

ABSTRACT
Variants in KCNMA1, encoding the voltage- and calcium-activated K+ (BK) channel, are associated with human neurological disease. The effects of gain-of-function (GOF) and loss-of-function (LOF) variants have been predominantly studied on BK channel currents evoked under steady-state voltage and Ca2+ conditions. However, in their physiological context, BK channels exist in partnership with voltage-gated Ca2+ channels and respond to dynamic changes in intracellular Ca2+ (Ca2+i). In this study, an L-type voltage-gated Ca2+ channel present in the brain, CaV1.2, was co-expressed with wild type and mutant BK channels containing GOF (D434G, N999S) and LOF (H444Q, D965V) patient-associated variants in HEK-293T cells. Whole-cell BK currents were recorded under CaV1.2 activation using buffering conditions that restrict Ca2+i to nano- or micro-domains. Both conditions permitted wild type BK current activation in response to CaV1.2 Ca2+ influx, but differences in behavior between wild type and mutant BK channels were reduced compared to prior studies in clamped Ca2+i. Only the N999S mutation produced an increase in BK current in both micro- and nano-domains using square voltage commands and was also detectable in BK current evoked by a neuronal action potential within a microdomain. These data corroborate the GOF effect of N999S on BK channel activity under dynamic voltage and Ca2+ stimuli, consistent with its pathogenicity in neurological disease. However, the patient-associated mutations D434G, H444Q, and D965V did not exhibit significant effects on BK current under CaV1.2-mediated Ca2+ influx, in contrast with prior steady-state protocols. These results demonstrate a differential potential for KCNMA1 variant pathogenicity compared under diverse voltage and Ca2+ conditions.
KEYWORDS: KCa1.1, calcium-activated potassium channel, channelopathy, CaV1.2, voltage-gated calcium channels, CACNA1C
Introduction
Channelopathy disorders are caused by gene mutations producing pathological deficits in ion channel function. In humans, KCNMA1 gene mutations underlie a rare neurological disorder associated with dysfunction of BK channels. KCNMA1 transcripts are widely expressed in the central nervous system, and the KCNMA1-linked channelopathy disorder has multiple brain manifestations, including dyskinesia, epilepsy, developmental delay, and intellectual disability [1–3]. Patient-associated KCNMA1 variants have been designated as gain-of-function (GOF) or loss-of-function (LOF) in BK channel activity based on experiments conducted under steady-state voltage and clamped the intracellular free calcium concentration ([Ca2+]i) in HEK293 and CHO cells. How BK channel dysfunction in vivo produces neurological disease is still under investigation [1,4–12].
BK channels are members of the voltage-activated potassium channel family. They are activated by depolarizing transmembrane voltage and by intracellular Ca2+ through the direct binding of calcium to two intracellular sites within the C-terminal gating ring [13]. These properties make BK channel activation responsive to dynamic Ca2+ signaling in neurons at physiological membrane potentials [14], as [Ca2+]i in neurons increases from resting conditions of <100 nM to as high as 700 nM upon stimulation [15]. Neuronal BK channel opening typically depends on local [Ca2+]i provided by multiple types of voltage-gated Ca2+ channels (CaV) and Ca2+-permeable channels [14,16]. While physical details of BK-CaV interactions are not fully elucidated, the evidence suggests that BK channels interact with the CaV α1 subunit and CaV auxiliary subunits [17,18] in a manner that varies with cell type and excitable signaling [14].
CaV-mediated BK channel activation occurs within diffusion-restricted membrane domains: microdomains (10–100 nm) and nanodomains (<10 nm) [14,19,20]. These domains can be investigated experimentally by recording BK channel currents under ethylene glycol tetraacetic acid (EGTA) or 1,2-bis(o-aminophenoxy) ethane-N,N,N,N-tetraacetic acid (BAPTA) buffering conditions. EGTA is a slow Ca2+ chelator that restricts Ca2+ diffusion over longer distances. BAPTA has a similar binding affinity to EGTA, but greater on-rate, restricting Ca2+ diffusion over shorter distances [21]. Single-molecule localization experiments revealed that voltage-activated CaV1.3 channels closely cluster within 10–20 nM of BK channels in rat hippocampal and sympathetic neurons and in heterologous tsA-201 cells [22]. Models developed from electrophysiological data estimate that BK channels in these clusters could encounter 20 µM Ca2+i under endogenous conditions or in the presence of EGTA buffering (microdomain conditions) and >5 µM Ca2+i under BAPTA (nanodomain) buffering conditions [16,23](Figure 1a). BK activation via CaV coupling is rapid (within a millisecond), regulated by the specific BK and CaV subunits present, and differentially sensitive to EGTA and BAPTA under different cellular conditions [14,18,22–31]. Despite the specificity of CaV-mediated BK channel activation in neuronal signaling, the details of BK-CaV function in neurological disease is not well studied.
