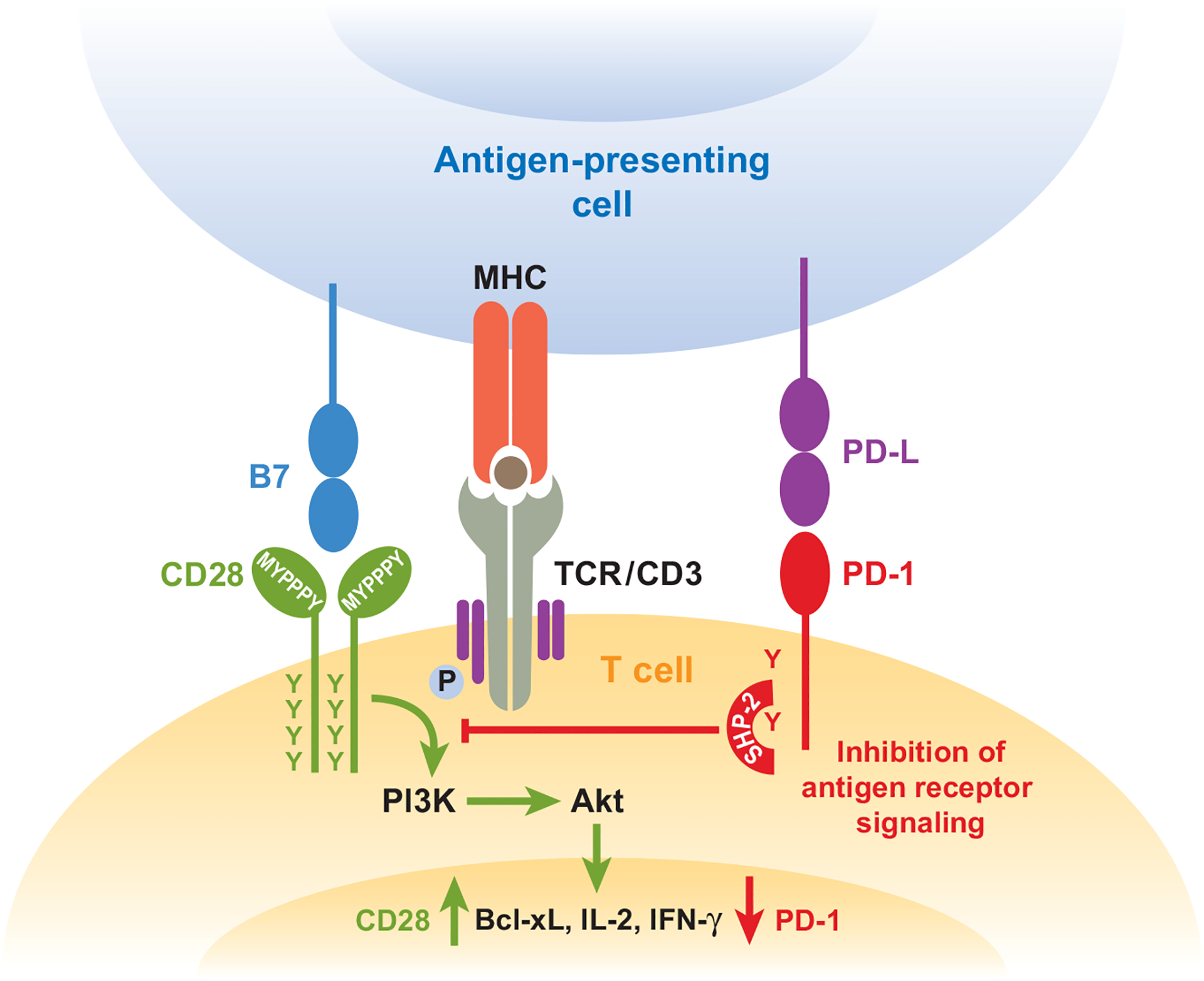
Ligation of PD-1 dampens TCR signaling but can be overcome by CD28 costimulation. PD-1 engagement on the cell surface leads to phosphorylation of PD-1 cytoplasmic tyrosines and increases SHP-2 association with the ITSM of PD-1. Recruitment of SHP-2 dephosphorylates signaling through the PI3K pathway and downstream signals through Akt. PD-1 ultimately decreases induction of cytokines, such as IFN-γ, and cell survival proteins, such as Bcl-xL. When signaling through CD28 is delivered at the same time as PD-1 and TCR ligation, inhibitory effects can be overcome, and cytokine production and cell survival is enhanced.
PD-1 ligation also reduces Erk activation, but this effect can be overcome through cytokine receptor signaling, particularly cytokines that activate STAT5, such as IL-2, IL-7, and IL-15 (37). SHP-2 positively regulates Erk phosphorylation by interacting with Gab2 after IL-2R ligation (42). Both activation of Erk, which is specifically inhibited by PD-1 ligation, and activation of STAT5, which can overcome PD-1 inhibition, are demonstrated to be involved in the anti-apoptotic and proliferative function of IL-2 (43). This suggests a model whereby the association of PD-1 with SHP-2 serves not only to dephosphorylate signaling intermediates, but also perhaps to sequester SHP-2 from its positive signaling role in Erk activation.
Reverse Signaling Through PD-L1 and PD-L2
PD-L1 and PD-L2 not only may influence responses by engaging PD-1 and modifying TCR or BCR signaling, but also may deliver signals into PD-L1- or PD-L2-expressing cells. The first indication that PD-L2 may transmit signals into DCs came from studies using a novel, naturally occurring human IgM antibody (sHIgM12), isolated from a patient with Waldenstrom’s macroglobulinemia, that binds both mouse and human PD-L2 (44). Although DCs treated with sHIgM12 do not upregulate MHC II or B7 costimulatory molecules, they produce greater amounts of proinflammatory cytokines, particularly TNF-α and IL-6, and stimulate naive T cell proliferation. Treatment of mice with the sHIgM12 enhances resistance to transplanted B16 melanoma and rapidly induces potent tumor-specific CTL (45–47). sHIgM12 also completely blocked the development of airway inflammatory disease in a mouse model of allergic asthma (48, 49). A recombinantly expressed IgM antibody was generated from sHIgM12 that recapitulates observations with the patient-derived antibody, arguing for a specific effect through PD-L2 ligation (50). Further studies are needed to determine how PD-L2 signals into the DC and how these signals influence immunity or tolerance.
Additional evidence for reverse signaling through PD-L1 or PD-L2 on DCs comes from studies of bone marrow–derived DCs cultured with soluble PD-1-Ig fusion protein containing the extracellular domain of mouse PD-1 fused to the constant region of human IgG (51). This soluble PD-1 (sPD-1) inhibited DC activation and increased IL-10 production independent of 2,3-dioxygenase (IDO). These effects could be prevented by preneutralization of sPD-1 with anti-PD-1, suggesting that the acquisition of this suppressive DC phenotype is PD-1-specific and occurs via PD-L1 or PD-L2. These findings are consistent with previous studies in which sPD-1 induced IL-10 production by CD4 T cells (52).
Bidirectional signaling of PD-1 and PD-L may help clarify some of the contradictory results of studies analyzing the PD-1:PD-L pathway. As is discussed in the next section, the discovery of B7-1 as an additional ligand for PD-L1 also may help explain differences observed in functional studies of PD-1 and PD-L1. The identification of bidirectional interactions between B7-1 and PD-L1 that inhibit T cell responses further underscores the importance of reverse signaling through PD-L1 and demonstrates that PD-L1 can signal into T cells.
B7-1:PD-L1 Interactions Inhibit T Cell Responses
A number of lines of evidence have suggested a receptor for PD-L1 or PD-L2, aside from PD-1. B7-1 has recently been identified as a binding partner for PD-L1 (11). Surface plasmon resonance studies demonstrate specific and unique interaction between PD-L1 and B7-1, with an affinity (~1.7 μM) intermediate between the affinities of B7-1 for CD28 (4 μM) and CTLA-4 (0.2 μM), and PD-L1 for PD-1 (0.5 μM). B7-2 does not bind to PD-L1 or PD-L2, and PD-L2 does not bind to B7-1. Chemical cross-linking studies indicate that PD-L1 and B7-1 can interact through their IgV-like domains. These domains are known to be important for interactions of B7-1 and PD-L1 with their previously known receptors. On B7-1, there is a partial overlap between the B7-1:PD-L1 interface and that of B7-1:CTLA-4. Likewise, on PD-L1, the PD-L1:B7-1 interface overlaps at least partially with the putative PD-L1:PD-1 interface (17).
The substantial overlap of B7-1:PD-L1 regions of interaction with those of their previously identified binding partners suggests that blocking antibodies might interrupt B7-1:PD-L1 binding as well as B7-1:CTLA-4 or PD-L1:PD-1 binding interactions. Adhesion assays tested the capacity of anti-B7-1 and anti-PD-L1 monoclonal antibodies (mAbs) to interfere with PD-L1 and B7-1 binding to B7-1 or PD-L1 300.19 transfectants, respectively. Two classes of B7-1 antibodies could be identified: one that blocks both B7-1:PD-L1 and B7-1:CTLA-4 binding and another that only blocks the B7-1:CTLA-4 interaction (Table 1). Four categories of PD-L1 antibodies have been identified: those that block only PD-1, those that block only B7-1, those that block both, and those that block neither PD-1 nor B7-1 interactions.
Table 1.
Commonly used mAbs that display neutralizing activity against PD-L1:B7-1, PD-L1:PD-1, or B7-1:CTLA-4
| PD-1:PD-L1 | B7-1:PD-L1 | |
| αPD-L1 | ||
| 10F.9G2 | ++ | + |
| 10F.2H11 | − | + |
| CD28, CTLA-4:B7-1 | B7-1:PD-L1 | |
| αB7-1 | ||
| 1G10 | + | + |
| 16–10A1 | ++ | − |
B7-1:PD-L1 interactions can induce an inhibitory signal into T cells. Ligation of PD-L1 on CD4 T cells by B7-1, or ligation of B7-1 on CD4 T cells by PD-L1, delivers a functionally significant, inhibitory signal. T cells lacking the previously known receptors for B7-1 (i.e., T cells lacking CD28 and CTLA-4) showed decreased proliferation and cytokine production when stimulated by anti-CD3 plus B7-1-coated beads. When T cells lacking all the receptors for B7-1 (i.e., T cells lacking CD28, CTLA-4, and PD-L1) were employed, T cell proliferation and cytokine production were no longer inhibited by anti-CD3 plus B7-1-coated beads, indicating that B7-1 acts specifically through PD-L1 on the T cell in the absence of CD28 and CTLA-4. Similarly, T cells lacking the previously known receptor for PD-L1 (PD-1) showed decreased proliferation and cytokine production when stimulated in the presence of anti-CD3 plus PD-L1-coated beads, demonstrating the inhibitory effect of PD-L1 ligation of B7-1 on T cells. When T cells lacking all known receptors for PD-L1 (i.e., T cells lacking PD-1 and B7-1) were used, T cell proliferation was no longer impaired by anti-CD3 plus PD-L1-coated beads. Thus, PD-L1 can exert an inhibitory effect on T cells either through B7-1 or PD-1.
