

Abstract
Surgery is the mainstay of treatment for meningioma, the most common primary intracranial tumor, but improvements in meningioma risk stratification are needed and indications for postoperative radiotherapy are controversial. Here we develop a targeted gene expression biomarker that predicts meningioma outcomes and radiotherapy responses. Using a discovery cohort of 173 meningiomas, we developed a 34-gene expression risk score and performed clinical and analytical validation of this biomarker on independent meningiomas from 12 institutions across 3 continents (N=1856), including 103 meningiomas from a prospective clinical trial. The gene expression biomarker improved discrimination of outcomes compared to all other systems tested (N=9) in the clinical validation cohort for local recurrence (5-year area under the curve [AUC] 0.81) and overall survival (5-year AUC 0.80). The increase in area under the curve compared to the standard of care, World Health Organization 2021 grade, was 0.11 for local recurrence (95% confidence interval [CI] 0.07–0.17, P<0.001). The gene expression biomarker identified meningiomas benefiting from postoperative radiotherapy (hazard ratio 0.54, 95% CI 0.37–0.78, P=0.0001) and suggested postoperative management could be refined for 29.8% of patients. In sum, our results identify a targeted gene expression biomarker that improves discrimination of meningioma outcomes, including prediction of postoperative radiotherapy responses.
Introduction
Meningiomas comprise 39.7% of primary intracranial tumors and are the only brain tumors that are more common in women, Black, and elderly patients, who are underrepresented in brain tumor clinical trials1,2. Meningioma treatments are largely restricted to surgery and radiotherapy, and systemic therapies remain ineffective or experimental3,4. Historically, the World Health Organization (WHO) has graded meningiomas according to histological features such as mitotic count5. Most WHO grade 1 meningiomas can be effectively treated with surgery or radiotherapy, but many WHO grade 2 or grade 3 meningiomas are resistant to treatment and cause significant neurological morbidity and mortality3. Moreover, some WHO grade 1 meningiomas develop recurrences that cannot be predicted from histological features, and some WHO grade 2 or grade 3 meningiomas are unexpectedly well controlled with surgery and radiotherapy. In recognition of the controversies surrounding meningioma risk stratification and treatment, the NRG BN-003 and EORTC 1308 Phase III clinical trials randomize patients with primary WHO grade 2 meningiomas to postoperative surveillance or postoperative radiotherapy after gross total resection6. The only multicenter prospective studies of meningioma radiotherapy that have reported data are RTOG 0539 and EORTC 22042, and these Phase II clinical trials provide safety and non-randomized data based on clinical criteria that do not predict radiotherapy responses in most retrospective series7–10. Thus, there are unmet needs for improved risk stratification and prediction of postoperative radiotherapy responses for patients with meningiomas.
In 2021, the WHO revised meningioma grading criteria to incorporate rare hotspot TERT promoter mutations and homozygous deletion of CDKN2A/B alongside traditional histological features11. The WHO 2021 update reflects a growing understanding of the molecular landscape of meningiomas from diverse bioinformatic studies. DNA sequencing12–15, copy number variant (CNV) analyses16–18, RNA sequencing19,20, or DNA methylation profiling21–24 have been used to classify meningiomas based on recurring somatic short variants12–15, chromosome gains or losses16–18, differentially expressed genes19,20, or DNA methylation probes23, families24, groups22, or subgroups21. Integrated systems have been proposed based on (1) CNVs, CDKN2A/B status, and histological features (integrated grade)16, (2) CNVs, DNA methylation families, and histological features (integrated score)17, or (3) CNVs, DNA methylation profiling, RNA sequencing, and DNA sequencing which reveal biological groups and subgroups of meningiomas that are concordant with results from DNA methylation profiling or RNA sequencing alone18,21,22. It is unknown which of these diverse classification system(s) may optimize risk stratification or predict postoperative radiotherapy responses for patients with meningiomas.
Knowledge of biological pathways underlying diverse cancers has generated robust targeted gene expression biomarkers that are recommended for risk stratification and prediction of treatment response by the National Comprehensive Cancer Network (NCCN)25–30. A small pilot study suggested that targeted gene expression profiling may be useful for meningioma risk stratification31, but an optimized gene expression biomarker, as well as the analytical validity, clinical validity, generalizability, and potential impact of this approach on postoperative meningioma management were unknown.
Here we use knowledge of biological pathways underlying meningiomas from bioinformatic studies11–18,20–22,31,32 to develop a 34-gene expression biomarker that is prognostic for clinical outcomes in a single-institution discovery cohort. We perform clinical and analytical validation of the gene expression biomarker using independent meningiomas from a large multicenter retrospective cohort, and compare biomarker performance across contemporary meningioma classification systems and clinical contexts using a total of 4898 bioinformatic assays (Fig. 1a, b). We provide investigator-blinded, independent validation of the gene expression biomarker using a multicenter prospective cohort of meningiomas from patients enrolled on RTOG 0539. In sum, our results reveal the gene expression biomarker provides additional information for meningioma outcomes compared to recent classification systems, including prediction of postoperative radiotherapy responses.
Figure 1. Study design and gene expression biomarker characteristics.

a, Study design and numbers of meningiomas used for gene expression biomarker development, analytical validation (Extended Data Fig. 4 and Extended Data Table 4), clinical validation, and comparison across classification systems. See Supplementary Methods, Table 1, and Extended Data Table 1–9 for additional details. Numbers in black text denote intersecting (e.g. 173, 332) or non-intersecting (e.g. 714, 534, 103) meningiomas used for each analysis. Color-matched numbers in adjacent text annotations denote the total number of meningiomas (whether intersecting or non-intersecting) that were used for each analysis. Superscript numbers correspond to manuscripts reporting comparator meningioma classification systems in the References. b, Upset plot of 4898 bioinformatic assays (horizontal) performed across 1856 unique meningiomas (vertical) to define and compare molecular classification systems in this study. c and d, Distribution of continuous (c) or discrete (d) gene expression risk scores in principal component space. Dots represents individual meningiomas from the training, multicenter retrospective, and prospective clinical validation cohorts (N=1142). e, Loading scores for the 34 genes comprising the gene expression biomarker. A simplified color scheme shows genes associated with higher risk in red and genes associated with lower risk in blue in the first 2 principal components. Further information regarding the biological significance of these genes can be found in the Supplementary Methods and Supplement Table 3.
Results
Gene expression biomarker development
Targeted gene expression profiling of 173 meningiomas in the discovery cohort (Table 1 and Extended Data Table 1, 2) resulted in a 34-gene expression biomarker and continuous risk score between 0 and 1 that was converted into discrete low, intermediate, and high risk groups for Kaplan Meier analyses (Fig. 1c–e, Extended Data Fig. 1, and Extended Data Table 3). The gene expression biomarker was well distributed across intracranial meningioma locations and recurring somatic short variants, and was prognostic for local freedom from recurrence (LFFR) and overall survival (OS) (Extended Data Fig. 2, 3). The gene expression biomarker model, risk score, and cutoffs were locked and applied without alteration to multicenter retrospective and prospective validation cohorts from 12 institutions (Table 1 and Extended Data Table 4–15).
Table 1.
Discovery and clinical validation cohort characteristics.
| Discovery | Retrospective clinical validation | Prospective clinical validation | |
|---|---|---|---|
| Meningiomas - no. | 173 | 866 | 103 |
| Patients - no. | 166 | 801 | 103 |
| Females - no. (%) | 112 (67.5) | 543 (68.7) | 68 (66.0) |
| Median age (IQR) - yr. | 57.0 (45–65.1) | 58.9 (48.6–67.6) | 57 (49–65) |
| Setting - no. (%) | |||
| Primary | 143 (82.7) | 635 (80.1) | 81 (78.6) |
| Recurrent | 30 (17.3) | 153 (19.4) | 22 (21.4) |
| Not available | 0 (0.0) | 78 (9.0) | 0 (0.0) |
| Extent of resection - no. (%) | |||
| Gross total | 110 (63.6) | 541 (69.8) | 70 (68.0) |
| Subtotal | 63 (36.4) | 234 (30.2) | 17 (16.5) |
| Not available | 0 (0.0) | 91 (10.5) | 16 (15.5)* |
| WHO grade - no. (%) ** | |||
| 1 | 83 (50.0) | 499 (57.6) | 51 (49.5) |
| 2 | 65 (37.6) | 240 (27.7) | 37 (35.9) |
| 3 | 25 (14.4) | 127 (14.7) | 15 (14.6) |
| Gene expression risk score - no. (%) | |||
| Low | 63 (36.4) | 2 52 (29.1) | 39 (37.9) |
| Intermediate | 72 (41.6) | 406 (46.9) | 46 (44.7) |
| High | 38 (22.0) | 208 (24.1) | 18 (17.5) |
| Postoperative radiotherapy - no. (%) | 33 (19.1) | 147 (17.3) | 63 (61.1) |
| Median follow up (IQR) - yr. | 8.1 (3.9–11.9) | 5.2 (2.3–8.7) | 8.4 (5.1–9.3) |
| Local recurrence - no. (%) | 61 (35.3) | 253 (29.2) | 29 (28.2)*** |
| Death – no. (%) | 46 (26.6) | 190 (21.9) | 21(20.4) |
The discovery cohort was comprised of frozen meningiomas from a single institution (UCSF) (Supplementary Data Table 2). The non-overlapping retrospective clinical validation cohort was comprised of frozen (N=572) and FFPE meningiomas (N=294) from 6 institutions: consecutive meningiomas from The University of Hong Kong (Supplementary Data Table 5), and non-consecutive meningiomas from Northwestern University (Supplementary Data Table 6), UCSF (Supplementary Data Table 7), Baylor College of Medicine (Supplementary Data Table 8), Heidelberg University and Medical University of Vienna (Supplementary Data Table 9). The non-overlapping prospective clinical validation cohort was comprised of FFPE meningiomas from RTOG 0539 (Supplementary Data Table 13), a completed prospective clinical trial of postoperative radiotherapy or postoperative observation for patients with meningiomas.
Some recurrent meningiomas from patients enrolled on RTOG 0539 received radiotherapy without repeat surgery.
WHO 2016 grade based on histological criteria.
The events from RTOG 0539 were defined as progression or death, and outcomes for this cohort are reported as progression free survival or overall survival.
Gene expression biomarker analytical validation
Analytical validity, including reproducibility over time and across laboratories, paired frozen and FFPE meningioma samples, and different approaches for gene expression quantification was established using the multicenter analytical validation cohort (N=1219 meningiomas, 8 institutions) (Fig. 1a, Extended Data Fig. 4, and Extended Data Table 4). Test-retest conditions, different centers, and paired frozen/FFPE meningiomas generated concordant gene expression risk scores (Extended Data Fig. 4a, b) that were tractable and discriminatory for meningioma outcomes when RNA sequencing or microarray approaches were used to assess the 34-gene signature (Extended Data Fig. 4c–g). With regard to cross platform interoperability, we found DNA methylation profiling was only able to predict the gene expression risk score we report to a moderate degree (Supplementary Methods)
Gene expression biomarker clinical validation
In the multicenter retrospective clinical validation cohort (N=866 meningiomas, 6 institutions, N=572 frozen, N=294 FFPE) (Fig. 1a and Extended Data Fig. 4–9), the gene expression biomarker achieved a concordance index (c-index) of 0.78 for LFFR and 0.78 for OS (Extended Data Fig. 5). The gene expression biomarker delineated clinically meaningful low, intermediate, or high risk groups with 5-year LFFR of 92.2% (95% confidence interval [CI], 88.3–96.2%), 72.6% (95% CI 67.8–77.8%), and 19.4% (95% CI 13.5–28.0%), respectively (Fig. 2a), and remained well-calibrated in meningiomas from individual clinical validation institutions (Extended Data Fig. 5a). The gene expression biomarker was prognostic for LFFR and OS among meningiomas presenting in primary or recurrent settings, after gross total resection (GTR) or subtotal resection (STR), across WHO grades using histological (WHO 2016)5 or histological and molecular criteria (WHO 2021)11, and remained independently prognostic on multivariate analysis incorporating meningioma setting (primary or recurrent), extent of resection, and WHO grade (Fig. 2b, Extended Data Fig. 5b, and Extended Data Table 10, 11). The gene expression biomarker was prognostic for LFFR and OS within strata from other meningioma molecular classification systems based on DNA methylation probes23, groups22, subgroups18,21, or families24, or based on gene expression types19, integrated score17, or integrated grade16 (Extended Data Fig. 5c), and remained independently prognostic on multivariate analyses incorporating each of the 9 other meningioma classification systems (Extended Data Table 10, 11).