Figure 1.

BK channel activation by Ca2+ influx through CaV1.2 channels. (a) CaV1.2 and BK channel subunits in 2 mM BAPTA and 10 mM EGTA delimited buffering domains. (b) Two-step voltage protocol used to elicit whole-cell CaV1.2 currents (conditioning step), followed by BK currents (test step). (c) Total current from HEK-293T cells co-expressing CaV1.2 and BK channels recorded in 10 mM EGTA. (d) Inward CaV1.2 current isolated by addition of 100 nM paxilline to block BK current. (e) Outward BK channel currents obtained by subtracting (d) from (c). Dotted line represents the zero current level.
CaV1.2 is a widely expressed L-type voltage gated Ca2+ channel that influences central neuronal excitability [32] and was previously shown to partner with BK channels [24,27,28,33]. In heterologous cells, CaV1.2 channels comprised CaV1.2 α1 (CACNA1C), α2δ1, and β1b operate between −30 mV and +60 mV, with a peak activation at 0 mV that overlaps with BK channel voltage sensitivity [27], and mutations in BK and CaV1.2 channels have some overlapping neurological dysfunctions, including epilepsy, developmental delay, and intellectual disability [1,34]. The consequences of KCNMA1 channelopathy variants that alter BK channel gating properties have not been investigated under BK-CaV1.2 channel coupled activation. In this study, four KCNMA1 variants previously studied under clamped Ca2+ conditions (two GOF and two LOF) were investigated in BK channels activated under CaV1.2 channel Ca2+ influx.
In recordings made in clamped Ca2+, BK channels containing the well-studied KCNMA1 channelopathy mutations D434G and N999S (BKD434G and BKN999S) exhibit GOF behavior, shifting voltage-dependent activation toward more hyperpolarized potentials over a range of [Ca2+]i from 0–100 μM [4,12,35–42]. Along with shifting the conductance-voltage (G-V) relationship, the D434G and N999S mutations also alter BK channel kinetics, causing faster activation and slower deactivation [4,12,36–42]. BKN999S channels show a greater shift in the G-V curve and a faster channel activation compared to BKD434G channels [4,37]. The LOF KCNMA1 variants H444Q and D965V (BKH444Q and BKD965V) produce channels with G-V relationships shifted toward more depolarized membrane potentials by 23 mV and 40 mV, respectively, and decrease activation kinetics compared to wild type BK channels [4,12,43]. BKH444Q also exhibits faster deactivation kinetics [12,43]. The effect of these four representative mutations was investigated by co-expressing BKWT, BKN999S, BKD434G, BKH444Q, or BKD965V channels with CaV1.2 channels in HEK-293 cells and recording whole-cell currents under voltage-clamp. GOF and LOF activities in CaV1.2-activated BK currents were compared to previous functional studies in clamped Ca2+ conditions to assess the congruency.