Taken together, these results demonstrate a specific and significant bidirectional interaction between B7-1 and PD-L1 that inhibits T cell responses (Figure 3). The identification of the PD-L1:B7-1 interaction necessitates a reassessment of the roles of B7-1 and PD-L1 in regulating the activation and inhibition of immune responses, as well as their therapeutic manipulation. Differences in anti-PD-L1-blocking antibody specificities may help explain distinct functional outcomes of blockade of PD-1, PD-L1, and PD-L2 in disease models in vivo. For example, many in vivo studies (discussed below) have found a greater effect of anti-PD-L1 blockade compared with anti-PD-1 or anti-PD-L2 blockade, and this has been attributed to differences in the expression of PD-L1 and PD-L2 or to the half-life or affinities of the antibodies. The 10F.9G2 anti-PD-L1 mAb that has been used in many in vivo studies blocks PD-L1:B7-1 as well as PD-L1:B7-1 interactions (Table 1). Anti-PD-L1 antibodies that block solely PD-L1:PD-1 or PD-L1:B7-1 interactions should have reduced effects on T cell activation as compared with the dual-specific anti-PD-L1 mAb. Further studies are needed to compare the functional effects of anti-PD-L1 mAb that block solely PD-L1:PD-1 or PD-L1:B7-1 interactions with mAbs that block both interactions.
Figure 3.
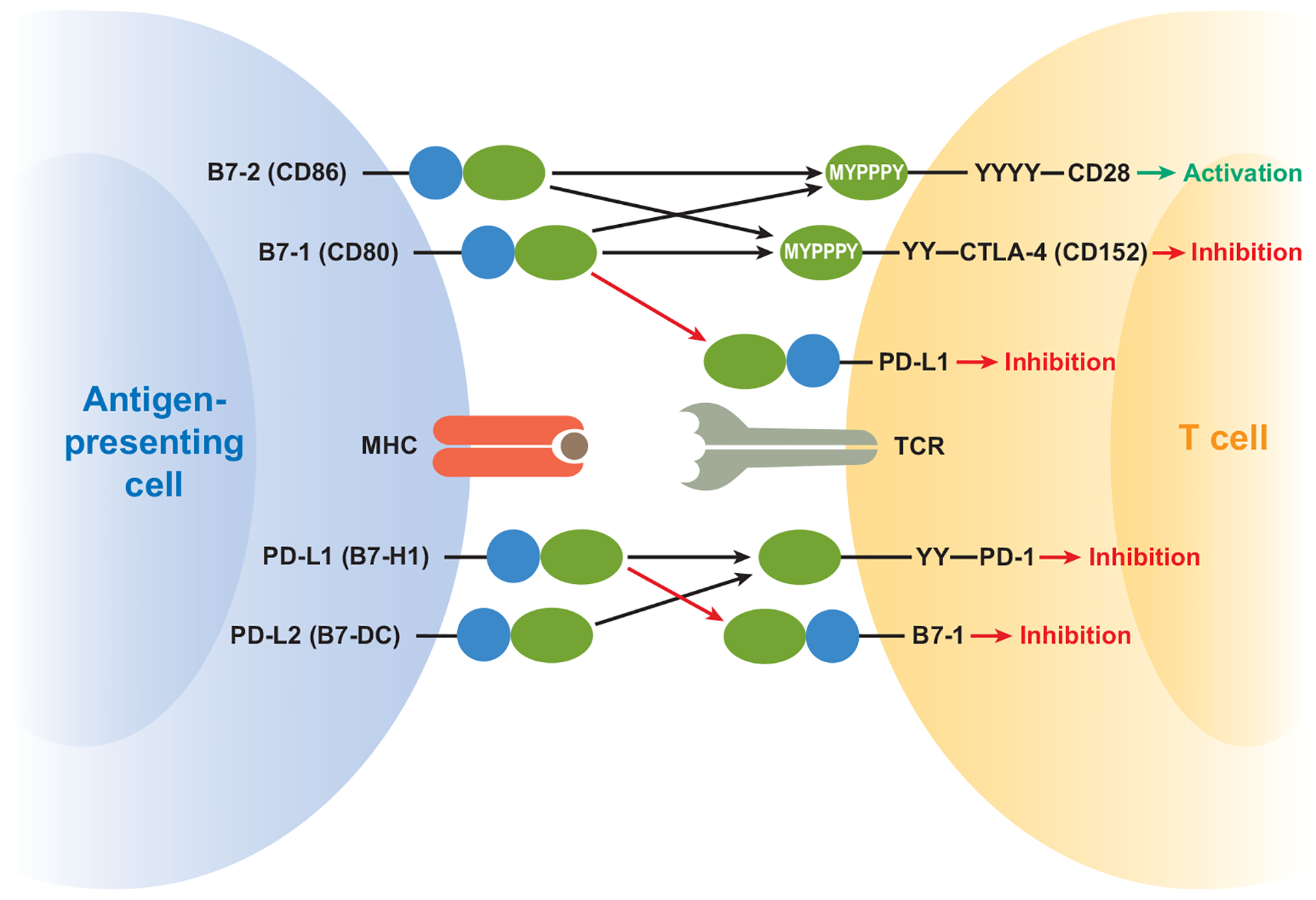
B7-1:PD-L1 interaction expands pathways in the B7:CD28 family. Recent data demonstrate that PD-L1 and B7-1 productively interact on T cells and can deliver bidirectional inhibitory signals. The identification of these costimulatory molecules as binding partners increases our understanding of the interactions that can occur on T cells and APCs and raises the possibility that PD-L1:B7-1 binding may not only deliver signals when ligated, but may also serve to segregate binding away from previously identified receptors (PD-1, CD28, CTLA-4). IgV-like regions are depicted in blue and IgC-like regions in green, while tyrosine-containing signaling motifs are depicted by Ys.
The direct interaction between B7-1 and PD-L1 also compels a revised view of the interactions among molecules within the B7:CD28 family in regulating T cell activation and tolerance and gives increased significance to B7-1 and PD-L1 on T cells. There are limited studies of PD-L1 function on T cells, but studies of PD-L1−/− T cells indicate that PD-L1 on T cells can downregulate T cell cytokine production (53). Because both PD-L1 and B7-1 are expressed on T cells, B cells, DCs, and macrophages, there is the potential for bidirectional interactions between B7-1 and PD-L1 on these cell types. In addition, PD-L1 on nonhematopoietic cells may interact with B7-1 as well as PD-1 on T cells to regulate cells.
In the sections below, we summarize our current understanding of the roles of PD-1 and its ligands in disease models and discuss the outcomes of PD-L1 manipulation in light of the new PD-L1:B7-1 interaction. We also consider the therapeutic potential of manipulation of PD-1 and its ligands.
AUTOIMMUNITY AND PERIPHERAL TOLERANCE
The first indication that the PD-1 pathway plays a critical role in autoimmunity came from the phenotype of Pdcd1−/− mice. Aged Pdcd1−/− C57BL/6 mice develop a mild glomerulonephritis with low frequency (5). Pdcd1−/− Balb/c mice develop a dilated cardiomyopathy owing to the production of an autoantibody against cardiac troponin (6, 54). Autoimmunity is accelerated by PD-1 deficiency on autoimmune-prone backgrounds. These findings broadly support a role for PD-1 in the induction and/or maintenance of tolerance. Subsequent work has examined the mechanisms by which PD-1 and its ligands can control self-reactive T cell responses.
PD-1 and PD-L1 provide inhibitory signals that regulate both central and peripheral tolerance in multiple ways. PD-1 is expressed on maturing thymocytes in the course of central tolerance induction. PD-L1 is expressed broadly on the thymic cortex and on thymocytes themselves, whereas PD-L2 expression is limited to the thymic medulla (31, 55). CD4−CD8− (DN) thymocytes start to express PD-1 as they undergo TCRβ rearrangement and begin to display functional pre-TCRs on the cell surface (19). PD-1:PD-L1 interactions inhibit positive selection during the DN to CD4+CD8+ (DP) maturational stage (56). PD-1 signaling modifies positive selection signaling thresholds, and loss of either PD-1 or PD-L1 increases DP thymocyte cell numbers (57). PD-1 also can contribute to negative selection (58) and has been identified as a candidate gene in a microarray analysis of aberrant central tolerance in nonobese diabetic (NOD) mice (59). Together, these findings point to a role for PD-1 and PD-L1 in central tolerance induction.
Self-reactive T cells that escape negative selection are controlled in the periphery by mechanisms of peripheral tolerance. Initial interactions between T cells and APCs, such as DCs, can modify potentially self-reactive responses by the display of self-antigen on resting DCs. PD-1 has an important role in controlling the outcome of initial encounters between naive self-reactive T cells and DCs by inhibiting responses of self-reactive T cells. Emerging evidence suggests that immature DCs tolerize T cells, and loss of PD-1 on antigen-specific T cells increases CD8 T cell responses to antigen-bearing resting DCs (60).
Studies in mouse models of autoimmunity and tolerance have revealed that PD-1:PD-L interactions not only are important in the initial phase of activation and expansion of self-reactive T cells, but also influence self-reactive T cell effector function upon antigen reencounter. In the NOD mouse model of autoimmune T cell–mediated diabetes, PD-L1 is upregulated in the pancreas on islet cells (31), and loss or blockade of PD-1 or PD-L1 leads to rapid and exacerbated diabetes with accelerated insulitis and proinflammatory cytokine production by T cells (61–63). In a model of antigen-specific therapy in which administration of antigen-coupled fixed splenocytes induces tolerance and reverses diabetes in NOD mice, PD-1:PD-L1 interactions were required for both the induction and maintenance of CD4 T cell tolerance (64). Notably, blockade of PD-1 or PD-L1 reversed anergy in islet-antigen-specific T cells, whereas CTLA-4 blockade did not break tolerance, indicating a unique function for PD-1:PD-L1 interactions in maintaining T cell anergy. Bone marrow chimera experiments have demonstrated that PD-L1 expression on non–bone marrow–derived cells, including islet cells, inhibits T cell effector function in tissues (63, 65, 66). These studies also suggest a key role for the PD-1:PD-L1 pathway in limiting immune-mediated tissue damage caused by pathogenic T cells upon antigen reencounter in the periphery. Collectively, these findings demonstrate that PD-1: PD-L1 interactions regulate both the initiation and progression of autoimmune diabetes in NOD mice and identify PD-1:PD-L1 interactions as key mediators of T cell tolerance in tissues.
In the experimental autoimmune encephalomyelitis (EAE) model of human multiple sclerosis (MS), PD-1 and its ligands also control self-reactive T cells. PD-1, PD-L1, and PD-L2 are all expressed on cellular infiltrates within the meninges during active EAE disease in C57BL/6 mice (31). PD-L1 is expressed in the CNS on inflammatory cells as well as on astrocytes and vascular endothelial cells. PD-L1 is specifically induced on CD11b+ APCs by IL-12 (67) and on microglial cells by IFN-γ (68). Initial studies described a role for PD-1 and PD-L2 using neutralizing antibody treatment (69). Anti-PD-1 or anti-PD-L2 mAb administration during the induction of EAE accelerated disease onset and severity, increased CNS inflammatory infiltrates, and led to increased myelin oligodendrocyte glycoprotein (MOG)-reactive T cells and antibodies. Subsequent studies (70) using blocking antibodies in different mouse strains, such as Balb/c, or gene-deficient animals (53, 71) suggest that PD-1 and PD-L1, but not PD-L2, are predominantly responsible for regulating the severity of disease in most mouse strains. The strain-dependent effects of PD-1 and its ligands in influencing disease severity in EAE cannot be explained by expression of PD-L1 or PD-L2 on APCs or inflammatory cells (70). Cytokine production is important in the pathogenesis of EAE, and Pdcd1−/− and Cd274−/− T cells secrete increased amounts of inflammatory cytokines, including IL-17 and IFN-γ, in recall responses to myelin antigen (71). Adoptive transfer studies emphasize the critical function for PD-L1 in limiting myelin-reactive pathogenic effector T cells and show that PD-L1 both on the transferred T cell and in the recipient restrains encephalitogenic T cell responses (53).
