

Abstract
More than 25 years after its discovery, the post-transcriptional gene regulation mechanism termed RNAi is now transforming pharmaceutical development, proved by the recent FDA approval of multiple small interfering RNA (siRNA) drugs that target the liver. Synthetic siRNAs that trigger RNAi have the potential to specifically silence virtually any therapeutic target with unprecedented potency and durability. Bringing this innovative class of medicines to patients, however, has been riddled with substantial challenges, with delivery issues at the forefront. Several classes of siRNA drug are under clinical evaluation, but their utility in treating extrahepatic diseases remains limited, demanding continued innovation. In this Review, we discuss principal considerations and future directions in the design of therapeutic siRNAs, with a particular emphasis on chemistry, the application of informatics, delivery strategies and the importance of careful target selection, which together influence therapeutic success.
Introduction
RNAi is an intrinsic post-transcriptional gene regulation mechanism that has been harnessed for therapeutic development since its discovery in Caenorhabditis elegans (Box 1). Double-stranded small interfering RNAs (siRNAs) can trigger the RNAi process to induce sequence-specific gene silencing. Upon cellular uptake, the guide strand of the siRNA is assembled into an RNA-induced silencing complex (RISC) capable of searching and degrading complementary mRNAs, thus aborting target protein translation. The current clinical success of N-acetylgalactosamine (GalNAc)-conjugated siRNAs targeting the liver, with multiple approved drugs and many clinical trials in process, required a long path of innovation. The technology progress from bench to clinic can be defined by three major milestones: animal proof of concept (POC) (statistically significant silencing of the target at an acceptable dosing and safety range in vivo); clinical POC (demonstration of robust safety and disease target modulation at a tolerable dose in humans); and regulatory approval (Table 1).
Box 1. Twenty-five years of RNAi from discovery to clinic utility.
The discovery by Andrew Fire, Craig Mello and colleagues in 1998 that double-stranded RNAs can trigger RNAi to catalytically degrade complementary mRNA transcripts in Caenorhabditis elegans212 not only provided researchers with powerful tools but also sparked excitement for pharmaceutical development213–216. RNAi was later demonstrated in mammalian cells by Elbashir et al.217 and Caplen et al.218 in 2001. Merely a year after, RNAi activity was shown in mice by McCaffrey et al.219 using hydrodynamic delivery. Many pioneering works have greatly contributed to the advancement of therapeutic small interfering RNAs (siRNAs) towards in vivo implementation2,81,82,220–229. These early efforts facilitated our understanding of the potential of RNAi in various applications, including cell-targeted delivery, disease allele gene-selective targeting and antiviral therapies.
The concept of applying RNAi-induced gene silencing for therapeutic development appeared simple and compelling — essentially, the ability to turn off any molecular target by simply rewriting the siRNA base sequence. Early clinical programmes proceeded rapidly, using unmodified or slightly modified siRNAs. The hope was that sufficient compound would be taken up by tissues of interest to provide therapeutic benefit. The evidence of RNAi in humans from systemic delivery was first reported in 2010 by Davis et al.104. However, the first-generation compounds showed either marginal clinical efficacy or unacceptable toxicity. The realization that siRNAs faced serious hurdles, such as immunogenicity230–234 and off-targeting effects69,235,236, quickly led to doubt in the technology, as pharmaceutical companies cut funding and abandoned their RNAi platforms237,238. By the early 2010s, inflated expectations had dampened enthusiasm in the field of siRNA drug development.
Learning from failures, we now have a much clearer understanding of the underlying biological mechanisms at play and the necessity for significant chemical innovation to evolve siRNAs towards drug candidates with acceptable pharmacokinetic and pharmacodynamic (PK–PD) properties and disease-modifying potential. This knowledge has greatly facilitated the clinical implementation of siRNAs with favourable efficacy and safety profiles in the liver239. In 2018, a major milestone was reached as the FDA historically approved the first siRNA drug patisiran for the treatment of hereditary transthyretin amyloidosis9,105. Today, siRNA drugs have entered the market14,240 and many others are in late-stage clinical validation, being tested to treat various diseases, including neurodegenerative, muscular and ocular disorders. The field is once again prospering, underscored by a notable shift in focus from rare genetic disorders to prevalent diseases. This is highlighted by the recent positive clinical results of zilebesiran in treating hypertension241,242, as well as the approval of inclisiran (2020) in the European Union (EU) and (2021) in the USA for the treatment of hypercholesterolaemia — a major cause of atherosclerotic cardiovascular disease243,244. Additional siRNA drugs for the liver and extrahepatic tissues are expected to be available soon, as robust results from advanced clinical programmes and progress in innovating delivery technologies have been seen. GalNAc, N-acetylgalactosamine. Figure adapted with permission from ref. 21, Springer Nature Limited.

Table 1 |.
Delivery technologies enabling therapeutic RNAi in various tissues
| Technology | Platform | Tissue | Animal proof of concept | Clinical proof of concept | Date of approval/development stage |
|---|---|---|---|---|---|
| LNP | Formulation | Liver | ~2000s: various LNP-mediated siRNA delivery platforms had been explored in vivo | 2012–2013: phase I results of patisiran ALP-TTR01 (1st gen. LNP) and ALP-TTR02 (2nd gen. LNP) show effective serum reduction of TTR protein | Regulatory approval 2018: 1st drug: patisiran (ALP-TTR02, i.v. infusion) for polyneuropathy of hereditary TTR-mediated amyloidosis |
| GalNAc 1st gen. (STC) | Sugar conjugate | Liver | ~2014: multivalent GalNAc-conjugated siRNA shows robust and prolonged gene silencing in mouse liver | 2013–2016: revusiran elicits significant reduction of serum TTR | Phase III; revusiran toxicity (discontinued) |
| GalNAc 2nd gen. (ESC) | Sugar conjugate | Liver | 2014 TIDES meeting: ESC-GalNAc improves potency and durability in NHP | ESC-GalNAc is applied in all advanced clinical programmes | Regulatory approval 2019: givosiran 2020: lumasiran 2020 (EU), 2021: inclisiran 2022: vutrisiran 2023: nedosiran (GalXC platform, stabilization similar to ESC) |
| GalNAc 3rd gen. (ESC+ with glycol nucleic acid modification) | Sugar conjugate | Liver | 2022: local administration of C16-conjugated siRNAs enable potent and durable gene silencing in CNS, eye and lung in rodents and NHP | 2023: phase I trial shows promising data for ALN-APP targeting APP for the treatment of Alzheimer disease and cerebral amyloid angiopathy | 2023: phase II study of zilebesiran demonstrates encouraging efficacy and tolerability profile in adult patients with mild-to-moderate hypertension |
| C16 | Lipophilic conjugate | CNS Eye Lung | 2022: local administration of C16-conjugated siRNAs enable potent and durable gene silencing in CNS, eye and lung in rodents and NHP | 2023: phase I trial shows promising data for ALN-APP targeting APP for the treatment of Alzheimer disease and cerebral amyloid angiopathy | Phase I |
| Cholesterol | Lipophilic conjugate | Placenta | 2018: cholesterol-conjugated siRNA enables placental reduction of sFLIT in pregnant mice and NHP, ameliorating clinical signs of pre-eclampsia | NA | 2018: preclinical (discontinued) |
| PC–DCA | Lipophilic conjugate | Placenta | 2022: PC–DCA conjugation increases siRNA accumulation and silencing efficacy of sFLIT in placenta of pregnant mice | 2023: PC–DCA–siRNA under phase I trial shows uneventful safety in healthy volunteers | Phase I |
| DCA | Lipophilic conjugate | Muscle Heart Skin | 2019–2023: DCA conjugation allows enhanced siRNA local retention and systemic distribution to multiple extrahepatic tissues with functional gene silencing 2022–2023: DCA–siRNA offers enhanced local retention and functional gene silencing in rodent and pig skin |
NA | Preclinical IND-enabling |
| Anti-TfR1 antibody | Protein conjugate | Muscle | 2021: AOC 1001 (anti-TfR1 siRNA) effectively reduces levels of DMPK mRNA in muscle of NHP | 2023: phase I/II trial of AOC 1001 treating myotonic dystrophy type 1 demonstrates functional improvement, DMPK reduction, splicing improvements and safety | 2024: phase III expected |
| Centyrins | Protein conjugate | Muscle | 2022: CD71 centyrin–siRNA shows robust muscle gene modulation in Pompe disease mouse model | NA | 2023: phase I trial of ABX1100 initiated for the treatment of Pompe disease |
| Centyrins | Protein conjugate | Tumour | 2021: EGFR centyrin–siRNA conjugates show effective tumour delivery in mouse models | NA | Preclinical |
| Divalent scaffold | Size/structure | CNS lung | 2019: intracerebroventricular injection of divalent siRNA supports potent and sustained gene silencing in the brain of mice and NHP for months 2022: intratracheal delivery of divalent siRNA enables effective suppression of SARS-CoV-2 in mouse model of pulmonary infection |
NA | Preclinical |
| Tetravalent scaffold | Size/structure | Eye | 2023: intravitreal injection induces potent and multi-month gene silencing in rodent and pig eyes | NA | Preclinical |
Representative siRNA delivery platforms that show promising in vivo proof of concept (POC). The timeline of major POC events reflects the most recognized research publications or conference reports that aim to provide a general overview of the technology progress. All events listed are discussed in the main text with references cited. APP, amyloid precursor protein; C16, 2′-O-hexadecyl; CNS, central nervous system; DCA, docosanoic acid; DMPK, myotonin-protein kinase; ESC, enhanced stabilization chemistry; EU, European Union; GalNAc, N-acetylgalactosamine; gen., generation; i.v., intravenous; LNP, lipid nanoparticle; NA, not applicable; NHP, non-human primate; PC, phosphocholine; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; siRNA, small interfering RNA; STC, standard template chemistry; TfR1, transferrin receptor 1; TTR, transthyretin.
In liver, lipid nanoparticles (LNPs) were first used to deliver patisiran, currently the only siRNA drug on the market that uses this technology platform. The animal POC studies of LNP-mediated siRNA delivery were explored in the early 2000s by multiple groups1–7. It took several technology generations to reach an acceptable clinical POC (2012–2013)8 and a few more years to attain regulatory approval (2018)9. The dominant platform for liver delivery today is GalNAc conjugation. The first animal POC was reported in 2014 (ref. 10), and clinical success required two reiterations of the technology. The first iteration, using Alnylam’s ‘standard template chemistry’ (STC) platform, achieved clinical POC with revusiran11, but had to be discontinued owing to unexpected toxicity in the phase III trial12. The clinical efficacy and safety of GalNAc technology was later demonstrated in the next-generation products using the ‘enhanced stabilization chemistry’ (ESC) platform, in which the necessity for additional siRNA stabilization was realized. Givosiran was the first compound in the category to receive regulatory approval (2019). The recent development of an advanced ESC strategy (ESC+), which incorporates a glycol nucleic acid (GNA) modification13, further improved the safety profile of GalNAc–siRNAs (discussed later in ‘Medicinal chemistry of siRNA design’). Aspects of the chemistry, structure and function of the currently approved siRNA drugs were recently reviewed14. More recently, the siRNA drug nedosiran (see Related links) – developed using a chemical stabilization concept similar to ESC but with a different GalNAc conjugation strategy (discussed in a later section) – received FDA approval for the treatment of primary hyperoxaluria type 1 by targeting lactate dehydrogenase A in hepatocytes.
So far, there have been no approved siRNA drugs to target extrahepatic tissues, but animal POC studies have demonstrated promising efficacy in multiple tissues. Several delivery technologies and strategies – for example, lipophilic conjugates, protein-antibody conjugates and manipulation of the size and structure of the siRNA molecule itself – are in development and show robust efficacy in various animal tissues with some platforms moving towards clinical translation (Table 1). For central nervous system (CNS) and muscle, efficacy has also been achieved in humans. In the near future, some technologies that have shown robust animal data might be translated into the development of drug classes in these tissues. The RNAi drug development field is very active and rapidly advancing. Every aspect of the diverse preclinical and clinical evaluations cannot be covered here, and much of the mechanistic details of RNAi biology and RNAi therapeutics have been reviewed elsewhere15. Instead, this Review will assess fundamental aspects of siRNA drug development – siRNA chemistry, the application of informatics, delivery strategies and target and indication selection – that may encourage future innovation and clinical translation of RNAi-based drugs.
Medicinal chemistry of siRNA design
Advances in nucleic acid chemistry have greatly accelerated the clinical development of oligonucleotide-based drugs. These approved products encompass a range of therapeutic modalities, including antisense oligonucleotides (ASOs), splice-switching oligonucleotides (SSOs), aptamers and siRNAs14. Although these modalities use various mechanisms of target recognition and gene modulation, their molecular configurations highlight the importance of chemical modifications in improving drug-like properties and long-term durability of oligonucleotides.
For siRNAs, initial biodistribution is usually completed within a few hours after injection. The first obstacle for productive delivery is the metabolic degradation and clearance during absorption and circulation. With sufficient accumulation in target tissues, cellular uptake of siRNA occurs primarily through membrane endocytosis. Internalized compounds become entrapped within endosomal and lysosomal compartments, creating an intracellular drug depot that gradually releases siRNA into the cytoplasm for RISC loading, offering prolonged durability on target silencing (Fig. 1). Endosomal escape is a rate-limiting step in the onset of silencing16, but slow release is the basis for the multi-month durability of GalNAc-conjugated siRNA drugs in the clinic. Strategies have been explored to improve the endosomal escape rate17; successful strategies might be advantageous for siRNA therapies designed to treat viral infections or cancers that require a rapid onset of silencing. For therapies in which only short-term RNAi activity is desired, designing partially modified compounds with degradable chemical composition is straightforward.
Fig. 1 |. Mechanism of action of siRNA drugs.
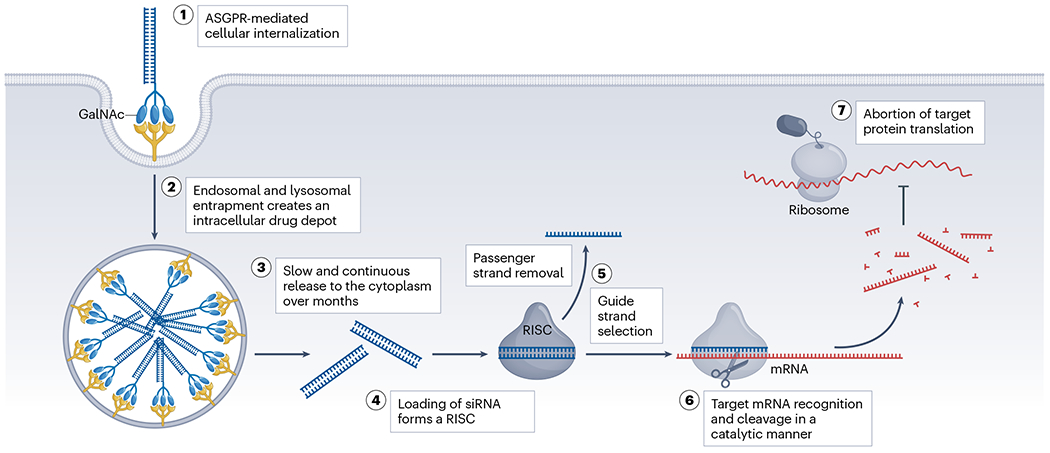
Effective gene silencing of small interfering RNA (siRNA) drugs requires efficient cellular internalization, endosomal escape, RNA-induced silencing complex (RISC) loading, target recognition and cleavage. N-acetylgalactosamine (GalNAc)–siRNAs exhibit multi-month durability owing to the rapid asialoglycoprotein receptor (ASGPR)-mediated membrane endocytosis and slow release from the intracellular drug depot after internalization. ASGPRs are highly expressed in hepatocytes and have a high recycling rate (minutes) for GalNAc-conjugated oligonucleotide internalization. Entrapment of chemically stabilized siRNAs in endosomal and lysosomal compartments can serve as an intracellular drug depot to support long-term durability. After being released into the cytoplasm, the siRNA needs to be assembled into a RISC to enable guide strand selection and subsequent recognition and cleavage of complementary mRNA substrates. Argonaute 2 (Ago2) protein is the catalytic component of RISC with RNA-guided endonuclease activity to mediate target cleavage. Ab, antibody.
Necessity for chemical modifications
Unmodified siRNAs exhibit poor metabolic stability and are susceptible to immediate degradation (>50% within a minute) in vivo18. Double-stranded siRNAs can also trigger innate immune responses19 and cause off-target effects20. These issues were largely resolved by introducing certain 2′-ribose and terminal backbone modifications, including 2′-O-methyl (2′-OMe), 2′-fluoro (2′-F) and phosphorothioate (PS)21 (Fig. 2a). These three modifications – now used in nearly all advanced siRNA designs – were originally applied in the early 1990s to enhance drug-like properties of ASOs (for example, fomivirsen, approved in 1998)22,23 and aptamers (for example, pegaptanib, approved in 2004)24. Nevertheless, nucleotide modifications were not a primary focus in the initial development of siRNA therapeutics. In hindsight, it is perhaps unsurprising that early trials using unmodified or slightly modified siRNA compounds yielded limited efficacy and dose-limiting toxicity.
Fig. 2 |. Medicinal chemistry of siRNA design.