Methods
Cell culture and transfection
Mutations were introduced into the WT BK channel cDNA in pcDNA3.1+ (GenBank MG279689; Supplemental Table 1). HEK-293T cells (CRL-11268, ATCC, Manassas, VA, USA) were maintained in DMEM media (Cat. #11995-065, Gibco, Life Technologies Corp., Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Cat. #100-106, GeminiBio, West Sacramento, California, USA), 1% penicillin/streptomycin (Cat. #400-109, GeminiBio, West Sacramento, California, USA) and 1% L-glutamine (Cat. #25-005-Cl, Mediatech Inc., Manassas, VA, USA) in a humidified incubator at 37°C with 5% CO2. Cells were transfected at 60–70% confluency with BKWT, BKD434G, BKN999S, BKH444Q or BKD965V (see Supplemental Table 1 for residue numbering), and human CaV1.2α (Cacna1c, CAA84341.1), rat CaVβ1b (Cacnb1, CAA43665.1) and rat CaVα2δ1 (Cacna2d1, AAG28164.1) using Fugene HD (Fugent LLC Middleton, Wisconsin, USA) at 0.8:1:1:1 ratio of cDNA and a 1:6 ratio of transfection reagent (μg/μL). BK channel, CaV1.2α, and CaVα2δ1/β1b plasmids were prepared from 2–3, 5, and 2 independent plasmid preparations, respectively, and the numbers of independent transfections per condition were as follows: in EGTA (BKWT, n = 8; BKD434G, 9; BKN999S, 8; BKH444Q, 6; and BKD965V, 4) and in BAPTA (BKWT, n = 9; BKD434G, 8; BKN999S, 8; BKH444Q, 4; and BKD965V, 6). BKWT was recorded alongside each mutation within the same week of data collection. After 24 hours, cells were washed with complete media containing Ca2+-free minimum essential medium (Cat. #11380-037, Gibco, Life Technologies Corp., Grand Island, NY, USA) in place of DMEM. After 24–48 hours, cells were re-plated onto pre-treated glass coverslips with poly-L-lysine (Cat. #P4832, Sigma-Aldrich, St. Louis, MO, USA). Experimental recordings were performed 48–72 hours post-transfection.
Electrophysiological recordings
Macroscopic BK and Ca2+ currents were recorded in whole-cell voltage-clamp mode at 22–25°C with a MultiClamp 700B amplifier using electrodes (3–6 MΩ) filled with intracellular solution (123 mM K-methanesulfonate, 9 mM NaCl, 10 mM EGTA, 9 mM HEPES, 2 mM Mg-ATP and 2 mM Na2-ATP, pH 7.3 (300–310 mOsm/kg)). BAPTA (5 mM) was substituted for EGTA in some internal solutions as specified in figure legends. The bath solution was composed of (125 mM NaCl, 1.2 mM MgCl2, 1.25 mM NaH2PO4, 3.5 mM KCl, 2.5 mM CaCl2, 10 mM HEPES and 10 mM D-glucose, pH 7.4 (~300 mOsm/kg)). The access resistance was <15 MΩ, and seal resistance was compensated 60–80%. Cells where Rs error > 20% were not included.
BK K+ and CaV1.2 Ca2+currents were isolated from total cell currents by bath application of 100 nM paxilline (Pax, Alomone Labs, Jerusalem, Israel, #P-450) as described in Figure 1b-d Pax was dissolved in DMSO (1000X) and focally applied to the bath at the concentrations listed above. Macroscopic currents were elicited from a holding potential of −90 mV in a two-part voltage protocol stepping for 50 ms from −100 to +60 mV in 20-mV increments followed by a second step to +60 mV for 50 ms. Ca2+ currents were assessed from the peak of the Ca2+ current from the first step, and K+ currents were evaluated from the peak current from the second step of the subtracted BK channel current. Action potential-evoked currents were elicited from a holding potential of −90 mV following by an action potential voltage command derived from a previously recorded granule neuron waveform (baseline membrane potential, −47 mV; half-width, 1.9 ms; peak, +46 mV; AHP, −52 mV) [4].
Currents were sampled at 50 kHz and filtered online at 10 kHz with a P/5 leak subtraction protocol. Representative traces were post-hoc filtered at 2 kHz. Voltage values were adjusted for the liquid junction potential (10 mV). The Ca2+ current levels were obtained from the peak inward current elicited from the first voltage step after paxilline application, and the BK current was obtained from the peak outward current elicited from the second voltage step of the subtracted current. Currents were normalized to cell capacitance, and current density-voltage plots were constructed by plotting the current densities for each as a function of the voltage of the first step of the protocol, which elicits Ca2+ influx.
Statistics
All data were tested for normality with the Shapiro-Wilk test and either parametric or non-parametric statistical tests were performed. For parametric tests, one-way ANOVA with Bonferroni’s post-hoc test was performed. For non-parametric tests, the Kruskal-Wallis testand Dunn’s multiple comparisons test were performed. Statistical significance was determined at p < 0.05 using Prism v10, and significant p values are presented in the figure legends. Data are reported as group mean ± SEM.