Figure 2. Gene expression biomarker discrimination of meningioma outcomes.

a, Kaplan Meier curves for LFFR or OS in the multicenter retrospective clinical validation cohort stratified by gene expression risk score (N=866 meningiomas, 6 institutions, N=854 with LFFR data, N=863 with OS data). P values shown are from a two-sided Log-rank test. Low, intermediate, and high gene expression risk scores were associated with 5-year LFFR of 92.2% (95% CI, 88.3–96.2%), 72.6% (95% CI 67.8–77.8%), and 19.4% (95% CI 13.5–28.0%), and 5-year OS of 95.3% (95% CI 92.9–97.8%), 83.3% (95% CI 79.3–87.5%), and 44.3% (95% CI 35.6–55.1%), respectively. b, Forest plot of hazard ratios (HR) with 95% confidence intervals (CI) for local recurrence or death in the multicenter retrospective clinical validation cohort for each 0.1 increase in gene expression risk score across meningioma settings (primary or recurrent), extent of resection (EOR), WHO grades, or multivariate analysis. N refers to the number of meningiomas included within each strata for which the hazard ratio is reported. For multivariate analysis, the variables shown in parentheses were included as covariates, and N refers to the number of meningiomas included in multivariate analysis. P values are from a two-sided Wald’s test without multiple comparisons adjustment.
Comparison across meningioma classification systems based on molecular18,19,21–24, molecular and histological16,17, or WHO criteria5,11 using pairwise model combinations33 revealed the gene expression biomarker provided additional prognostic information for LFFR and OS in combination with each of the 9 other systems tested (Fig. 3a and Extended Data Fig. 6a). No other meningioma classification system provided additional prognostic information for LFFR in combination with the gene expression biomarker (Fig. 3a and Extended Data Fig. 6b, c), and only WHO 2021 grade provided additional prognostic information for OS (Fig. 3a). The gene expression biomarker achieved the lowest Brier error score over time for LFFR across meningioma classification systems and had an error score that was comparable to WHO 2021 grade and integrated grade over time for OS (Fig. 3b). The gene expression biomarker achieved the highest 5-year area under the curve (AUC) for LFFR (0.81) and OS (0.80) across meningioma classification systems, with a delta-AUC for LFFR of +0.07 (95% CI 0.02–0.12, P<0.001) compared to the next best performing system (integrated grade), and a delta-AUC for LFFR of +0.11 (95% CI 0.07–0.17, P<0.001) compared to the current standard of care (WHO 2021 grade) (Fig. 3c). To translate these findings into clinical practice, nomograms were generated for prediction of 5-year LFFR or OS based on meningioma gene expression risk score, setting (primary or recurrent), extent of resection, and WHO grade (Fig. 4 and Extended Data Fig. 7).
Figure 3. Gene expression biomarker comparisons to other meningioma classification systems.
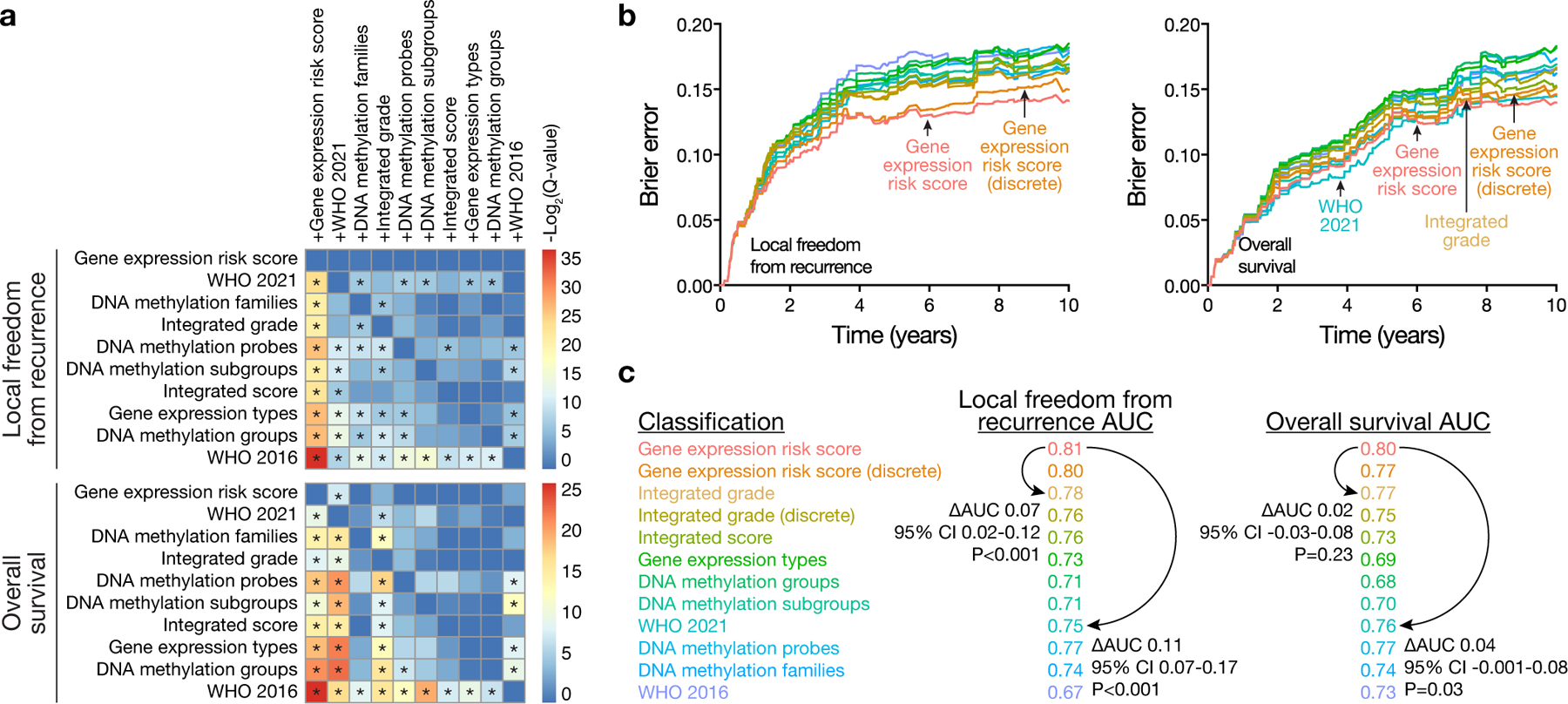
a, Heatmap of −Log2-transformed two-sided P-values with false-discovery-rate (FDR) correction (Q-values) for pairwise likelihood-ratio tests33 of improvements in Cox regression models for LFFR or OS. Meningioma classification systems in columns (e.g. +Gene expression risk score) were combined with meningioma classification systems in rows. The performance of combined models was assessed using 290 consecutive meningiomas from The University of Hong Kong validation cohort with available data to define all 10 meningioma classification systems tested. Asterixis denote Benjamini-Hochberg corrected Q<0.01. Combination with the gene expression risk score improved all other models tested for both LFFR and OS (first column). Conversely, no models improved the gene expression risk score for LFFR (first row, top heatmap), and only WHO 2021 grade provided improvement for OS (first row, bottom heatmap). These findings were additionally validated using multivariate analyses (Supplement Data Table 10, 11) and Kaplan Meier analyses (Extended Data Fig. 5, 6). b, Brier error curves over time for LFFR or OS in the same retrospective validation cohort as in a. The gene expression biomarker achieved the lowest Brier error score over time for LFFR across meningioma classification systems and had an error score that was comparable to WHO 2021 grade and integrated grade over time for OS. c, 5-year time dependent area under the receiver operating characteristic (AUC) for all meningioma classification systems tested. AUC values reflect the performance of each system in all multicenter retrospective clinical validation cohort meningiomas (N=866 meningiomas, 6 institutions) with available data to define each system tested (Supplement Data Table 4, 10, 11). Pairwise comparisons were performed for select systems using bootstrap delta-AUC. P values shown are 1-sided bootstrap P values. The gene expression biomarker achieved the highest 5-year AUC for LFFR and OS across meningioma classification systems, with a delta-AUC for LFFR of +0.07 (95% CI 0.02–0.12, P<0.001) compared to the next best performing system (integrated grade), and a delta-AUC of +0.11 for LFFR (95% CI 0.07–0.17, P<0.001) and +0.04 for OS (95% CI −0.001–0.08, P=0.03) compared to the current standard of care (WHO 2021 grade). As was the case for AUC calculations, the number of meningiomas included in each delta-AUC comparison varied depending on the number of meningiomas in the multicenter retrospective clinical validation cohort with available data to define the systems tested in each comparison (Supplement Data Table 4, 10, 11). Unless specified (as in a.), P values are not corrected for multiple comparisons.
Figure 4. Gene expression biomarker nomograms for meningioma outcomes.

a, Nomograms are shown for prediction of 5-year LFFR (left) or OS (right) based on gene expression risk score, setting (primary or recurrent), extent of resection, and WHO 2021 grade using data from the multicenter retrospective clinical validation cohort. Similar nomograms based on WHO 2016 grade are available in Extended Data Fig. 7. To use the nomograms, use a straight-edge to draw a vertical line between the variable of interest and the points scale at the top of the nomogram to determine the contribution in points to the total score for each variable. Add up the points from each variable, and then draw a vertical line from the total points scale at the bottom of the nomogram to the 5-year outcome scale to determine the estimated outcome.
Biomarker prediction of radiotherapy responses
To incorporate the gene expression biomarker into a clinical framework consistent with contemporary NCCN and European Association of Neuro-Oncology (EANO) guidelines4,34, meningiomas treated with surgical monotherapy in the multicenter retrospective clinical validation cohort were stratified by extent of resection and gene expression risk score, resulting in a range of clinical subgroups spanning the spectrum of recurrence risk from 5-year LFFR of 96.1% for gene expression low risk meningiomas with GTR, to 9.8% for gene expression high risk meningiomas with STR (Fig. 5a). Based on these combined biomarker/surgical strata, favorable and unfavorable meningiomas were distinguished using (1) gene expression low risk with any resection, or gene expression intermediate risk with GTR (favorable), versus (2) gene expression intermediate risk with STR, or gene expression high risk with any resection (unfavorable) (Fig. 5a).
Figure 5. Gene expression biomarker prediction of meningioma radiotherapy responses and prognostic validation in samples from a prospective clinical trial.

a, Kaplan Meier curves for LFFR for meningiomas in the multicenter retrospective clinical validation cohort that were treated with surgical monotherapy, stratified by extent of resection and the gene expression risk score. 5-year LFFR was 96.1% for gene expression low risk meningiomas with GTR, 80.3% for gene expression low risk with STR, 80.5% for gene expression intermediate risk with GTR, 54.9% for gene expression intermediate risk with STR, 30.0% for gene expression high risk with GTR, and 9.8% for gene expression high risk with STR. Meningiomas were grouped as favorable (N=442) or unfavorable (N=210) as shown if they had >80% or <80% 5-year LFFR, respectively, for subsequent analyses. P value shown is from a two-sided Log-rank test. b, Kaplan Meier curves for LFFR of favorable versus unfavorable primary WHO grade 2 meningiomas in the multicenter retrospective clinical validation cohort that received postoperative radiotherapy (RT) or underwent postoperative observation (Obs). Unfavorable primary WHO grade 2 meningiomas benefitted from postoperative radiotherapy (HR 0.33, 95% CI 0.14–0.76, two-sided Log-rank P=0.009), while favorable primary WHO grade 2 meningiomas did not (two-sided Log-rank P=0.88). c, Kaplan Meier curves for LFFR of favorable versus unfavorable propensity matched meningiomas in the multicenter retrospective clinical validation cohort that received postoperative radiotherapy or underwent postoperative observation. P values shown are from two-sided Log-rank tests. Cases were first stratified by favorable versus unfavorable criteria, and then matched based on gene expression risk score, extent of resection, and WHO grade (Supplement Data Table 12). Unfavorable propensity matched meningiomas benefitted from postoperative radiotherapy (HR 0.54, 95% CI 0.37–0.78, P=0.0001), while favorable propensity matched meningiomas did not (P=0.42). d, Sankey plot of RTOG 0539 clinical risk groups versus gene expression biomarker risk groups in the multicenter retrospective clinical validation cohort (left) or the multicenter prospective clinical validation cohort from RTOG 0539 itself (right). Compared to clinical risk groups used for postoperative radiotherapy stratification in RTOG 0539, the gene expression biomarker re-classified 52.0% (N=416, Supplement Data Table 15) of retrospective validation cohort meningiomas, and 39.8% (N=41, Supplement Data Table 15) of RTOG 0539 meningiomas. Reclassified meningiomas were better stratified by gene expression risk (Fig. 8d). e, Kaplan Meier curves for PFS of patients enrolled on RTOG 0539, stratified by meningioma gene expression risk score. P values are from 2-sided Log-rank tests. 5-year PFS was 92.0%, 76.5%, and 38.6% for gene expression low, intermediate, and high risk groups, respectively (c-index 0.73). f. Panel shows Kaplan Meier curves for OS of patients enrolled on RTOG 0539, stratified by meningioma gene expression risk score. 5-year OS was 94.7%, 85.7%, and 63.0% for gene expression low, intermediate, and high risk groups, respectively (c-index 0.73). P value is from a two-sided Log-rank test. Unless specified, P values are not corrected for multiple comparisons.