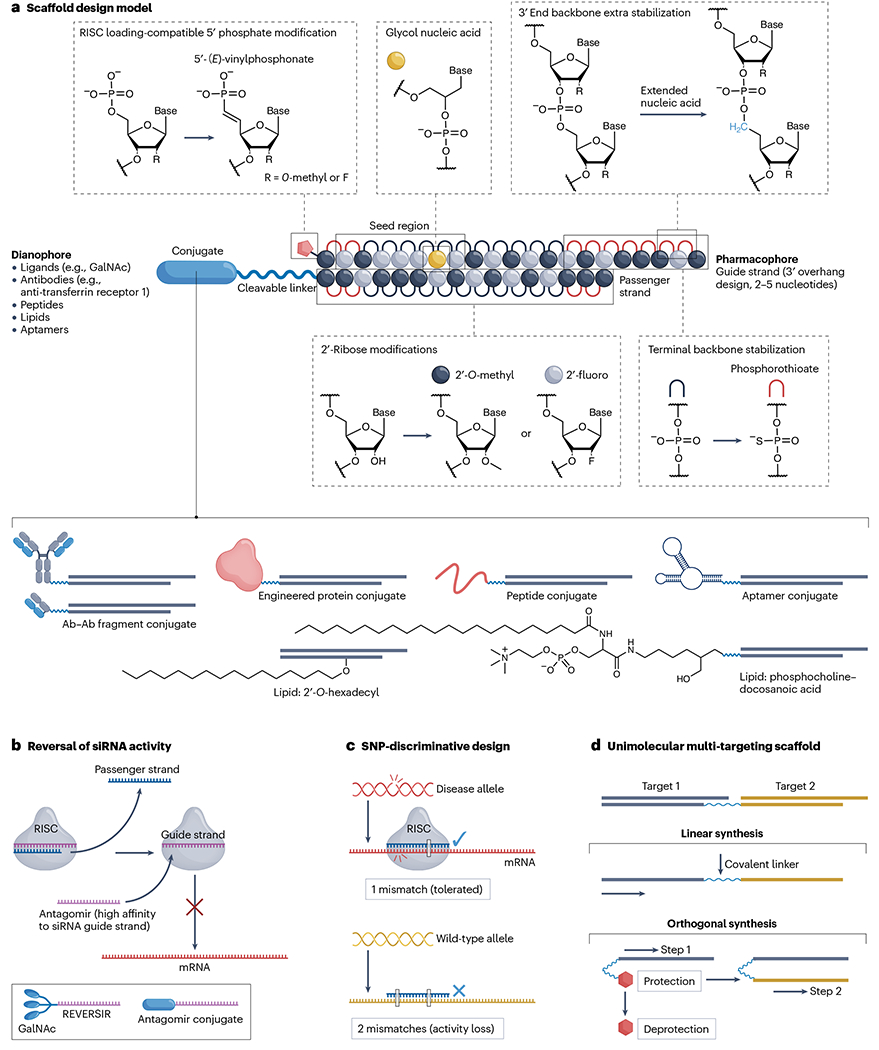
A schematic of small interfering RNA (siRNA) nucleotide configuration, targeting entity conjugation, chemical modification and fine-tuning strategies for a specific therapeutic purpose. a, Scaffold design model for conjugated siRNAs. Pharmacophore: base sequence that determines siRNA specificity against target mRNA transcript; dianophore: targeting entities that dictate the tissue distribution profile of the siRNA; seed region: nucleotides 2–8 from the 5′-end of the guide strand; cleavable linker: nuclease-labile phosphodiester bonds or other designs. b, Antagomir (or anti-microRNA) design to reverse undesired siRNA activity. An antagomir is a single-stranded oligonucleotide that typically uses an identical delivery strategy to its target siRNA to inactivate RNA-induced silencing complex (RISC)-loaded guide strand activity in the same tissue or cell type. c, Guide strand mismatch design to improve SNP-discriminative targeting on the disease-causing allele while reducing activity against the wild-type allele. d, Synthetic strategies for unimolecular siRNA scaffold to incorporate multiple targeting sequences (dual-targeting design shown). GalNAc, N-acetylgalactosamine. Part a adapted from ref. 156, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/).
For an extended period, the prevailing approach – LNP-mediated delivery – involved only partial modification of siRNAs to improve stability21. Partially modified compounds (for example, patisiran) are fully functional in the context of LNP formulation, driven by the mechanism of fast internalization and endosomal release of lipid-encapsulated drug molecules. Lipid formulations protect siRNA from nuclease degradation and enable faster onset of silencing, but when being used to deliver partially modified compounds they have limited durability in vivo. For conjugate-mediated delivery to achieve multi-month durability without LNP formulation, compounds must survive prolonged exposure to the highly aggressive, nuclease-rich endosomal and lysosomal environments25. Extensive modification of the siRNA scaffold is therefore essential for long-term stability. Indeed, full stabilization of the scaffold by incorporating modifications at every nucleotide position and several terminal backbone modifications profoundly improves siRNA accumulation and potency in tissues26.
Basic scaffold stabilization
To prevent metabolic degradation during tissue distribution and intracellular entrapment, scaffold optimization is required to modulate siRNA interactions with cellular processes, mostly 2′-hydroxyl (2′-OH)-mediated hydrolysis, endonucleases, as well as 5′ and 3′ exonucleases. Various chemical strategies were developed early for oligonucleotide-based therapeutics, including modifications of nucleobases, ribose sugars and backbones14. Studies revealed that a selectively small set of RISC-compatible modifications (2′-OMe, 2′-F and PS) are sufficient to confer nuclease resistance on the siRNA duplex18,27–29 and can also reduce immunogenicity and off-targeting effects. 2′-OMe and 2′-F modifications reduce 2′-OH vulnerability, while maintaining the pre-organized A-form helix of the guide strand, which is necessary for successful RISC recognition and loading30. 2′-OMe and 2′-F also enhance the affinity of target interactions, likely owing to more favourable pre-organization of the backbone in a 3′-endo conformation31.
The bulky 2′-OMe is generally well tolerated, but its effect on activity depends in part on sequence and position. For example, 2′-OMe at positions 2 and 14 of the guide strand consistently reduces activity32. The smaller 2′-F is tolerated in most positions of an siRNA; moreover, 2′-F enhances hydrophobicity and improves siRNA cellular uptake and potency28,33. Simultaneously, because 2′-F is not a natural modification, there was some concern that it might be incorporated into mitochondrial DNA by polymerases and cause toxicity, The exact cause of clinical toxicity of revusiran (see Related links) – the first generation of STC GalNAc-conjugated siRNA – during the phase III trial remains unknown. Detailed toxicology studies provide proof that long-term administration of 2′-F modified siRNAs is safe34. Although the levels of detectable 2′-F metabolites were significantly higher in revusiran than in its next-generation ESC compound, the only differential structural finding was enlarged mitochondria in hepatocytes and certain myocytes. Although the levels of detectable 2′-F degradation products were marginal, the hypothesis that the administration of an extremely high dose (~25 g per year) of oligonucleotides and highly inefficient, but detectable incorporation of 2′-F metabolites into mitochondrial DNA might be involved, cannot be completely ruled out.
The most commonly used fully 2′-modified siRNA configuration was initially introduced as an alternating 2′-OMe and 2′-F scaffold pattern27. 2′-OMe provides a substantially higher level of stabilization, and indeed more methyl-rich configurations result in better efficacy and durability in vivo32. The 5′ nucleotide of the guide strand does not interact with the target and is predominantly modified with 2′-OMe uridine, which provides an optimal fit in the Argonaute 2 (Ago2) MID domain35. Increasing the 2′-OMe content through the remaining positions of the guide strand enhances durability in vivo, but also introduces challenges. The extreme example of the methyl-rich scaffold uses compounds in which all but two positions (positions 2 and 14 of the guide strand) are 2′-OMe modified36. This scaffold provides high stability in vivo, but it is compatible with only a limited number of targeting sequences.
When modified siRNAs are delivered in vivo, long-term efficacy relies on the continuous release of the drug from endosomal and lysosomal compartments. Upon release into the cytoplasm, they compete with naturally occurring microRNAs (miRNAs) to be loaded into de novo synthesized Ago2 (ref. 37). Thus, even a minor reduction in RISC-entering ability might negatively affect long-term biological efficacy. Indeed, identification of optimal siRNA configurations requires iterative screening of sequence and modification patterns. Interestingly, while the pharmacokinetic and pharmacodynamic (PK–PD) profile of the siRNA scaffold is believed to be mainly defined by the general chemical configuration21, the exact clinical durability varies significantly between approved drugs (1- to 6-month dosing intervals), at least in part owing to differences in the 2′-OMe and 2′-F patterns used. Although the exact modification patterns may vary, recently approved drugs and clinical programmes predominantly adopted fully stabilized scaffolds.
Exonuclease stabilization
Replacing 2′-OH groups with 2′-OMe and 2′-F modifications is a proven strategy to protect siRNA scaffolds against endonuclease-mediated cleavage. However, this approach is insufficient to provide stability against 5′ and 3′ exonucleases, which are generally effective in degrading both RNA and DNA and do not rely on 2′-OH recognition. Incorporating PS modifications, usually at the ends of both the guide and passenger strands, is currently the dominant strategy for terminal stabilization. Indeed, the simple addition of two PS-modified backbone linkages at the 5′ ends provides orders of magnitude improvement in exonuclease stability and is the primary difference between Alnylam’s STC and ESC platforms32,38. The first-generation GalNAc-conjugated siRNAs, such as revusiran, lacked these simple additional stabilizations, and the weekly 500 mg doses of revusiran in clinical trials resulted in cumulative toxicity39.
The 3′ end of the passenger strand of siRNA is commonly conjugated to targeting entities such as the tri-antennary GalNAc or small-molecule lipids, providing some protection from 3′ exonuclease-mediated degradation. In currently approved siRNA drugs, no PS modifications are incorporated at the 3′ end of the passenger strand. This design may be acceptable for certain 3′-conjugated siRNAs, but for internal or 5′-conjugated siRNAs, stabilizing the 3′ end is essential for protection against 3′ exonucleases. The PS-free design at the 3′ end might help to release the siRNA from membranes (for example, where the conjugate is tightly bound) for RISC loading, but it might also reduce the durability of the siRNA. Introducing a cleavable linker40 (for example, the labile phosphodiester bond) after PS stabilization of the 3′ end might be a strategy to improve both conjugate release and 3′ stability (Fig. 2a). Notably, nedosiran uses a longer variant of the passenger strand, in which the 3′ sequence forms a loop from which four nucleotides are linked via their 2′ positions to GalNAc moieties for hepatocyte delivery. In general, the modifications and structure motifs at the 3′ end of the passenger strand can be tailored to the purpose.
The 3′ end of the guide strand is more susceptible to 3′ exonuclease degradation, particularly in scaffolds with a longer overhang design than the typical dinucleotide overhang. Increasing the number of PS modifications in the single-stranded overhang region can enhance siRNA cellular uptake and potency41. As PS modifications do not completely eliminate the vulnerability of siRNA to exonucleases, developing strategies to further stabilize the backbone might improve siRNA in vivo durability.
5′-Terminal modification of guide strand
The 5′-terminal phosphate of the guide strand is crucial for efficient RISC loading and interaction with the MID domain of the Ago2 protein42,43. The currently approved siRNA drugs lack chemical incorporation of the 5′-terminal phosphate or its analogues, with the exception of nedosiran, which uses a methyl-protected 4′-phosphate analogue. These drugs likely rely on intracellular phosphorylation of the 5′-hydroxyl for RISC loading. Interestingly, in liver, the effects of the terminal phosphate are observed for some compounds but not for others44. The differences might be explained in part by the sequence effect on efficiency of intracellular phosphorylation. For extrahepatic delivery, in which overall accumulation is lower, chemical introduction of phosphatase-resistant 5′-phosphate analogues significantly improves siRNA potency45.
The most commonly used metabolically stable analogue of 5′-terminal phosphate is 5′-(E)-vinylphosphonate (5′-VP), which substitutes the bridging oxygen with carbon in a fixed stereo configuration optimal for RISC loading (Fig. 2a). This modification was originally described in the context of single-stranded siRNA variants46–48 and is currently a key component in most conjugated siRNA designs, enabling prolonged durability for extrahepatic silencing49–52. The 5′-VP-mediated enhancement of in vivo efficacy is likely due to two main factors. First is the metabolic stabilization conferred by the phosphonate bond, which is not cleaved by phosphatase. Second is the enhanced fit of the 5′-VP into the Ago2 MID domain53, resulting in an energetically favourable configuration. This can partially provide an added advantage to artificial siRNAs in competition with naturally generated miRNAs for Ago2 loading. However, the 5′-VP does not add significantly to the stabilization of phosphorothioates against 5′ exonucleases. The development of 5′-terminal phosphate modifications to enhance resistance to phosphatase and 5′ exonuclease, as well as to facilitate Ago2 loading, is an important direction54.
Guide strand design for RISC selection
Tuning the global scaffold of siRNA has significant implications for its functionality. The optimal configuration of the scaffold, achieved through 2′-OMe, 2′-F, PS and 5′-VP modifications, enables the efficient formation of RISC, guide strand loading and target recognition. Advanced designs to improve guide strand selection by RISC complex have mostly adopted asymmetrical scaffolds, meaning that the modification pattern of the guide strand differs in length (with 2- to 5-nucleotide overhangs at the 3′ end) compared with the passenger strand (Fig. 2a). In some scaffolds, the guide strand is significantly longer than the passenger strand, generating an extended 3′ end overhang that may also enhance tissue distribution in a mechanism similar to that of ASOs55. Asymmetry56 can be chemically introduced to ensure the selective recognition of the guide strand and prevent the passenger strand from entering the RISC57, thus reducing passenger strand-mediated off-target effects. Another variable is the length of siRNA; longer (~27 nucleotides) asymmetrical variants58,59 require processing of the scaffold into the optimal length by an endogenous RNAi pathway enzyme called Dicer before RISC loading60,61. Many strategies have been explored to improve guide strand selection, including shortening the asymmetrical duplex62, introducing paired 2′-OMe63, including a 5′-morpholino analogue64 on the 5′ end of the passenger strand, 5′-O-methylation of the passenger strand65, terminal bridging of the siRNA duplex66,67 and other chemical approaches68.
Fine-tuning siRNA chemistries
Advanced chemical configurations typically include a fully modified duplex with a combination of 2′-OMe, 2′-F, PS and 5′-VP modifications, applicable for most sequences. In addition to the four core chemistries, other strategies can selectively enhance particular siRNA properties.
Off-target effects from the passenger strand can be blocked by strand asymmetry and specific structural modifications, but guide strand-mediated off-target silencing through seed complementarity can induce undesired effects69. Off-target effects are typically derived from seed complement frequency70 and high affinity to the targets71. Although the exact off-target profile is hard to predict and is often cell-type and species specific, the overall ‘specificity’ of a compound can be partially predicted bioinformatically and validated experimentally70. In many cases, in vivo off-target signatures are barely detectable49,72, with only a handful of targets showing marginal effects. Nevertheless, off-target effects can drive in vivo toxicity73. In these cases, the introduction of modifications in the seed region to reduce affinity to off-target sites can significantly improve specificity and reduce toxicity while maintaining the primary target activity.
Various strategies can be used to minimize off-targeting effects. Early studies explored the introduction of a 2′-OMe at position 2 of the guide strand, which reduced off-targeting effects through two potential mechanisms63. First, 2′-OMe modifications at positions 1 and 2 make the 5′ terminus a poor substrate for intracellular phosphorylation. This significantly reduces the efficiency of RISC loading of the modified strand, thus limiting passenger strand off-targeting. Second, the modification of the 2′-hydroxyl of ribose with a methyl group in the context of a chemically phosphorylated guide strand enables efficient RISC loading, but restricts miRNA-mediated target recognition. This limitation provides better discrimination between full-length complementarity-based target cleavage and seed interaction-driven off-targeting. Position 2 of the guide strand is the primary hydroxyl contact maintained in the RISC crystal structure, and disruption of this interaction is likely to have a strong negative impact on the seed-mediated off-targeting. More advanced chemical modifications, such as GNA, in the seed region (for example, at position 6 or 7 of the guide strand; Fig. 2a) substantially reduces seed-mediated off-targeting with minimal impact on primary target activity74–76. Indeed, the GNA modification has been applied to several of the latest clinical candidates, including Alnylam’s VIR-2218 for the treatment of chronic hepatitis B virus infection.
Many clinically relevant scaffold configurations use only PS modifications for backbone stabilization. There is additional space to further advance chemistries to increase backbone stability. Extension of the phosphate backbone by inserting an extra carbon, named extended nucleic acid (exNA) (Fig. 2a), was recently demonstrated to profoundly increase stability against 3′ exonucleases. Indeed, replacing only two PS modifications with exNAs at the 3′ end of the guide strand significantly improves extrahepatic tissue accumulation and efficacy77. Interestingly, delayed clearance of exNA-modified siRNAs indicates that 3′ exonuclease degradation during the distribution phase significantly limits efficacy.
Currently, the most advanced siRNA scaffolds can achieve high potency and multi-month durability in on-target silencing, which are the most attractive features of this class of medicine. However, the potent and prolonged pharmacological effects may raise safety concerns, for example, by causing unexpected on-target side effects. Such siRNA-induced side effects can be mitigated by introducing a single-stranded oligonucleotide with high affinity for the seed region of the guide strand. These oligonucleotides, termed antagomirs (or anti-miRs)78, bind to the loaded guide strand in RISC, thus blocking its target recognition and silencing activity (Fig. 2b). Indeed, REVERSIR (GalNAc-modified antagomir) can effectively reverse siRNA activity in GalNAc–siRNA-treated animals79. This concept is not clinically realized and, so far, has not been requested by regulatory agencies. Another variant of this idea can be used to improve tissue selectivity. GalNAc enables selective delivery of siRNA to hepatocytes, but other classes of conjugate are significantly less selective. As liver and kidney are the main tissues responsible for oligonucleotide clearance, some extent of liver and/or kidney exposure is always observed, regardless of the conjugate and route of administration. For example, REVERSIR could be used to improve CNS-selective targeting by reducing liver clearance-related siRNA activity80.