Another important mechanism of peripheral tolerance involves regulatory T cells, which can suppress activated T cell proliferation and cytokine production. Both PD-1 and PD-L1 are highly expressed on these populations and may play a role in regulatory T cell function (72). A number of studies suggest that PD-L1 may be important for inducing regulatory T cell populations, although the mechanism is not yet clear (73). Experiments in colitis models support the argument for a role of PD-1:PD-L1 on a regulatory cell population and identified a regulatory subpopulation of CD4+CD25−PD-1+ T cells that can inhibit the development of colitis (74). PD-L1 is important for in vitro inhibition by another suppressive population of CD4+DX5+ T cells (75).
A role for PD-1:PD-L in humans is suggested by polymorphisms in PDCD1 that have been associated with human autoimmune diseases, including systemic lupus erythematosus (SLE), type 1 diabetes, rheumatoid arthritis, Grave’s disease, and MS (76). Most of these polymorphisms are found in conserved regions in intronic sequences. One intronic single nucleotide polymorphism (G7146A) in PDCD1 is located in a binding site for Runx1 (AML-1), a transcription factor with an important regulatory role in hematopoiesis (77). This polymorphism may alter PD-1 mRNA stability or expression level and is associated with reduced PD-1-mediated inhibition of IFN-γ production in German patients with MS (78). A recent study suggests that PDCD1 genetic variation may influence the risk and expression of SLE, and effects of PD-1 polymorphisms vary according to ethnic background, similar to the effects of mouse PD-1 deficiency in different genetic backgrounds (79). A polymorphism in PDCD1LG2 has been described that correlates with SLE (80), but no polymorphisms in CD274 have been linked to human autoimmune diseases. In addition, autoantibodies to PD-L1 that correlate with active disease have been found in rheumatoid arthritis patients and may contribute to disease progression by dysregulating T cell responses (52).
It remains to be determined if the signals that hold autoimmunity in check are generated through interactions between PD-1 and PD-L1 or between PD-L1 and B7-1. In the NOD mouse model, Cd80−/− NOD mice develop autoimmunity more rapidly and with higher incidence than NOD mice (81), although less quickly than Pdcd1−/− and Cd274−/− NOD mice. B7-1-blocking antibodies significantly accelerate diabetes onset in NOD mice and induce diabetes in normally resistant male NOD mice, but the two anti-B7-1 mAbs appear to differ in their functional effects. The 16–10A1 mAb (which blocks B7-1:CTLA-4 but not B7-1:PD-L1 interactions) caused more rapid diabetes onset than did the 1G10 mAb (which blocks both B7-1:CTLA-4 and B7-1:PD-L1 interactions) (82). Further studies are needed to test the role of PD-L1:B7-1 interactions and to understand the mechanisms by which PD-L1 promotes peripheral tolerance.
In view of its important role in autoimmune disease, the PD-1:PD-L pathway has become a new therapeutic target. Therapies that increase the expression of PD-L and trigger PD-1 may ameloriate autoimmune diseases. These approaches are only beginning to be evaluated in animal models, but the results appear promising. DCs genetically modified to overexpress PD-L1 and MOG in the context of MHC II dramatically ameliorate clinical EAE and reduce severity of CNS inflammation (83). A recombinant adenovirus expressing full-length mouse PD-L1 partially protects against the development of nephritis in lupus-prone mice (84). IFN-β, an immune-modulatory treatment for MS, can upregulate PD-L1 on APC in vitro and in MS patients in vivo, suggesting that IFN-β may exert its anti-inflammatory effects in part via upregulation of PD-L1 expression (26).
INFECTIOUS DISEASE AND MICROBIAL PATHOGENESIS
PD-1 and its ligands have important roles in regulating immune defenses against microbes that cause acute and chronic infections. The PD-1:PD-L pathway appears to be a key determinant of the outcome of infection, regulating the delicate balance between effective antimicrobial immune defenses and immune-mediated tissue damage. For example, Pdcd1−/− mice clear an adenovirus infection more rapidly but develop more severe hepatocellular injury than WT mice (85). In a mouse model of herpes stromal keratitis, a blocking anti-PD-L1 mAb exacerbated keratitis, increasing HSV-1-specific effector CD4 T cell expansion and IFN-γ production and survival (86). These studies suggest that the PD-1:PD-L pathway limits the potentially detrimental consequences of vigorous antipathogen effector T cells.
A number of microorganisms that cause chronic infection appear to have exploited the PD-1:PD-L pathway to evade the immune responses and establish persistent infection. Studies in the lymphocytic choriomeningitis virus (LCMV) model of chronic viral infection were the first to show a role for the PD-1:PD-L pathway during chronic infection (87). Viruses that cause chronic infections can render virus-specific T cells non-functional and thereby silence the antiviral T cell response (88). Functional dysregulation, or exhaustion, of CD8 T cells is an important reason for ineffective viral control during chronic infections and is characteristic of chronic LCMV infection in mice, as well as of HIV, HBV, HCV, and HTLV infection in humans and SIV infection in primates. There appears to be a hierarchical, progressive loss of function within the phenotype of exhausted virus-specific CD8 T cells, with cytotoxicity and IL-2 production lost first, followed by effector cytokine production (88).
PD-1 is upregulated upon activation, and a functionally significant high level of expression is maintained by exhausted CD8 T cells in mice chronically infected with LCMV (87). In vivo administration of antibodies that blocked PD-1:PD-L1 interactions enhanced T cell responses. Most importantly, PD-1 and PD-L1 blockade led to a substantial reduction in viral burden. In persistently infected mice that lack CD4 T cell help, blockaded of this pathway restored and maintained the ability of “helpless” CD8 T cells to proliferate, secrete cytokines, kill infected cells, and decrease viral load. Taken together, these studies demonstrate that the PD-1:PD-L pathway contributes to T cell dysfunction and lack of viral control in chronic LCMV infection, and they suggest a novel strategy for treating chronic viral infections.
Because of the critical role of PD-1:PD-L1 interactions in LCMV, there has been interest in extending this work to chronic viral infection in humans. Several groups have shown that PD-1 expression is high on HIV-specific (23, 89, 90), HBV-specific (91, 92), and HCV-specific T cells (93). PD-L1 is also upregulated on peripheral blood CD14+ monocytes and myeloid DCs in patients with chronic HBV infection (94, 95), and on CD14+ cells and T cells in HIV patients (96). Blocking PD-1:PD-L interactions in vitro reverses the exhaustion of HIV-specific, HBV-specific (92), HCV-specific, and SIV-specific (97) CD8 and CD4 T cells and restores proliferation and cytokine production (23, 89, 90, 93). A mechanistic understanding of PD-1 upregulation during chronic viral infection is at an early stage. Recent work shows that the HCV core, a nucleocapsid protein, can upregulate PD-1 and PD-L1 expression on healthy donor T cells and that upregulation of PD-1 is mediated by interaction of the HCV core with the complement receptor C1QBP (98).
PD-1 also may serve as a useful marker on virus-specific CD8 T cells to indicate the degree of T cell exhaustion and disease severity. The level of PD-1 protein per cell is important in regulating T cell dysfunction. For example, the level and percentage of PD-1 expression on HIV-specific CD8 T cells correlates with viral load, declining CD4 counts, and decreased capacity of CD8 T cells to proliferate in response to HIV antigen in vitro. Similarly, there was a direct correlation between PD-1 expression on HIV-specific CD4 T cells and viral load (99). Long-term nonprogressors have functional HIV-specific memory CD8 T cells with markedly lower PD-1 expression, in contrast to typical progressors who express significantly upregulated PD-1, which correlates with reduced CD4 T cell number, decreased HIV-specific effector memory CD8 T cell function, and elevated plasma viral load (100).
The PD-1:PD-L pathway also may play a key role in the chronicity of bacterial infections. Helicobacter pylori causes chronic gastritis and gastroduodenal ulcers and is a risk factor for development of gastric cancer. During H. pylori infection, T cell responses are insufficient to clear infection, leading to persistent infection. Gastric epithelial cells express MHC class II molecules and are thought to have important APC function during H. pylori infection. Following exposure to H. pylori in vitro or in vivo, PD-L1 also is upregulated on human gastric epithelial cells. Anti-PD-L1 blocking antibodies enhance T cell proliferation and IL-2 production in cultures of gastric epithelial cells exposed to H. pylori and CD4 T cells, suggesting that PD-L1 may play an important role in inhibiting T cell responses during H. pylori infection (101). PD-L1 is up-regulated in gastric mucosal biopsies from H. pylori–infected individuals, who show a marked increase in the CD4+CD25hiFoxP3+ cell population. Naive T cells cultured with H. pylori–exposed gastric epithelial cells can develop into functional CD4+CD25hiFoxP3+ regulatory T cells (102). Blocking PD-L1 on in gastric epithelial cells with anti-PD-L1 mAb or specific small interfering RNA (siRNA) prevented generation of the regulatory T cells, suggesting that PD-L1 may promote T cell suppression and persistent infection by controlling the dynamic between regulatory and effector T cells during H. pylori infection.
Parasitic worms also have exploited the PD-1:PD-L pathway to induce macrophages with strong suppressive function. During Taenia crassiceps infection in mice, PD-L1 and PD-L2 are upregulated on activated macrophages, and a high percentage of CD4 T cells express PD-1. Blockade of PD-L1, PD-L2, or PD-1 significantly decreased suppression of in vitro T cell proliferation by macrophages from Taenia-infected mice (103). Similarly, during Schistosoma mansoni infection in mice, macrophages express high levels of PD-L1 and more modest levels of PD-L2. Anti-PD-L1 completely abrogated the ability of these macrophages to suppress T cell proliferation in vitro, whereas anti-PD-L2 had no effect. PD-L1 expression on macrophages from infected mice declines after 12 weeks of infection, correlating with a break in T cell anergy (104). Thus, an emerging theme is that PD-L1 and PD-L2 can mediate the suppressive functions of macrophages during parasite infections.
PD-L1 and PD-L2 have distinct roles in the immune response to the protozoan parasite Leishmania mexicana. Cd274−/− 129Sv mice showed resistance to L. mexicana, whereas Pdcd1lg2−/− mice developed exacerbated disease with increased parasite burdens. Cd274−/− mice exhibited a diminished Th2 response, which may explain the increased resistance of Cd274−/− mice. Pdcd1lg2−/− mice exhibited a marked increase in L. mexicana–specific IgM and IgG2a, which may contribute to the exacerbated disease observed in Pdcd1lg2−/− mice. Increased parasite-specific IgG production may suppress the healing response through FcγR ligation on macrophages. Further studies are needed to determine whether these distinct outcomes of infection reflect impaired PD-1 signaling into T cells, B cells, and/or macrophages (105).
The key roles of the PD-1:PD-L pathway in reducing T cell responses during chronic viral infections propel development of strategies to manipulate the interaction of PD-1 and its ligands to restore antiviral T cell responses during chronic viral infections. This pathway may have evolved to limit immune-mediated damage to the host during infection by turning off pathogen-specific T cells. We must better understand the immunoregulatory roles of this pathway to determine how to modulate it to activate effective pathogen-specific T cells while minimizing the risk of autoimmunity and immunopathology. Notably, anti-PD-L1 mAb had greater effects than anti-PD-1 mAb in the chronic LCMV model; the anti-PD-L1 mAb used in these studies blocks both PD-L1:PD-1 and PD-L1:B7-1 interactions. The therapeutic potential of manipulating PD-1 and PD-L1 to enhance immune responses during chronic infection gives impetus to analyzing the relative effects of blocking PD-1:PD-L1 interactions versus B7-1:PD-L1 interactions during chronic infection.