In clinical practice, meningiomas with unfavorable histological features or STR are often treated with postoperative radiotherapy based on retrospective data4,6,34. NRG BN-003 and EORTC 1308 represent important prospective studies of radiotherapy for meningioma, but these trials were initiated before the development of biomarkers for meningioma risk stratification and do not incorporate biomarkers potentially elucidating postoperative radiotherapy responses, as defined by a reduced risk of recurrence. In the multicenter retrospective clinical validation cohort, the gene expression biomarker remained prognostic for primary meningioma outcomes among patients receiving fractionated postoperative radiotherapy (Extended Data Fig. 8a), and also among patients with primary WHO grade 2 meningiomas with GTR who may have been eligible for NRG BN-003 or EORTC 1308 (Extended Data Fig. 8b). However, in the absence of biomarker stratification, primary WHO grade 2 meningiomas with GTR did not benefit from postoperative radiotherapy in the multicenter retrospective clinical validation cohort (Extended Data Fig. 8c). Thus, to determine if the gene expression biomarker could predict meningioma radiotherapy responses, primary WHO grade 2 meningiomas were stratified based on favorable versus unfavorable biomarker/surgical criteria (Fig. 5a), which revealed that unfavorable primary WHO grade 2 meningiomas benefitted from postoperative radiotherapy (HR 0.33, 95% CI 0.14–0.76, P=0.009) but favorable primary WHO grade 2 meningiomas did not (P=0.88) (Fig. 5b). Applying the same biomarker/surgical strata across all WHO grades in the multicenter retrospective clinical validation cohort with propensity matching based on gene expression risk score, extent of resection, and WHO grade revealed that unfavorable meningiomas benefitted from postoperative radiotherapy (HR 0.54, 95% CI 0.37–0.78, P=0.0001) but favorable meningiomas did not (P=0.42) (Fig. 5c and Extended Data Table 12).
RTOG 0539 was a Phase II multicenter prospective trial that enrolled patients with meningiomas from 78 institutions into 3 clinical risk groups: (1) low clinical risk comprised of primary WHO grade 1 meningiomas after any resection, (2) intermediate clinical risk comprised of recurrent WHO grade 1 meningiomas after any resection, or primary WHO grade 2 meningiomas after GTR, and (3) high clinical risk comprised of WHO grade 3 meningiomas after any resection, recurrent WHO grade 2 meningiomas after any resection, and primary WHO grade 2 meningiomas after STR. Intermediate and high clinical risk patients enrolled on RTOG 0539 received postoperative radiotherapy8,9, and low clinical risk patients underwent postoperative surveillance7. To determine how the gene expression biomarker could potentially refine postoperative management, meningiomas in the multicenter retrospective clinical validation cohort were assigned to RTOG 0539 clinical risk groups and compared across assignments to gene expression biomarker risk groups. The gene expression biomarker improved discrimination of meningioma outcomes across clinical groups used for postoperative radiotherapy stratification in RTOG 0539 (Extended Data Fig. 8d) and re-classified 52.0% (Extended Data Table 15) of meningiomas compared to clinical criteria, including downstaging 21.3% of intermediate clinical risk patients who would have received postoperative radiotherapy on RTOG 0539 (Fig. 5d). Using favorable versus unfavorable biomarker/surgical strata that predict radiotherapy responses (Fig. 5a–c), these data suggest that postoperative management could have been refined for 29.8% of patients in the multicenter retrospective clinical validation cohort compared to clinical criteria from RTOG 0539.
Validation in samples from a prospective clinical trial
Investigator-blinded, independent validation of the gene expression biomarker was performed using meningiomas and clinical data that were prospectively collected from patients enrolled on RTOG 0539 itself (N=103) (Table 1 and Extended Data Table 13). In comparison to clinical risk groups used to allocate patients to postoperative radiotherapy or postoperative surveillance on this study, the gene expression biomarker re-classified 39.8% of meningiomas from RTOG 0539 (Fig. 5d and Extended Data Table 15), including downstaging 30.3% of intermediate clinical risk patients who received postoperative radiotherapy. The gene expression biomarker was prognostic for progression free survival (PFS) and OS in patients from RTOG 0539 (Fig. 5d, e) and was well calibrated with 5-year PFS of 92.0%, 76.5%, and 38.6% for low, intermediate, and high risk groups, respectively. Moreover, the gene expression biomarker remained independently prognostic on multivariate analysis incorporating meningioma setting (primary or recurrent), extent of resection, and WHO grade using data from RTOG 0539 (Extended Data Table 14).
Discussion
Here we use targeted gene expression profiling to develop and validate a polygenic biomarker that provides additional information for meningioma outcomes compared to other classification systems, including prediction of postoperative radiotherapy responses. The gene expression biomarker we report is independently prognostic across all clinical, histological, and molecular contexts tested5,16,17,19,21–24, including WHO 2021 grade11, the current standard of care. When incorporated into clinical risk groups defined by contemporary trials7–9 that are consistent with consensus NCCN and EANO guidelines4,34, the gene expression biomarker potentially refines postoperative management for 29.8% of patients.
DNA methylation profiling21–24,35, CNV analysis16–18, DNA sequencing12–15, and RNA sequencing18–20,22 have improved understanding of meningioma biology. Unsupervised bioinformatic analyses paired with mechanistic and functional approaches have identified molecular groups and subgroups of meningiomas with distinct biologic drivers, therapeutic vulnerabilities, and clinical outcomes18,19,21,22. Supervised bioinformatic models incorporating clinical endpoints have refined risk stratification for meningioma local recurrence16,17,24,35. The gene expression biomarker reported here provides additional prognostic information for local recurrence and overall survival when combined with all unsupervised or supervised meningioma molecular classification systems tested. These findings are concordant with pan-cancer analyses examining gene expression, CNV, DNA methylation, protein expression, and DNA sequencing data in 10,884 patients, which suggest gene expression encodes the greatest prognostic information across cancer types28. Efforts to reduce molecular classification of meningioma to one or several immunohistochemical stains have thus far not been reproducible36, and such qualitative or semi-quantitative protein expression biomarkers may not fully capture the quantitative signal of a gene expression-based continuous risk score. Inspection of the genes comprising the biomarker reported here across the cellular architecture of meningiomas from single-cell RNA sequencing22 reveals that some biomarker genes are enriched in meningioma cells, while other biomarker genes are expressed by the tumor microenvironment (Extended Data Fig. 9). Moreover, the meningioma genes of interest from the literature that we interrogated using targeted gene expression profiling display a range of pairwise correlations in the samples from this study (Extended Data Table 16), and the functional interactions among these genes (which include a long non-coding RNA) or among the cell types in meningiomas, or the relationship(s) of these genes to meningioma histological variants11, are incompletely understood. Genes included in the biomarker (Extended Data Table S3) comprised genes involved in cell cycle and epigenetic regulation (CDK6, CDKN2A, CDC20, CKS2, CHEK1, EZH2, MYBL1) or mitotic stability (KIF20A), genes located on recurrent CNVs (LINC02593 on chromosome 1p, TMEM30B on 14q, USF1 on 1q, ARID1B on 6q), immune related genes (CCL21, CD3E), or previously identified prognostic markers in meningioma such as PGR, IGF2, COL1A1.
Current indications for postoperative radiotherapy for patients with meningiomas are controversial, particularly for patients with primary WHO grade 2 meningiomas who are randomized to postoperative surveillance or postoperative radiotherapy on NRG BN-003 and EORTC 1308 after GTR3,6. Conflicting retrospective series have variably reported a benefit9,37–44 or no benefit from radiotherapy in this setting45–53, which has fueled debate and inspired these international Phase III clinical trials of radiotherapy for patients with meningiomas. The gene expression biomarker reported here improves risk stratification for primary WHO grade 2 meningiomas and may identify favorable WHO grade 2 meningiomas where postoperative radiotherapy could be safely omitted in favor of close surveillance. The gene expression biomarker also identifies primary WHO grade 1 meningiomas with elevated risk of recurrence (Extended Data Fig. 8e). Most meningiomas are WHO grade 1 and are often considered benign, but we found that 6.4% of primary WHO grade 1 meningiomas in the multicenter retrospective clinical validation cohort were classified as gene expression high risk (N=27), with 5-year LFFR of 43.0%. Of these, only 1 patient received postoperative radiotherapy (3.7%). The gene expression biomarker also identified 59 primary WHO grade 1 meningiomas (13.9%) with subtotal resection in the multicenter retrospective clinical validation cohort as intermediate risk, and this unfavorable combination was associated with 5-year LFFR of 65.1%. Of these, only 3 patients (5.1%) received postoperative radiotherapy. In sum, 20.3% of primary WHO grade 1 meningiomas in the multicenter retrospective clinical validation cohort (N=86 of 423) were re-classified as unfavorable using biomarker/surgical strata, and the overwhelming majority of these patients did not receive radiotherapy (95.3%) although may have benefitted from postoperative treatment.
Previous meningioma molecular classification studies have largely not reported overall survival outcomes. A prospective trial of trabectedin in 90 patients with recurrent WHO grade 2 or grade 3 meningiomas examined DNA methylation families in multivariate analysis without including WHO grade as a covariate, and found meningiomas in the malignant DNA methylation family had worse overall survival compared to non-malignant families, although all families (including benign and intermediate) experienced poor outcomes54. The data we present using meningiomas from RTOG 0539 demonstrate the gene expression biomarker was prognostic for overall survival both before and after adjusting for WHO grade on multivariate analysis, and that outcomes remained well-calibrated in this prospective, investigator-blinded validation cohort. For patients with meningiomas, prospective trials such as these will be critical to distinguish conventionally higher risk cases that may safely undergo postoperative surveillance (Extended Data Fig. 8f, g), elucidate which biomarker(s) could be used for stratification (Extended Data Fig. 8h, i), and determine whether the timing of postoperative radiotherapy or other interventions improves overall survival (Extended Data Fig. 8j). As clinical trials develop, we do not anticipate targeted gene expression profiling will obviate longstanding and robust meningioma classification systems, such as WHO grade11, or more recent classification systems that are tractable across multiple brain tumor types, such as DNA methylation profiling, which elucidates biological drivers and vulnerabilities to molecular therapy for meningiomas21,22,55. We found DNA methylation profiling was only able to predict the gene expression risk score to a moderate degree (Supplementary Methods). Thus, if incorporated alongside other meningioma classification systems and clinical factors such as extent of resection that are already in widespread use, the gene expression biomarker reported here may offer additional benefit to patients with the most common primary intracranial tumor1, particularly in terms of postoperative radiotherapy response.