Most of the siRNAs in the clinic target both alleles of a disease-causing gene. In some genetically defined disorders, selective targeting of a mutant allele to unleash the wild-type allele is desired. Therefore, it is beneficial to design compounds81–84 that can efficiently discriminate between the mutant and wild-type alleles, which often differ in only a single nucleotide. Typically, a series of consecutive complementary guide strands around the SNP site of the mutant allele transcript are first screened to identify whether any of the designed siRNAs complementary to the SNP position are active. A single mismatch to the wild-type allele often fails to provide sufficient selectivity for the mutant allele; therefore, incorporation of an additional mismatch is necessary (Fig. 2c). A single mismatch between the mutant allele and the guide strand (in a tolerated position) and two mismatches between the wild-type allele and the guide can provide 50- to 80-fold discrimination83. Selectivity can be further enhanced by chemically constraining the backbone with an internucleotide (E)-vinylphosphonate (iE-VP) modification next to the allele-discriminating mismatch85. In the iE-VP modification, a bridging oxygen of a phosphodiester bond is substituted with a double bond in a locked stereo conformation, which supports proper guide strand target recognition, but prevents guide strand flexibility to accommodate the mismatch. Although the design of allele-specific siRNAs is feasible for many sites, it can often come at the cost of lower potency compared with some non-selective compounds, thus potentially requiring more frequent dosing.
Another interesting concept is the development of a unimolecular scaffold for multi-gene targeting. In this case, a ‘passenger’ strand carries two or more different sequences and is annealed to the corresponding guide strands. The resulting configuration can also be conjugated and can efficiently silence multiple genes simultaneously in vivo86. Chemically, there are two strategies to generate this type of molecule: linear synthesis, whereby passenger strands are separated by a linker; and the use of orthogonal protection groups, whereby the strands are synthesized in a step-wise manner (Fig. 2d). The utility of this approach remains to be established clinically. In general, as PK–PD properties of the siRNAs are driven mainly by the molecular architecture, incorporating multiple siRNAs into a defined scaffold can provide a viable alternative for multi-gene modulation. Indeed, although this approach requires synthesis of two or more compounds and a slightly more complex preclinical development plan, the robust scientific rationale for co-administration justifies the complexity. Notably, the FDA has previously permitted the administration of a pool of more than one siRNA as a single product in a few clinical trials: NCT00882180, NCT04669808 and NCT05881993 (clinicaltrials.gov). This pooling approach provides more flexibility, including the ability to differentially tune the ratio of the compounds and thus, the relative levels of modulation. In addition, the chemistry, manufacturing and controls (CMC) plan for manufacturing of the smaller, less-complex entities is easier.
In silico design and biological screening
The design of siRNA drugs requires thorough analysis of genetic information, including mRNA expression profile, presence of SNPs, sequence specificity and homology. Informatics can help to predict siRNA functionality, specificity and cross-species targetability before experimental validation, eventually increasing the success rate for identification of siRNA leads. However, this approach faces several challenges (Box 2). Multiple factors can affect therapeutic siRNA efficacy; some are easy to predict with high confidence and others require extensive experimental screening. A combined approach is usually necessary to identify the optimal siRNA sequence. Below, we discuss key factors that influence the overall performance of bioinformatics in predicting hyper-functional siRNAs.
Box 2. Challenges of computational design.
Functional small interfering RNA (siRNA) hits can be predicted from algorithms. However, for certain genes, the overall chance of identifying potent hits is significantly lower than for other genes. This gap between computational prediction and actual biological activity is mainly due to the complexity of target biology and cellular environments. Factors such as masking effects on the siRNA targeting sites owing to the presence of secondary or tertiary structures245,246 or RNA-binding proteins need to be considered, as these elements can profoundly limit the accessibility of siRNA for target site recognition.
With unmodified siRNA datasets, linear regression can provide reasonable predictive power and indeed many early works in the field used this methodology247–250. The application of more advanced machine learning methods can slightly increase the predictive power, and among various decision trees, the random forest251 is the commonly used approach to enhance predictive power94. Although it is relatively easy to algorithmize the ability of the siRNA sequence to load into the RNA-induced silencing complex (RISC) and cleave the mRNA target in ideal circumstances, other target-specific factors such as the aforementioned masking effects from higher-order mRNA structures246,252,253 and interactions with RNA-binding proteins254,255 are hard to model. For example, only a small fraction of functional siRNA hits targeting viral sequences in a context of reporter-based validation (that is, viral genes constructed in an expression vector) are active in blocking the viral infection in natural conditions52.
The lack of perfect correlation between the ability of RISC entry and its activity in modulation of the native target is highly target related. For some targets, only a small fraction of RISC-competent compounds are active in the context of native mRNAs256. In contrast, for many targets, specifically for housekeeping genes, the subcellular localization and accessibility of transcripts are not significant factors affecting siRNA activity and typically there is a strong correlation between the readouts of reporter and native targeting. For genes with a significant fraction of the transcripts localized in the nucleus257,258, especially those expressed in neurons259,260, or targets involved in immune responses261, cytoplasmic accessibility in specific cell types could be a major limiting factor. Novel methods, including semi-supervised feature extraction262 and other artificial intelligence-type elements, may provide a viable path to improvement of the predictive power of the algorithms in these complex circumstances. Currently, the limited predictive power of informatics needs to be compensated by extensive experimental screening of a large panel of siRNA candidates.
Unmodified versus modified siRNA
Extensive chemical stabilization is essential to maximize the durability of siRNA in vivo, but modification patterns affect siRNA functionality. A significant fraction of active non-modified siRNAs do not tolerate extensive modification. Currently, a cellular screen is usually performed in the context of a fully modified scaffold of one configuration. Identified leads may be further optimized by fine-tuning the modification pattern to the sequence. Although the correlation between the efficacy of unmodified and modified siRNA is relatively low, the data generated with one chemical modification pattern can be, at least partially, translated into a different chemical or structural scaffold.
Most publicly available siRNA prediction algorithms are based on unmodified siRNA datasets, which limits their ability to predict modified siRNAs. For example, one of the earliest robust algorithms was developed by machine learning trained on a large set of unmodified siRNAs87. When the algorithm developed with the Huesken dataset was applied to the chemically modified siRNAs, it had limited predictive power88. Conversely, algorithms developed on the basis of modified siRNAs do not show significant predictive power for non-modified compounds. The difference is likely due to the different factors driving efficacy. For example, thermodynamic bias56,89 – also called strand asymmetry – is a principal component of efficacy for unmodified siRNAs, but in modified siRNAs asymmetry is usually chemically defined and thus contributes less. Primary screening must be conducted using modification patterns that resemble clinically applicable scaffolds. Most laboratories and companies now use a preferred modification pattern as a starting point and then optimize the chemistry for maximal potency. The final choice of the pattern is often defined by the lead compound sequence and the target tissue.
siRNA potency in vitro versus in vivo
Identification of an siRNA that silences a preferred target in vitro is often an easy task, and many commercial resources provide off-the-shelf solutions. Unfortunately, optimal clinical efficacy of a compound is highly dependent on potency, which can vary significantly depending on chemical modifications and delivery entities. Generally, compounds with better half-maximal inhibitory concentration (IC50) values in vitro tend to show enhancement of in vivo activity, but the correlation between in vitro and in vivo efficacy is not always straightforward. Many unpublished cases exist in which highly potent siRNAs in vitro exhibit minimal in vivo efficacy, as the complexity of the in vivo environment can profoundly impact siRNA behaviour. The precise relationship between in vitro, in vivo and clinical potency can be difficult to resolve, as available data are often not well matched, especially in compounds for which the sequence, modification pattern and dosing regimen are changed. Hyper-functional sequences are well documented, but a fundamental understanding of the factors that drive hyper-functionality and informatics of their prediction is not well established. For example, mRNA local structure, the presence of miRNA binding sites or RNA-binding proteins might be contributing factors.
Another crucial factor that could influence the efficacy of siRNA is prevalent adenosine methylation, particularly N6-methyladenosine (m6A), in target mRNA transcripts, which have a significant role in various cellular processes90. Recent reports suggest that the presence of m6A at miRNA binding sites in target mRNAs enhances miRNA-mediated silencing91,92. Most of the current clinical leads target 3′ untranslated regions (UTRs), which are enriched for miRNA binding sites, offering higher chances to enhance local context for gene silencing. Although many approaches have been explored to improve prediction accuracy, the correlation between computational outputs and actual activity remains limited mainly owing to biological complexities, demanding continued innovations. The use of machine learning methods93,94 to help identify patterns or correlations between sequences, modifications and efficacy would advance the development of functional siRNA informatics. This will require large-scale, high-quality datasets derived from biological validation under consistent experimental conditions.
Specificity
Target specificity is another key factor to consider. Compounds should be selected with at least one – preferably more – mismatch to potential off-target sites. In addition, factors such as seed complement frequency and the presence of natural miRNA seed95, or sequence factors that affect CMC, all contribute to design. Seed-based off-targeting is a major challenge in the application of genome-wide RNAi-based screening. Indeed, in vitro, each siRNA will cleave the target mRNA but may also silence other mRNAs through the seed-based complementarity in their 3′ UTRs69,70,96. The exact identity of off-targets will depend on cell type and species69, but whether an siRNA has a few or many off-target sites can be partially predictable70 by informatics and is consistent between species.
Interestingly, off-target miRNA-like regulation by siRNAs seems to be less of a problem in vivo than in vitro. Although siRNAs have been shown to cause toxicity in vivo by seed-based off-targeting73, most do not49,72. Apparent differences in the magnitude of off-targeting effects observed in vitro versus in vivo might be due to reported differences in the molecular weight and intracellular localization of RISC97. Indeed, only a high-molecular-weight RISC is capable of miRNA-like function and is enriched in actively dividing cells in vitro; its presence in vivo is highly variable between tissues and cell types.
Cross-species targeting
siRNAs with cross-species targetability can streamline and expedite the translation of drug candidates from preclinical development to the clinic. Identification of such siRNAs requires analysis of the homology of target sequences between humans and species of interest98. Databases such as NCBI BLAST, Ensembl and The UCSC Genome Browser are commonly used for the initial alignment of mRNA transcripts of different species. Regions of sequence identity are typically preferred targets for cross-species siRNA design and will serve as the basis for subsequent analyses. When there is limited identity, ‘cross-species’ siRNAs can be developed by carrying out additional screenings in human cells to identify hits that are partially complementary (with a few mismatches) to the mRNA of the species of interest as a first step. These human-active compounds indicate sites in the other species that, although not perfectly conserved by sequence, are nevertheless accessible to the RNAi machinery. Human-active siRNA sequences can then be reprogrammed to fully match the target sites in the species of interest for testing. Informatics and biological validation will still be needed to confirm efficacy and identify potential off-target effects. This approach may help in the development of closely related ‘tool’ compounds for human siRNA preclinical development. In other cases, tool compounds with different sequences but an identical scaffold configuration to the active human compound may be developed.
Oftentimes, there is a degree of variation in transcriptomics between humans and experimental species. Making the prediction to minimize off-target effects in both species is challenging. This is, in part, due to the limited availability of well-annotated sequencing information, necessitating open access to more validated datasets. Sometimes cross-species siRNAs effectively silence human targets but show little-to-no activity in other species or vice versa99,100. In such cases, target silencing is typically validated in vitro using human cell- or tissue-based models, whereas molecular toxicity can be assessed in animal species, even in the absence of target engagement. A separate ‘tool’ compound with a similar or different sequence but an identical scaffold to the human lead can be used to assess the safety of on-target silencing in vivo. The biological mechanisms that mediate cross-species differences in activity remain poorly understood and could be multifactorial. Computational tools to fully address these challenges are not yet available, and their development represents a highly important direction.
Delivery principles
The design principles of optimized chemical scaffolds are similar for all delivery strategies. Without delivery conjugates, fully chemically stabilized siRNAs would essentially be ineffective. More than 90% of the systemically injected siRNA (which is smaller than the renal clearance threshold of 40–60 kDa101) would be rapidly eliminated by the kidney, resulting in low bioavailability102,103. Moreover, in the absence of serum protein binding and targeting entities, compounds would not be efficiently distributed through target tissues or taken up by cells.
Functional delivery of siRNA to target tissues or cell types is affected by various factors, including route of administration, biological barriers, extravasation, tissue or cellular uptake and endosomal escape. LNPs and conjugates are clinically approved approaches for siRNA delivery. The development of LNPs has a long history with many chemistries available. LNP-mediated delivery represents one of the earliest methods to demonstrate effective gene silencing in humans8,104 and is the basis of the first approved siRNA drug, patisiran9,105. Significant advances have recently been achieved in delivering nucleic acid-based therapeutics (for example, mRNA, CRISPR–Cas gene-editing systems and siRNA) to extrahepatic tissues, including lung, spleen and solid tumours, with improved selectivity to certain cell types106–110; this will potentially open new avenues for the functional delivery of siRNAs to these tissues using the established LNP platforms. Currently, conjugate-mediated delivery is the dominant platform for siRNA delivery in the clinic.
Current tissues amenable to delivery are expanding following the success of LNP- and GalNAc-mediated liver delivery. For non-liver tissues, the central nervous system (CNS), eye, lung, muscle and skin show promising progress; additional tissues, including kidney, heart, fat, placenta and pancreas, are under active exploration. This expansion is mainly attributed to recent innovations in delivery platforms and modes of administration. Whereas selective delivery of siRNA to the liver is straightforward with a GalNAc conjugate, selective delivery to CNS, eye, lung and skin is achieved via local administration. Systemic delivery is used to reach muscle, kidney, heart, fat, placenta and pancreas; although this approach is not selective, siRNAs preferentially accumulate in some cell types in these tissues at acceptable dose levels.
Systemic delivery
Conjugate-mediated delivery.
Conjugation of targeting entities to siRNA enables targeted or preferential tissue delivery without complex formulation. GalNAc-conjugated siRNA binds to the surface asialoglycoprotein receptor (ASGPR) to prompt hepatocyte uptake through endocytosis16. Currently, five products that use GalNAc for delivery (that is, givosiran, lumasiran, inclisiran, vutrisiran and nedosiran) are on the market. Among them, vutrisiran targets transthyretin in the liver for the treatment of hereditary transthyretin-mediated amyloidosis and is given every 3 months through subcutaneous injection. Patisiran targets the same gene but is formulated in LNP for intravenous (i.v.) infusion every 3 weeks. Although the two drugs show comparable clinical efficacy111, the convenient and less-frequent dosing regimen of vutrisiran will likely increase its popularity.
GalNAc-mediated delivery to hepatocytes represents a unique case, whereby together the natural filtering function of the liver, high blood flow, fenestrated endothelium and fast recycling of ASGPRs16 result in robust efficacy and durability. Notably, GalNAc conjugation also enables the delivery of various other types of oligonucleotide to the liver, making this approach applicable to a wide range of conditions112. Currently, several targeting conjugates, including antibodies, engineered proteins, peptides, aptamers and lipids, are under development for siRNA extrahepatic delivery (Fig. 2a).
Extrahepatic delivery.
Systemic delivery of siRNA to non-liver tissues is conceptually complex and requires a better understanding of the principles that drive clearance and biodistribution (Fig. 3a). The biodistribution of siRNA can be influenced by routes of administration, nuclease stability and physiological features of clearance organs. Liver and kidney are the main clearance organs for systemically injected siRNAs. The high rate of blood flow, discontinuous endothelium of the liver and the natural filtration function of the kidney eliminate most of the injected compounds before they reach sufficient systemic accumulation (Fig. 3b).
Fig. 3 |. Strategies for altering pharmacokinetic profiles to improve siRNA systemic distribution and promote extrahepatic efficacy.

a, Systemic distribution is a multistep process that requires optimization of small interfering RNA (siRNA) molecular configuration to improve the efficiency. b, Liver and kidney are two major clearance organs that limit systemic distribution. High blood flow rate and discontinuous membrane of liver capillaries lead to a high level of accumulation of systemically injected compounds. Kidney filtration is a natural process of siRNA clearance with molecular size below the molecular weight threshold. The overall hydrophobicity and hydrophilicity ratio of the siRNA scaffold has a significant impact on tissue clearance mode, with more hydrophobic siRNA being cleared in the liver and more hydrophilic siRNA being filtered by the kidney. c, Conjugation enables improvement of the area under the curve (AUC) of plasma concentration upon systemic administration. d, Pharmacokinetic (PK) modifiers increase the scaffold overall molecular size that drives AUC improvement. e, Valency of the siRNA has a significant impact on molecular size to reduce systemic clearance. f, Targeting entities that associate with serum lipoproteins and albumin during circulation can take advantage of the natural processes to improve the siRNA PK profile for systemic distribution. HDL, high-density lipoprotein; i.v., intravenous; LDL, low-density lipoprotein; MW, molecular weight; s.c., subcutaneous; VLDL, very low-density lipoprotein.
In general, the overall hydrophobicity-to-hydrophilicity ratio of the scaffold defines the siRNA clearance mode. Highly hydrophobic compounds are cleared more in the liver, whereas highly hydrophilic compounds are cleared more by the kidney. Interestingly, unconjugated siRNAs can efficiently accumulate in kidney proximal tubule epithelial cells103,113,114. Achieving accumulation in kidney is relatively easy, but most of the compound is tissue-entrapped and nonproductive. Indeed, target silencing in the kidney requires almost two orders of magnitude higher accumulation (>100 μg g−1) of siRNA than tissues such as muscle and the CNS, where typically 1–2 μg g−1 is sufficient49,103. Thus, for the kidney, additional chemical advances, such as enhanced stability and endosomal escape, are needed to improve efficacy. The kidney is an organ of interest for many renal disorders; successful delivery of siRNAs to additional cell types, such as glomerular endothelial cells and podocytes, would unlock unprecedented opportunities115,116.