THE ROLE OF PD-1:PD-L IN TRANSPLANTATION
Negative costimulatory pathways play a key role in regulating rejection of transplanted allogeneic tissues (106–108), and several important lines of evidence demonstrate that PD-1:PD-L1 interactions control engraftment of solid organs and graft-versus-host disease (GVHD). Redundancy among negative costimulatory pathways is clearly important for controlling alloreactive T cells, and several studies have demonstrated unique roles for the PD-1:PD-L and CTLA-4:B7 pathways.
Both PD-1 and PD-L1 are significantly upregulated on alloreactive T cells in transplant recipients (109, 110). The inducible expression of PD-L1 in tissues, such as the heart and pancreatic islets, as well as constitutive expression on other tissues such as the cornea, also may control alloreactive immune responses. Studies in heart transplant models have demonstrated that PD-L1 is upregulated on cardiac allografts as early as one day after heart transplant (111). PD-1 and PD-L2 are induced much later in the response, presumably because they are expressed primarily by infiltrating cells. PD-1 upregulation occurs after the onset of GVHD, and PD-L1 is extensively expressed on most cells in GVHD target organs.
In heart (112), corneal (113), and skin transplant models (109), administration of PD-L1, but not PD-L2, blocking antibodies accelerated transplant rejection. The absence of a significant effect of PD-L2 blocking antibody in transplant models suggests several possibilities. First, PD-L1 and PD-L2 may play different roles in tolerance induction by tolerogenic DCs. In support of this view, there is evidence of differential expression of these molecules on tolerogenic DCs in a transplant setting, but there has been no direct test of their respective roles on DCs during rejection (114, 115).
In addition, the importance of PD-L1 as a ligand, either on tissues or on lymphocytes themselves, may be central to graft tolerance (63). Recent studies have investigated the role of the PD-1:PD-L pathway in acquired transplantation tolerance using a fully MHC mismatch cardiac transplant model, in which tolerance can be induced by CTLA-4-Ig (116). To examine the role of PD-L1 on the donor versus the recipient in the acquired tolerance model of CTLA-4-Ig, Cd274−/− mice were used as graft donors or recipients of cardiac allografts and given CTLA-4-Ig treatment. Cd274−/− recipients of WT cardiac allografts had accelerated graft rejection compared with WT recipients given CTLA-4-Ig therapy. Cd274−/− cardiac allografts were accepted by WT recipients treated with CTLA-4-Ig, yet histologic examination showed evidence of severe chronic rejection and vasculopathy. These data indicate that PD-L1 in the graft protects from local pathology and chronic rejection, whereas PD-L1 expression in the recipient immune system is required for induction and/or maintenance of transplantation tolerance after CTLA-4-Ig therapy. These findings suggest that PD-1 agonists may be useful for promoting transplant tolerance and preventing local inflammation that leads to graft arterial disease (GAD) and chronic rejection.
In fully MHC class I and II mismatched allografts, acute rejection occurs within days, and there is a strong induction of PD-1 expression on T cells. Administration of PD-L1 blocking antibodies significantly hastens rejection in the full mismatch cardiac transplant model (112). The role of PD-1 in regulating alloresponses also appears to depend on the strength of TCR signaling. In heart transplant models in which only MHC class I or II are mismatched, chronic rejection in the form of GAD is induced. GAD is accompanied by mild PD-1 and PD-L1 upregulation (117), and blockade in the setting of MHC class I or II mismatch does not hasten rejection (110).
PD-1 can exert its negative regulatory effect in the presence or absence of positive CD28 costimulation. When fully MHC class I and II mismatched hearts are transplanted into Cd28−/− recipients, positive signals transduced through CD28 are eliminated, which doubles the time to rejection. When PD-1 or PD-L1 blocking antibodies are used in Cd28−/− recipients, rejection is accelerated, indicating that PD-1:PD-L1 interactions are important in acute rejection in the absence of CD28 stimulation. Additionally, PD-1 and CTLA-4 effects are not redundant, as evidenced by findings in GVHD, where blocking antibodies to PD-1 augment disease induction and coadministration with anti-CTLA-4 substantially accelerates GVHD (118).
One mechanism by which PD-L1 blockade may accelerate graft rejection is through prevention of T cell apoptosis. PD-L1, but not PD-1 or PD-L2, blocking antibodies induce accelerated rejection in a skin transplant model using allospecific TCR transgenic T cells (109). PD-L1 blocking antibody inhibited in vivo apoptosis of alloreactive TCR transgenic T cells, whereas a PD-1 blocking antibody did not. Similar results were obtained in a corneal transplant mode, where only PD-L1, but not PD-1 or PD-L2, blocking antibodies induced rapid rejection (113). This suggests a PD-1-independent role for PD-L1 in promoting tolerance through inducing apoptosis of alloreactive T cells. The mechanism of apoptosis induction, whether it is TCR-dependent or -independent, and whether B7-1 is involved have yet to be determined.
Induction of allograft tolerance is dependent on the balance of regulatory and effector T cells, and PD-1 and PD-L1 have been implicated in controlling both regulatory T cell induction as well as increased effector T cell generation in transplant models. PD-L1 blockade can inhibit allospecific effector responses in vitro (119), as well as inhibit the induction of allogeneic regulatory T cells by vascular endothelium (73). The acceleration of graft rejection observed after PD-L1 neutralizing antibody administration may depend on regulatory T cells. Administration of anti-PD-L1 on the day of transplantation or on day 60 after transplantation led to cardiac graft rejection in CTLA-4-Ig-treated recipients, with an increase in lymphocyte production of inflammatory cytokines and significant decreases in regulatory T cell numbers within the allograft. Similar studies in both a skin allograft and a GVHD model have also suggested that PD-1:PD-L1 interactions on regulatory and effector T cells may be important (109, 119). No acceleration of rejection was observed when anti-PD-L1 treatment was given in the absence of CD25+ T cells. The loss of PD-1 and PD-L1 through genetic deletion or antibody blockade also has been demonstrated to increase effector cell generation and cytokine production (110, 118, 119). These studies suggest that PD-L1 may regulate transplantation tolerance by controlling the balance between regulatory and pathogenic effector cells, limiting the expansion of alloreactive effector cells in the periphery.
ALLERGY AND ASTHMA
Asthma and allergic responses constitute inappropriately vigorous immune responses to innocuous stimuli. B7 family members are implicated in the regulation of allergy and asthma and are involved in the priming events, helper T cell differentiation, and effector function that underlie the pathology of these diseases. Although a picture of the definitive role of PD-1 and its ligands in asthma and allergy has yet to emerge, most data point to PD-L2 as a critical molecule in these diseases. PD-L2 is abundantly expressed in DCs in the lung in a mouse model of asthma. In a mouse ovalbumin-induced allergic asthma model, PD-L2 treatment during challenge but not sensitization resulted in an increase in airway hyperresponsiveness and in Th2 cytokine production (120). There was no effect of either PD-1 or PD-L1 mAbs in either the priming or effector phase. These results suggest that the effector responses of T cells in asthma may be influenced by PD-L2, but not PD-1. Use of sHIgM12 mAb, which was previously described as inducing reverse signaling through PD-L2, in a mouse model of allergic asthma completely blocked the development of airway inflammatory disease, and sHIgM12-treated DCs could mediate these same protective effects when transferred into hyperimmune mice (48, 49). Finally, treatment with PD-L2-Fc prior to sensitization and challenge exacerbated T cell proliferation and cytokine production and increased eosinophilia in vivo but had the opposite effect in vitro, where it inhibited T cell proliferation and cytokine production (121). PD-L2 blockade, but not PD-1 or PD-L1 blockade, diminished eosinophil migration to the conjunctiva in a mouse model of allergic conjunctivitis (122). Among the studies that have tested the function of PD-1 and its ligands in allergy and asthma, most have utilized different reagents. Further work is required to understand the role of PD-1 and its ligands in allergic diseases, yet a consistent finding has been the implication of PD-L2 in these processes.
TUMOR IMMUNITY
Tumors express antigens that can be recognized by host T cells, but immunologic clearance of tumors is rare. Part of this failure is due to immune suppression by the tumor microenvironment. PD-L1 expression on many tumors is a component of this suppressive milieu and may act in concert with other immunosuppressive signals. PD-L1 expression has been shown in situ on a wide variety of solid tumors including breast, lung, colon, ovarian, melanoma, bladder, liver, salivary, stomach, gliomas, thyroid, thymic epithelial, head, and neck (55, 123–131). Immunohistochemical staining of normal and malignant breast tissue is contrasted in Figure 4. In addition, PD-1 expression is upregulated on tumor infiltrating lymphocytes, and this may also contribute to tumor immunosuppression (58). In ovarian cancer, PD-L1 expression is inversely correlated with intraepithelial, but not stromal, infiltrating CD8 T cells, suggesting that PD-L1 inhibits the intratumor migration of CD8 T cells (124). Expression on tumor cell lines is generally lower in vitro, and expression is increased upon adoptive transfer into animals. Translation of PD-L1 mRNA is enhanced by loss of PTEN and the ensuing activation of Akt, a common event in tumorigenesis (29). Most importantly, studies relating PD-L1 expression on tumors to disease outcome show that PD-L1 expression strongly correlates with unfavorable prognosis in kidney, ovarian, bladder, breast, gastric, and pancreatic cancer but not small cell lung cancer (124, 126–131). In addition, these studies suggest that higher levels of PD-L1 expression on tumors may facilitate advancement of tumor stage and invasion into deeper tissue structures.
Figure 4.