This study should be interpreted in the context of its limitations. First, clinical data in the discovery and multicenter validation cohorts were obtained retrospectively, suggesting our results are susceptible to biases inherent to retrospective research. To address this limitation, we provide additional investigator-blinded, independent validation using meningiomas and clinical data that were prospectively collected from patients enrolled on RTOG 0539. Second, pathology and radiology reviews were performed independently at each institution for meningiomas in the retrospective discovery and validation cohorts. Nevertheless, inter-observer concordance for meningioma WHO grade and imaging characteristics are high56–58, and any heterogeneity in clinical review across independent cohorts may better represent the heterogeneity intrinsic to routine clinical practice than might be anticipated from central review. To further address this limitation, the meningiomas from RTOG 0539 that were included in this study underwent central pathology and radiology review7–9,58. To establish the impact of the biomarker we report on routine clinical practice, widely available technology with established assay cutoffs and robust, standardized practices for biomarker calculation will be critical. Moreover, further prospective validation is needed to confirm our findings, potentially with benchmarking against other molecular classification systems for meningiomas such as integrated grade, which in our data was the next best performing system. Prospective clinical trials where patients are stratified to postoperative radiotherapy versus observation based on gene expression risk score may be warranted if NRG BN-003 or EORTC 1308 do not show a benefit to postoperative radiotherapy for patients with WHO grade 2 meningiomas after GTR. More broadly, our discovery of a biomarker that identifies meningiomas benefiting from radiotherapy suggests that biopsy-based gene expression risk scores may be useful for guiding observation versus definitive radiotherapy for incidental meningiomas or other meningiomas that may be managed non-operatively. In contrast to many other tumors, the paradigm of biopsy-based management has not been historically applied to meningiomas, but with identification of meningioma biomarkers that shed light on therapeutic vulnerabilities, longstanding clinical approaches to meningiomas may be revisited.
Methods
Study design
A discovery cohort comprised of 173 retrospective meningiomas with well-annotated clinical follow up data from a single institution was used to identify and optimize a 34-gene expression biomarker and risk score (Fig. 1a, Extended Data Fig. 1, and Extended Data Table 1–3). The performance of the gene expression biomarker was validated in 3 cohorts. First, the analytical validity of the gene expression biomarker was tested in a retrospective analytical validation cohort comprised of 1219 meningiomas from 8 international institutions, some of which had sparse or absent clinical follow up data (Fig. 1a and Extended Data Table 4). Meningiomas from the discovery cohort, which had matched RNA sequencing, were also used for analytical validation of orthogonal approaches for gene expression quantification (Fig. 1a and Extended Data Table 4). Second, the clinical validity and performance of the gene expression biomarker in comparison to other meningioma classification systems were tested in an independent retrospective clinical validation cohort comprised of 866 meningiomas with well-annotated clinical follow up data from 6 international institutions (Fig. 1a, Table 1, and Extended Data Table 5–9), some of which were also used for analytical validation (Extended Data Table 4). There was no overlap among meningiomas used to identify and optimize the gene expression biomarker in the discovery cohort (Extended Data Table 2) and meningiomas used for clinical validation (Extended Data Table 4–9). C-index, log-rank test, Brier error score, time-dependent area under the receiver operant curve (AUC), delta-AUC, the Kaplan Meier method, multivariate analysis (Extended Data Table 10, 11), and propensity matching (Extended Data Table 12) were used to compare gene expression biomarker performance across contemporary molecular and histological classification systems and clinical contexts. Third, a prospectively collected cohort of 103 meningiomas from patients enrolled on RTOG 0539 were used for investigator-blinded, independent clinical validation (Fig. 1a, Table 1, and Extended Data Table 13, 14). In total, 4898 genomic assays were performed and analyzed across 1856 unique meningiomas to define and compare molecular classification systems (Fig. 1b). Details on data collection, tissue and nucleic acid processing, genomic assays, pathology review, imaging review, statistical analyses, and prognostic and predictive validation (Extended Data Fig. 1) are reported in the Supplementary Methods. This study complied with ethical regulations and was approved by the UCSF Institutional Review Board (13-12587, 17-22324, 17-23196 and 18-24633), by The University of Hong Kong (HKU) Institutional Review Board (UW 07-273 and UW 21-112), and by relevant Institutional Review Boards at all included institutions. As part of routine clinical care, all patients who were included in this study signed a waiver of informed consent to contribute deidentified data to research projects.
Targeted gene expression profiling and analysis
Targeted gene expression profiling was performed using a hybridization and barcode-based panel (Nanostring nCounter) with internal negative and spike-in positive controls59 (Supplementary Methods). Positive-control normalized gene counts were standardized by normalization to the geometric mean count of 7 meningioma-specific housekeeping genes (Extended Data Table 3). Log2 transformed gene expression values were used for all subsequent analyses. Meningioma related genes of interest (Extended Data Table 1) were selected based on prognostic or biological significance in the literature11–18,20–22,31,32 (Supplementary Methods), and feature selection was performed using a LASSO regularized Cox regression model with the c-index of LFFR in the discovery cohort as the target endpoint (Extended Data Table 2). An optimized set of 34 genes was identified within 1 standard error of the model achieving maximal c-index (Extended Data Fig. 1a and Extended Data Table 3), resulting in a highly discriminatory set of linearly rescaled risk scores between 0 and 1 (Fig. 1c–e and Extended Data Fig. 2). To further reduce over-fitting and to facilitate re-calibration of the model for data derived from frozen or formalin-fixed and paraffin embedded (FFPE) meningiomas, or for data derived from orthogonal approaches for gene expression quantification such as RNA sequencing, bootstrap aggregation was used to train 500 ridge-regression sub-models using normalized and log2-transformed gene counts as input and discovery cohort risk scores as target variables60.
Gene expression risk score cutoffs were determined using a nested procedure in the discovery cohort and applied without alteration to validation cohorts (Fig. 1d). An initial cutoff was determined in the discovery cohort using the maximally selected rank statistic. The subsets above and below this threshold were again split by maximally selected rank statistic. The lowest risk score group was considered low risk (LFFR cutoff ≤0.3760769, overall survival [OS] cutoff ≤0.4206913), and the highest risk score group was considered high risk (LFFR cutoff >0.5651741, OS cutoff >0.6453035). The intervening risk score groups were combined as intermediate risk (LFFR cutoff (0.3760769, 0.5651741], OS cutoff (0.4206913, 0.6453035]). All model training, calibration, and cutoff determination was performed in the discovery cohort (N=173).
Reproduction of molecular classification systems in validation cohort meningiomas
Assignment of validation cohort meningiomas to DNA methylation groups22 or DNA methylation subgroups21 (WCC, AC, CHGL, HNV, STM, DRR), DNA methylation families24 or integrated score17 (SLNM, FS), or gene expression types19 (JCB, ASH, AH, TK, AJP) was performed independently by investigators who developed each of these classification systems. Integrated grade16 was assigned using CNVs derived from DNA methylation profiles and histological features under supervision of investigators who developed this classification system (SS, WLB). DNA methylation probe risk scores were estimated by training a LASSO regularized Cox regression model with LFFR as the endpoint in the discovery cohort using β-values of 283 unfavorable CpG loci23. The resulting continuous risk score was converted into low, intermediate, and high risk groups using the same nested procedure described for the gene expression risk score above. All meningioma classification system assignments were performed by investigators who were blinded to clinical outcomes and other molecular characteristics of the meningiomas included in this study (Fig. 1a).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1. Prognostic and predictive gene expression biomarker development.

a, C-index for LFFR models based on meningioma Nanostring targeted gene expression profiling plotted against the natural-log of the lambda parameter during algorithm training. Center and error bars shows the mean C-index +/− estimated SEM. Candidate model performance was estimated using 10-fold cross validation in the UCSF discovery cohort (N=173). An optimal gene set (N=34 genes, dotted lines, Extended Data Table 3) was selected within 1 standard error of the model achieving maximal c-index to reduce over-fitting. The number of genes in each model is displayed at the top of the graph. In order to further reduce over-fitting, improve calibration and stability, and facilitate risk score calculations using FFPE meningiomas or gene expression quantification from RNA sequencing or microarrays (Extended Data Fig. 4), bootstrap aggregation60 was used to train 500 ridge-regression sub-models using the normalized and log-transformed gene counts as inputs and discovery cohort risk scores between 0 and 1 as target variables. In brief, this procedure nominates a bootstrap aggregated risk score defined as the arithmetic mean across sub-model risk scores. b, Log2 values for Nanostring counts or RNA sequencing data (transcripts per million, TPM) for the 34 genes comprising the gene expression risk score in the frozen meningiomas from the UCSF discovery cohort (N=173), revealing high concordance with R2=0.81 (two sided F-test P<2.2×10−16). Similarly, a non-regularized Cox model for LFFR using RNA sequencing TPMs for the same 34 genes also achieved excellent performance in the UCSF discovery cohort (N=173 meningiomas, LFFR c-index 0.89 ± 0.02, OS c-index 0.84 ± 0.02), and outperformed 10,000 randomly sampled sets of 34 genes (one-sided bootstrap P<0.0001). c, Limitations to using RNA sequencing for targeted gene expression biomarker discovery, in comparison to using RNA sequencing for targeted gene expression biomarker validation or implementation (as described for b and as shown in Extended Data Fig. 4). The distribution of univariate LFFR Cox model two-sided unadjusted Wald test P-values for all RNA sequencing genes in the UCSF discovery cohort are shown (N=58,830 genes, N=173 meningiomas). A background uniform distribution is evident, with a peak towards lower P-values. Between P=0.0 and 0.2, at least 6904 of 17437 P-values (40%) could be expected to be false positives rather than related to true biological significance. Spike-in experiments and simulations report false discovery rates across bioinformatic methods and experimental conditions for observational whole transcriptomic approaches between 10–75%, depending on the ground-truth prevalence61. d, Log10 coefficient magnitudes (red for positive coefficients, blue for negative coefficients) versus log2 transformed P-values from individual Cox models from the discovery cohort RNA sequencing are shown, demonstrating challenges with feature selection using sparse observations in high dimensional space. Despite the limitations in using RNA sequencing to discover clinical biomarkers, we show RNA sequencing can be used to validate and implement the 34-gene expression biomarker that was developed using Nanostring targeted gene expression profiling (Extended Data Fig. 4).
Extended Data Fig. 2. Gene expression biomarker characteristics in the discovery cohort.