Results
The goal of this study was to assess the BK current produced by channels containing one of four KCNMA1 channelopathy associated mutations under dynamic activation by Ca2+ entry through voltage-gated CaV1.2 channels. Wild type (BKWT) or mutant (BKD434G, BKN999S, BKH444Q or BKD965V) channels were co-expressed with CaV1.2 channels in HEK-293T cells. The auxiliary subunits CaVβ1b and CaVα2δ1 were expressed along with the CaVα1 subunit due to their copurification from rat brain with both CaVα1 and BKα, role in enhancing expression levels, requirement for normal gating properties, and high prevalence in neurons [24,32,44,45].
Macroscopic CaV1.2 and BK channel currents were recorded in the whole-cell voltage-clamp configuration, using a physiological K+ gradient and 2.5 mM [Ca2+]ext. We employed a two-part voltage step protocol to activate the currents (Figure 1b). The first part comprised depolarizing voltage steps (−100 mV to +60 mV, Δ20 mV, 50 ms) to activate CaV1.2 channels and initiate Ca2+ influx (conditioning step). The second part was a test step to +60 mV (50 ms) to activate BK channels at a voltage that does not result in significant inward Ca2+ current (ECa2+ = +130 mV, assuming a maximum [Ca2+]i of 100 nM). Using this protocol, total whole-cell currents consisting of inward Ca2+ current through CaV1.2 channels, followed by outward K+ current through BK channels, were elicited (Figure 1c). BK currents were pharmacologically isolated from CaV1.2 Ca2+ currents by application of 100 nM paxilline, a membrane permeant BK channel inhibitor (Figure 1d–e). These results show the sequential activation of CaV1.2 Ca2+ currents, followed by BK currents.
BK-CaV.2 channel currents under microdomain conditions
To assess the activation of wild type and mutant BK-CaV1.2 channels within a microdomain, whole-cell macroscopic current recordings were made in 10 mM intracellular EGTA buffering conditions [16,23]. Current density-voltage plots (I-Vs) were constructed from cells co-expressing BKWT channels with CaV1.2 channels (Figure 2a). BKWT-CaV1.2 channel currents showed the inward Ca2+ current peaked at 0 mV. The outward K+ current, from the second step in the voltage protocol to +60 mV, activated at depolarized voltages and peaked at +40 mV (Figure 2a, left panel). Ca2+ currents were assessed from the peak current density of the inward current from the first step, and K+ currents were evaluated from the peak outward current density of the subtracted BK channel current from the second step (Figure 2b).
Figure 2.

CaV1.2 and BK channel currents from cells co-expressing BKWT, BKD434G, BKN999S, BKH444Q and BKD965V in 10 mM EGTA. (a) Current versus conditioning step voltage relationships for CaV1.2 and BKWT (N = 17), BKD434G (N = 13), BKN999S (N = 12), BKH444Q (N = 17) and BKD965V (N = 12) channel currents plotted as a function of the first voltage step of the protocol which elicits Ca2+ influx. Representative traces are displayed in Supplemental Figure 1. (b) Peak CaV1.2 and BK channel current levels from (a). BKN999S currents were larger (p = 0.0034), and BKD965V currents were smaller (p = 0.0122), than BKWT. Expanded y-axis view of BK current levels shown in Supplemental Figure 3. CaV1.2 (BKN999S) currents were reduced compared to CaV1.2 (BKWT; p = 0.0003). (c) Normalized current ratios (IBK/ICav) were increased for CaV1.2 (BKD434G; p = 0.0572) and CaV1.2 (BKN999S; p < 0.0001) compared to CaV1.2 (BKWT). BKWT data on the right-hand side of the split x-axis is replotted for ease of comparison to BKN999S. in B-C panels, values are plotted as individual measurements with average and s.e.m. p values < 0.05 were considered significant.
Next, we tested BKD434G, BKN999S, BKH444Q, and BKD965V mutant channels to determine if their activity under CaV1.2 activation was consistent with prior GOF and LOF designations derived from clamped [Ca2+]i recordings [4,12,36,37,40]. Since both GOF channels exhibit faster activation compared to wild type, BKD434G and BKN999S channels were predicted to display increased BK current under CaV1.2 channel-mediated activation compared to BKWT. Moreover, given the larger G-V shift and faster activation kinetics compared to BKD434G, BKN999S was predicted to have a greater impact on BK-CaV elicited K+ current. Conversely, LOF channels activate slower than the wild type, and the delayed activation is anticipated to decrease BKH444Q and BKD965V channel currents under these conditions.