The liver-to-kidney ratio related to siRNA clearance mode appears to be more complicated when it involves other factors, such as conjugate variants and molecular size, which can all impact the in vivo behaviours of the siRNA. Below, based on the available literature and unpublished data, we propose several models of plasma clearance kinetics typically observed in siRNA therapeutic designs.
Unconjugated siRNAs are quickly cleared from the bloodstream after i.v. injection. With conjugation, their systemic distribution can be enhanced with an improved area under the clearance curve (Fig. 3c). By comparison with subcutaneous injection, i.v. injection results in a higher maximal plasma concentration (Cmax), but it is associated with faster clearance38,117. Designs that allow the incorporation of PK modifiers can increase the overall size of an siRNA; approaching a molecular weight of 40–60 kDa can lead to a significant reduction in renal clearance102 (Fig. 3d). Likewise, multivalent scaffold designs (Box 3) can significantly influence siRNA clearance kinetics owing to the multiplication of the molecular weight of monomeric compounds, thereby may also reduce kidney filtration (Fig. 3e). A primary mechanism by which hydrophobic conjugates improve the plasma half-life of siRNAs is binding to serum lipoproteins118, such as high-density lipoprotein (HDL) and low-density lipoprotein (LDL). This trafficking mechanism enhances the delivery of siRNAs to tissues enriched with lipoprotein receptors. Serum albumin – the most abundant protein in the blood – can also serve as an excellent natural carrier of siRNA for systemic distribution119. Therefore, conjugates or molecular characteristics that promote siRNA association with lipoproteins and albumin can greatly benefit extrahepatic delivery (Fig. 3f).
Box 3. Multivalent scaffolds for local siRNA delivery.
Multivalent scaffolds, designed to covalently link two or more small interfering RNAs (siRNAs) that target the same gene, can be chemically made in a relatively straightforward synthetic scheme. In optimal designs, branched linkers are used to simultaneously elongate multiple passenger strands, followed by annealing of the guide strands. The direct impact of these scaffolds is the increase in molecular weight, which alters the pharmacokinetic properties of the siRNA construct. As discussed, when injected systemically, the high-valency nature of the siRNAs can help to reduce renal clearance and promote systemic distribution. When delivered locally, this property could also reduce clearance from systemic absorption, improve siRNA retention and enable widespread tissue distribution.
The effect of multi-valency (that is, di-, tri- and tetravalent scaffolds) on siRNA has been systematically evaluated in a recent study for lung delivery to treat severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) viral infection52. Intratracheal administration of the multivalent compounds showed robust tissue distribution and target modulation in a mouse model of SARS-CoV-2 infection. The optimized multivalent scaffolds could be reprogrammed to target other genes. As almost any siRNAs validated as functional could be formulated to generate aerosols for use with inhalers or nebulizers263, numerous opportunities exist to enable non-invasive lung delivery in the treatment of pulmonary diseases.
More recently, intravitreal administration of di-, tri- and tetravalent scaffolds into rodent and porcine eyes has been systematically studied144. A single intravitreal injection of the tetravalent siRNA targeting photoreceptor cell-expressed genes demonstrated safe and multi-month durability. The technology platform may open up opportunities to treat various retinal diseases264–266 with a yearly or twice-a-year dosing frequency. The observed effects in lung and eyes are attributed to the increase in molecular weight of the high-valency designs. These designs could carry a payload of two to four siRNAs within a single molecular entity, thus dramatically reducing clearance and promoting distribution in the tested tissues. Although multivalent scaffolds have shown robust efficacy in the lung and eyes, their therapeutic potential has not been fully realized as these scaffolds could be conjugated to targeting entities to further manipulate the PK properties, which remains to be tested in future studies.
Indeed, lipid conjugates, such as docosanoic acid (DCA) and phosphocholine (PC)–DCA, profoundly improve siRNA systemic distribution, demonstrating efficacy in several extrahepatic tissues, including muscle, heart and fat103. The level of extrahepatic accumulation is superior to that of cholesterol conjugation. How the structure of the lipid conjugate affects distribution is not fully elucidated, but the higher level of DCA-driven extrahepatic uptake is likely due to a combination of lipoprotein association, preferential endothelial interaction and trans-vascularization properties103,118. In addition to demonstrating better extrahepatic uptake, this class of compounds affords improved safety profiles; whereas cholesterol-modified compounds are toxic at doses approximating 100 mg kg−1, saturated lipid-conjugated compounds are safe at this dosing level50. Further chemical engineering of lipid conjugates can enhance efficacy or safety. Several strategies are being used, including increasing valency, modification of the conjugate head group120 and building complex artificial architectures. Several PC–DCA-conjugated siRNAs are currently being evaluated for the modulation of sFLT1 in a phase I clinical trial to treat pre-eclampsia36,121. A class of hydrophobic siRNAs conjugated to the 2′-O-hexadecyl (C16) lipid is widely used both preclinically and clinically. Although this conjugate shows limited systemic efficacy, it demonstrated robust clinical results upon local administration into the CNS, eye and lung (discussed in the following section)51.
Other types of conjugate that demonstrate robust systemic extrahepatic efficacy include antibodies or antibody fragments122–124 and engineered proteins125. These conjugates also significantly increase the overall size of the siRNA construct, thus promoting systemic distribution by reducing renal clearance, similar to PK-modifier and multivalent designs.
The most advanced clinical programme of the antibody-siRNA conjugate platform uses a monoclonal antibody to the transferrin receptor 1 (TfR1) for skeletal, cardiac and smooth muscle delivery. In a preclinical study, i.v. administration of a single 6 mg kg−1 dose of the anti-TfR1–siRNA targeting the SSB mRNA (encodes small RNA-binding exonuclease protection factor La) resulted in up to 75% target downregulation123. A phase I/II study in human patients using anti-TfR1–siRNA to target the myotonin-protein kinase (DMPK) for the treatment of myotonic dystrophy type 1 demonstrated robust improvement in disease outcome (see Related links).
Centyrins are a class of small, engineered human protein derivatives that are being harnessed for siRNA extrahepatic delivery125. The stability and favourable in vivo properties of this type of targeting entity can be achieved by varying certain amino acids within select structural regions. The centyrin–siRNA conjugate platform has shown promising systemic delivery properties. An advanced clinical programme that recently moved to phase I trial uses centyrin-conjugated siRNA (ABX1100) for muscle delivery to silence the glycogen synthase 1 (GYS1) gene for the treatment of Pompe disease (see Related links). Led by the strong promise of this strategy, multiple other pipelines are currently under development to further advance the technology.
Peptide conjugates are expected to expand as an siRNA delivery platform. Peptides can be chemically synthesized, which may support less-complex CMC development. Glucagon-like peptide 1 (GLP1), a ligand of the GLP1 receptor, has been successfully used to deliver ASOs into pancreatic islet cells126. This early work resulted in significant effort in exploring peptide chemical space for extrahepatic delivery of siRNA. Although there is limited information available publicly, many academic laboratories and pharmaceutical companies are working on focused and unbiased selection strategies to further explore the potential of peptide conjugation as a promising strategy for siRNA delivery.
Aptamers were among the first conjugates to be explored for delivery of siRNAs and other oligonucleotide-based therapeutics24,127–134, but they have not gained significant traction in the translational space, possibly owing to limited durability observed in early attempts. More recently, however, aptamers have yielded notable advancements in animal models of disease, especially for targeting certain biomarkers involved in oncology135,136 and thus hold potential for future clinical translation.
Another strategy to improve systemic delivery involves the use of nanoconstructs as siRNA carriers137–142. Molecularly defined nanoconstructs with appropriate shape and size can be designed to incorporate siRNA strands with high programmability. Most studies have used only unmodified or partially modified siRNA configurations, and in vivo characterization of these constructs is often less explored. Certain high-payload designs to incorporate large numbers of siRNAs may trigger undesired immune responses. Thus, to harness nanoconstructs for therapeutic applications in vivo, future exploratory efforts are needed to advance our understanding of how to avoid potential immunogenicity and improve payload release.
Local delivery
Local administration of optimized siRNAs into the CNS, eye, lung and skin has shown robust efficacy49,52,99,143,144. Direct delivery of compounds to these relatively enclosed or easy-to-access tissues enhances retention and ensures an adequate supply of compounds for cellular uptake. As noted above, molecular characteristics of siRNAs – for example, hydrophobicity and molecular size – also reduce clearance and promote uptake. For example, conjugation of a C16 lipid chain to a 2′-ribose on the passenger strand (Fig. 4a) enables safe, potent and multi-month silencing in CNS, eye and lung of rodents and non-human primates (NHPs)143. In practice, when siRNAs are administered locally, various designs can be tested to optimize scaffold configurations. Indeed, different scaffolds can generate distinct cell-type uptake profiles51,52,99,145. Thus, determining where a particular scaffold accumulates can help to achieve selectivity, which in turn requires knowledge of the cell-type expression profiles of disease targets. Currently, there are numerous ongoing activities in the field that are expected to expand the utility of siRNA drugs through local delivery.
Fig. 4 |. Local delivery of siRNAs.
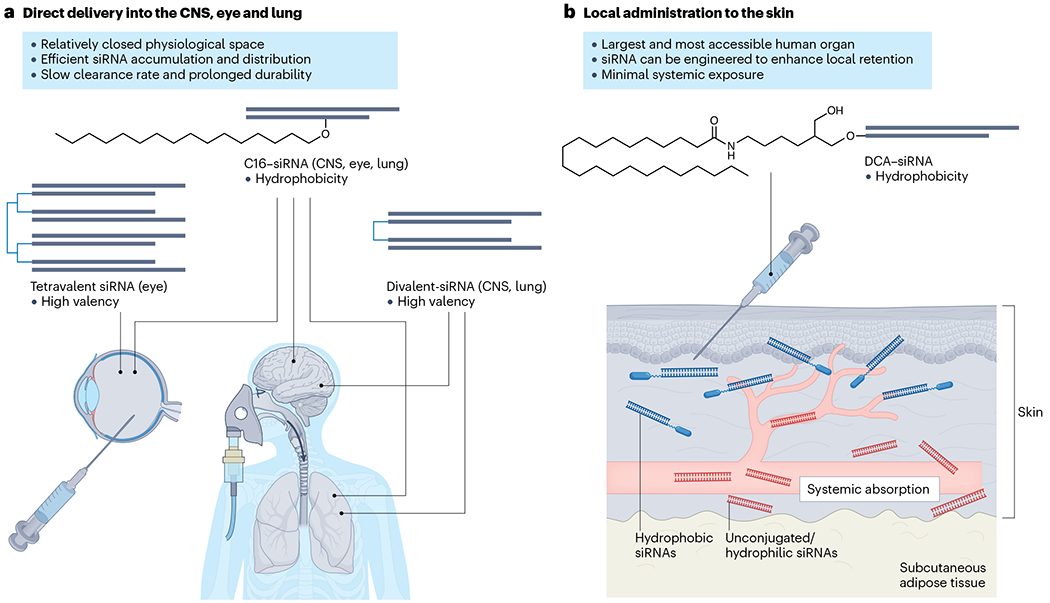
a, Representative examples of optimized small interfering RNA (siRNA) configurations for local delivery into accessible tissues including central nervous system (CNS), eye and lung. Lipophilic conjugates such as 2′-O-hexadecyl (C16) supports effective delivery into multiple tissues attributed to the increase in hydrophobicity of the siRNA. The high-valency design allows the manipulation of size and structure of the siRNA scaffold, which provides enhanced distribution and reduced clearance properties for certain in vivo applications. b, Skin is the largest and most accessible human tissue for siRNA local administration (transdermal and intradermal). Systemic exposure of siRNA could be minimized through increasing the hydrophobicity (thus better skin retention) of the scaffold to treat localized skin diseases. Unconjugated or hydrophilic conjugates have limited local retention and cellular uptake, and thus lead to more systemic absorption. Topical application of siRNA molecules crossing the outermostskin layer (stratum corneum) poses inherent challenges that require innovations in delivery formulations. DCA, docosanoic acid.
Central nervous system.
Delivering siRNA therapeutics to the CNS is feasible via direct injection into brain regions or infusion into the cerebrospinal fluid (CSF). The dosing regimen is an important consideration in delivering siRNA to the CNS; indeed, the brain and spinal cord have a diverse composition, and siRNA often accumulates unequally, perhaps related to the flow of CSF146. For example, direct injection of divalent siRNA led to a highly local accumulation, which might prove useful for delivering antitumour therapies. Infusion of divalent siRNA into CSF supported widespread distribution and function, with high levels of accumulation in regions with high CSF exposure (for example, hippocampus, thalamus, cortex and spinal cord) and lower levels of accumulation in deeper regions (for example, caudate and putamen). Repetitive administration may help to reduce this unequal distribution. Thus CSF infusion of therapeutic siRNAs might be suitable to treat neurodegenerative disorders that affect cortex, hippocampus and spinal cord involvement, such as amyotrophic lateral sclerosis147 and Alzheimer disease148.
Skin.
Skin represents the largest and most accessible human organ for local siRNA delivery. Topical delivery of large molecules such as siRNAs across the outermost layer of the skin barrier (that is, the stratum corneum) is challenging. Although many earlier studies using various topical application methods have been conducted149–155, their clinical translation is limited, particularly when complex formulation is involved. Intradermal injection – commonly used in the clinic to administer therapeutics for localized dermatological indications – is a viable method for siRNA delivery. Hydrophobic conjugates can significantly improve retention of siRNA in the skin after local injection103 (Fig. 4b) and support functional modulation of disease pathways in rodent and ex vivo human skin99,156. Future work to test intradermal injection of the optimized siRNA platforms in pig models, which better resemble human skin, may provide insights into siRNA PK–PD properties and facilitate clinical translation.
Ex vivo transplantation.
Delivery of siRNAs to ex vivo organs for transplantation is a potentially exciting application157,158. Machine perfusion delivery of siRNAs that silence genes involved in ischaemia–reperfusion injury159–161 can help to preserve transplant quality. Delivery of siRNAs that silence immune factors may help to reduce fast graft rejection, but additional strategies are likely needed to prevent rejection in the long term. This is definitely an area to watch.
Clinical development considerations
SiRNA drug development differs significantly from that of traditional small molecules and biologics. The main difference is that the PK properties and target specificity of a siRNA can be independently optimized21. The PK properties are defined by the overall chemical scaffold and conjugation (dianophore), whereas the target specificity is primarily defined by the guide strand sequence (pharmacophore) (Fig. 2a). Therefore, the pipeline demands chemical optimization to improve delivery and bioinformatics to improve target specificity. Although computational design can profoundly accelerate siRNA drug discovery, as discussed, it sometimes faces challenges (Box 2). Notably, the success rate of design and development of a siRNA drug candidate towards clinical validation, on average, is substantially higher than that achieved with conventional pipelines. Below, we highlight key considerations for siRNA drug development.
Clinical indication and target selection
The successful development of an siRNA drug for a clinical indication requires selection of a relevant target. The optimized siRNA delivery platform should align with the tissue in which the target is expressed (Fig. 5), and any potential on-target but off-tissue silencing should be safe. For example, when treating liver-related indications using GalNAc-conjugated siRNAs, the causative disease target should be expressed by hepatocytes. For neurological indications, several optimized platforms (for example, C16 and divalent siRNAs) can support a broad range of cell-type distribution in the CNS. Currently, multi-target modulation is gaining popularity, as many complex diseases are often caused by the dysregulation of more than one gene162–164 (Box 4).
Fig. 5 |. Clinical indication and target selection for RNAi modulation.
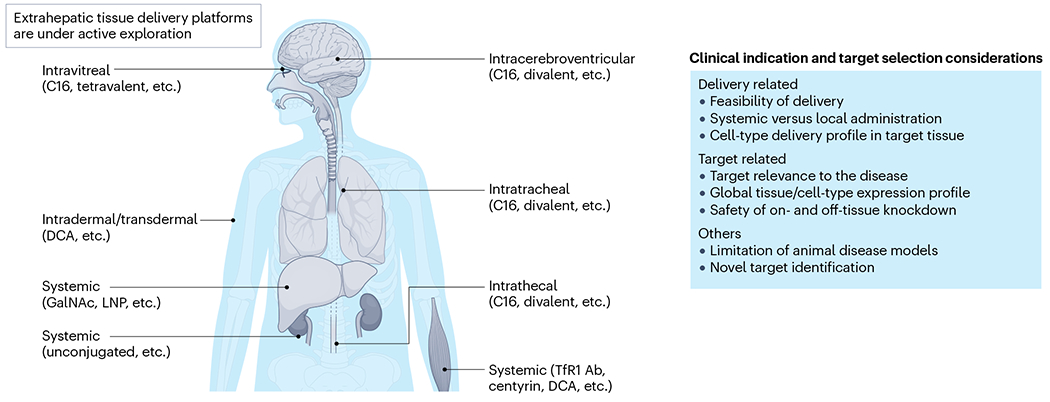
Clinical indication and target selection should be aligned with the feasibility of current optimized delivery platforms (locally or systemically) to target tissues or cell types. The listed small interfering RNA (siRNA) conjugates or designs are approved or at least have shown robust animal or clinical proof of concept (discussed in the main text). Certain tissues such as muscle can be delivered to by either intramuscular or systemic injections depending on the platform. Systemic administration of unconjugated siRNAs to kidney is mainly through clearance mode mechanism, the compounds are mostly accumulated in proximal tubule epithelia; further technology innovation may expand the deliverable cell types in the kidney. Ab, antibody; C16, 2′-O-hexadecyl; DAC, docosanoic acid.