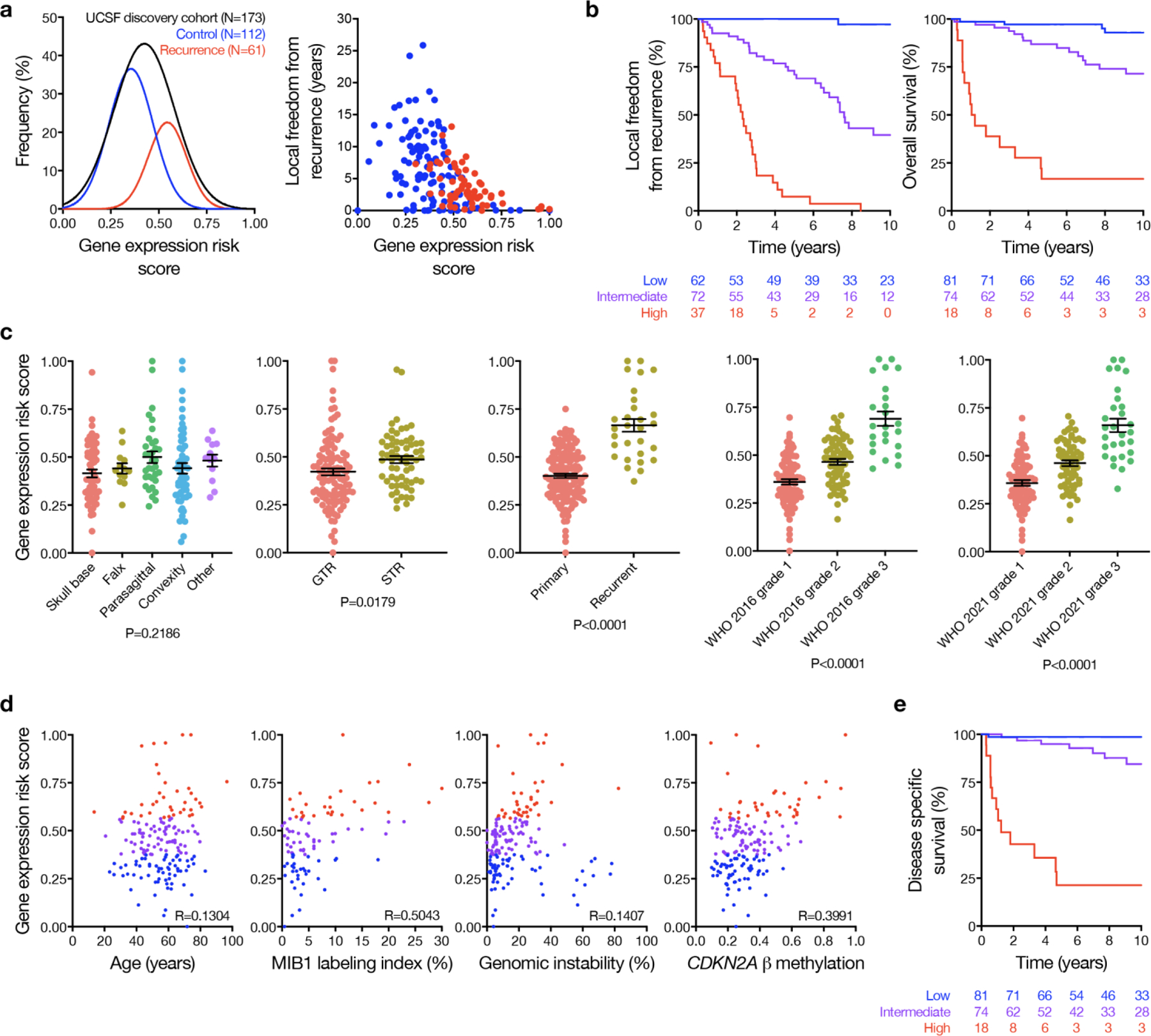
a, Gaussian fits (left) to the distribution of gene expression risk scores in the UCSF discovery cohort, stratified by cases with local recurrence (red) or without local recurrence (blue) on clinical follow up. The gene expression risk score was also plotted against the time to censorship or local recurrence (right), and higher risk score correlated with greater risk of local recurrence and shorter time to recurrence. b, Kaplan Meier curves for LFFR or OS in the UCSF discovery cohort stratified by the gene expression risk score. c, Gene expression risk score distributions stratified by clinical characteristics in the UCSF discovery cohort. Mean +/− standard error measurements are shown for gene expression risk scores stratified by tumor location (skull base, N=60; falx, N=13; parasagittal, N=34; convexity, N=54; other, N=10), extent of resection (GTR, N=110; STR, N=63), setting (primary, N=144; recurrent, N=29), WHO 2016 grade (grade 1, N=83; grade 2, N=66; grade 3, N=24), and WHO 2021 grade (grade 1, N=81; grade 2, N=61; grade 3, N=29). There was no significant difference across meningioma locations (ANOVA, two-sided unadjusted P=0.22), but gene expression risk scores were higher among subtotally resected meningiomas (t-test two-sided P=0.02), recurrent meningiomas (t-test two-sided P<0.0001), and were stratified by WHO 2016 or 2021 grade (P<0.0001). Convexity meningiomas arise adjacent to the cerebral convexity underlying the calvarium, while parasagittal meningiomas abut or involve the parasagittal sinus along the calvarial midline, falx meningiomas involve the falx without extending superiorly to the parasagittal sinus, and skull base meningiomas arise adjacent to the bones of the skull base. d, UCSF discovery cohort gene expression risk score scatter plots across clinical or molecular variables associated with meningioma biology or outcomes (blue, low risk; purple, intermediate risk; red, high risk). There was no clear association between patient age and gene expression risk score, but risk score was loosely correlated with MIB1 labeling index40, genomic instability as defined by the proportion of non-centromeric, non-acrocentric chromosomes affected by copy number gain or loss62, and DNA methylation of the CDKN2A locus21. P values shown are from a two-sided, unadjusted F-test. Thus, the gene expression biomarker correlated with surrogate markers of aggressive meningiomas. e, Disease specific survival among patients in the UCSF discovery cohort stratified by gene expression risk score.
Extended Data Fig. 3. Gene expression biomarker across somatic short variants in the discovery cohort.

Targeted DNA sequencing of recurrent somatic short variants was performed on 171 meningiomas from the UCSF discovery cohort (98.8%). a, Oncoplot distribution of identified pathogenic short somatic variants with variant allele frequency (VAF) of at least 5.0% (N=98 variants, median VAF 38.0%, interquartile range [IQR] 29–43%, median sequencing depth 551.5, IQR 354–856). Consistent with prior reports, variants in NF2 were most common (N=67, 39.2%), followed by TRAF7 (N=10, 5.8%) and AKT1 (N=8, 4.7%). A minority of meningiomas (N=16, 9.4%) were identified without alteration of NF2 or loss of chromosome 22q, but with a characteristic pathogenic variant in one of the following genes: TRAF7, AKT1, PIK3CA, SMARCB1, SMARCE1, SMO, SUFU, KLF4, or POLR2A. The majority of these were WHO 2021 grade 1 meningiomas (N=9, 60.0%), and were associated with favorable histologic characteristics and outcomes (median MIB1 labeling index 2.0%, range 0.5–4.0, 5-year LFFR 90.9%). TERT promoter C228T and C250T hotspot mutations were not identified in the discovery cohort. BAP1 mutations were rare (N=5, 2.9%) and correlated with high histological grade and poor outcomes (N=3 [60.0%] WHO 2016 grade 2 or 3, 5-year LFFR 40.0%). Homozygous CDKN2A/B loss, derived from meningioma DNA CNVs (Supplementary Methods), was identified in 10 meningiomas from the UCSF discovery cohort (5.8%, 80.0% WHO 2016 grade 3, 20.0% WHO 2016 grade 2) and was associated with poor outcomes (5-year LFFR 14.2%). These findings were supported by targeted DNA sequencing of recurrent somatic short variants in 35 consecutive clinical validation cohort meningiomas from The University of Hong Kong using the same approach. b, Same oncoplot from the UCSF discovery cohort as in a, but ordered by VAF instead of gene expression risk score.
Extended Data Fig. 4. Gene expression biomarker characteristics in the analytical validation cohort.

a, Gene expression risk score concordance across multiple conditions and replicates. Test-retest conditions (combined N=44, R=0.94, P<0.0001) were comprised of varying probe batches (N=10, R=0.98, P<0.0001), within probe batch testing of technical replicates (N=12, R=0.98, P<0.0001), and test-retest conditions for meningiomas with serial RNA extraction on the same FFPE block or frozen tumor chunk at least 4 weeks apart (N=22, R=0.94, P<0.001). Gene expression risk scores on paired frozen/FFPE meningiomas also demonstrated high concordance (N=90, R=0.88, P<0.001), and FFPE gene expression risk scores provided excellent discrimination of outcomes across FFPE clinical validation datasets, including a prospective clinical trial (Fig. 2, 3, 5 and Extended Data Fig. 5, 6, 8). b, Principal component analysis on FFPE gene expression risk scores from meningiomas processed at multiple laboratories spanning academic institutions (Northwestern University, San Francisco Veterans Administration) or Clinical Laboratory Improvement Amendments (CLIA) certified private industry (Canopy Biosciences), demonstrating no laboratory batch effects. c, Publicly available microarray and clinical data were used to test the performance of the gene expression risk score on a non-Nanostring platform (N=33 of 34 genes available). No paired microarray/Nanostring data was available to train a calibration model, which precluded direct comparison. Thus, the RNA sequencing calibration model described below (and described in further detail in the Supplementary Methods) was adapted to microarray data as an exploratory analysis, yielding prognostic risk groups as shown in the Kaplan Meier plot (P=0.0014, Log-rank test). d, Concordance of gene expression risk scores derived from RNA sequencing or Nanostring targeted gene expression profiling on the same meningiomas (N=469 meningiomas, R=0.89, F-test two-sided unadjusted P<0.0001). e, Distribution of gene expression risk scores (mean +/− SEM is shown) derived from RNA sequencing of cohorts overlapping (UCSF, The University of Hong Kong, N=502) or non-overlapping (Caris Life Sciences, Heidelberg University, Brigham and Women’s Hospital, University Hospital Magdeburg, Children’s Brain Tumor Network, Baylor College of Medicine, N=640) with the discovery or clinical validation cohorts, comprising 1142 unique meningiomas. Gene expression risk scores remained well distributed across all datasets, including RNA sequencing of pediatric meningiomas (Children’s Brain Tumor Network, N=29), meningiomas with KLF4 or AKT1 somatic short variants63 (University Hospital Magdeburg, N=31), or FFPE (N=428) or frozen (N=718) meningiomas, and demonstrated similar stratification by 2016 WHO histological grade as with Nanostring targeted gene expression profiling analyses (Extended Data Fig. 2c). f, Principal component analysis of gene expression risk scores across RNA sequencing cohorts after correction for batch effects using the COMBAT64 pipeline in the sva package in R. g, LFFR or OS stratified by gene expression risk scores from RNA sequencing of cohorts with available clinical data (UCSF discovery, The University of Hong Kong, and Baylor College of Medicine).
Extended Data Fig. 5. Gene expression biomarker characteristics in the clinical validation cohort.

A, Kaplan Meier curves for LFFR stratified by gene expression risk score (blue lines, low risk; purple lines, intermediate risk; red lines, high risk) for individual clinical validation cohorts, including The University of Hong Kong (Frozen N=339; c-index=0.80; low risk N=122, 5-year LFFR 95.1%; intermediate risk N=151, 5-year LFFR 73.6%; high risk N=66, 5-year LFFR 19.5%), Northwestern University (Frozen and FFPE N=180; c-index=0.74; low risk N=42, 5-year LFFR 90.0%; intermediate risk N=98, 5-year LFFR 76.0%; high risk N=42, 5-year LFFR 21.4%), UCSF WHO grade 2 or grade 3 (FFPE N=158; c-index=0.78; low risk N=24, 5-year LFFR 87.4%; intermediate risk N=69, 5-year LFFR 77.5%; high risk N=65, 5-year LFFR 22.0%), Baylor College of Medicine (Frozen N=116; c-index=0.77; low risk N=35, 5-year LFFR 90.0%; intermediate risk N=61, 5-year LFFR 63.0%; high risk N=20, 5-year LFFR 0.0%), and Heidelberg University plus the Medical University of Vienna (FFPE N=61 with LFFR data; c-index=0.76; low risk N=24, 5-year LFFR 80.4%; intermediate risk N=23, 5-year LFFR 48.1%; high risk N=14, 5-year LFFR 19.3%). The gene expression risk score remained well calibrated across multiple independent clinical validation cohorts comprising both frozen and FFPE meningiomas. When assessed separately within each independent retrospective cohort site, the gene expression risk score remained independently prognostic in multivariate analysis combining the risk score with WHO 2016 grade (P<0.001 in all cases, two-sided unadjusted Wald test P-value). B, Kaplan Meier curves for LFFR in clinical validation cohort meningiomas stratified by gene expression risk score within WHO 2021 grades, demonstrating that the gene expression biomarker remained discriminatory across WHO 2021 grade 1 (low risk N=114, intermediate risk N=127, high risk N=26), WHO 2021 grade 2 (low risk N=7, intermediate risk N=17, high risk N=26), and WHO 2021 grade 3 meningiomas (low risk N=2, intermediate risk N=46, high risk N=98). Shown are two-sided unadjusted Log-rank P-values. C, Forest plots of hazard ratios (HR) with 95% confidence intervals (CI) for local recurrence (left) or death (right) for each 0.1 increase in gene expression risk score are shown (center and error bars denote the hazard ratio and 95% confidence interval). The gene expression biomarker was prognostic across all molecular classification systems tested for both recurrence and survival. HRs according to gene expression risk score across meningioma settings, extent of resection (EOR), and WHO grades from Fig. 2b are re-presented for ease of comparison to HRs in molecular classification systems. P values shown are from two-sided unadjusted Wald’s tests. Overall, in the retrospective clinical validation cohort, LFFR Harrel’s c-index/Uno’s c-index for the gene expression biomarker was 0.78/0.77 (N=854), while LFFR c-index was 0.68/0.66 for WHO 2016 grade (N=854), 0.72/0.71 for WHO 2021 grade (N=462), 0.72/0.73 for integrated score (N=398), 0.73/0.73 for integrated grade (N=460), 0.68/0.69 for DNA methylation groups (N=460), 0.69/0.70 for DNA methylation subgroups (N=460), 0.74/0.73 for DNA methylation probes (N=455), and 0.70/0.71 for gene expression type (N=389). OS Harrel’s c-index/Uno’s c-index for the gene expression biomarker was 0.78/0.78 (N=863), while OS c-index was 0.72/0.72 for WHO 2016 grade (N=863), 0.74/0.73 for WHO 2021 grade (N=463), 0.73/0.72 for integrated score (N=410), 0.75/0.75 for integrated grade (N=460), 0.66/0.66 for DNA methylation groups (N=460), 0.68/0.68 for DNA methylation subgroups (N=460), 0.73/0.74 for DNA methylation probes (N=455), and 0.70/0.67 for gene expression type (N=386).