Ca2+ currents peaked around 0 mV (Figure 2a) and the levels were not significantly different between CaV1.2 channels co-expressed with BKWT (−46 ± 9 pA/pF) and CaV1.2 co-expressed with BKD434G, BKH444Q or BKD965V (−29 ± 5 pA/pF, −25 ± 2 pA/pF and −50 ± 14 pA/pF, respectively; Figure 2b). However, Ca2+ current from CaV1.2 (BKN999S) was significantly decreased compared to the wild type, averaging −17 ± 3 pA/pF. As BKN999S is the most severe GOF mutation [37], the observed difference between CaV1.2 (BKN999S) and CaV1.2 (BKWT) current densities may stem from limitations in clamping the particularly large currents induced by BKN999S upon Ca2+ influx, causing more seals to break in this condition compared to others. It is possible that successful recordings were easier to obtain from cells with smaller CaV1.2 currents. It is also possible that these differences reflect differences in CaV1.2 expression levels, although this difference was not observed consistently for CaV1.2 (BKN999S) across study conditions.
CaV1.2-activated BK channel currents peaked at +40 mV (Figure 2a–b). BKWT current density averaged 12 ± 3 pA/pF. The current density of the GOF mutant BKN999S was significantly larger than BKWT at 109 ± 30 pA/pF, despite the lower Ca2+ current densities described above, while the current density of the LOF mutant BKD965V was significantly smaller than BKWT at 3 ± 1 pA/pF. However, the current densities of BKD434G (25 ± 7 pA/pF) and BKH444Q (5 ± 1 pA/pF) were not significantly different from BKWT, and these respective measurements are plotted on an expanded scale in Supplemental Figure S3. Thus, the more severe GOF N999S and LOF D965V mutations both led to detectable differences in BK channel current density and in the expected directions, under CaV1.2 activation. Because these differences are similar to what has been observed for BKN999S and BKD965V under clamped calcium [1], it is likely that these differences arise from altered channel activity of these mutants.
In order to assess the current through wild type and mutant BK channels as a function of the available Ca2+ influx, the BK channel current was normalized to the absolute value of the peak inward CaV1.2 Ca2+ current shown in Figure 2b. In this analysis, GOF mutations should be more sensitive to Ca2+ influx than the wild type, resulting in a higher normalized current ratio (IBK/ICav), while LOF mutations should be less sensitive, resulting in a lower ratio. IBKWT/ICav averaged 0.3 ± 0.01, while IBKN999S/ICav was ~28 times greater at 8.4 ± 1.8. This large increase was not solely due to the voltage-dependent activation of BKN999S channels, as no significant BK current was detected when BKN999S channels were expressed alone in HEK-293T cells and activated using the same voltage protocols (data not shown). BKD434G also showed a trend toward an increase in the IBK/ICav ratio (0.9 ± 0.1). The normalized IBK/ICav was not significantly changed with either LOF mutation compared to BKWT. The BK channel current was also normalized to the maximum value of CaV1.2 Ca2+ total charge, which demonstrated identical statistically significant results as normalization to the peak inward CaV1.2 current (data not shown).
These data demonstrate that the GOF variant (N999S) shows a clear increase in BK channel current under voltage-gated CaV1.2 Ca2+ influx within an EGTA-buffered microdomain. The D434G, H444Q, and D965V patient variants did not exhibit distinguishable GOF or LOF behavior under these conditions, in contrast to what has been previously observed in clamped [Ca2+]i recordings [4,12,35–40,43]. This result suggests that the dynamic diffusion of Ca2+ from CaV1.2 to BK channels within an EGTA-delimited microdomain can influence the extent of mutant BK channel phenotypes.
BK-CaV.2 channel currents under nanodomain conditions
While EGTA permits Ca2+ diffusion across tens to hundreds of nanometers, BAPTA restricts Ca2+ diffusion to a significantly smaller nanodomain around CaV1.2. Channels within nanodomains are tightly functionally coupled, typically due to co-localization proximity or direct interactions within neurons [14]. We next asked if the changes in current from CaV1.2-activated mutant BK channels persisted within nanodomains by recording currents in 2 mM BAPTA using the same two pulse voltage protocol. Under BAPTA conditions, CaV1.2 Ca2+ currents peaked between 0–20 mV (Figure 3a) and were smaller than the inward currents recorded in EGTA. CaV1.2 (BKWT) current density averaged −13 ± 3 pA/pF (Figure 3b). There was no significant difference in CaV1.2 Ca2+ currents when CaV1.2 was co-expressed with BKD434G, BKN999S or BKH444Q. However, CaV1.2 currents were significantly increased (−32 ± 8 pA/pF) when co-expressed with BKD965V compared to BKWT.