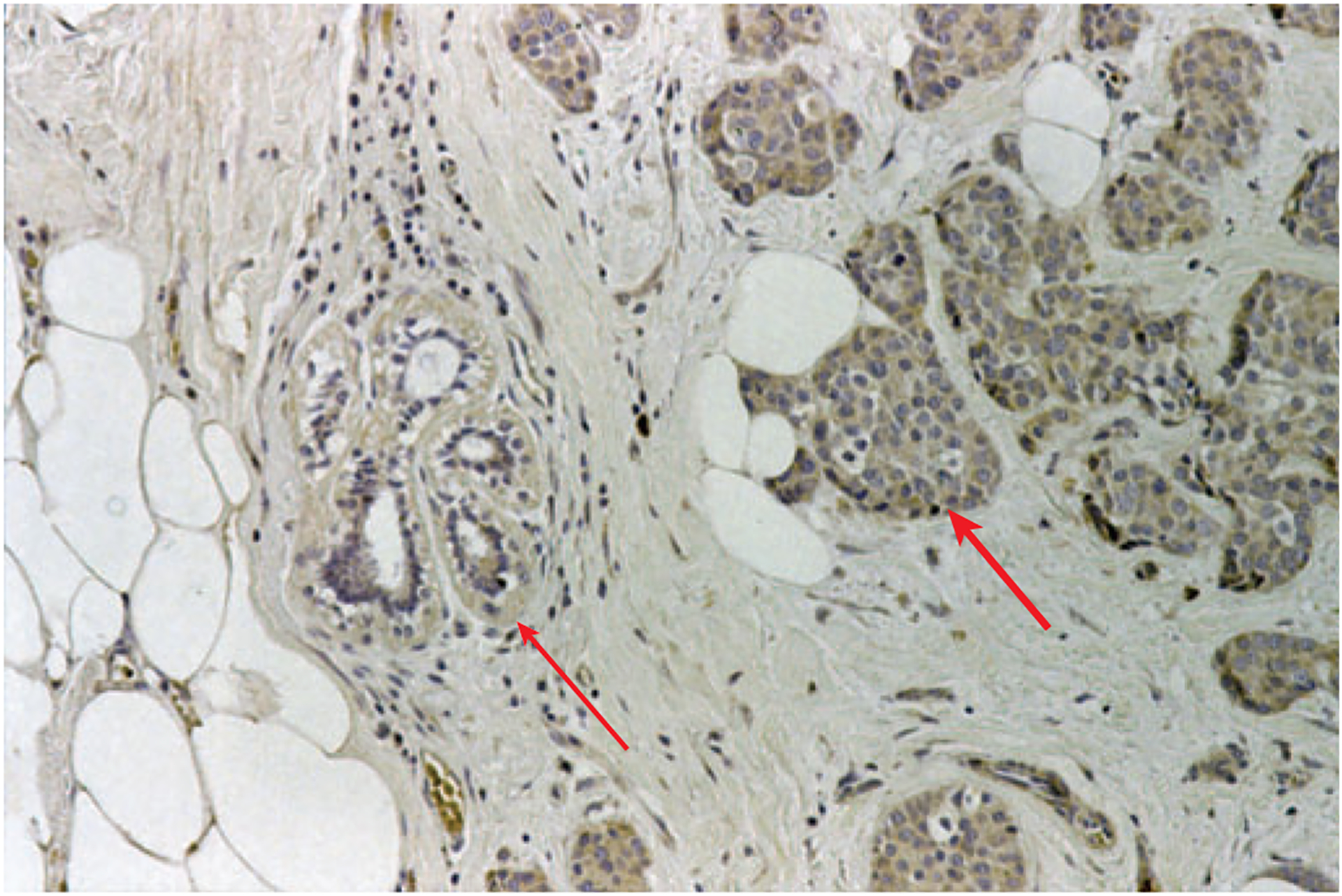
PD-L1 is upregulated on breast tumor, but not normal, cells Immunohistochemical staining (brown) of a breast ductal carcinoma showing high expression of PD-L1 on neoplastic tissue (thick arrow) and low expression on adjacent normal tissue (thin arrow) (figure kindly provided by David Dorfman, Brigham and Women’s Hospital). Note that lymphocytes are more abundant in normal tissue areas than in cancer cell nests.
The PD-1 pathway may also play a role in hematologic malignancies. PD-1 or PD-L1 are rarely expressed on B cell malignancies (55), but PD-L2 has been identified by microarray analysis as being highly expressed in mantle cell lymphomas (132). PD-L1 is expressed on multiple myeloma cells but not on normal plasma cells. T cell expansion in response to myeloma cells is enhanced in vitro by PD-L1 blockade (28). PD-L1 is expressed on some primary T cell lymphomas, particularly anaplastic large cell T lymphomas (55). PD-1 is highly expressed on the T cells of angioimmunoblastic lymphomas, and PD-L1 is expressed on the associated follicular dendritic cell network (133). In nodular lymphocyte-predominant Hodgkin lymphoma, the T cells associated with lymphocytic and/or histiocytic (L&H) cells express PD-1. Microarray analysis using a readout of genes induced by PD-1 ligation suggests that tumor-associated T cells are responding to PD-1 signals in situ in Hodgkin lymphoma (134). PD-1 and PD-L1 are expressed on CD4 T cells in HTLV-1-mediated adult T cell leukemia and lymphoma (135). These tumor cells are hyporesponsive to TCR signals, and PD-1 blockade increased their expression of TNF-α, but not IFN-γ.
Studies in animal models demonstrate that PD-L1 on tumors inhibits T cell activation and lysis of tumor cells and in some cases leads to increased tumor-specific T cell death (123, 136). Treatment with anti-PD-L1 or injection of tumor cells into Pdcd1−/− mice augments antitumor responses, as measured by cytotoxicity and cytokine production (136–138). Treatment with anti-PD-L1 in vivo delays, but does not halt, tumorigenesis of PD-L1-expressing mouse myeloma cell lines (137) PD-L1 blockade can improve the outcome of immunotherapy. PD-L1 expression on the immunogenic tumor P815 causes resistance to immunotherapy, but blockade with anti-PD-L1 restores the response to anti-CD137 therapeutic mAb (136). Treatment with anti-PD-L1 mAb enhances T cell immunotherapy, and administration of anti-PD-L1 with activated T cells augments rejection of a PD-L1-expressing squamous cell carcinoma (125).
Tumor-associated APCs can also utilize the PD-1:PD-L pathway to control antitumor T cell responses. PD-L1 expression on a population of tumor-associated myeloid DCs is upregulated by tumor environmental factors, and PD-L1 mAb blockade enhanced DC-mediated T cell activation (139). Plasmacytoid DCs in the tumor-draining lymph node of B16 melanoma express IDO, which strongly activates the suppressive activity of regulatory T cells. The suppressive activity of IDO-treated regulatory T cells required cell contact with IDO-expressing DCs and was abrogated by PD-L blockade (140).
A fully human PD-1 mAb has been developed and is in Phase 1 clinical trials for cancer. Preclinical studies show that PD-1 is expressed on tumor-specific human T cells following vaccination with tumor peptide antigen. Blockade of PD-1 with this mAb during in vitro stimulation with melanoma peptide increased the numbers and effector activity of tumor-specific human T cells (141). Both Th1 and Th2 cytokine production were increased. PD-1 blockade did not change the percentage of apoptotic antigen-specific human T cells, indicating that the increase in number was due to increased proliferation, not decreased death. In mice, a triple treatment therapy of anti-PD-L1 blockade, depletion of CD4 T cells (primarily regulatory T cells), and irradiated tumor cell vaccination induced complete elimination of large established renal cancer cell (RENCA) tumors with long-lasting tumor-specific immunity (142), further suggesting that this pathway is a promising target for therapeutic intervention.
PD-1 AND IMMUNOPATHOLOGY
A number of studies point to an important role for PD-L1 in limiting immunopathology. Following infection with LCMV clone 13, WT mice develop a chronic infection, whereas Cd274−/− mice die (87). Blockade or elimination of PD-L1 in mouse models of autoimmunity leads to exacerbated autoimmunity associated with severe inflammation and tissue damage (61, 63–66, 69). Bone marrow chimera studies point to an important role for PD-L1 on non–bone marrow–derived cells in limiting effector T cell responses and immunopathology.
The expression of PD-L1 on vascular endothelial cells has led to the hypothesis that PD-L1 on endothelial cells may regulate the activation of T cells that contact the vessel wall, the extravasation of T cells into tissue, and/or limit detrimental consequences of immunopathology. Blockade of PD-L1 on vascular endothelial cells enhances IFN-γ production and cytolytic activity of CD8 T cells in vitro (143). Cd274−/−Pdcd1lg2−/− mice developed severely increased atherosclerotic lesion burden, suggesting that PD-L1 also may play a significant role in inflammatory diseases in which vascular endothelium and T cells are important for pathogenesis (144).
Fibroblastic reticular cells (FRC) express high levels of PD-L1, which can be upregulated during LCMV clone 13 infection (145). Elegant confocal, electron, and intravital microscopy studies demonstrate that the FRC network can regulate T cell access to the paracortex within the lymph node and regulate movement within the LN. In vivo administration of blocking PD-L1 mAb during LCMV clone 13 infection induced significant CD8 T cell–mediated damage to the splenic stroma, demonstrating an essential role for this pathway in minimizing virus-induced immunopathology. These studies suggest that PD-L1 on FRC may contribute to viral persistence during chronic infection. Further studies are needed to test whether expression of PD-L1 by FRC may contribute to the influence of FRC on T cell trafficking.
CONCLUDING REMARKS
PD-1:PD-L interactions exert a vital and diverse range of immunoregulatory roles in T cell activation, tolerance, and immune-mediated tissue damage. How do PD-1 and its ligands exert their inhibitory effects? One of their most significant functions is controlling potentially pathogenic effector T cells, yet PD-1:PD-L can also dampen early activation events when naive T cells encounter antigen in lymph nodes. PD-1 and PD-L1 are also expressed on regulatory T cells and may control their suppression of effector T cells. Recent studies indicate that PD-L1 and PD-L2 can signal bidirectionally by engaging PD-1 on T cells and by delivering signals into PD-L-expressing cells. These bidirectional interactions between PD-1 and its ligands, along with the identification of B7-1 as an additional binding partner for PD-L1, may help explain the seemingly contradictory results seen with reagents developed to manipulate this pathway. The discovery of B7-1:PD-L1 interactions also reveals additional ways by which PD-L1 exerts its inhibitory functions.
Because both PD-L1 and B7-1 are expressed on T cells, B cells, DCs, and macrophages, there is the potential for bidirectional interactions between B7-1 and PD-L1 on these cell types. Emerging evidence demonstrates a unique and critical role for PD-L1 on nonhematopoetic cells for mediating tissue tolerance as well as protecting tissues from the detrimental consequences of overaggressive effector responses. Microbes and tumors appear to have exploited this pathway to evade eradication by the immune system. These distinctive functions provide therapeutic opportunities for developing PD-1/PD-L antagonists to boost antimicrobial and antitumor immunity as well as agonists to control pathogenic T cells in autoimmune diseases and graft rejection. The fundamental and therapeutic importance of the PD-1:PD-L pathway gives impetus to further investigation of its functions.
ACKNOWLEDGMENTS
This work was supported by grants from National Institutes of Health and the Foundation for the National Institutes of Health through the Grand Challenges in Global Health Initiative (to A.H.S. and G.J.F.) and the National Multiple Sclerosis Society (to A.H.S.). Because of space restrictions, we were able to cite only a fraction of the relevant literature and apologize to colleagues whose contributions may not be appropriately acknowledged in this review.
DISCLOSURE STATEMENT
A.H.S. and G.J.F. have patents on PD-1 ligands and grants on PD-1 and PD-1 ligands.