Extended Data Fig. 6. Molecular classification comparisons in the clinical validation cohort.

Kaplan Meier curves are shown for LFFR (shown are two-sided unadjusted Log-rank P-values) in clinical validation cohort meningiomas stratified by molecular risk groups (blue lines, low risk; purple lines, intermediate risk; red lines, high risk) using the gene expression biomarker in a, or 2 contemporary supervised meningioma classification systems based on combined molecular and clinical features: integrated grade16 based on CNVs and mitoses in b, or integrated score17 based on CNVs, DNA methylation families24, and WHO 2016 grade in c. In a, the gene expression biomarker remained robustly discriminatory across integrated grade or integrated score risk groups, concordant with the independent prognostic value of the gene expression risk score on multivariate analyses (Extended Data Table 10, 11) and within groups from the 6 other molecular and/or histological classification systems tested (Fig. 3a). The converse was examined in b and c, where integrated grade was unable to discriminate outcomes across gene expression risk score groups, and integrated score had limited discriminatory power for intermediate and high gene expression risk score groups and was not discriminatory for low gene expression low risk groups.
Extended Data Fig. 7. Gene expression biomarker nomograms for meningioma outcomes.

A, Nomograms are shown for prediction of 5-year LFFR or OS based on gene expression risk score, extent of resection, setting, and WHO 2016 histologic grade. To use the nomograms, use a straight-edge to draw a vertical line between the variable of interest and the points scale at the top of the nomogram to determine the contribution in points to the total score for each variable. Add up the points from each variable, and then draw a vertical line from the total points scale at the bottom of the nomogram to the 5-year outcome scale to determine the estimated outcome. B, Calibration curves are shown for the models corresponding to the nomograms in Fig. 4a for LFFR (top) and OS (bottom) using the gene expression risk score, extent of resection, primary vs recurrent status, and WHO 2021 grade (and the addition of age for OS). The calibrate function from the rms package in R was used, with B=1000 iterations and N=75 samples per group. Center and error bars denote the predicted 5-year LFFR versus the observed 5-year LFFR calculated via the Kaplan Meier method, with a 95% confidence interval. c, Calibration curves corresponding to the nomograms in a, for LFFR (top) and OS (bottom) using 150 samples per group. Center and error bars denote the predicted 5-year LFFR versus the observed 5-year LFFR calculated via the Kaplan Meier method, with a 95% confidence interval. d, Time dependent AUC is shown for LFFR and OS for the retrospective clinical validation cohort (N=866) as a function of time.
Extended Data Fig. 8. Molecular classification systems and response to treatment.
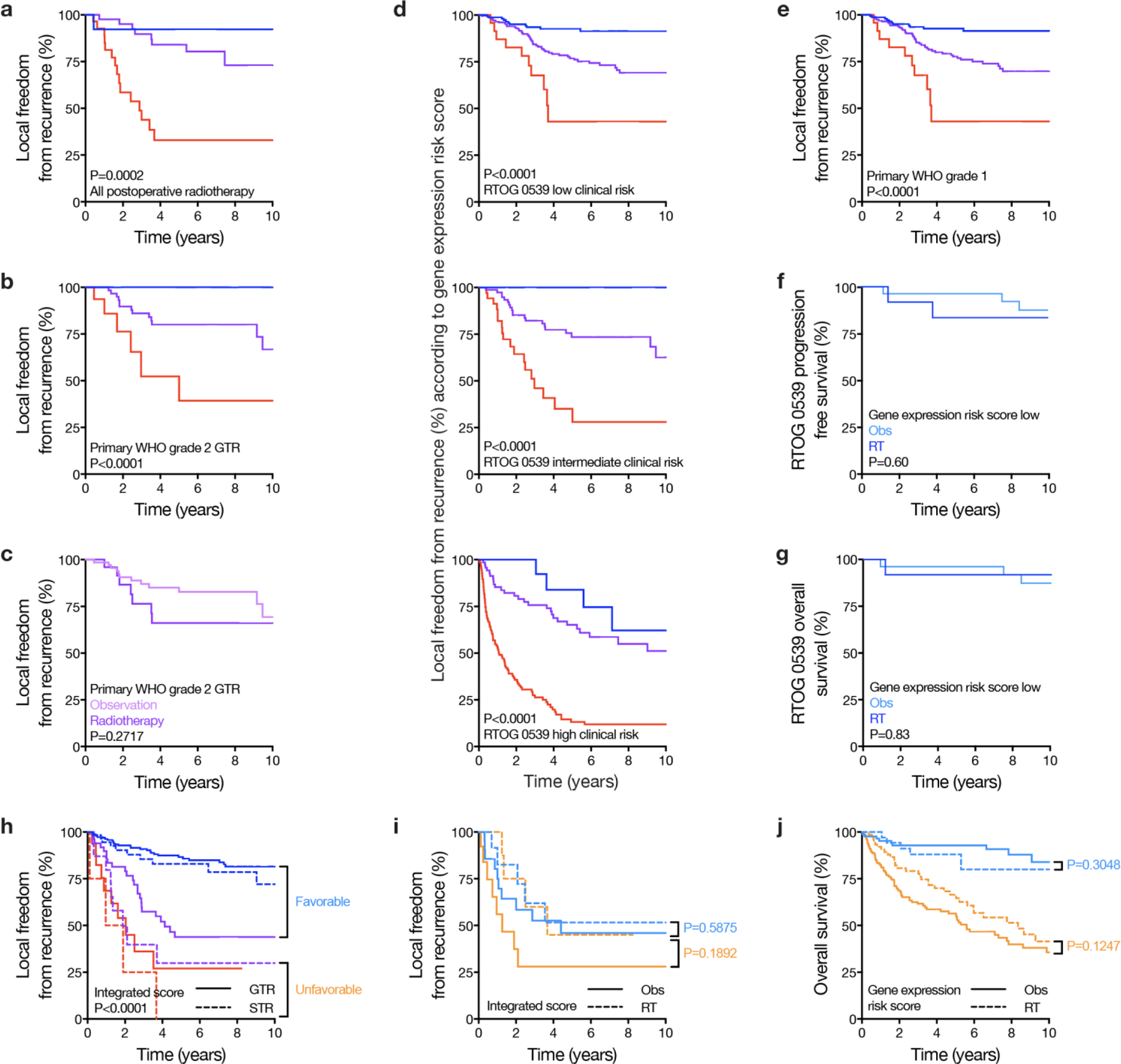
Kaplan Meier curves are shown for LFFR or OS (shown are two-sided unadjusted Log-rank P-values) in retrospective clinical validation cohort meningiomas or prospective RTOG 0539 meningiomas, stratified by gene expression risk score (blue lines, low risk; purple, intermediate risk lines; red lines, high risk), extent of resection, postoperative observation (Obs), or postoperative radiotherapy (RT). a, Primary retrospective clinical validation cohort meningiomas receiving postoperative external beam radiotherapy (N=89) stratified by gene expression risk score (low risk N=14, intermediate risk N=45, high risk N=30), revealing the gene expression biomarker remained prognostic among patients receiving postoperative radiotherapy. b, Primary WHO 2016 grade 2 meningiomas with GTR from the retrospective clinical validation cohort stratified by gene expression risk score (N=21 low risk, N=63 intermediate risk, N=18 high risk). The gene expression risk score remained prognostic among gross totally resected primary WHO grade 2 meningiomas (N=102, HR for local recurrence of 1.75 per 0.1 increase, 95% CI 1.18–2.59, P=0.0057). c, Primary WHO grade 2 meningiomas with GTR from the retrospective clinical validation cohort stratified by postoperative radiotherapy (N=28) or observation (N=74). Patients with meningiomas meeting these criteria were eligible for 2 Phase III randomized multi-institutional trials (NRG BN003 and ROAM-EORTC 1308) examining clinical outcomes with postoperative radiotherapy versus observation. Postoperative radiotherapy did not offer a benefit to patients with meningiomas meeting these criteria in the retrospective clinical validation cohort. d, Retrospective clinical validation cohort meningiomas stratified by gene expression risk score across RTOG 0539 clinical risk groups (low clinical risk, primary WHO grade 1 meningiomas; intermediate clinical risk, recurrent WHO grade 1 meningiomas or primary WHO grade 2 meningiomas status post GTR; high clinical risk, recurrent or STR WHO grade 2 meningiomas or WHO grade 3 meningiomas after any resection). The gene expression biomarker remained prognostic across RTOG 0539 low clinical risk (gene expression risk score low risk N=173, intermediate risk N=224, high risk N=27), RTOG 0539 intermediate clinical risk (gene expression risk score low risk N=32, intermediate risk N=80, high risk N=38), and RTOG 0539 high clinical risk groups (gene expression risk score low risk N=16, intermediate risk N=75, high risk N=128). e, Primary WHO grade 1 meningiomas from the retrospective clinical validation cohort (equivalent to RTOG 0539 low clinical risk meningiomas) stratified by gene expression risk score (N=173 low risk, N=224 intermediate risk, N=27 high risk, 5-year LFFR 92.7%, 77.3%, and 43.0% for low, intermediate, or high risk meningiomas, respectively). f and g, Prospective validation cohort meningiomas from RTOG 0539 identified as low risk by the gene expression biomarker stratified by postoperative radiotherapy (N=12 WHO 2016 grade 2 or 3 or recurrent WHO 2016 grade 1 meningiomas) or observation (N=27 primary WHO 2016 grade 1 meningiomas). These analyses showed favorable outcomes for prospectively collected meningiomas with low gene expression risk scores across clinical risk strata, consistent with findings from retrospective clinical validation cohort meningiomas. More broadly, these data support the hypothesis that the gene expression biomarker may be useful for identifying meningiomas where postoperative radiotherapy could be safely omitted, even in the setting of conventionally high risk clinical features. h, Meningiomas treated with surgical monotherapy from the retrospective clinical validation cohort stratified by integrated score17 (the only contemporary molecular classification system potentially providing additional prognostic information for LFFR within gene expression biomarker strata, Extended Data Fig. 6c) and extent of resection. Favorable (light blue) and unfavorable (yellow) groups were identified using the same criteria for identification of biomarker/surgical strata (Fig. 5a). I, Favorable and unfavorable strata based on integrated score were unable to identify meningiomas benefitting from postoperative radiotherapy even after propensity matching on integrated score, extent of resection, and WHO 2016 grade. j, OS in the same meningiomas as Fig. 5c (i.e. propensity matched favorable and unfavorable retrospective clinical validation cohort meningiomas based on biomarker/surgical strata), demonstrating a trend towards benefit with postoperative radiotherapy for unfavorable meningiomas.
Extended Data Fig. 9. Biomarker risk score gene distribution in tumor versus microenvironment cell types from meningioma single-cell RNA sequencing.

a, Single-cell RNA sequencing uniform manifold approximation and projection (UMAP) of 57,114 transcriptomes from 8 human meningioma samples and 2 human dura samples shaded by cell clusters that were defined using cell signature gene sets, cell cycle analysis, and differentially expressed cluster marker genes, as previously reported22. Image is reproduced with permission. b, Feature plots showing normalized biomarker risk score gene expression across reduced dimensionality clusters of meningioma and tumor microenvironment cells. 33 of 34 biomarker genes were available for analysis in meningioma single-cell RNA sequencing data from a, although several were sparsely captured in single-cell RNA data (a known limitation that can be overcome using bulk RNA sequencing or Nanostring hybridization targeted gene expression profiling).