Box 4. Multi-targeting siRNA drugs.
Many complex diseases are often caused by the dysreguiation of more than one gene162–164. Targeting multiple genes is likely necessary for the treatment of these diseases, particularly cancers and neurological disorders196,267–269. The ability to simultaneously modulate several genes with flexibility in target selection, including targeting those previously considered ‘undruggable’, is highly attractive. This can be achieved through unimolecular multi-targeting small interfering RNA (siRNA) designs capable of carrying different siRNA sequences86,270–272. Such designs allow the modulation of genes from the same pathway or different pathways, potentially benefiting clinical treatment outcomes. In this direction, there are many opportunities to further advance multi-targeting siRNA designs, especially for those capable of targeting three or more genes and possessing drug-like properties suitable for clinical translation.
One of the key advantages of multi-targeting siRNA drugs over combination therapies is the predictable biodistribution of drug molecules targeting different genes as a whole. This advantage can greatly benefit preclinical development by simplifying the characterization of in vivo pharmacokinetic properties. Additionally, the unprecedented programmability of the unimolecular multi-targeting siRNA scaffolds allows accommodation of virtually any combination of targets of interest. The concept could be applied to treat neurodegenerative diseases, such as Alzheimer disease, that are associated with overactive neuroinflammation, as well as inflammatory diseases that involve dysregulation of multiple genes.
Currently, dual-gene targeting scaffolds have been developed and been demonstrated to have successful in vivo activity by several biopharmaceutical and academic laboratories, each using different designs86,273 (see Related links ‘The Alnylam GEMINI platform’). More complex scaffolds capable of incorporating three or more different siRNAs are under active exploration, with some successful outcomes that have not yet been published. The modulation of multiple genes using a single molecular construct might represent the next generation of innovations in siRNA drug development.
Target selection and validation.
Definition of the underlying cause of a disease is essential for target selection. Although many genes are ‘identified’ as disease targets by analysing upregulated biomarkers in disease tissues or animal models, often, the knockdown or inhibition of these genes does not translate into therapeutic efficacy. This is either because the targets are irrelevant (bystanders in the dysregulated pathways) or compensating pathways exist for the pathology. Thus, the validity of a disease target must be substantiated by strong evidence for its role in driving the disease. Unfortunately, the molecular characteristics of many human diseases are challenging to model in animals. Target validation can benefit from more innovative approaches and better access to biopsy samples from human patients. Advances in genetics, multi-omics and artificial intelligence for target prediction, based on large numbers of complex human datasets, have shown strong promise165–168.
The fundamental factor that governs target selection is the ability to deliver siRNA to the target tissue with robust efficacy and acceptable safety. As discussed, delivery to the liver is fully validated, and robust animal data exist for other tissues, such as CNS, lung, eye, muscle and skin. A second key factor is understanding the distribution and clearance profile. Although selective delivery and silencing has been achieved for the liver, selective delivery to other organs remains a challenge. For example, most muscle-targeting conjugates (for example, antibodies, DCA, and so forth) would also deliver to and induce target silencing in heart and liver. For therapeutic applications, therefore, developers should ensure the target is preferentially expressed in the intended tissue or that off-tissue modulation is benign.
If a disease target is highly upregulated, potentially surpassing the capacity of the RNAi machinery, an alternative strategy might be to target upstream signalling factors that interact with that target. Another approach might be to include additional targets using a multi-targeting siRNA, or a combination therapy with other drug modalities, to achieve potential synergistic effects. Simultaneously, target selection also requires consideration of the influence of mRNA turnover rates on siRNA efficacy. Studies show that mRNAs with high turnover rates can be more resistant to RNAi-mediated silencing169.
The proliferation rate of target-expressing cell types can also dramatically affect siRNA potency and durability. Fast-dividing cells dilute the cellular concentration of internalized siRNAs over time. Thus, RNAi activity may be reduced in actively dividing cells (for example, in tumour cells170 and epidermal keratinocytes171), requiring repetitive dosing to maintain silencing efficacy. In contrast, siRNA durability typically lasts longer in cell types in a non-dividing state, such as neurons172.
Another crucial consideration is clinical validity. For a long period of time, it was believed that siRNAs could not compete with small molecules and biologics; thus any targets for which there was already an effective drug were not considered. However, the potential for twice a year or yearly dosing frequency, which supports practical convenience and patient compliance, is now making siRNA a highly attractive alternative modality. The most obvious example is inclisiran, which targets proprotein convertase subtilisin/kexin type 9 (PCSK9) and lowers dysregulated cholesterol levels, although there are already several approved biologics on the market173.
The PK–PD of siRNAs can offer advantages over small molecules. For example, mismatch repair factor MutS homologue 3 (MSH3) promotes somatic repeat expansion in the Huntington disease gene (HTT), encoding hungtingtin174, and silencing MSH3 might offer a strategy to prevent expansion of HTT repeats. Silencing a factor required for maintenance of genomic stability raises concerns about potential genotoxicity and immunogenicity, but disease-causing MSH3 mutations are mostly related to the carcinogenesis of colorectal cancers175,176. The ability to deliver MSH3-silencing siRNAs to the CNS but not to the colon100 provides a competitive advantage over small molecules, which would generally act in many tissues, including the colon. The mismatch repair pathway is most active in dividing cells177, so silencing MSH3 in the brain is less likely to be a problem in neurons, but it could be a problem in microglia. This hypothesis requires testing in future studies.
On-tissue target selectivity.
Selective delivery to target tissues through systemic administration is intrinsically difficult. Although mostly cleared in liver and kidney, many siRNA designs accumulate sufficiently in certain tissues to induce functional gene silencing. To avoid potential side effects from off-tissue silencing, a useful strategy is to focus on therapeutic targets that are preferentially expressed in one of the siRNA-deliverable tissues (see The Human Protein Atlas database (see Related links) for target expression information). For example, myostatin (encoded by MSTN) is a myokine that is primarily produced in skeletal muscle and acts on myocytes to inhibit muscle growth. Therapeutic modulation of myostatin may be beneficial for treating muscular dystrophies178–180. An illustrative POC study is the use of a hydrophobic conjugate DCA to systemically deliver myostatin-targeting compounds to muscle to increase growth50. The accumulation of DCA-conjugated siRNAs in multiple tissues is a minor concern103, because myostatin is preferentially expressed in the muscle, with little or no expression in other tissues. In the case of universally expressed genes across multiple tissues, systemic targeting may be tolerable, but it requires careful evaluation through safety studies, as off-tissue target silencing could potentially carry liabilities.
In vitro screening and lead identification
Screening siRNAs in a disease-relevant cell model that expresses the target transcripts is ideal. However, difficulties often exist in practice, including the limited availability of suitable cell lines and the low expression level of the target. Additional challenges include handling cell lines that require complex procedures or are resistant to siRNA transfection and passive delivery methods. An alternative strategy is to use a reporter system to identify functional hits, followed by biological validation in a more complex model system52,121. As discussed, reporter systems might not correlate with native targets and will require additional screening and validation.
Both the IC50 values and the maximal silencing efficacy are important for lead selection from functional hits. Ideally, an optimal lead should demonstrate superior maximal silencing efficacy and a low IC50 value. In cases where these two factors are not aligned, an evaluation is needed to find the compromise. In general, in vitro to in vivo translation is not absolute, thus several in vitro leads should be evaluated in vivo, but this requires more laborious work. Importantly, comparing IC50 values determined under disparate experimental conditions is not advised for selecting the lead compound, as IC50 values are significantly influenced by many factors, including cell line type and delivery method.
Lead optimization
Lead optimization is a crucial step for bridging in vitro screening and in vivo validation. Scaffold and nucleotide modifications (for example, 2′-OMe, 2′-F or PS backbone) can significantly affect siRNA activity, so any major changes during optimization must be tested to compare activity against the parental compound. The focus of lead optimization is mostly on conjugation, valency and the addition of extra stabilization modifications, to improve in vivo delivery, potency and durability. Typically, lead variants are systematically generated and evaluated in parallel to identify the best configuration for further development. Although this process can be carried out in vitro depending on the purpose (for example, RISC compatibility), in vivo evaluation is preferred to better understand the therapeutic behaviour in the complex biological context. Lead compounds should be meticulously purified and quality-controlled. A quick in vivo validation of the optimized lead to confirm target silencing would be helpful before committing to comprehensive and resource-intensive preclinical safety and efficacy studies.
Preclinical evaluation
Ideally, the efficacy of the lead compound should be evaluated in a relevant disease model. However, the lack or limited availability of animal models for many human diseases poses significant challenges. Additionally, the relevance of an animal model – that is, whether it accurately resembles the human disease characteristics181–183 – is essential, particularly for inflammatory disorders and cancers where the exact cause of the disease might be unknown. In the case of other diseases, such as Huntington disease184,185 and myotonic dystrophy186, in which the underlying mechanisms are clearly defined at the genetic level, siRNA design and preclinical evaluation are more straightforward, and often more advantageous, than conventional therapeutic modalities, especially when humanized transgenic models are established. Whereas the efficacy and safety evaluation of many siRNA drug candidates typically requires a distinct reiteration process, PK studies and dosing regimen optimization will generally follow a more standardized process.
Clinical validation
Significant progress has been made in the past two decades in understanding how to evolve drug-like properties of siRNA molecules, which has helped many preclinical programmes to succeed to clinical trials. Despite favourable preclinical safety profiles of siRNAs in rodents and NHPs187, uncertainties may persist at clinical validation, as seen in the phase III trial of revusiran, which resulted in unexpected mortality12. The exact cause (or causes) of failure remain unclear despite extensive investigations, as discussed in detail above34,39, and emphasize the need for thorough safety evaluation at all stages of siRNA drug development.
Of note, the short timeline of siRNA lead identification and preclinical development supports high feasibility of clinical trials for individual patients with rare genetic diseases or advanced-stage cancers that fail to respond to existing treatments. These N-of-1 studies represent exceptional opportunities for fast-track clinical validation and development of siRNA drugs, thus benefiting the treatment of additional patients188. This field is rapidly progressing, as shown by the recent efforts to advance ASO drug development189 (see Related links ‘FDA takes steps to provide clarity on developing new drug products in the age of individualized medicine’ and ‘FDA takes new steps aimed at advancing development of individualized medicines to treat genetic diseases’).
Future directions
We are witnessing a notable shift of pharmaceutical development towards nucleic acid-based medicines. siRNAs stand out with distinct advantages over conventional drugs, including their ability to silence targets previously considered ‘undruggable’, their shorter development timeline and their prolonged clinical durability (Box 5).
Box 5. Advantages of siRNAs over conventional drugs.
Certain proteins83,274,275, protein isoforms (encoded by mRNA isoforms of the same gene)121 and regulatory RNAs276 were once considered ‘undruggable’ when there was a need for discriminative targeting in a disease state. These clinically meaningful targets offer great therapeutic opportunities but pose significant challenges for conventional drug modalities. This is mainly because many protein targets lack proper ligand binding sites or have limited properties for allosteric inhibition by small molecules, and intracellular targets are inefficient for therapeutic intervention using biologics. Additionally, modulation of regulatory RNAs is also challenging using small molecules and biologics. Thus, the ability of small interfering RNA (siRNA) drugs to expand the druggability of many well-defined targets at the RNA level through a simple base-pairing mechanism could profoundly influence the future of pharmaceutical development.
The ‘informational’ nature of RNAi-based drug design could significantly shorten the development timeline, especially with the streamlined platforms for high-throughput chemical synthesis, in vitro screening and in vivo validation. Typically, the identification of an siRNA drug lead candidate requires only 1–2 months, a time-frame rarely seen in the process of identification and medicinal chemistry optimization of small-molecule drug candidates. After establishing a tissue delivery platform, it becomes possible to develop a class of siRNA drugs that target many more genes within the same tissue. On average, the overall cost in an siRNA drug pipeline is significantly lower than in conventional drug pipelines when considering the short development timeline and high clinical translatability. In addition, the infrequent dosing regimen and multi-month durability of siRNA drugs will undoubtedly reduce the consumer pricing of future siRNA drugs, making them more affordable to patients.
Recent advances in nucleic acid chemistry have allowed further improvement in the durability of siRNA drugs. One or two yearly clinical visits would dramatically relieve the burden on the health-care system and encourage full patient compliance. Additional innovations to further enhance the clinical durability and potency of siRNA drugs are conceivable, considering that many advances used in the currently approved siRNA drugs, once thought difficult, were realized by applying only simple chemical modifications after understanding the fundamental principles at play. For treating many chronic diseases, including ageing-related disorders that once required lifelong repetitive dosing, the potential cumulative side effects from long-term medications may not be a significant concern with siRNA drugs.
As the chemical configurations to enable safe and effective tissue delivery are defined, the cost and time for the development of follow-up drugs that work in the same tissue will be profoundly reduced. We have seen this revolution happen in the development of siRNA drugs that are delivered to the liver. With several technology generations, the latest GalNAc-conjugated siRNAs now support up to 12 months of durability, with unprecedented efficacy and safety profiles. Validation of the tissue delivery platform enables development of a large variety of siRNA drugs, with limitations mostly driven by the biological understanding of the target involvement and disease relevance.
The natural evolution of siRNA drugs that are delivered to the liver included platform development, clinical exploration and target validation for genetically defined rare diseases, extending to highly prevalent disorders (for example, hypercholesterolaemia and hypertension) and many other targets13. Interestingly, RNAi-based drugs have the potential to replace certain small molecules and biologics, owing to their convenient, less-frequent dosing. Gene-editing approaches may eventually outcompete RNAi-based approaches for the treatment of genetically well-defined diseases for which reversal of the therapy would never be desired. Although next-generation gene-editing technologies may provide a one-and-done treatment solution, the durability and safety of siRNA drugs could be a prerequisite for the clinical acceptance of gene-editing approaches. There are several unique advantages of RNAi-based therapies over conventional modalities, making them particularly promising in the following areas.
Isoform-specific modulation
Post-transcriptional processes such as alternative splicing are inherent cellular mechanisms that enable the production of multiple regulatory and coding RNA transcript isoforms from a single gene190,191. This process may result in regulatory RNA and protein isoforms that differ in biological function and pathological roles, which have been suggested in certain cancers and neurodegenerative disorders192–196. Owing to the lack of characterization of structural properties in RNA or protein isoforms, specific modulation of them through conventional small molecules and biologics is highly challenging. In contrast, oligonucleotide-based approaches offer solutions to precisely target disease-relevant isoforms on a post-transcriptional level, to achieve selectivity and improve safety121,195.
The design of isoform-specific siRNAs requires that the target transcript possesses a unique sequence stretch that distinguishes it from non-targeted isoforms (Fig. 6a). Thus, validation of the disease-causative isoform and the difference in the use of alternative exons or UTRs for that specific isoform is needed. Although current genetic databases serve as a valuable starting point, using emerging and powerful tools such as long-read sequencing to ascertain the isoform expression profile in a disease state would greatly add confidence to the design197. As this field is moving forwards, the ability of siRNA drugs to selectively modulate a disease-causative isoform would be unprecedented.
Fig. 6 |. Promising future applications of siRNA drugs.
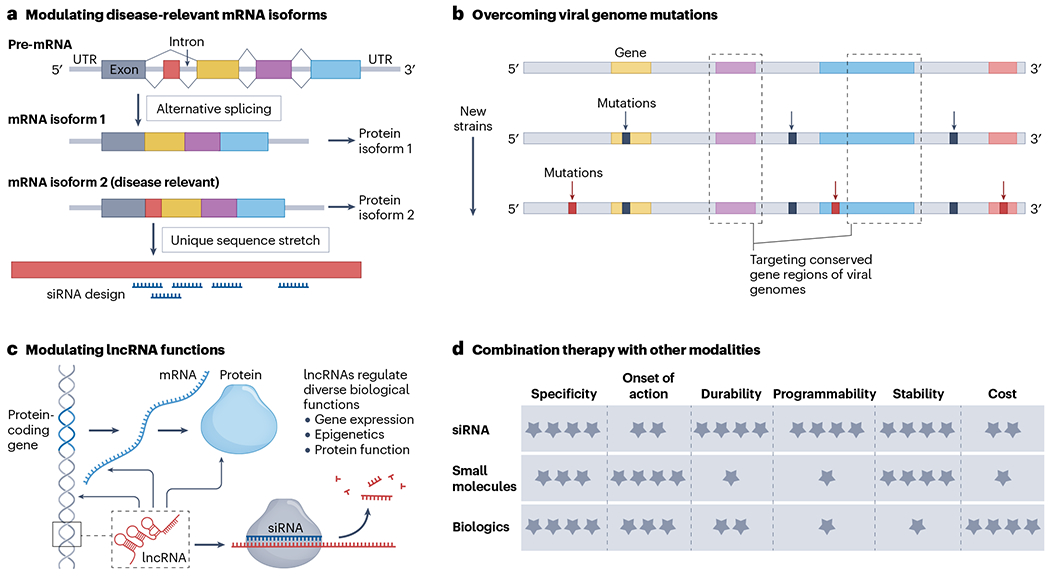
a, RNA splicing is a common post-transcriptional processing mechanism that may result in multiple regulatory or protein-coding transcript isoforms. The action of mechanism of RNAi potentiates the design of small interfering RNA (siRNA) drugs to precisely modulate transcript isoforms, particularly those that are disease relevant. b, Mutations in viral genomes impose substantial challenges for antiviral drugs and vaccines. The discovery pipeline of siRNA drugs not only enables targeting of conserved gene regions of viral genomes to overcome the challenges but also allows rapid design of new variants of antiviral drugs for future strains. c, The expanding knowledge of long non-coding RNAs (lncRNAs) indicate their role in regulation of diverse biological functions. siRNA drugs may be designed to modulate certain lncRNAs that are involved in disease progression. d, Combination of a siRNA drug with another drug modality can benefit the treatment outcome of certain diseases. This benefit could be from the perspectives of both target modulation and clinical practice to combine the advantages of each specific drug modality. UTR, untranslated region.