Glossary
- ITIM
immunoreceptor tyrosine-based inhibitory motif
- ITSM
immunoreceptor tyrosine-based switch motif
- sHIgM12
naturally occurring human anti-PD-L2 IgM antibody derived from a patient with Waldenstrom’s macroglobulinemia
LITERATURE CITED
- 1.Lafferty KJ, Cunningham AJ. 1975. A new analysis of allogeneic interactions. Aust. J. Exp. Biol. Med. Sci 53:27–42 [DOI] [PubMed] [Google Scholar]
- 2.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. 1995. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 3:541–47 [DOI] [PubMed] [Google Scholar]
- 3.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, et al. 1995. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 270:985–88 [DOI] [PubMed] [Google Scholar]
- 4.Ishida Y, Agata Y, Shibahara K, Honjo T. 1992. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 11:3887–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. 1999. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 11:141–51 [DOI] [PubMed] [Google Scholar]
- 6.Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, et al. 2001. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 291:319–22 [DOI] [PubMed] [Google Scholar]
- 7.Dong H, Zhu G, Tamada K, Chen L. 1999. B7-H1, a third member of the B7 family, costimulates T-cell proliferation and interleukin-10 secretion. Nat. Med 5:1365–69 [DOI] [PubMed] [Google Scholar]
- 8.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, et al. 2000. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med 192:1027–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, et al. 2001. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol 2:261–68 [DOI] [PubMed] [Google Scholar]
- 10.Tseng SY, Otsuji M, Gorski K, Huang X, Slansky JE, et al. 2001. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J. Exp. Med 193:839–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. 2007. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 27:111–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nielsen C, Ohm-Laursen L, Barington T, Husby S, Lillevang ST. 2005. Alternative splice variants of the human PD-1 gene. Cell. Immunol 235:109–16 [DOI] [PubMed] [Google Scholar]
- 13.Ueda H, Howson JM, Esposito L, Heward J, Snook H, et al. 2003. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 423:506–11 [DOI] [PubMed] [Google Scholar]
- 14.Wan B, Nie H, Liu A, Feng G, He D, et al. 2006. Aberrant regulation of synovial T cell activation by soluble costimulatory molecules in rheumatoid arthritis. J. Immunol 177:8844–50 [DOI] [PubMed] [Google Scholar]
- 15.He XH, Xu LH, Liu Y. 2005. Identification of a novel splice variant of human PD-L1 mRNA encoding an isoform-lacking Igv-like domain. Acta Pharmacol. Sin 26:462–68 [DOI] [PubMed] [Google Scholar]
- 16.He XH, Liu Y, Xu LH, Zeng YY. 2004. Cloning and identification of two novel splice variants of human PD-L2. Acta Biochim. Biophys. Sin 36:284–89 [DOI] [PubMed] [Google Scholar]
- 17.Wang S, Bajorath J, Flies DB, Dong H, Honjo T, Chen L. 2003. Molecular modeling and functional mapping of B7-H1 and B7-DC uncouple costimulatory function from PD-1 interaction. J. Exp. Med 197:1083–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang X, Schwartz JC, Guo X, Bhatia S, Cao E, et al. 2004. Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity 20:337–47 [DOI] [PubMed] [Google Scholar]
- 19.Nishimura H, Agata Y, Kawasaki A, Sato M, Imamura S, et al. 1996. Developmentally regulated expression of the PD-1 protein on the surface of double-negative (CD4−CD8−) thymocytes. Int. Immunol 8:773–80 [DOI] [PubMed] [Google Scholar]
- 20.Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. 2004. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol 173:945–54 [DOI] [PubMed] [Google Scholar]
- 21.Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, et al. 1996. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol 8:765–72 [DOI] [PubMed] [Google Scholar]
- 22.Raimondi G, Shufesky WJ, Tokita D, Morelli AE, Thomson AW. 2006. Regulated compartmentalization of programmed cell death-1 discriminates CD4+CD25+ resting regulatory T cells from activated T cells. J. Immunol 176:2808–16 [DOI] [PubMed] [Google Scholar]
- 22a.Pentcheva-Hoang T, Chen L, Pardoll DM, Allison JP. 2007. Programmed death-1 concentration at the immunological synapse is determined by ligand affinity and availability. Proc. Natl. Acad. Sci. USA 104:17765–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petrovas C, Casazza JP, Brenchley JM, Price DA, Gostick E, et al. 2006. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J. Exp. Med 203:2281–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, et al. 2002. Expression of programmed death 1 ligands by murine T cells and APC. J. Immunol 169:5538–45 [DOI] [PubMed] [Google Scholar]
- 25.Eppihimer MJ, Gunn J, Freeman GJ, Greenfield EA, Chernova T, et al. 2002. Expression and regulation of the PD-L1 immunoinhibitory molecule on microvascular endothelial cells. Microcirculation 9:133–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schreiner B, Mitsdoerffer M, Kieseier BC, Chen L, Hartung HP, et al. 2004. Interferon-β enhances monocyte and dendritic cell expression of B7-H1 (PD-L1), a strong inhibitor of autologous T-cell activation: relevance for the immune modulatory effect in multiple sclerosis. J. Neuroimmunol 155:172–82 [DOI] [PubMed] [Google Scholar]
- 27.Lee SJ, Jang BC, Lee SW, Yang YI, Suh SI, et al. 2006. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-γ-induced upregulation of B7-H1 (CD274). FEBS Lett. 580:755–62 [DOI] [PubMed] [Google Scholar]
- 28.Liu J, Hamrouni A, Wolowiec D, Coiteux V, Kuliczkowski K, et al. 2007. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-γ and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood 110:296–304 [DOI] [PubMed] [Google Scholar]
- 29.Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, et al. 2007. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med 13:84–88 [DOI] [PubMed] [Google Scholar]
- 30.Zhong X, Tumang JR, Gao W, Bai C, Rothstein TL. 2007. PD-L2 expression extends beyond dendritic cells/macrophages to B1 cells enriched for VH11/VH12 and phosphatidylcholine binding. Eur. J. Immunol 37:2405–10 [DOI] [PubMed] [Google Scholar]
- 31.Liang SC, Latchman YE, Buhlmann JE, Tomczak MF, Horwitz BH, et al. 2003. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur. J. Immunol 33:2706–16 [DOI] [PubMed] [Google Scholar]
- 32.Loke P, Allison JP. 2003. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc. Natl. Acad. Sci. USA 100:5336–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carter L, Fouser LA, Jussif J, Fitz L, Deng B, et al. 2002. PD-1:PD-L inhibitory pathway affects both CD4+ and CD8+ T cells and is overcome by IL-2. Eur. J. Immunol 32:634–43 [DOI] [PubMed] [Google Scholar]
- 34.Nurieva R, Thomas S, Nguyen T, Martin-Orozco N, Wang Y, et al. 2006. T-cell tolerance or function is determined by combinatorial costimulatory signals. EMBO J. 25:2623–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, et al. 2004. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3ζ signalosome and downstream signaling to PKCθ. FEBS Lett. 574:37–41 [DOI] [PubMed] [Google Scholar]
- 36.Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. 2001. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. USA 98:13866–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bennett F, Luxenberg D, Ling V, Wang IM, Marquette K, et al. 2003. Program death-1 engagement upon TCR activation has distinct effects on costimulation and cytokine-driven proliferation: attenuation of ICOS, IL-4, and IL-21, but not CD28, IL-7, and IL-15 responses. J. Immunol 170:711–18 [DOI] [PubMed] [Google Scholar]
- 38.Egen JG, Allison JP. 2002. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity 16:23–35 [DOI] [PubMed] [Google Scholar]
- 39.Saunders PA, Hendrycks VR, Lidinsky WA, Woods ML. 2005. PD-L2:PD-1 involvement in T cell proliferation, cytokine production, and integrin-mediated adhesion. Eur. J. Immunol 35:3561–69 [DOI] [PubMed] [Google Scholar]
- 40.Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, et al. 2005. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol 25:9543–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shlapatska LM, Mikhalap SV, Berdova AG, Zelensky OM, Yun TJ, et al. 2001. CD150 association with either the SH2-containing inositol phosphatase or the SH2-containing protein tyrosine phosphatase is regulated by the adaptor protein SH2D1A. J. Immunol 166:5480–87 [DOI] [PubMed] [Google Scholar]
- 42.Arnaud M, Crouin C, Deon C, Loyaux D, Bertoglio J. 2004. Phosphorylation of Grb2-associated binder 2 on serine 623 by ERK MAPK regulates its association with the phosphatase SHP-2 and decreases STAT5 activation. J. Immunol 173:3962–71 [DOI] [PubMed] [Google Scholar]
- 43.Van Parijs L, Refaeli Y, Lord JD, Nelson BH, Abbas AK, Baltimore D. 1999. Uncoupling IL-2 signals that regulate T cell proliferation, survival, and Fas-mediated activation-induced cell death. Immunity 11:281–88 [DOI] [PubMed] [Google Scholar]
- 44.Nguyen LT, Radhakrishnan S, Ciric B, Tamada K, Shin T, et al. 2002. Cross-linking the B7 family molecule B7-DC directly activates immune functions of dendritic cells. J. Exp. Med 196:1393–98 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Radhakrishnan S, Nguyen LT, Ciric B, Ure DR, Zhou B, et al. 2003. Naturally occurring human IgM antibody that binds B7-DC and potentiates T cell stimulation by dendritic cells. J. Immunol 170:1830–38 [DOI] [PubMed] [Google Scholar]
- 46.Radhakrishnan S, Nguyen LT, Ciric B, Flies D, Van Keulen VP, et al. 2004. Immunotherapeutic potential of B7-DC (PD-L2) cross-linking antibody in conferring antitumor immunity. Cancer Res. 64:4965–72 [DOI] [PubMed] [Google Scholar]
- 47.Heckman KL, Schenk EL, Radhakrishnan S, Pavelko KD, Hansen MJ, Pease LR. 2007. Fast-tracked CTL: rapid induction of potent antitumor killer T cells in situ. Eur. J. Immunol 37:1827–35 [DOI] [PubMed] [Google Scholar]
- 48.Radhakrishnan S, Iijima K, Kobayashi T, Rodriguez M, Kita H, Pease LR. 2004. Blockade of allergic airway inflammation following systemic treatment with a B7-dendritic cell (PD-L2) cross-linking human antibody. J. Immunol 173:1360–65 [DOI] [PubMed] [Google Scholar]
- 49.Radhakrishnan S, Iijima K, Kobayashi T, Kita H, Pease LR. 2005. Dendritic cells activated by cross-linking B7-DC (PD-L2) block inflammatory airway disease. J. Allergy Clin. Immunol 116:668–74 [DOI] [PubMed] [Google Scholar]
- 50.Van Keulen VP, Ciric B, Radhakrishnan S, Heckman KL, Mitsunaga Y, et al. 2006. Immunomodulation using the recombinant monoclonal human B7-DC cross-linking antibody rHIgM12. Clin. Exp. Immunol 143:314–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuipers H, Muskens F, Willart M, Hijdra D, van Assema FB, et al. 2006. Contribution of the PD-1 ligands/PD-1 signaling pathway to dendritic cell-mediated CD4+ T cell activation. Eur. J. Immunol 36:2472–82 [DOI] [PubMed] [Google Scholar]