Supplementary Material
Acknowledgements
The authors thank John de Groot, Shawn Hervey-Jumper, Catherine Park, Susan Chang, and Nicole Willmarth for providing comments, and Fei Jiang for providing biostatistical feedback during the conception of this study. This study was supported by NIH grants P50 CA097257 (WCC, DRR, MSB), P50 CA221747 (KM, JPC, CMH, STM), U01 CA180868 and U10 CA180822 (MYCP, MW, SLP, MPM), R01 CA262311 (DRR), F32 CA213944 (STM), F30 CA246808 and T32 GM007618 (AC), a UCSF Catalyst Program Award (WCC, DRR), the Northwestern Medicine Malnati Brain Tumor Institute of the Lurie Cancer Center (KM, JPC, CMH, STM), the UCSF Wolfe Meningioma Program Project (DRR, MWM), and the Trenchard Family Charitable Fund (DRR). This work was also supported by the Northwestern University NUSeq Core Facility and a Conquer Cancer Herman H. Freckman, MD, Endowed Young Investigator Award from the American Society of Clinical Oncology (WCC), and by the Helen Diller Family Cancer Center Physician Scientist Program in Clinical Oncology program (WCC, 5K12 CA260225-03). WCC is a Chan Zuckerberg Biohub – San Francisco Physician-Scientist Fellow. Any opinions, findings, and conclusions expressed in this material are those of the author(s) and do not necessarily reflect those of Conquer Cancer or the American Society of Clinical Oncology.
Footnotes
Competing interests statement
MP has received honoraria for lectures, consultation, or advisory board participation from the following for-profit companies: Bayer, Bristol-Myers Squibb, Novartis, Gerson Lehrman Group (GLG), CMC Contrast, GlaxoSmithKline, Mundipharma, Roche, BMJ Journals, MedMedia, Astra Zeneca, AbbVie, Lilly, Medahead, Daiichi Sankyo, Sanofi, Merck Sharp & Dome, Tocagen, Adastra, Gan & Lee Pharmaceuticals. WCC and DRR are the inventors on patent PCT/US 21/70288 describing the use of targeted gene expression profiling to predict meningioma outcomes and radiotherapy responses. The remaining authors declare no competing interests.
Data and code availability
Raw data from targeted gene expression panels are deposited in the NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE222054. Raw DNA methylation data from the UCSF WHO grade 2 or grade 3 validation cohort and the RTOG 0539 validation cohort are available under accession number GSE221029. Raw amplicon and targeted exome sequencing data from discovery and validation cohort meningiomas are deposited in the NCBI Sequencing Reads Archive (https://www.ncbi.nlm.nih.gov/sra) under project numbers PRJNA916225 and PRJNA916253. Matrices containing TPM data from RNA sequencing cohorts used for analytical validation are deposited along with code on github (https://github.com/william-c-chen/Meningioma_GE_Biomarker). Accession numbers and publications containing previously reported data are available in Extended Data Table 4. The publicly available GRCh38 (hg38), CRCh37.p13 (hg19), and Kallisto index v10 datasets were used in this study. Source data are provided with this paper.
References
- 1.Ostrom QT, Price M, Neff C, et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2015–2019. Neuro Oncol 2022;24(Suppl 5):v1–v95. doi: 10.1093/neuonc/noac202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reihl SJ, Patil N, Morshed RA, et al. A population study of clinical trial accrual for women and minorities in neuro-oncology following the NIH Revitalization Act. Neuro Oncol 2022;24(8):1341–1349. doi: 10.1093/neuonc/noac011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brastianos PK, Galanis E, Butowski N, et al. Advances in multidisciplinary therapy for meningiomas. Neuro Oncol 2019;21:I18–I31. doi: 10.1093/neuonc/noy136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldbrunner R, Stavrinou P, Jenkinson MD, et al. EANO guideline on the diagnosis and management of meningiomas. Neuro Oncol 2021;23(11):1821–1834. doi: 10.1093/neuonc/noab150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 2016;131(6):803–820. doi: 10.1007/s00401-016-1545-1 [DOI] [PubMed] [Google Scholar]
- 6.Chen WC, Perlow HK, Choudhury A, et al. Radiotherapy for meningiomas. J Neurooncol 2022;160(2):505–515. doi: 10.1007/s11060-022-04171-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rogers CL, Pugh SL, Vogelbaum MA, et al. Low-risk meningioma: Initial outcomes from NRG Oncology/RTOG 0539. Neuro Oncol 2023;25(1):137–145. doi: 10.1093/neuonc/noac137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rogers CL, Won M, Vogelbaum MA, et al. High-risk Meningioma: Initial Outcomes From NRG Oncology/RTOG 0539. Int J Radiat Oncol Biol Phys Published online 2020. doi: 10.1016/j.ijrobp.2019.11.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rogers L, Zhang P, Vogelbaum MA, et al. Intermediate-risk meningioma: Initial outcomes from NRG Oncology RTOG 0539. J Neurosurg 2018;129(1):35–47. doi: 10.3171/2016.11.JNS161170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weber DC, Ares C, Villa S, et al. Adjuvant postoperative high-dose radiotherapy for atypical and malignant meningioma: A phase-II parallel non-randomized and observation study (EORTC 22042–26042). Radiotherapy and Oncology. Published online 2018. doi: 10.1016/j.radonc.2018.06.018 [DOI] [PubMed] [Google Scholar]
- 11.Louis DN, Perry A, Wesseling P, et al. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro Oncol 2021;23(8):1231–1251. doi: 10.1093/neuonc/noab106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Youngblood MW, Duran D, Montejo JD, et al. Correlations between genomic subgroup and clinical features in a cohort of more than 3000 meningiomas. J Neurosurg 2020;133(5):1345–1354. doi: 10.3171/2019.8.JNS191266 [DOI] [PubMed] [Google Scholar]
- 13.Clark VE, Erson-Omay EZ, Serin A, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science (1979). 2013;339(6123):1077–1080. doi: 10.1126/science.1233009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sahm F, Schrimpf D, Olar A, et al. TERT Promoter Mutations and Risk of Recurrence in Meningioma. J Natl Cancer Inst 2016;108(5). doi: 10.1093/jnci/djv377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guyot A, Duchesne M, Robert S, et al. Analysis of CDKN2A gene alterations in recurrent and non-recurrent meningioma. J Neurooncol Published online 2019. doi: 10.1007/s11060-019-03333-6 [DOI] [PubMed] [Google Scholar]
- 16.Driver J, Hoffman SE, Tavakol S, et al. A molecularly integrated grade for meningioma. Neuro Oncol 2022;24(5):796–808. doi: 10.1093/neuonc/noab213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maas SLN, Stichel D, Hielscher T, et al. Integrated Molecular-Morphologic Meningioma Classification: A Multicenter Retrospective Analysis, Retrospectively and Prospectively Validated. Journal of Clinical Oncology. 2021;39(34):3839–3852. doi: 10.1200/JCO.21.00784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nassiri F, Liu J, Patil V, et al. A clinically applicable integrative molecular classification of meningiomas. Nature. 2021;597(7874):119–125. doi: 10.1038/s41586-021-03850-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patel AJ, Wan YW, Al-Ouran R, et al. Molecular profiling predicts meningioma recurrence and reveals loss of DREAM complex repression in aggressive tumors. Proc Natl Acad Sci U S A 2019;116(43):21715–21726. doi: 10.1073/pnas.1912858116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vasudevan HN, Braunstein SE, Phillips JJ, et al. Comprehensive Molecular Profiling Identifies FOXM1 as a Key Transcription Factor for Meningioma Proliferation. Cell Rep 2018;22(13):3672–3683. doi: 10.1016/j.celrep.2018.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choudhury A, Chen WC, Lucas CHG, et al. Hypermitotic meningiomas harbor DNA methylation subgroups with distinct biological and clinical features. Neuro Oncol 2023;25(3):520–530. doi: 10.1093/neuonc/noac224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choudhury A, Magill ST, Eaton CD, et al. Meningioma DNA methylation groups identify biological drivers and therapeutic vulnerabilities. Nat Genet 2022;54(5):649–659. doi: 10.1038/s41588-022-01061-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olar A, Wani KM, Wilson CD, et al. Global epigenetic profiling identifies methylation subgroups associated with recurrence-free survival in meningioma. Acta Neuropathol 2017;133(3):431–444. doi: 10.1007/s00401-017-1678-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sahm F, Schrimpf D, Stichel D, et al. DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol 2017;18(5):682–694. doi: 10.1016/S1470-2045(17)30155-9 [DOI] [PubMed] [Google Scholar]
- 25.Sparano JA, Gray RJ, Makower DF, et al. Adjuvant Chemotherapy Guided by a 21-Gene Expression Assay in Breast Cancer. New England Journal of Medicine. 2018;379(2):111–121. doi: 10.1056/nejmoa1804710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu JC, Tosoian JJ, Qi J, et al. Clinical Utility of Gene Expression Classifiers in Men With Newly Diagnosed Prostate Cancer. JCO Precis Oncol 2018;(2):1–15. doi: 10.1200/po.18.00163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spratt DE, Zhang J, Santiago-Jiḿenez M, et al. Development and validation of a novel integrated clinical-genomic risk group classification for localized prostate cancer. Journal of Clinical Oncology. Published online 2018. doi: 10.1200/JCO.2017.74.2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith JC, Sheltzer JM. Genome-wide identification and analysis of prognostic features in human cancers. Cell Rep 2022;38(13):110569. doi: 10.1016/j.celrep.2022.110569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schaeffer EM, Srinivas S, Antonarakis ES, et al. NCCN Clinical Practice Guidelines in Oncology: Prostate Cancer Version 2. 2022. National Comprehensive Cancer Network. [Google Scholar]
- 30.Gradishar WJ, Moran MS, Abraham J, et al. Breast Cancer, Version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2022;20(6):691–722. doi: 10.6004/jnccn.2022.0030 [DOI] [PubMed] [Google Scholar]
- 31.Chen WC, Vasudevan HN, Choudhury A, et al. A prognostic gene-expression signature and risk score for meningioma recurrence after resection. Neurosurgery. 2021;88(1):202–210. doi: 10.1093/neuros/nyaa355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patel AJ, Wan YW, Al-Ouran R, et al. Molecular profiling predicts meningioma recurrence and reveals loss of DREAM complex repression in aggressive tumors. Proc Natl Acad Sci U S A 2019;116(43):21715–21726. doi: 10.1073/pnas.1912858116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vuong QH. Likelihood Ratio Tests for Model Selection and Non-Nested Hypotheses. Econometrica. 1989;57(2):307. doi: 10.2307/1912557 [DOI] [Google Scholar]
- 34.Nabors LB, Portnow J, Ahluwalia M, et al. Central nervous system cancers, version 3.2020. JNCCN Journal of the National Comprehensive Cancer Network. 2020;18(11):1537–1570. doi: 10.6004/JNCCN.2020.0052 [DOI] [PubMed] [Google Scholar]
- 35.Nassiri F, Mamatjan Y, Suppiah S, et al. DNA methylation profiling to predict recurrence risk in meningioma: Development and validation of a nomogram to optimize clinical management. Neuro Oncol 2019;21(7):901–910. doi: 10.1093/neuonc/noz061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Näslund O, Lipatnikova A, Dénes A, et al. Meningioma classification by immunohistochemistry: A replicability study. Brain and Spine. 2023;3:101711. doi: 10.1016/j.bas.2022.101711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bray DP, Buster BE, Quillin JW, Press RH, Eaton BR, Olson JJ. Adjuvant Radiotherapy in Grade II, Atypical Meningioma of the Skull Base. J Neurol Surg B Skull Base. 2022;83(4):418–422. doi: 10.1055/s-0041-1735879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bray DP, Quillin JW, Press RH, et al. Adjuvant Radiotherapy Versus Watchful Waiting for World Health Organization Grade II Atypical Meningioma: A Single-Institution Experience. Neurosurgery. 2021;88(5):E435–E442. doi: 10.1093/neuros/nyaa580 [DOI] [PubMed] [Google Scholar]