Viral infections
siRNAs could have an important role in rapid response to emerging viral pathogens and pandemics. The ability to rapidly develop antiviral drugs is an unmet need exemplified by the recent coronavirus disease 2019 (COVID-19) pandemic and its subsequent variants. The lung and liver are currently promising tissues for applying siRNAs, such as for the treatment of viral pneumonia and hepatitis198. Indeed, multiple anti-hepatitis B siRNAs have advanced to clinical validation199. siRNAs could be ideal therapeutic modalities for treating viral infectious diseases. First, siRNAs are designed exclusively against viral RNA components while minimizing off-target effects on host RNAs. Second, by targeting the conserved regions of viral genomes that are essential to maintain viability, siRNAs could cover multiple existing and future strains52,200 (Fig. 6b). Validation of the efficacy of antiviral siRNAs often faces challenges in establishing proper animal models. Although humanized rodent models are valuable for evaluating therapeutic outcomes, the virus infection mechanisms and antiviral responses in NHP models, although costly, closely resemble those of the human immune system, making them the most accurate model for this purpose201.
Modulation of long non-coding RNAs
A large fraction of the human genome does not encode proteins but instead produces non-coding RNAs (ncRNAs) that have regulatory roles in gene expression and protein functions202. Accumulating evidence reveals that ncRNAs, including long non-coding RNAs (lncRNAs), could serve as therapeutic targets for certain diseases203–208. The existing definition of lncRNA includes a diverse group of RNAs, characterized by size in excess of 200 nucleotides. Given the diversity of lncRNAs and their various roles in regulation of biological functions, therapeutic modulation of lncRNAs represents a less-explored but opportunistic direction for drug developers. Although currently there are limited drugs targeting lncRNAs that have achieved clinical productivity, siRNAs and other oligonucleotide-based therapeutics are well suited for this purpose and have high potential to stand out in the future (Fig. 6c).
Combination therapy
Combining siRNAs with other drug modalities, such as small molecules or biologics, may synergistically improve clinical outcomes in treating complex diseases. Practically, combination therapy may also benefit patients by leveraging the advantages of each type of drug modality (Fig. 6d). An ongoing study is the combination of Alnylam’s cemdisiran209 (ALN-CC5; siRNA that targets complement C5 component) with Regeneron’s pozelimab (anti-C5 monoclonal antibody) for the treatment of paroxysmal nocturnal haemoglobinuria210,211. The two companies also announced a broad collaboration (see Related links ‘Regeneron and Alnylam announce a broad collaboration’) that might involve other development programmes. The concept of combination therapy is not entirely new, but often requires certain scientific and regulatory issues that may arise during the co-development of two or more therapeutic agents to be addressed, as per FDA requirements. The clinical success of combination therapies incorporating siRNAs is yet to be determined, but the future potential is easily foreseeable.
Conclusion
RNAi technologies have undergone a remarkable journey, spanning almost the first quarter of the twenty-first century, from RNAi mechanism discovery in C. elegans, demonstration of efficacy in mammalian cells, to the development of a new class of human medicines, especially to treat liver diseases. The current six approved products are based on rational design and entered the market within a relatively short time-frame, highlighting the exceptional productivity of RNAi drug development pipelines. Several advanced clinical programmes, using lipid and antibody–protein conjugates, have shown promising disease-modifying efficacy, which may lead to the next wave of siRNA drug approvals. As molecular design and extrahepatic delivery platforms continue to advance, we anticipate a major expansion in the utility of siRNA drugs across various indications in the near future.
Acknowledgements
The authors acknowledge the nucleic acid–medicine community who contributed to the technology innovations, and whose excellent work could not be included here owing to lack of space. This work was supported by the NIH (grant nos. R35 GM131839 and R01 NS104022 to A.K.; and K99 AR082987 to Q.T.). The authors thank M. B. Dziewietin for her administrative assistance and D. Conte at RNA Therapeutics Institute for help in revising and proofreading this manuscript.
Related links
Nedosiran: https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/215842s000lbl.pdf
Revusiran: https://www.alnylam.de/wp-content/uploads/2017/08/Revusiran-RNAi-Roundtable_FINAL2_08092017.pdf
A phase I/II study: https://www.aviditybiosciences.com/wp-content/uploads/2023/04/230330-AAN-2023-MARINA-Prelim-Data-Oral-Presentation_v6.0_FINAL.pdf
ABX1100: https://www.arobiotx.com/aro-receives-fda-orphan-drug-designation-for-abx1100
FDA Takes New Steps Aimed at Advancing Development of Individualized Medicines to Treat Genetic Diseases: https://www.fda.gov/news-events/press-announcements/fda-brief-fda-takes-new-steps-aimed-advancing-development-individualized-medicines-treat-genetic
FDA Takes Steps to Provide Clarity on Developing New Drug Products in the Age of Individualized Medicine: https://www.fda.gov/news-events/press-announcements/fda-takes-steps-provide-clarity-developing-new-drug-products-age-individualized-medicine
Regeneron and Alnylam announce a broad collaboration: https://investor.regeneron.com/news-releases/news-release-details/regeneron-and-alnylam-announce-broad-collaboration-discover
The Alnylam GEMINI platform: https://capella.alnylam.com/wp-content/uploads/2022/05/Theile_TIDES-2022.pdf
The Human Protein Atlas database: https://www.proteinatlas.org
Footnotes
Competing interests
A.K. is a founder of Atalanta Therapeutics and Comanche Biopharma; serves on the Scientific Advisory Board of Aldena Therapeutics, Prime Medicine, Alltrna, Advirna and Evox Therapeutics. A.K. and Q.T. are Listed as inventors of RNAi technology patents and patent applications.
References
- 1.Morrissey DV et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat. Biotechnol 23, 1002–1007 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Zimmermann TS et al. RNAi-mediated gene silencing in non-human primates. Nature 441, 111–114 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Frank-Kamenetsky M et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc. Natl Acad. Sci. USA 105, 11915–11920 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akinc A et al. Development of lipidoid–siRNA formulations for systemic delivery to the liver. Mol. Ther 17, 872–879 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whitehead KA, Langer R & Anderson DG Knocking down barriers: advances in siRNA delivery. Nat. Rev. Drug. Discov 8, 129–138 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tao W et al. Noninvasive imaging of lipid nanoparticle–mediated systemic delivery of small-interfering RNA to the liver. Mol. Ther 18, 1657–1666 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Semple SC et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol 28, 172–176 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Coelho T et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis.N. Engl. J. Med 369, 819–829 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Hoy SM Patisiran: first global approval. Drugs 78, 1625–1631 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Nair JK et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc 136, 16958–16961 (2014). [DOI] [PubMed] [Google Scholar]; This article reports the use of GalNAc conjugates for targeted delivery of siRNA to hepatocytes.
- 11.Zimmermann TS et al. Clinical proof of concept for a novel hepatocyte-targeting GalNAc-siRNA conjugate. Mol. Ther 25, 71–78 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garber K Alnylam terminates revusiran program, stock plunges. Nat. Biotechnol 34, 1213–1214 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Maraganore J Reflections on Alnylam. Nat. Biotechnol 40, 641–650 (2022). [DOI] [PubMed] [Google Scholar]
- 14.Egli M & Manoharan M Chemistry, structure and function of approved oligonucleotide therapeutics. Nucleic Acids Res. 51, 2529–2573 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article provides a comprehensive review on the chemistry and function of approved oligonucleotide-based drugs by early 2023.
- 15.Setten RL, Rossi JJ & Han S-P The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov 18, 421–446 (2019). [DOI] [PubMed] [Google Scholar]; This review provides an excellent resource for understanding the basic biology of RNAi therapeutics.
- 16.Dowdy SF Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol 35, 222–229 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Dowdy SF, Setten RL, Cui X-S & Jadhav SG Delivery of RNA therapeutics: the great endosomal escape! Nucleic Acid. Ther 32, 361–368 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; References 16 and 17 discuss the fundamental challenges of cellular uptake and endosomal escape of siRNAs.
- 18.Layzer JM et al. In vivo activity of nuclease-resistant siRNAs. RNA 10, 766–771 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gantier MP & Williams BRG The response of mammalian cells to double-stranded RNA. Cytokine Growth Factor Rev. 18, 363–371 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scacheri PC et al. Short interfering RNAs can induce unexpected and divergent changes in the levels of untargeted proteins in mammalian cells. Proc. Natl Acad. Sci. USA 101, 1892–1897 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khvorova A & Watts JK The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol 35, 238–248 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This review discusses chemical modifications that evolve ASO and siRNA therapeutics towards clinical application.
- 22.De Smet MD, Meenken C & Van Den Horn GJ Fomivirsen—a phosphorothioate oligonucleotide for the treatment of CMV retinitis. Ocul. Immunol. Inflamm 7, 189–198 (1999). [DOI] [PubMed] [Google Scholar]
- 23.Kurreck J Antisense technologies. Improvement through novel chemical modifications. Eur. J. Biochem 270, 1628–1644 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Lee J-H et al. A therapeutic aptamer inhibits angiogenesis by specifically targeting the heparin binding domain of VEGF165. Proc. Natl Acad. Sci. USA 102, 18902–18907 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown CR et al. Investigating the pharmacodynamic durability of GalNAc–siRNA conjugates. Nucleic Acids Res. 48, 11827–11844 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hassler MR et al. Comparison of partially and fully chemically-modified siRNA in conjugate-mediated delivery in vivo. Nucleic Acids Res. 46, 2185–2196 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article demonstrates that full chemical modification is essential for improving siRNA in vivo efficacy.
- 27.Allerson CR et al. Fully 2′-modified oligonucleotide duplexes with improved in vitro potency and stability compared to unmodified small interfering RNA. J. Med. Chem 48, 901–904 (2005). [DOI] [PubMed] [Google Scholar]; This paper compares the potency of fully 2′-modified siRNA with unmodified siRNA.
- 28.Manoharan M et al. Unique gene-silencing and structural properties of 2′-fluoro-modified siRNAs. Angew. Chem. Int. Ed 50, 2284–2288 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jahns H et al. Stereochemical bias introduced during RNA synthesis modulates the activity of phosphorothioate siRNAs. Nat. Commun 6, 6317 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schirle NT, Sheu-Gruttadauria J & MacRae IJ Structural basis for microRNA targeting. Science 346, 608–613 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manoharan M 2′-Carbohydrate modifications in antisense oligonucleotide therapy: importance of conformation, configuration and conjugation. Biochim. Biophys. Acta Gene Struct. Expr 1489, 117–130 (1999). [DOI] [PubMed] [Google Scholar]
- 32.Foster DJ et al. Advanced siRNA designs further improve in vivo performance of GalNAc-siRNA conjugates. Mol. Ther 26, 708–717 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blidner RA, Hammer RP, Lopez MJ, Robinson SO & Monroe WT Fully 2′-deoxy-2′-fluoro substituted nucleic acids induce RNA interference in mammalian cell culture. Chem. Biol. Drug Des 70, 113–122 (2007). [DOI] [PubMed] [Google Scholar]
- 34. Janas MM et al. Safety evaluation of 2′-deoxy-2′-fluoro nucleotides in GalNAc-siRNA conjugates. Nucleic Acids Res. 47, 3306–3320 (2019). This article investigates the safety of 2′-F nucleotides in GalNAc–siRNAs.
- 35.Schirle NT & MacRae IJ The crystal structure of human argonaute2. Science 336, 1037–1040 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davis SM et al. Chemical optimization of siRNA for safe and efficient silencing of placental sFLT1. Mol. Ther. Nucleic Acids 29, 135–149 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grimm D et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 441, 537–541 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Nair JK et al. Impact of enhanced metabolic stability on pharmacokinetics and pharmacodynamics of GalNAc-siRNA conjugates. Nucleic Acids Res. 45, 10969–10977 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Judge DP et al. Phase 3 multicenter study of revusiran in patients with hereditary transthyretin-mediated (hATTR) amyloidosis with cardiomyopathy (ENDEAVOUR). Cardiovasc. Drugs Ther 34, 357–370 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Biscans A et al. The chemical structure and phosphorothioate content of hydrophobically modified siRNAs impact extrahepatic distribution and efficacy. Nucleic Acids Res. 48, 7665–7680 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ly S, Echeverria D, Sousa J & Khvorova A Single-stranded phosphorothioated regions enhance cellular uptake of cholesterol-conjugated siRNA but not silencing efficacy. Mol. Ther. Nucleic Acids 21, 991–1005 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y, Sheng G, Juranek S, Tuschl T & Patel DJ Structure of the guide-strand-containing argonaute silencing complex. Nature 456, 209–213 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frank F, Sonenberg N & Nagar B Structural basis for 5′-nucleotide base-specific recognition of guide RNA by human AGO2. Nature 465, 818–822 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Parmar R et al. 5′-(E)-vinylphosphonate: a stable phosphate mimic can improve the RNAi activity of siRNA-GalNAc conjugates. ChemBioChem 17, 985–989 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Haraszti RA et al. 5′-Vinylphosphonate improves tissue accumulation and efficacy of conjugated siRNAs in vivo. Nucleic Acids Res. 45, 7581–7592 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lima WF et al. Single-stranded siRNAs activate RNAi in animals. Cell 150, 883–894 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Yu D et al. Single-stranded RNAs use RNAi to potently and allele-selectively inhibit mutant Huntingtin expression. Cell 150, 895–908 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prakash TP et al. Identification of metabolically stable 5′-phosphate analogs that support single-stranded siRNA activity. Nucleic Acids Res. 43, 2993–3011 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alterman JF et al. A divalent siRNA chemical scaffold for potent and sustained modulation of gene expression throughout the central nervous system. Nat. Biotechnol 37, 884–894 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes the effect of siRNA valency on improving tissue distribution profile and potency in the CNS.
- 50.Biscans A et al. Docosanoic acid conjugation to siRNA enables functional and safe delivery to skeletal and cardiac muscles. Mol. Ther 29, 1382–1394 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brown KM et al. Expanding RNAi therapeutics to extrahepatic tissues with lipophilic conjugates. Nat. Biotechnol 40, 1500–1508 (2022). [DOI] [PubMed] [Google Scholar]; This paper reports on the application of lipophilic conjugates to enhance the utility of siRNA in extrahepatic tissues.
- 52.Hariharan VN et al. Divalent siRNAs are bioavailable in the lung and efficiently block SARS-CoV-2 infection. Proc. Natl Acad. Sci. USA 120, e2219523120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elkayam E et al. siRNA carrying an (E)-vinylphosphonate moiety at the 5′ end of the guide strand augments gene silencing by enhanced binding to human Argonaute-2. Nucleic Acids Res. 45, 3528–3536 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guenther DC et al. Role of a “magic” methyl: 2′-Deoxy-2′-α-F-2′-α-β-C-methyl pyrimidine nucleotides modulate RNA interference activity through synergy with 5′-phosphate mimics and mitigation of off-target effects. J. Am. Chem. Soc 144, 14517–14534 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koller E et al. Mechanisms of single-stranded phosphorothioate modified antisense oligonucleotide accumulation in hepatocytes. Nucleic Acids Res. 39, 4795–4807 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schwarz DS et al. Asymmetry in the assembly of the RNAi enzyme complex. Cell 115, 199–208 (2003). [DOI] [PubMed] [Google Scholar]
- 57.Sano M et al. Effect of asymmetric terminal structures of short RNA duplexes on the RNA interference activity and strand selection. Nucleic Acids Res. 36, 5812–5821 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim D-H et al. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat. Biotechnol 23, 222–226 (2005). [DOI] [PubMed] [Google Scholar]; This paper describes a siRNA variant that requires Dicer processing to enable RNAi activity.
- 59.Snead NM et al. Molecular basis for improved gene silencing by Dicer substrate interfering RNA compared with other siRNA variants. Nucleic Acids Res. 41, 6209–6221 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zamore PD, Tuschl T, Sharp PA & Bartel DP RNAi. Cell 101, 25–33 (2000). [DOI] [PubMed] [Google Scholar]
- 61.Bernstein E, Caudy AA, Hammond SM & Hannon GJ Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409, 363–366 (2001). [DOI] [PubMed] [Google Scholar]
- 62.Chang CI et al. Asymmetric shorter-duplex siRNA structures trigger efficient gene silencing with reduced nonspecific effects. Mol. Ther 17, 725–732 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jackson AL et al. Position-specific chemical modification of siRNAs reduces “off-target” transcript silencing. RNA 12, 1197–1205 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Datta D et al. Rational optimization of siRNA to ensure strand bias in the interaction with the RNA-induced silencing complex. Chem. Commun 59, 6347–6350 (2023). [DOI] [PubMed] [Google Scholar]
- 65.Chen PY et al. Strand-specific 5′-O-methylation of siRNA duplexes controls guide strand selection and targeting specificity. RNA 14, 263–274 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shibata A et al. Terminal bridging of siRNA duplex at the ribose 2′ position controls strand bias and target sequence preference. Mol. Ther. Nucleic Acids 32, 468–477 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rossi JJ Bridging siRNA strands for better function. Mol. Ther. Nucleic Acids 33, 209 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Varley AJ & Desaulniers J-P Chemical strategies for strand selection in short-interfering RNAs. RSC Adv. 11, 2415–2426 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jackson AL et al. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA 12, 1179–1187 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Anderson EM et al. Experimental validation of the importance of seed complement frequency to siRNA specificity. RNA 14, 853–861 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bartel DP MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ferguson CM et al. Silencing Apoe with divalent-siRNAs improves amyloid burden and activates immune response pathways in Alzheimer’s disease. Alzheimers Dement. 10.1002/alz.13703 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Janas MM et al. Selection of GalNAc-conjugated siRNAs with limited off-target-driven rat hepatotoxicity. Nat. Commun 9, 723 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schlegel MK et al. Overcoming GNA/RNA base-pairing limitations using isonucleotides improves the pharmacodynamic activity of ESC+ GalNAc-siRNAs. Nucleic Acids Res. 49, 10851–10867 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schlegel MK et al. From bench to bedside: improving the clinical safety of GalNAc-siRNA conjugates using seed-pairing destabilization. Nucleic Acids Res. 50, 6656–6670 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Egli M, Schlegel MK & Manoharan M Acyclic (S)-glycol nucleic acid (S -GNA) modification of siRNAs improves the safety of RNAi therapeutics while maintaining potency. RNA 29, 402–414 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article introduces GNA modification at the seed region of the guide strand, which could mitigate RNAi-mediated off-targeting effects.