- 52.Dong H, Strome SE, Matteson EL, Moder KG, Flies DB, et al. 2003. Costimulating aberrant T cell responses by B7-H1 autoantibodies in rheumatoid arthritis. J. Clin. Invest 111:363–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, et al. 2004. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc. Natl. Acad. Sci. USA 101:10691–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Okazaki T, Tanaka Y, Nishio R, Mitsuiye T, Mizoguchi A, et al. 2003. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat. Med 9:1477–83 [DOI] [PubMed] [Google Scholar]
- 55.Brown JA, Dorfman DM, Ma FR, Sullivan EL, Munoz O, et al. 2003. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J. Immunol 170:1257–66 [DOI] [PubMed] [Google Scholar]
- 56.Nishimura H, Honjo T, Minato N. 2000. Facilitation of beta selection and modification of positive selection in the thymus of PD-1-deficient mice. J. Exp. Med 191:891–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Keir ME, Latchman YE, Freeman GJ, Sharpe AH. 2005. Programmed death-1 (PD-1): PD-ligand 1 interactions inhibit TCR-mediated positive selection of thymocytes. J. Immunol 175:7372–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blank C, Brown I, Marks R, Nishimura H, Honjo T, Gajewski TF. 2003. Absence of programmed death receptor 1 alters thymic development and enhances generation of CD4/CD8 double-negative TCR-transgenic T cells. J. Immunol 171:4574–81 [DOI] [PubMed] [Google Scholar]
- 59.Zucchelli S, Holler P, Yamagata T, Roy M, Benoist C, Mathis D. 2005. Defective central tolerance induction in NOD mice: genomics and genetics. Immunity 22:385–96 [DOI] [PubMed] [Google Scholar]
- 60.Probst HC, McCoy K, Okazaki T, Honjo T, van den Broek M. 2005. Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat. Immunol 6:280–86 [DOI] [PubMed] [Google Scholar]
- 61.Ansari MJ, Salama AD, Chitnis T, Smith RN, Yagita H, et al. 2003. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J. Exp. Med 198:63–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T. 2005. Establishment of NOD-Pdcd1−/− mice as an efficient animal model of type I diabetes. Proc. Natl. Acad. Sci. USA 102:11823–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, et al. 2006. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med 203:883–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fife BT, Guleria I, Gubbels Bupp M, Eagar TN, Tang Q, et al. 2006. Insulin-induced remission in new-onset NOD mice is maintained by the PD-1-PD-L1 pathway. J. Exp. Med 203:2737–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grabie N, Gotsman I, Dacosta R, Pang H, Stravrakis G, et al. 2007. Endothelial PD-L1 regulates CD8+ T cell mediated injury in the heart. Circulation 116:2062–71 [DOI] [PubMed] [Google Scholar]
- 66.Keir ME, Freeman GJ, Sharpe AH. 2007. PD-1 regulates self-antigen specific responses in lymph nodes and tissues. J. Immunol 179:5064–70 [DOI] [PubMed] [Google Scholar]
- 67.Cheng X, Zhao Z, Ventura E, Gran B, Shindler KS, Rostami A. 2007. The PD-1/PD-L pathway is up-regulated during IL-12-induced suppression of EAE mediated by IFN-γ. J. Neuroimmunol 185:75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Magnus T, Schreiner B, Korn T, Jack C, Guo H, et al. 2005. Microglial expression of the B7 family member B7 homolog 1 confers strong immune inhibition: implications for immune responses and autoimmunity in the CNS. J. Neurosci 25:2537–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Salama AD, Chitnis T, Imitola J, Ansari MJ, Akiba H, et al. 2003. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J. Exp. Med 198:71–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu B, Guleria I, Khosroshahi A, Chitnis T, Imitola J, et al. 2006. Differential role of programmed death-ligand 1 and programmed death-ligand 2 in regulating the susceptibility and chronic progression of experimental autoimmune encephalomyelitis. J. Immunol 176:3480–89 [DOI] [PubMed] [Google Scholar]
- 71.Carter LL, Leach MW, Azoitei ML, Cui J, Pelker JW, et al. 2007. PD-1/PD-L1, but not PD-1/PD-L2, interactions regulate the severity of experimental autoimmune encephalomyelitis. J. Neuroimmunol 182:124–34 [DOI] [PubMed] [Google Scholar]
- 72.Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. 2003. CD4+CD25+ regulatory cells from human peripheral blood express very high levels of CD25 ex vivo. Novartis Found. Symp 252:67–88 [DOI] [PubMed] [Google Scholar]
- 73.Krupnick AS, Gelman AE, Barchet W, Richardson S, Kreisel FH, et al. 2005. Murine vascular endothelium activates and induces the generation of allogeneic CD4+25+Foxp3+ regulatory T cells. J. Immunol 175:6265–70 [DOI] [PubMed] [Google Scholar]
- 74.Totsuka T, Kanai T, Makita S, Fujii R, Nemoto Y, et al. 2005. Regulation of murine chronic colitis by CD4+CD25− programmed death-1+ T cells. Eur. J. Immunol 35:1773–85 [DOI] [PubMed] [Google Scholar]
- 75.Hornung M, Farkas SA, Sattler C, Schlitt HJ, Geissler EK. 2006. DX5+ NKT cells induce the death of colitis-associated cells: involvement of programmed death ligand-1. Eur. J. Immunol 36:1210–21 [DOI] [PubMed] [Google Scholar]
- 76.Okazaki T, Honjo T. 2007. PD-1 and PD-1 ligands: from discovery to clinical application. Int. Immunol 19:813–24 [DOI] [PubMed] [Google Scholar]
- 77.Prokunina L, Castillejo-Lopez C, Oberg F, Gunnarsson I, Berg L, et al. 2002. A regulatory polymorphism in PDCD1 is associated with susceptibility to systemic lupus erythematosus in humans. Nat. Genet 32:666–69 [DOI] [PubMed] [Google Scholar]
- 78.Kroner A, Mehling M, Hemmer B, Rieckmann P, Toyka KV, et al. 2005. A PD-1 polymorphism is associated with disease progression in multiple sclerosis. Ann. Neurol 58:50–57 [DOI] [PubMed] [Google Scholar]
- 79.Thorburn CM, Prokunina-Olsson L, Sterba KA, Lum RF, Seldin MF, et al. 2007. Association of PDCD1 genetic variation with risk and clinical manifestations of systemic lupus erythematosus in a multiethnic cohort. Genes Immun. 8:279–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang SC, Lin CH, Ou TT, Wu CC, Tsai WC, et al. 2007. Ligands for programmed cell death 1 gene in patients with systemic lupus erythematosus. J. Rheumatol 34:721–25 [PubMed] [Google Scholar]
- 81.Yadav D, Fine C, Azuma M, Sarvetnick N. 2007. B7-1 mediated costimulation regulates pancreatic autoimmunity. Mol. Immunol 44:2616–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lenschow DJ, Ho SC, Sattar H, Rhee L, Gray G, et al. 1995. Differential effects of anti-B7-1 and anti-B7-2 monoclonal antibody treatment on the development of diabetes in the nonobese diabetic mouse. J. Exp. Med 181:1145–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hirata S, Senju S, Matsuyoshi H, Fukuma D, Uemura Y, Nishimura Y. 2005. Prevention of experimental autoimmune encephalomyelitis by transfer of embryonic stem cell-derived dendritic cells expressing myelin oligodendrocyte glycoprotein peptide along with TRAIL or programmed death-1 ligand. J. Immunol 174:1888–97 [DOI] [PubMed] [Google Scholar]
- 84.Ding H, Wu X, Wu J, Yagita H, He Y, et al. 2006. Delivering PD-1 inhibitory signal concomitant with blocking ICOS costimulation suppresses lupus-like syndrome in autoimmune BXSB mice. Clin. Immunol 118:258–67 [DOI] [PubMed] [Google Scholar]
- 85.Iwai Y, Terawaki S, Ikegawa M, Okazaki T, Honjo T. 2003. PD-1 inhibits antiviral immunity at the effector phase in the liver. J. Exp. Med 198:39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jun H, Seo SK, Jeong HY, Seo HM, Zhu G, et al. 2005. B7-H1 (CD274) inhibits the development of herpetic stromal keratitis (HSK). FEBS Lett. 579:6259–64 [DOI] [PubMed] [Google Scholar]
- 87.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, et al. 2006. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439:682–87 [DOI] [PubMed] [Google Scholar]
- 88.Wherry EJ, Ahmed R. 2004. Memory CD8 T-cell differentiation during viral infection. J. Virol 78:5535–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, et al. 2006. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443:350–54 [DOI] [PubMed] [Google Scholar]
- 90.Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, et al. 2006. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med 12:1198–202 [DOI] [PubMed] [Google Scholar]
- 91.Boettler T, Panther E, Bengsch B, Nazarova N, Spangenberg HC, et al. 2006. Expression of the interleukin-7 receptor α chain (CD127) on virus-specific CD8+ T cells identifies functionally and phenotypically defined memory T cells during acute resolving hepatitis B virus infection. J. Virol 80:3532–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Boni C, Fisicaro P, Valdatta C, Amadei B, Di Vincenzo P, et al. 2007. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J. Virol 81:4215–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Urbani S, Amadei B, Tola D, Massari M, Schivazappa S, et al. 2006. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J. Virol 80:11398–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chen L, Zhang Z, Chen W, Zhang Z, Li Y, et al. 2007. B7-H1 up-regulation on myeloid dendritic cells significantly suppresses T cell immune function in patients with chronic hepatitis B. J. Immunol 178:6634–41 [DOI] [PubMed] [Google Scholar]
- 95.Geng L, Jiang G, Fang Y, Dong S, Xie H, et al. 2006. B7-H1 expression is upregulated in peripheral blood CD14+ monocytes of patients with chronic hepatitis B virus infection, which correlates with higher serum IL-10 levels. J. Viral Hepat 13:725–33 [DOI] [PubMed] [Google Scholar]
- 96.Trabattoni D, Saresella M, Biasin M, Boasso A, Piacentini L, et al. 2003. B7-H1 is up-regulated in HIV infection and is a novel surrogate marker of disease progression. Blood 101:2514–20 [DOI] [PubMed] [Google Scholar]
- 97.Velu V, Kannanganat S, Ibegbu C, Chennareddi L, Villinger F, et al. 2007. Elevated expression levels of inhibitory receptor programmed death 1 on simian immunodeficiency virus-specific CD8 T cells during chronic infection but not after vaccination. J. Virol 81:5819–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yao ZQ, King E, Prayther D, Yin D, Moorman J. 2007. T cell dysfunction by hepatitis C virus core protein involves PD-1/PDL-1 signaling. Viral Immunol. 20:276–87 [DOI] [PubMed] [Google Scholar]
- 99.D’Souza M, Fontenot AP, Mack DG, Lozupone C, Dillon S, et al. 2007. Programmed death 1 expression on HIV-specific CD4+ T cells is driven by viral replication and associated with T cell dysfunction. J. Immunol 179:1979–87 [DOI] [PubMed] [Google Scholar]
- 100.Zhang JY, Zhang Z, Wang X, Fu JL, Yao J, et al. 2007. PD-1 up-regulation is correlated with HIV-specific memory CD8+ T-cell exhaustion in typical progressors but not in long-term nonprogressors. Blood 109:4671–78 [DOI] [PubMed] [Google Scholar]
- 101.Das S, Suarez G, Beswick EJ, Sierra JC, Graham DY, Reyes VE. 2006. Expression of B7-H1 on gastric epithelial cells: its potential role in regulating T cells during Helicobacter pylori infection. J. Immunol 176:3000–9 [DOI] [PubMed] [Google Scholar]
- 102.Beswick EJ, Pinchuk IV, Das S, Powell DW, Reyes VE. 2007. Expression of the programmed death ligand 1, B7-H1, on gastric epithelial cells after Helicobacter pylori exposure promotes development of CD4+ CD25+ FoxP3+ regulatory T cells. Infect. Immun 75:4334–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Terrazas LI, Montero D, Terrazas CA, Reyes JL, Rodriguez-Sosa M. 2005. Role of the programmed Death-1 pathway in the suppressive activity of alternatively activated macrophages in experimental cysticercosis. Int. J. Parasitol 35:1349–58 [DOI] [PubMed] [Google Scholar]