- 39.Byun HK, Chang WI, Lee JH, et al. Adjuvant Radiotherapy Versus Surveillance for Grade 2 Intracranial Meningiomas: A Multi-Institutional Propensity Score-Matched Study. Front Oncol 2022;12:877244. doi: 10.3389/fonc.2022.877244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen WC, Magill ST, Wu A, et al. Histopathological features predictive of local control of atypical meningioma after surgery and adjuvant radiotherapy. J Neurosurg Published online 2019. doi: 10.3171/2017.9.JNS171609 [DOI] [PubMed] [Google Scholar]
- 41.Dohm A, McTyre ER, Chan MD, et al. Early or late radiotherapy following gross or subtotal resection for atypical meningiomas: Clinical outcomes and local control. J Clin Neurosci 2017;46:90–98. doi: 10.1016/j.jocn.2017.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kent CL, Mowery YM, Babatunde O, et al. Long-Term Outcomes for Patients With Atypical or Malignant Meningiomas Treated With or Without Radiation Therapy: A 25-Year Retrospective Analysis of a Single-Institution Experience. Adv Radiat Oncol 2022;7(3):100878. doi: 10.1016/j.adro.2021.100878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pan PC, Pisapia DJ, Ramakrishna R, et al. Outcomes following upfront radiation versus monitoring in atypical meningiomas: 16-year experience at a tertiary medical center. Neurooncol Adv 2021;3(1):vdab094. doi: 10.1093/noajnl/vdab094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rydzewski NR, Lesniak MS, Chandler JP, et al. Gross total resection and adjuvant radiotherapy most significant predictors of improved survival in patients with atypical meningioma. Cancer. 2018;124(4):734–742. doi: 10.1002/cncr.31088 [DOI] [PubMed] [Google Scholar]
- 45.Barrett OC, Hackney JR, McDonald AM, Willey CD, Bredel M, Fiveash JB. Pathologic Predictors of Local Recurrence in Atypical Meningiomas Following Gross Total Resection. Int J Radiat Oncol Biol Phys 2019;103(2):453–459. doi: 10.1016/j.ijrobp.2018.09.019 [DOI] [PubMed] [Google Scholar]
- 46.Graffeo CS, Leeper HE, Perry A, et al. Revisiting Adjuvant Radiotherapy After Gross Total Resection of World Health Organization Grade II Meningioma. World Neurosurg 2017;103:655–663. doi: 10.1016/j.wneu.2017.04.095 [DOI] [PubMed] [Google Scholar]
- 47.Keric N, Kalasauskas D, Freyschlag CF, et al. Impact of postoperative radiotherapy on recurrence of primary intracranial atypical meningiomas. J Neurooncol 2020;146(2):347–355. doi: 10.1007/s11060-019-03382-x [DOI] [PubMed] [Google Scholar]
- 48.Momin AA, Soni P, Shao J, et al. Adjuvant radiation versus observation with salvage radiation after gross-total resection of WHO grade II meningiomas: a propensity score-adjusted analysis. J Neurosurg Published online October 8, 2021:1–8. doi: 10.3171/2021.4.JNS21559 [DOI] [PubMed] [Google Scholar]
- 49.Pan PC, Pisapia DJ, Ramakrishna R, et al. Outcomes following upfront radiation versus monitoring in atypical meningiomas: 16-year experience at a tertiary medical center. Neurooncol Adv 2021;3(1):vdab094. doi: 10.1093/noajnl/vdab094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rebchuk AD, Alam A, Hounjet CD, et al. Survival and Recurrence Outcomes Following Adjuvant Radiotherapy for Grade 2 Intracranial Meningiomas: 13-Year Experience in a Tertiary-Care Center. World Neurosurg 2022;161:e748–e756. doi: 10.1016/j.wneu.2022.02.088 [DOI] [PubMed] [Google Scholar]
- 51.Sun SQ, Kim AH, Cai C, et al. Management of atypical cranial meningiomas, part 1: predictors of recurrence and the role of adjuvant radiation after gross total resection. Neurosurgery. 2014;75(4):347–354; discussion 354–5; quiz 355. doi: 10.1227/NEU.0000000000000461 [DOI] [PubMed] [Google Scholar]
- 52.Wang F, Xu D, Liu Y, et al. Risk factors associated with postoperative recurrence in atypical intracranial meningioma: analysis of 263 cases at a single neurosurgical centre. Acta Neurochir (Wien) 2019;161(12):2563–2570. doi: 10.1007/s00701-019-04073-2 [DOI] [PubMed] [Google Scholar]
- 53.Zeng Q, Tian Z, Gao Q, et al. Effectiveness of Postoperative Radiotherapy in Patients with Atypical Meningiomas After Gross Total Resection: Analysis of 260 Cases. World Neurosurg 2022;162:e580–e586. doi: 10.1016/j.wneu.2022.03.053 [DOI] [PubMed] [Google Scholar]
- 54.Preusser M, Silvani A, Le Rhun E, et al. Trabectedin for recurrent WHO grade 2 or 3 meningioma: A randomized phase II study of the EORTC Brain Tumor Group (EORTC-1320-BTG). Neuro Oncol 2022;24(5):755–767. doi: 10.1093/neuonc/noab243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Capper D, Jones DTW, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555(7697):469–474. doi: 10.1038/nature26000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morin O, Chen WC, Nassiri F, et al. Integrated models incorporating radiologic and radiomic features predict meningioma grade, local failure, and overall survival. Neurooncol Adv 2019;1(1). doi: 10.1093/noajnl/vdz011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Magill ST, Vasudevan HN, Seo K, et al. Multiplatform genomic profiling and magnetic resonance imaging identify mechanisms underlying intratumor heterogeneity in meningioma. Nat Commun 2020;11(1). doi: 10.1038/s41467-020-18582-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rogers CL, Perry A, Pugh S, et al. Pathology concordance levels for meningioma classification and grading in NRG Oncology RTOG Trial 0539. Neuro Oncol 2016;18(4):565–574. doi: 10.1093/neuonc/nov247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Geiss GK, Bumgarner RE, Birditt B, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol 2008;26(3):317–325. doi: 10.1038/nbt1385 [DOI] [PubMed] [Google Scholar]
- 60.Breiman L Bagging predictors - Springer. Mach Learn Published online 1996. [Google Scholar]
- 61.Baik B, Yoon S, Nam D. Benchmarking RNA-seq differential expression analysis methods using spike-in and simulation data. PLoS One. 2020;15(4). doi: 10.1371/journal.pone.0232271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bi WL, Greenwald NF, Abedalthagafi M, et al. Genomic landscape of high-grade meningiomas. NPJ Genom Med 2017;2(1). doi: 10.1038/s41525-017-0014-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Von Spreckelsen N, Waldt N, Poetschke R, et al. KLF4 K409Q-mutated meningiomas show enhanced hypoxia signaling and respond to mTORC1 inhibitor treatment. Acta Neuropathol Commun 2020;8(1). doi: 10.1186/s40478-020-00912-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The SVA package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28(6):882–883. doi: 10.1093/bioinformatics/bts034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chang CW, Cheng WC, Chen CR, et al. Identification of human housekeeping genes and Tissue-Selective genes by microarray Meta-Analysis. PLoS One. 2011;6(7). doi: 10.1371/journal.pone.0022859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pfister C, Tatabiga MS, Roser F. Selection of suitable reference genes for quantitative real-time polymerase chain reaction in human meningiomas and arachnoidea. BMC Res Notes. 2011;4. doi: 10.1186/1756-0500-4-275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.De Ferrari L, Aitken S. Mining housekeeping genes with a Naive Bayes classifier. BMC Genomics. 2006;7. doi: 10.1186/1471-2164-7-277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Breiman L Bagging predictors - Springer. Mach Learn. Published online 1996. [Google Scholar]
- 69.Sievers P, Hielscher T, Schrimpf D, et al. CDKN2A/B homozygous deletion is associated with early recurrence in meningiomas. Acta Neuropathol 2020;140(3):409–413. doi: 10.1007/s00401-020-02188-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Koelsche C, Sahm F, Capper D, et al. Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta Neuropathol 2013;126(6):907–915. doi: 10.1007/s00401-013-1195-5 [DOI] [PubMed] [Google Scholar]
- 71.Youngblood MW, Miyagishima DF, Jin L, et al. Associations of meningioma molecular subgroup and tumor recurrence. Neuro Oncol 2021;23(5):783–794. doi: 10.1093/neuonc/noaa226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li MM, Datto M, Duncavage EJ, et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. Journal of Molecular Diagnostics. 2017;19(1):4–23. doi: 10.1016/j.jmoldx.2016.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Paik S, Shak S, Tang G, et al. A Multigene Assay to Predict Recurrence of Tamoxifen-Treated, Node-Negative Breast Cancer. New England Journal of Medicine. 2004;351(27):2817–2826. doi: 10.1056/nejmoa041588 [DOI] [PubMed] [Google Scholar]
- 74.Kalinsky K, Barlow WE, Gralow JR, et al. 21-Gene Assay to Inform Chemotherapy Benefit in Node-Positive Breast Cancer. New England Journal of Medicine. 2021;385(25):2336–2347. doi: 10.1056/nejmoa2108873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cardoso F, van ‘t Veer L, Poncet C, et al. MINDACT: Long-term results of the large prospective trial testing the 70-gene signature MammaPrint as guidance for adjuvant chemotherapy in breast cancer patients. Journal of Clinical Oncology. 2020;38(15_suppl):506–506. doi: 10.1200/jco.2020.38.15_suppl.506 [DOI] [Google Scholar]
- 76.Sjöström M, Fyles A, Liu FF, et al. Development and Validation of a Genomic Profile for the Omission of Local Adjuvant Radiation in Breast Cancer. J Clin Oncol 2023;41(8):1533–1540. doi: 10.1200/JCO.22.00655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Therneau T A Package for Survival Analysis in S. R package version. Survival (Lond) 2012;(R package version 2.37–7):1–3. [Google Scholar]
- 78.Lin H, Zelterman D. Modeling Survival Data: Extending the Cox Model. In: Technometrics. Vol 44.; 2002:85–86. doi: 10.1198/tech.2002.s656 [DOI] [Google Scholar]
- 79.Simon N, Friedman J, Hastie T, Tibshirani R. Regularization paths for Cox’s proportional hazards model via coordinate descent. J Stat Softw 2011;39(5):1–13. doi: 10.18637/jss.v039.i05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mogensen UB, Ishwaran H, Gerds TA. Evaluating Random Forests for Survival Analysis Using Prediction Error Curves. J Stat Softw 2012;50(11):1–23. doi: 10.18637/jss.v050.i11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ho DE, Imai K, King G, Stuart EA. MatchIt: Nonparametric preprocessing for parametric causal inference. J Stat Softw 2011;42(8):1–28. doi: 10.18637/jss.v042.i08 [DOI] [Google Scholar]
- 82.Saha-Chaudhuri P Time-dependent ROC curve estimation from censored survival data. 2013;1:2–6. [Google Scholar]
- 83.Harrell FE Jr. rms: Regression Modeling Strategies. Published online 2018:1–246. [Google Scholar]
- 84.Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw 2010;33(1):1–22. doi: 10.18637/jss.v033.i01 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Uno H, Cai T, Pencina MJ, D’Agostino RB, Wei LJ. On the C-statistics for evaluating overall adequacy of risk prediction procedures with censored survival data. Stat Med 2011;30(10):1105–1117. doi: 10.1002/sim.4154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Efron B Bootstrap Methods: Another Look at the Jackknife. The Annals of Statistics. 2007;7(1). doi: 10.1214/aos/1176344552 [DOI] [Google Scholar]
- 87.Ledell E, Petersen M, Van Der Laan M. Computationally efficient confidence intervals for cross-validated area under the ROC curve estimates. Electron J Stat 2015;9(1):1583–1607. doi: 10.1214/15-EJS1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data from targeted gene expression panels are deposited in the NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE222054. Raw DNA methylation data from the UCSF WHO grade 2 or grade 3 validation cohort and the RTOG 0539 validation cohort are available under accession number GSE221029. Raw amplicon and targeted exome sequencing data from discovery and validation cohort meningiomas are deposited in the NCBI Sequencing Reads Archive (https://www.ncbi.nlm.nih.gov/sra) under project numbers PRJNA916225 and PRJNA916253. Matrices containing TPM data from RNA sequencing cohorts used for analytical validation are deposited along with code on github (https://github.com/william-c-chen/Meningioma_GE_Biomarker). Accession numbers and publications containing previously reported data are available in Extended Data Table 4. The publicly available GRCh38 (hg38), CRCh37.p13 (hg19), and Kallisto index v10 datasets were used in this study. Source data are provided with this paper.