- 77.Yamada K et al. Extended nucleic acid (exNA): a novel, biologically compatible backbone that significantly enhances oligonucleotide efficacy in vivo. Preprint at 10.1101/2023.05.26.542506 (2023). [DOI] [Google Scholar]
- 78.Krützfeldt J et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 438, 685–689 (2005). [DOI] [PubMed] [Google Scholar]
- 79.Zlatev I et al. Reversal of siRNA-mediated gene silencing in vivo. Nat. Biotechnol 36, 509–511 (2018). [DOI] [PubMed] [Google Scholar]
- 80.Ferguson et al. A combinatorial approach for achieving CNS-selective RNAi. Nucl. Acids Res 10.1093/nar/gkae100 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Miller VM et al. Allele-specific silencing of dominant disease genes. Proc. Natl Acad. Sci. USA 100, 7195–7200 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schwarz DS et al. Designing siRNA that distinguish between genes that differ by a single nucleotide. PLoS Genet. 2, e140 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Conroy F et al. Chemical engineering of therapeutic siRNAs for allele-specific gene silencing in Huntington’s disease models. Nat. Commun 13, 5802 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jongejan YK et al. Small interfering RNA-mediated allele-selective silencing of von Willebrand factor in vitro and in vivo. Blood Adv. 7, 6108–6119 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yamada K et al. Structurally constrained phosphonate internucleotide linkage impacts oligonucleotide-enzyme interaction, and modulates siRNA activity and allele specificity. Nucleic Acids Res. 49, 12069–12088 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Belgrad J et al. A programmable dual-targeting di-valent siRNA scaffold supports potent multi-gene modulation in the central nervous system. Preprint at 10.1101/2023.12.19.572404 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huesken D et al. Design of a genome-wide siRNA library using an artificial neural network. Nat. Biotechnol 23, 995–1001 (2005). [DOI] [PubMed] [Google Scholar]
- 88.Shmushkovich T et al. Functional features defining the efficacy of cholesterol-conjugated, self-deliverable, chemically modified siRNAs. Nucleic Acids Res. 46, 10905–10916 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Khvorova A, Reynolds A & Jayasena SD Functional siRNAs and miRNAs exhibit strand bias. Cell 115, 209–216 (2003). [DOI] [PubMed] [Google Scholar]; This article reveals that thermodynamic properties of siRNA can induce bias in duplex unwinding and guide strand selection.
- 90.Boulias K & Greer EL Biological roles of adenine methylation in RNA. Nat. Rev. Genet 24, 143–160 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Das Mandal S & Ray PS Transcriptome-wide analysis reveals spatial correlation between N6-methyladenosine and binding sites of microRNAs and RNA-binding proteins. Genomics 113, 205–216 (2021). [DOI] [PubMed] [Google Scholar]
- 92.Kanoria S, Rennie WA, Carmack CS, Lu J & Ding Y N6-methyladenosine enhances post-transcriptional gene regulation by microRNAs. Bioinform. Adv 2, vbab046 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee M Machine learning for small interfering RNAs: a concise review of recent developments. Front. Genet 14, 1226336 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Monopoli KR, Korkin D & Khvorova A Asymmetric trichotomous partitioning overcomes dataset limitations in building machine learning models for predicting siRNA efficacy. Mol. Ther. Nucleic Acids 33, 93–109 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Birmingham A et al. A protocol for designing siRNAs with high functionality and specificity. Nat. Protoc 2, 2068–2078 (2007). [DOI] [PubMed] [Google Scholar]; This protocol outlines the fundamental principles involved in designing specific and functional siRNAs.
- 96.Birmingham A et al. 3′ UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nat. Methods 3, 199–204 (2006). [DOI] [PubMed] [Google Scholar]
- 97.La Rocca G et al. In vivo, Argonaute-bound microRNAs exist predominantly in a reservoir of low molecular weight complexes not associated with mRNA. Proc. Natl Acad. Sci. USA 112, 767–772 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tang Q et al. Multispecies-targeting siRNAs for the modulation of JAK1 in the skin. Mol. Ther. Nucleic Acids 35, 102117 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tang Q et al. RNAi-based modulation of IFN-γ signaling in skin. Mol. Ther 30, 2709–2721 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.O’Reilly D et al. Di-valent siRNA-mediated silencing of MSH3 blocks somatic repeat expansion in mouse models of Huntington’s disease. Mol. Ther 31, 1661–1674 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schenk S, Schoenhals GJ, De Souza G & Mann M A high confidence, manually validated human blood plasma protein reference set. BMC Med. Genomics 1, 41 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Godinho BMDC et al. PK-modifying anchors significantly alter clearance kinetics, tissue distribution, and efficacy of therapeutics siRNAs. Mol. Ther. Nucleic Acids 29, 116–132 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Biscans A et al. Diverse lipid conjugates for functional extra-hepatic siRNA delivery in vivo. Nucleic Acids Res. 47, 1082–1096 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Davis ME et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 464, 1067–1070 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper presents the initial evidence of RNAi in humans after systemic administration using nanoparticles.
- 105.Adams D et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med 379, 11–21 (2018). [DOI] [PubMed] [Google Scholar]
- 106.Cheng Q et al. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat. Nanotechnol 15, 313–320 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang X et al. Preparation of selective organ-targeting (SORT) lipid nanoparticles (LNPs) using multiple technical methods for tissue-specific mRNA delivery. Nat. Protoc 18, 265–291 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dilliard SA & Siegwart DJ Passive, active and endogenous organ-targeted lipid and polymer nanoparticles for delivery of genetic drugs. Nat. Rev. Mater 8, 282–300 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Da Silva Sanchez AJ et al. Universal barcoding predicts in vivo ApoE-independent lipid nanoparticle delivery. Nano Lett. 22, 4822–4830 (2022). [DOI] [PubMed] [Google Scholar]
- 110.Huayamares SG et al. High-throughput screens identify a lipid nanoparticle that preferentially delivers mRNA to human tumors in vivo. J. Control. Rel 357, 394–403 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Polydefkis M et al. Comparison of efficacy outcomes with vutrisiran vs. patisiran in hATTR amyloidosis with polyneuropathy: post-hoc analysis of the HELIOS — a study (S14.003). Neurology 100 (17 Suppl. 2), S14.003 (2023). [Google Scholar]
- 112.Debacker AJ, Voutila J, Catley M, Blakey D & Habib N Delivery of oligonucleotides to the liver with GalNAc: from research to registered therapeutic drug. Mol. Ther 28, 1759–1771 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Van De Water FM et al. Intravenously administered short interfering RNA accumulates in the kidney and selectively suppresses gene function in renal proximal tubules. Drug. Metab. Dispos 34, 1393–1397 (2006). [DOI] [PubMed] [Google Scholar]
- 114.Thielmann M et al. Teprasiran, a small interfering RNA, for the prevention of acute kidney injury in high-risk patients undergoing cardiac surgery: a randomized clinical study. Circulation 144, 1133–1144 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kopp JB et al. Podocytopathies. Nat. Rev. Dis. Prim 6, 68 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Anders H-J, Kitching AR, Leung N & Romagnani P Glomerulonephritis: immunopathogenesis and immunotherapy. Nat. Rev. Immunol 23, 453–471 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Godinho BMDC et al. Pharmacokinetic profiling of conjugated therapeutic oligonucleotides: a high-throughput method based upon serial blood microsampling coupled to peptide nucleic acid hybridization assay. Nucleic Acid. Ther 27, 323–334 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Osborn MF et al. Hydrophobicity drives the systemic distribution of lipid-conjugated siRNAs via lipid transport pathways. Nucleic Acids Res. 47, 1070–1081 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fakih HH et al. Dendritic amphiphilic siRNA: selective albumin binding, in vivo efficacy, and low toxicity. Mol. Ther. Nucleic Acids 10.1016/j.omtn.2023.102080 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Biscans A, Ly S, McHugh N, Cooper DA & Khvorova A Engineered ionizable lipid siRNA conjugates enhance endosomal escape but induce toxicity in vivo. J. Control. Release 349, 831–843 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Turanov AA et al. RNAi modulation of placental sFLT1 for the treatment of preeclampsia. Nat. Biotechnol 36, 1164–1173 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nanna AR et al. Generation and validation of structurally defined antibody-siRNA conjugates. Nucleic Acids Res. 48, 5281–5293 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Malecova B et al. Targeted tissue delivery of RNA therapeutics using antibody-oligonucleotide conjugates (AOCs). Nucleic Acids Res. 51, 5901–5910 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes the use of antibody-oligonucleotide conjugates as an effective strategy for delivering siRNA into muscle tissues.
- 124.Sugo T et al. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Control. Rel 237, 1–13 (2016). [DOI] [PubMed] [Google Scholar]
- 125.Klein D et al. Centyrin ligands for extrahepatic delivery of siRNA. Mol. Ther 29, 2053–2066 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ämmälä C et al. Targeted delivery of antisense oligonucleotides to pancreatic β-cells. Sci. Adv 4, eaat3386 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.McNamara JO et al. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat. Biotechnol 24, 1005–1015 (2006). [DOI] [PubMed] [Google Scholar]
- 128.Chu TC Aptamer mediated siRNA delivery. Nucleic Acids Res. 34, e73 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhou J, Li H, Li S, Zaia J & Rossi J Novel cell type-specific aptamer-siRNA delivery system for HIV-1 therapy. Nat. Preced 10.1038/npre.2007.1299.1 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dassie JP et al. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat. Biotechnol 27, 839–846 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Neff CP et al. An aptamer-siRNA chimera suppresses HIV-1 viral loads and protects from helper CD4+ T cell decline in humanized mice. Sci. Transl. Med 3, 66ra6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wheeler LA et al. Inhibition of HIV transmission in human cervicovaginal explants and humanized mice using CD4 aptamer-siRNA chimeras. J. Clin. Invest 121, 2401–2412 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wheeler LA et al. Durable knockdown and protection from HIV transmission in humanized mice treated with gel-formulated CD4 aptamer-siRNA chimeras. Mol. Ther 21, 1378–1389 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhou J & Rossi J Aptamers as targeted therapeutics: current potential and challenges. Nat. Rev. Drug Discov 16, 181–202 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This review discusses aptamers as targeted therapeutics and siRNA delivery agents.
- 135.Zhang Y et al. Immunotherapy for breast cancer using EpCAM aptamer tumor-targeted gene knockdown. Proc. Natl Acad. Sci. USA 118, e2022830118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Xie S et al. Aptamer-based targeted delivery of functional nucleic acids. J. Am. Chem. Soc 145, 7677–7691 (2023). [DOI] [PubMed] [Google Scholar]
- 137.Lee H et al. Molecularly self-assembled nucleic acid nanoparticles for targeted in vivo siRNA delivery. Nat. Nanotechnol 7, 389–393 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fakhoury JJ, McLaughlin CK, Edwardson TW, Conway JW & Sleiman HF Development and characterization of gene silencing DNA cages. Biomacromolecules 15, 276–282 (2014). [DOI] [PubMed] [Google Scholar]
- 139.Chen Y-J, Groves B, Muscat RA & Seelig G DNA nanotechnology from the test tube to the cell. Nat. Nanotechnol 10, 748–760 (2015). [DOI] [PubMed] [Google Scholar]
- 140.Bujold KE, Fakih HH & Sleiman HF in RNA Interference and Cancer Therapy (ed. Dinesh Kumar L) 69–81. Methods in Molecular Biology vol. 1974 (Springer, 2019). [DOI] [PubMed] [Google Scholar]
- 141.Zhang H et al. DNA nanostructures coordinate gene silencing in mature plants. Proc. Natl Acad. Sci. USA 116, 7543–7548 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhang H et al. Engineering DNA nanostructures for siRNA delivery in plants. Nat. Protoc 15, 3064–3087 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.No authors listed. Lipophilic siRNA conjugates yield durable silencing in extrahepatic tissues. Nat. Biotechnol 40, 1439–1440 (2022). [DOI] [PubMed] [Google Scholar]
- 144.Cheng S-Y et al. Single intravitreal administration of a tetravalent siRNA exhibits robust and efficient gene silencing in rodent and swine photoreceptors. Mol. Ther. Nucleic Acids 35, 102088 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ng B et al. Intratracheal administration of siRNA triggers mRNA silencing in the lung to modulate T cell immune response and lung inflammation. Mol. Ther. Nucleic Acids 16, 194–205 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ferguson CM et al. Comparative route of administration studies using therapeutic siRNAs show widespread gene modulation in Dorset sheep. JCI Insight 6, e152203 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hardiman O et al. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim 3, 17071 (2017). [DOI] [PubMed] [Google Scholar]
- 148.Masters CL et al. Alzheimer’s disease. Nat. Rev. Dis. Prim 1, 15056 (2015). [DOI] [PubMed] [Google Scholar]
- 149.Hsu T & Mitragotri S Delivery of siRNA and other macromolecules into skin and cells using a peptide enhancer. Proc. Natl Acad. Sci. USA 108, 15816–15821 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zheng D et al. Topical delivery of siRNA-based spherical nucleic acid nanoparticle conjugates for gene regulation. Proc. Natl Acad. Sci. USA 109, 11975–11980 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Liang X et al. Skin delivery of siRNA using sponge spicules in combination with cationic flexible liposomes. Mol. Ther. Nucleic Acids 20, 639–648 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Dharamdasani V et al. Topical delivery of siRNA into skin using ionic liquids. J. Control. Rel 323, 475–482 (2020). [DOI] [PubMed] [Google Scholar]
- 153.Lee K et al. Study and evaluation of the potential of lipid nanocarriers for transdermal delivery of siRNA. Biotechnol. J 15, 2000079 (2020). [DOI] [PubMed] [Google Scholar]
- 154.Mandal A et al. Treatment of psoriasis with NFKBIZ siRNA using topical ionic liquid formulations. Sci. Adv 6, eabb6049 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Aldawsari M, Chougule MB & Babu RJ Progress in topical siRNA delivery approaches for skin disorders. Curr. Pharm. Des 21, 4594–4605 (2015). [DOI] [PubMed] [Google Scholar]
- 156.Tang Q et al. Rational design of a JAK1-selective siRNA inhibitor for the modulation of autoimmunity in the skin. Nat. Commun 14, 7099 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cui J et al. Ex vivo pretreatment of human vessels with siRNA nanoparticles provides protein silencing in endothelial cells. Nat. Commun 8, 191 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Brüggenwirth IMA & Martins PN RNA interference therapeutics in organ transplantation: the dawn of a new era. Am. J. Transplant 20, 931–941 (2020). [DOI] [PubMed] [Google Scholar]
- 159.Eltzschig HK & Eckle T Ischemia and reperfusion — from mechanism to translation. Nat. Med 17, 1391–1401 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.No authors listed. Buying time for transplants. Nat. Biotechnol 35, 801 (2017). [DOI] [PubMed] [Google Scholar]
- 161.Senior M Beating the organ clock. Nat. Biotechnol 36, 488–492 (2018). [DOI] [PubMed] [Google Scholar]
- 162.Theruvath J et al. Anti-GD2 synergizes with CD47 blockade to mediate tumor eradication. Nat. Med 28, 333–344 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gulhati P et al. Targeting T cell checkpoints 41BB and LAG3 and myeloid cell CXCR1/CXCR2 results in antitumor immunity and durable response in pancreatic cancer. Nat. Cancer 4, 62–80 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Richardson PG et al. Mezigdomide plus dexamethasone in relapsed and refractory multiple myeloma. N. Engl. J. Med 389, 1009–1022 (2023). [DOI] [PubMed] [Google Scholar]
- 165.Trajanoska K et al. From target discovery to clinical drug development with human genetics. Nature 620, 737–745 (2023). [DOI] [PubMed] [Google Scholar]
- 166.Wu T, Cooper SA & Shah VH Omics and AI advance biomarker discovery for liver disease. Nat. Med 28, 1131–1132 (2022). [DOI] [PubMed] [Google Scholar]
- 167.Liu S et al. Multi-organ landscape of therapy-resistant melanoma. Nat. Med 29, 1123–1134 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Theodoris CV et al. Transfer learning enables predictions in network biology. Nature 618, 616–624 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Larsson E, Sander C & Marks D mRNA turnover rate limits siRNA and microRNA efficacy. Mol. Syst. Biol 6, 433 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Judge AD et al. Confirming the RNAi-mediated mechanism of action of siRNA-based cancer therapeutics in mice. J. Clin. Invest 119, 661–673 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Gonzalez-Gonzalez E et al. siRNA siLencing of keratinocyte-specific GFP expression in a transgenic mouse skin model. Gene Ther. 16, 963–972 (2009). [DOI] [PubMed] [Google Scholar]
- 172.Omi K, Tokunaga K & Hohjoh H Long-lasting RNAi activity in mammalian neurons. FEBS Lett. 558, 89–95 (2004). [DOI] [PubMed] [Google Scholar]