- 104.Smith P, Walsh CM, Mangan NE, Fallon RE, Sayers JR, et al. 2004. Schistosoma mansoni worms induce anergy of T cells via selective up-regulation of programmed death ligand 1 on macrophages. J. Immunol 173:1240–48 [DOI] [PubMed] [Google Scholar]
- 105.Liang SC, Greenwald RJ, Latchman YE, Rosas L, Satoskar A, et al. 2006. PD-L1 and PD-L2 have distinct roles in regulating host immunity to cutaneous leishmaniasis. Eur. J. Immunol 36:58–64 [DOI] [PubMed] [Google Scholar]
- 106.Clarkson MR, Sayegh MH. 2005. T-cell costimulatory pathways in allograft rejection and tolerance. Transplantation 80:555–63 [DOI] [PubMed] [Google Scholar]
- 107.Vadivel N, Trikudanathan S, Chandraker A. 2007. Transplant tolerance through costimulation blockade—are we there yet? Front. Biosci 12:2935–46 [DOI] [PubMed] [Google Scholar]
- 108.Kean LS, Gangappa S, Pearson TC, Larsen CP. 2006. Transplant tolerance in nonhuman primates: progress, current challenges and unmet needs. Am. J. Transplant 6:884–93 [DOI] [PubMed] [Google Scholar]
- 109.Sandner SE, Clarkson MR, Salama AD, Sanchez-Fueyo A, Domenig C, et al. 2005. Role of the programmed death-1 pathway in regulation of alloimmune responses in vivo. J. Immunol 174:3408–15 [DOI] [PubMed] [Google Scholar]
- 110.Tao R, Wang L, Han R, Wang T, Ye Q, et al. 2005. Differential effects of B and T lymphocyte attenuator and programmed death-1 on acceptance of partially versus fully MHC-mismatched cardiac allografts. J. Immunol 175:5774–82 [DOI] [PubMed] [Google Scholar]
- 111.Ozkaynak E, Wang L, Goodearl A, McDonald K, Qin S, et al. 2002. Programmed death-1 targeting can promote allograft survival. J. Immunol 169:6546–53 [DOI] [PubMed] [Google Scholar]
- 112.Ito T, Ueno T, Clarkson MR, Yuan X, Jurewicz MM, et al. 2005. Analysis of the role of negative T cell costimulatory pathways in CD4 and CD8 T cell-mediated alloimmune responses in vivo. J. Immunol 174:6648–56 [DOI] [PubMed] [Google Scholar]
- 113.Hori J, Wang M, Miyashita M, Tanemoto K, Takahashi H, et al. 2006. B7-H1-induced apoptosis as a mechanism of immune privilege of corneal allografts. J. Immunol 177:5928–35 [DOI] [PubMed] [Google Scholar]
- 114.Selenko-Gebauer N, Majdic O, Szekeres A, Hofler G, Guthann E, et al. 2003. B7-H1 (programmed death-1 ligand) on dendritic cells is involved in the induction and maintenance of T cell anergy. J. Immunol 170:3637–44 [DOI] [PubMed] [Google Scholar]
- 115.Abe M, Wang Z, de Creus A, Thomson AW. 2005. Plasmacytoid dendritic cell precursors induce allogeneic T-cell hyporesponsiveness and prolong heart graft survival. Am. J. Transplant 5:1808–19 [DOI] [PubMed] [Google Scholar]
- 116.Tanaka K, Albin MJ, Yuan X, Yamaura K, Habicht A, et al. 2007. PD-L1 is required for peripheral transplantation tolerance and protection from chronic allograft rejection. J. Immunol 179:5204–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Koga N, Suzuki J, Kosuge H, Haraguchi G, Onai Y, et al. 2004. Blockade of the interaction between PD-1 and PD-L1 accelerates graft arterial disease in cardiac allografts. Arterioscler. Thromb. Vasc. Biol 24:2057–62 [DOI] [PubMed] [Google Scholar]
- 118.Blazar BR, Carreno BM, Panoskaltsis-Mortari A, Carter L, Iwai Y, et al. 2003. Blockade of programmed death-1 engagement accelerates graft-versus-host disease lethality by an IFN-γ-dependent mechanism. J. Immunol 171:1272–77 [DOI] [PubMed] [Google Scholar]
- 119.Kitazawa Y, Fujino M, Wang Q, Kimura H, Azuma M, et al. 2007. Involvement of the programmed death-1/programmed death-1 ligand pathway in CD4+CD25+ regulatory T-cell activity to suppress alloimmune responses. Transplantation 83:774–82 [DOI] [PubMed] [Google Scholar]
- 120.Matsumoto K, Inoue H, Nakano T, Tsuda M, Yoshiura Y, et al. 2004. B7-DC regulates asthmatic response by an IFN-γ-dependent mechanism. J. Immunol 172:2530–41 [DOI] [PubMed] [Google Scholar]
- 121.Oflazoglu E, Swart DA, Anders-Bartholo P, Jessup HK, Norment AM, et al. 2004. Paradoxical role of programmed death-1 ligand 2 in Th2 immune responses in vitro and in a mouse asthma model in vivo. Eur. J. Immunol 34:3326–36 [DOI] [PubMed] [Google Scholar]
- 122.Fukushima A, Yamaguchi T, Azuma M, Yagita H, Ueno H. 2006. Involvement of programmed death-ligand 2 (PD-L2) in the development of experimental allergic conjunctivitis in mice. Br. J. Ophthalmol 90:1040–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, et al. 2002. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat. Med 8:793–800 [DOI] [PubMed] [Google Scholar]
- 124.Hamanishi J, Mandai M, Iwasaki M, Okazaki T, Tanaka Y, et al. 2007. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl. Acad. Sci. USA 104:3360–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Strome SE, Dong H, Tamura H, Voss SG, Flies DB, et al. 2003. B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma. Cancer Res. 63:6501–5 [PubMed] [Google Scholar]
- 126.Inman BA, Sebo TJ, Frigola X, Dong H, Bergstralh EJ, et al. 2007. PD-L1 (B7-H1) expression by urothelial carcinoma of the bladder and BCG-induced granulomata: associations with localized stage progression. Cancer 109:1499–505 [DOI] [PubMed] [Google Scholar]
- 127.Konishi J, Yamazaki K, Azuma M, Kinoshita I, Dosaka-Akita H, Nishimura M. 2004. B7-H1 expression on nonsmall cell lung cancer cells and its relationship with tumor-infiltrating lymphocytes and their PD-1 expression. Clin. Cancer Res 10:5094–100 [DOI] [PubMed] [Google Scholar]
- 128.Nakanishi J, Wada Y, Matsumoto K, Azuma M, Kikuchi K, Ueda S. 2007. Overexpression of B7-H1 (PD-L1) significantly associates with tumor grade and postoperative prognosis in human urothelial cancers. Cancer Immunol. Immunother 56:1173–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nomi T, Sho M, Akahori T, Hamada K, Kubo A, et al. 2007. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin. Cancer Res 13:2151–57 [DOI] [PubMed] [Google Scholar]
- 130.Thompson RH, Gillett MD, Cheville JC, Lohse CM, Dong H, et al. 2004. Costimulatory B7-H1 in renal cell carcinoma patients: indicator of tumor aggressiveness and potential therapeutic target. Proc. Natl. Acad. Sci. USA 101:17174–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wu C, Zhu Y, Jiang J, Zhao J, Zhang XG, Xu N. 2006. Immunohistochemical localization of programmed death-1 ligand-1 (PD-L1) in gastric carcinoma and its clinical significance. Acta Histochem. 108:19–24 [DOI] [PubMed] [Google Scholar]
- 132.Rosenwald A, Wright G, Leroy K, Yu X, Gaulard RD, et al. 2003. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J. Exp. Med 198:851–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dorfman DM, Brown JA, Shahsafaei A, Freeman GJ. 2006. Programmed death-1 (PD-1) is a marker of germinal center-associated T cells and angioimmunoblastic T-cell lymphoma. Am. J. Surg. Pathol 30:802–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chemnitz JM, Eggle D, Driesen J, Classen S, Riley JL, et al. 2007. RNA-fingerprints provide direct evidence for the inhibitory role of TGFβ and PD-1 on CD4+ T cells in Hodgkin’s lymphoma. Blood 110:3226–33 [DOI] [PubMed] [Google Scholar]
- 135.Shimauchi T, Kabashima K, Nakashima D, Sugita K, Yamada Y, et al. 2007. Augmented expression of programmed death-1 in both neoplastic and non-neoplastic CD4+ T-cells in adult T-cell leukemia/lymphoma. Int. J. Cancer 121:2585–90 [DOI] [PubMed] [Google Scholar]
- 136.Hirano F, Kaneko K, Tamura H, Dong H, Wang S, et al. 2005. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 65:1089–96 [PubMed] [Google Scholar]
- 137.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. 2002. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 99:12293–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Iwai Y, Terawaki S, Honjo T. 2005. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int. Immunol 17:133–44 [DOI] [PubMed] [Google Scholar]
- 139.Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, et al. 2003. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat. Med 9:562–67 [DOI] [PubMed] [Google Scholar]
- 140.Sharma MD, Baban B, Chandler P, Hou DY, Singh N, et al. 2007. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J. Clin. Invest 117:2570–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wong RM, Scotland RR, Lau RL, Wang C, Korman AJ, et al. 2007. Programmed death-1 blockade enhances expansion and functional capacity of human melanoma antigen-specific CTLs. Int. Immunol 19:1223–34 [DOI] [PubMed] [Google Scholar]
- 142.Webster WS, Thompson RH, Harris KJ, Frigola X, Kuntz S, et al. 2007. Targeting molecular and cellular inhibitory mechanisms for improvement of antitumor memory responses reactivated by tumor cell vaccine. J. Immunol 179:2860–69 [DOI] [PubMed] [Google Scholar]
- 143.Rodig N, Ryan T, Allen JA, Pang H, Grabie N, et al. 2003. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur. J. Immunol 33:3117–26 [DOI] [PubMed] [Google Scholar]
- 144.Gotsman I, Grabie N, Dacosta R, Sukhova G, Sharpe A, Lichtman AH. 2007. Proatherogenic immune responses are regulated by the PD-1/PD-L pathway in mice. J. Clin. Invest 117:2974–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mueller SN, Matloubian M, Clemens DM, Sharpe AH, Freeman GJ, et al. 2007. Viral targeting of fibroblastic reticular cells contributes to immunosuppression and persistence during chronic infection. Proc. Natl. Acad. Sci. USA 104:15430–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Iwai Y, Okazaki T, Nishimura H, Kawasaki A, Yagita H, Honjo T. 2002. Microanatomical localization of PD-1 in human tonsils. Immunol. Lett 83:215–20 [DOI] [PubMed] [Google Scholar]
- 147.Tsuda M, Matsumoto K, Inoue H, Matsumura M, Nakano T, et al. 2005. Expression of B7-H1 and B7-DC on the airway epithelium is enhanced by double-stranded RNA. Biochem. Biophys. Res. Commun 330:263–70 [DOI] [PubMed] [Google Scholar]
- 148.Mataki N, Kikuchi K, Kawai T, Higashiyama M, Okada Y, et al. 2007. Expression of PD-1, PD-L1, and PD-L2 in the liver in autoimmune liver diseases. Am. J. Gastroenterol 102:302–12 [DOI] [PubMed] [Google Scholar]
- 149.Mazanet MM, Hughes CC. 2002. B7-H1 is expressed by human endothelial cells and suppresses T cell cytokine synthesis. J. Immunol 169:3581–88 [DOI] [PubMed] [Google Scholar]
- 150.Nakae S, Suto H, Iikura M, Kakurai M, Sedgwick JD, et al. 2006. Mast cells enhance T cell activation: importance of mast cell costimulatory molecules and secreted TNF. J. Immunol 176:2238–48 [DOI] [PubMed] [Google Scholar]
- 151.Augello A, Tasso R, Negrini SM, Amateis A, Indiveri F, et al. 2005. Bone marrow mesenchymal progenitor cells inhibit lymphocyte proliferation by activation of the programmed death 1 pathway. Eur. J. Immunol 35:1482–90 [DOI] [PubMed] [Google Scholar]
- 152.English K, Barry FP, Field-Corbett CP, Mahon BP. 2007. IFN-γ and TNF-α differentially regulate immunomodulation by murine mesenchymal stem cells. Immunol. Lett 110:91–100 [DOI] [PubMed] [Google Scholar]
- 153.Genomics Institute of the Novartis Research Foundation. Online database. GNF SymAtlas v1.2.4. http://expression.gnf.org