- 173.McDonagh M, Peterson K, Holzhammer B & Fazio S A systematic review of PCSK9 inhibitors alirocumab and evolocumab. Manag. Care Spec. Pharm 22, 641–653q (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Flower M et al. MSH3 modifies somatic instability and disease severity in Huntington’s and myotonic dystrophy type 1. Brain 142, 1876–1886 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Takahashi M, Koi M, Balaguer F, Boland CR & Goel A MSH3 mediates sensitization of colorectal cancer cells to cisplatin, oxaliplatin, and a poly(ADP-ribose) polymerase inhibitor. J. Biol. Chem 286, 12157–12165 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Adam R et al. Exome sequencing identifies biallelic MSH3 germline mutations as a recessive subtype of colorectal adenomatous polyposis. Am. J. Hum. Genet 99, 337–351 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Iyama T & Wilson DM DNA repair mechanisms in dividing and non-dividing cells. DNA Repair 12, 620–636 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Bogdanovich S et al. Functional improvement of dystrophic muscle by myostatin blockade. Nature 420, 418–421 (2002). [DOI] [PubMed] [Google Scholar]
- 179.Kota J et al. Follistatin gene delivery enhances muscle growth and strength in nonhuman primates. Sci. Transl. Med 1, 6ra15 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lee S-J Targeting the myostatin signaling pathway to treat muscle loss and metabolic dysfunction. J. Clin. Invest 131, e148372 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Van Der Worp HB et al. Can animal models of disease reliably inform human studies? PLoS Med. 7, e1000245 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Seok J et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl Acad. Sci. USA 110, 3507–3512 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Perlman RL Mouse models of human disease: an evolutionary perspective. Evol. Med. Public Health 2016, 170–176 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Gusella JF et al. A polymorphic DNA marker genetically linked to Huntington’s disease. Nature 306, 234–238 (1983). [DOI] [PubMed] [Google Scholar]
- 185.The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983 (1993). [DOI] [PubMed] [Google Scholar]
- 186.Brook JD et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 68, 799–808 (1992). [DOI] [PubMed] [Google Scholar]
- 187.Sutherland JE et al. Nonclinical safety profile of revusiran, a 1st-generation GalNAc-siRNA conjugate for treatment of hereditary transthyretin-mediated amyloidosis. Nucleic Acid Ther. 30, 33–49 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Gouda MA, Buschhorn L, Schneeweiss A, Wahida A & Subbiah V N-of-1 trials in cancer drug development. Cancer Discov. 13, 1301–1309 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Tran H et al. Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat. Med 28, 117–124 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Matlin AJ, Clark F & Smith CWJ Understanding alternative splicing: towards a cellular code. Nat. Rev. Mol. Cell Biol 6, 386–398 (2005). [DOI] [PubMed] [Google Scholar]
- 191.Nilsen TW & Graveley BR Expansion of the eukaryotic proteome by alternative splicing. Nature 463, 457–463 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Prinos P et al. Alternative splicing of SYK regulates mitosis and cell survival. Nat. Struct. Mol. Biol 18, 673–679 (2011). [DOI] [PubMed] [Google Scholar]
- 193.Schwerk J et al. RNA-binding protein isoforms ZAP-S and ZAP-L have distinct antiviral and immune resolution functions. Nat. Immunol 20, 1610–1620 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ray TA et al. Comprehensive identification of mRNA isoforms reveals the diversity of neural cell-surface molecules with roles in retinal development and disease. Nat. Commun 11, 3328 (2020).32620864 [Google Scholar]
- 195.Nikom D & Zheng S Alternative splicing in neurodegenerative disease and the promise of RNA therapies. Nat. Rev. Neurosci 24, 457–473 (2023). [DOI] [PubMed] [Google Scholar]
- 196.Bradley RK & Anczuków O RNA splicing dysregulation and the hallmarks of cancer. Nat. Rev. Cancer 23, 135–155 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Marx V Method of the year: long-read sequencing. Nat. Methods 20, 6–11 (2023). [DOI] [PubMed] [Google Scholar]
- 198.Shaffer C Mist begins to clear for lung delivery of RNA. Nat. Biotechnol 38, 1110–1112 (2020). [DOI] [PubMed] [Google Scholar]
- 199.Dusheiko G, Agarwal K & Maini MK New approaches to chronic hepatitis B. N. Engl. J. Med 388, 55–69 (2023). [DOI] [PubMed] [Google Scholar]
- 200.Ambike S et al. Targeting genomic SARS-CoV-2 RNA with siRNAs allows efficient inhibition of viral replication and spread. Nucleic Acids Res. 50, 333–349 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Estes JD, Wong SW & Brenchley JM Nonhuman primate models of human viral infections. Nat. Rev. Immunol 18, 390–404 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Dennis C The brave new world of RNA. Nature 418, 122–124 (2002). [DOI] [PubMed] [Google Scholar]
- 203.Matsui M & Corey DR Non-coding RNAs as drug targets. Nat. Rev. Drug. Discov 16, 167–179 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Slack FJ & Chinnaiyan AM The role of non-coding RNAs in oncology. Cell 179, 1033–1055 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Winkle M, El-Daly SM, Fabbri M & Calin GA Noncoding RNA therapeutics — challenges and potential solutions. Nat. Rev. Drug Discov 20, 629–651 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Statello L, Guo C-J, Chen L-L & Huarte M Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol 22, 96–118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Mattick JS et al. Long non-coding RNAs: definitions, functions, challenges and recommendations. Nat. Rev. Mol. Cell Biol 24, 430–447 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Balusu S et al. MEG3 activates necroptosis in human neuron xenografts modeling Alzheimer’s disease. Science 381, 1176–1182 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Gaya A et al. Results of a phase 1/2 study of cemdisiran in healthy subjects and patients with paroxysmal nocturnal hemoglobinuria. eJHaem 4, 612–624 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Devalaraja-Narashimha K et al. Pharmacokinetics and pharmacodynamics of pozelimab alone or in combination with cemdisiran in non-human primates. PLoS ONE 17, e0269749 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Griffin M Pozelimab/cemdisiran. Complement protein C5 inhibitor, treatment of complement-mediated diseases. Drugs Fut. 48, 93 (2023). [Google Scholar]
- 212.Fire A et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811 (1998). [DOI] [PubMed] [Google Scholar]; This paper discovers that double-stranded RNA can induce catalytic cleavage of target mRNAs in C. elegans.
- 213.Hannon GJ RNA interference. Nature 418, 244–251 (2002). [DOI] [PubMed] [Google Scholar]
- 214.Dorsett Y & Tuschl T siRNAs: applications in functional genomics and potential as therapeutics. Nat. Rev. Drug Discov 3, 318–329 (2004). [DOI] [PubMed] [Google Scholar]
- 215.Castanotto D & Rossi JJ The promises and pitfalls of RNA-interference-based therapeutics. Nature 457, 426–433 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Hannon GJ & Rossi JJ Unlocking the potential of the human genome with RNA interference. Nature 431, 371–378 (2004). [DOI] [PubMed] [Google Scholar]
- 217.Elbashir SM et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411, 494–498 (2001). [DOI] [PubMed] [Google Scholar]
- 218.Caplen NJ, Parrish S, Imani F, Fire A & Morgan RA Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proc. Natl Acad. Sci. USA 98, 9742–9747 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]; References 213 and 214 were among the pioneering studies that demonstrated RNAi in mammalian cells.
- 219.McCaffrey AP et al. RNA interference in adult mice. Nature 418, 38–39 (2002). [DOI] [PubMed] [Google Scholar]
- 220.Hamar P et al. Small interfering RNA targeting Fas protects mice against renal ischemia-reperfusion injury. Proc. Natl Acad. Sci. USA 101, 14883–14888 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Soutschek J et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 432, 173–178 (2004). [DOI] [PubMed] [Google Scholar]
- 222.Kumar P et al. T cell-specific siRNA delivery suppresses HIV-1 infection in humanized mice. Cell 134, 577–586 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Song E et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat. Biotechnol 23, 709–717 (2005). [DOI] [PubMed] [Google Scholar]
- 224.Palliser D et al. An siRNA-based microbicide protects mice from lethal herpes simplex virus 2 infection. Nature 439, 89–94 (2006). [DOI] [PubMed] [Google Scholar]
- 225.Li B et al. Using siRNA in prophylactic and therapeutic regimens against SARS coronavirus in Rhesus macaque. Nat. Med 11, 944–951 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Woodrow KA et al. Intravaginal gene silencing using biodegradable polymer nanoparticles densely loaded with small-interfering RNA. Nat. Mater 8, 526–533 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Song E et al. RNA interference targeting Fas protects mice from fulminant hepatitis. Nat. Med 9, 347–351 (2003). [DOI] [PubMed] [Google Scholar]
- 228.Dykxhoorn DM & Lieberman J Knocking down disease with siRNAs. Cell 126, 231–235 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.De Fougerolles A, Vornlocher H-P, Maraganore J & Lieberman J Interfering with disease: a progress report on siRNA-based therapeutics. Nat. Rev. Drug Discov 6, 443–453 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Sledz CA, Holko M, De Veer MJ, Silverman RH & Williams BRG Activation of the interferon system by short-interfering RNAs. Nat. Cell Biol 5, 834–839 (2003). [DOI] [PubMed] [Google Scholar]
- 231.Frantz S Studies reveal potential pitfalls of RNAi. Nat. Rev. Drug Discov 2, 763–764 (2003). [Google Scholar]
- 232.Marques JT & Williams BRG Activation of the mammalian immune system by siRNAs. Nat. Biotechnol 23, 1399–1405 (2005). [DOI] [PubMed] [Google Scholar]
- 233.Judge AD et al. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat. Biotechnol 23, 457–462 (2005). [DOI] [PubMed] [Google Scholar]
- 234.Hornung V et al. Sequence-specific potent induction of IFN-α by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat. Med 11, 263–270 (2005). [DOI] [PubMed] [Google Scholar]
- 235.Jackson AL et al. Expression profiling reveals off-target gene regulation by RNAi. Nat. Biotechnol 21, 635–637 (2003). [DOI] [PubMed] [Google Scholar]; This article demonstrates that siRNAs can induce off-targeting effects with limited target sequence similarity.
- 236.Karikó K, Bhuyan P, Capodici J & Weissman D Small interfering RNAs mediate sequence-independent gene suppression and induce immune activation by signaling through toll-like receptor 3. J. Immunol 172, 6545–6549 (2004). [DOI] [PubMed] [Google Scholar]; This paper demonstrates the role of Toll-like receptor 3 in mediating siRNA-induced immune activation.
- 237.Ledford H Drug giants turn their backs on RNA interference. Nature 468, 487–487 (2010). [DOI] [PubMed] [Google Scholar]
- 238.Schmidt C RNAi momentum fizzles as pharma shifts priorities. Nat. Biotechnol 29, 93–94 (2011). [DOI] [PubMed] [Google Scholar]
- 239.Check Hayden E RNA interference rebooted. Nature 508, 443–443 (2014). [DOI] [PubMed] [Google Scholar]
- 240.Amrite A, Fuentes E, Marbury TC & Zhang S Safety, pharmacokinetics, and exposure–response modeling of nedosiran in participants with severe chronic kidney disease. Clin. Pharmacol. Drug Dev 12, 1164–1177 (2023). [DOI] [PubMed] [Google Scholar]
- 241.Desai AS et al. Zilebesiran, an RNA interference therapeutic agent for hypertension. N. Engl. J. Med 389, 228–238 (2023). [DOI] [PubMed] [Google Scholar]; This article reports on the clinical efficacy of siRNA in treating the prevalent disease hypertension.
- 242.Fernández-Ruiz I Promising novel siRNA for the treatment of hypertension. Nat. Rev. Cardiol 20, 647–647 (2023). [DOI] [PubMed] [Google Scholar]
- 243.Raal FJ et al. IncLisiran for the treatment of heterozygous familial hypercholesterolemia. N. Engl. J. Med 382, 1520–1530 (2020). [DOI] [PubMed] [Google Scholar]; This article reports on the clinical efficacy of siRNA in treating the prevalent disease hypercholesterolaemia.
- 244.Lamb YN Inclisiran: first approval. Drugs 81, 389–395 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Long D et al. Potent effect of target structure on microRNA function. Nat. Struct. Mol. Biol 14, 287–294 (2007). [DOI] [PubMed] [Google Scholar]
- 246.Małecka EM & Woodson SA Ribosomes clear the way for siRNA targeting. Nat. Struct. Mol. Biol 27, 775–777 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; References 241 and 242 describe how mRNA secondary structure is a factor impacting siRNA/miRNA efficacy.
- 247.Vert J-P, Foveau N, Lajaunie C & Vandenbrouck Y An accurate and interpretable model for siRNA efficacy prediction. BMC Bioinformatics 7, 520 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Shabalina SA, Spiridonov AN & Ogurtsov AY Computational models with thermodynamic and composition features improve siRNA design. BMC Bioinformatics 7, 65 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Matveeva O et al. Comparison of approaches for rational siRNA design leading to a new efficient and transparent method. Nucleic Acids Res. 35, e63 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Ichihara M et al. Thermodynamic instability of siRNA duplex is a prerequisite for dependable prediction of siRNA activities. Nucleic Acids Res. 35, e123 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Breiman L Random forests. Mach. Learn 45, 5–32 (2001). [Google Scholar]
- 252.Brown KM, Chu C & Rana TM Target accessibility dictates the potency of human RISC. Nat. Struct. Mol. Biol 12, 469–470 (2005). [DOI] [PubMed] [Google Scholar]; This article reports on the effect of increased target accessibility in improving RISC potency.
- 253.Heale BSE siRNA target site secondary structure predictions using local stable substructures. Nucleic Acids Res. 33, e30 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Kedde M et al. RNA-binding protein Dnd1 inhibits microRNA access to target mRNA. Cell 131, 1273–1286 (2007). [DOI] [PubMed] [Google Scholar]
- 255.Kim S et al. The regulatory impact of RNA-binding proteins on microRNA targeting. Nat. Commun 12, 5057 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Davis SM Guidelines for designing therapeutic siRNAs. PhD Thesis, UMass Chan Medical School; (2023). [Google Scholar]
- 257.Bahar Halpern K et al. Nuclear retention of mRNA in mammalian tissues. Cell Rep. 13, 2653–2662 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Wegener M & Müller-McNicoll M Nuclear retention of mRNAs — quality control, gene regulation and human disease. Semin. Cell Dev. Biol 79, 131–142 (2018). [DOI] [PubMed] [Google Scholar]
- 259.Didiot M-C et al. Nuclear localization of Huntingtin mRNA is specific to cells of neuronal origin. Cell Rep. 24, 2553–2560.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Ferguson CM, Echeverria D, Hassler M, Ly S & Khvorova A Cell type impacts accessibility of mRNA to silencing by RNA interference. Mol. Ther. Nucleic Acids 21, 384–393 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes how the subcellular localization of mRNA transcripts in different cell types contributes to affecting siRNA efficacy.
- 261.Dawson MA et al. Nuclear JAK2. Blood 118, 6987–6988 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Narykov O, Johnson NT & Korkin D Predicting protein interaction network perturbation by alternative splicing with semi-supervised learning. Cell Rep. 37, 110045 (2021). [DOI] [PubMed] [Google Scholar]
- 263.Dua K et al. The potential of siRNA based drug delivery in respiratory disorders: recent advances and progress. Drug. Dev. Res 80, 714–730 (2019). [DOI] [PubMed] [Google Scholar]
- 264.Donoso LA et al. Autosomal dominant stargardt-like macular dystrophy. Surv. Ophthalmol 46, 149–163 (2001). [DOI] [PubMed] [Google Scholar]
- 265.Ayuso C & Millan JM Retinitis pigmentosa and allied conditions today: a paradigm of translational research. Genome Med. 2, 34 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Meng D, Ragi SD & Tsang SH Therapy in rhodopsin-mediated autosomal dominant retinitis pigmentosa. Mol. Ther 28, 2139–2149 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Samson N & Ablasser A The cGAS-STING pathway and cancer. Nat. Cancer 3, 1452–1463 (2022). [DOI] [PubMed] [Google Scholar]
- 268.Kaya T et al. CD8+ T cells induce interferon-responsive oligodendrocytes and microglia in white matter aging. Nat. Neurosci 25, 1446–1457 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Chen X et al. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature 615, 668–677 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Fitzgerald K, Frank-Kamenetsky M & Charisse K Dual targeting siRNA. US patent 9,187,746B2 (2015).
- 271.Lee SH, Mok H, Jo S, Hong CA & Park TG Dual gene targeted multimeric siRNA for combinatorial gene silencing. Biomaterials 32, 2359–2368 (2011). [DOI] [PubMed] [Google Scholar]
- 272.Brown JM et al. Ligand conjugated multimeric siRNAs enable enhanced uptake and multiplexed gene silencing. Nucleic Acid Ther. 29, 231–244 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Chareddy Y et al. P2.11-03 Synergistic co-targeting of MYC and KRAS in Lung cancer by novel Ligand-directed inverted chimeric RNAi molecules. J. Thorac. Oncol 18, S360–S361 (2023). [Google Scholar]
- 274.No authors listed. Undruggable KRAS — time to rebrand? Lancet Oncol. 22, 289 (2021). [DOI] [PubMed] [Google Scholar]
- 275.Kamerkar S et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 546, 498–503 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Stojic L et al. Transcriptional silencing of long noncoding RNA GNG12-AS1 uncouples its transcriptional and product-related functions. Nat. Commun 7, 10406 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]