Figure 3.

CaV1.2 and BK channel currents from cells co-expressing BKWT, BKD434G, BKN999S, BKH444Q and BKD965V in 2 mM BAPTA. (A) Current versus conditioning step voltage relationships for CaV1.2 and BKWT (N = 7), BKD434G (N = 10), BKN999S (N = 7), BKH444Q (N = 4) and BKD965V (N = 7) channel currents plotted as a function of the first voltage step of the protocol which elicits Ca2+ influx. Representative traces are displayed in Supplemental Figure 2. (B) Peak CaV1.2 and BK channel current levels from (A). BKN999S currents were larger than BKWT (p = 0.0247), and CaV1.2 (BKD965V) currents were increased compared to CaV1.2 (BKWT)(p = 0.0099). (C) Normalized current ratios (IBK/ICav) were increased for CaV1.2 (BKN999S; p = 0.0319) compared to CaV1.2 (BKWT).
CaV1.2-activated BKWT current density averaged 10 ± 2 pA/pF (Figure 3A–B). BKN999S current density was significantly larger than BKWT at 29 ± 5 pA/pF, as it was in EGTA buffering conditions (Figure 2). Some of this BKN999S current was detectable at hyperpolarized voltages in the absence of CaV1.2 expression (7.6 ± 1.2 pA/pF at −20 mV, n = 5), suggesting it results from the GOF effect of the N999S mutation. There were no statistically significant differences in BK current density between BKWT and BKD434G, BKH444Q, or BKD965V under 2 mM BAPTA buffering conditions. However, when BK channel current was normalized to the inward CaV1.2 Ca2+ current to assess BK channel current as a function of Ca2+ influx, the normalized IBKWT/ICav averaged 0.8 ± 0.2 within BAPTA nanodomains (Figure 3c), higher than IBKWT/ICav in EGTA (Figure 2c). Normalized IBKN999S/ICav current was ~4 times greater (3.5 ± 1) than IBKWT/ICav. BKD434G, BKH444Q or BKD965V showed no statistically significant difference in IBK/ICav compared to BKWT. Taken together, the data in Figures 2, 3 demonstrate that while the BK to CaV1.2 channel current ratio increases from a Ca2+ microdomain to a nanodomain, only the most severe GOF variant (N999S) produced an increase in both Ca2+ conditions. The GOF and LOF behaviors for the D434G, H444Q, or D965V variants shown in clamped [Ca2+]i conditions were not recapitulated under activation by CaV1.2 Ca2+ current in heterologous cells.
BK-CaV.2 channel currents elicited by action potentials
We next applied single action potential voltage commands to test whether the observed effects of channelopathy mutations on BK channel current persist under conditions that use physiologically relevant stimuli to open CaV1.2 channels. During an action potential, changes in the voltage dependence of activation, as well as activation rate, will affect the peak BK current levels. Action potential-evoked currents were elicited from a holding potential of −90 mV, followed by an action potential voltage command obtained from a previously recorded dentate granule neuron waveform [4]. CaV1.2 and BK channel currents were recorded under microdomain and nanodomain buffering conditions.
In 10 mM EGTA, we found that CaV1.2 (BKWT) current density averaged −35 ± 6 pA/pF, and there were no statistically significant differences between CaV1.2 (BKWT) current density and that of CaV1.2 (BKD434G), CaV1.2 (BKN999S), CaV1.2 (BKH444Q) and CaV1.2 (BKD965V) under these conditions (Figure 4a). Outward BKWT current density averaged 5 ± 2 pA/pF, and BKN999S current density was significantly larger than BKWT at 24 ± 7 pA/pF. BKD434G, BKH444Q, and BKD965V currents were not statistically different from BKWT (Figure 4b and Supplemental Figure S4). Normalized IBK/ICav averaged 0.2 ± 0.02 for BKWT (Figure 4c). IBKN999S/ICav was 1.3 ± 0.3, ~7 times greater than BKWT; the other human patient variants showed no statistically significant difference in the IBK/ICav ratio compared to BKWT.
Figure 4.
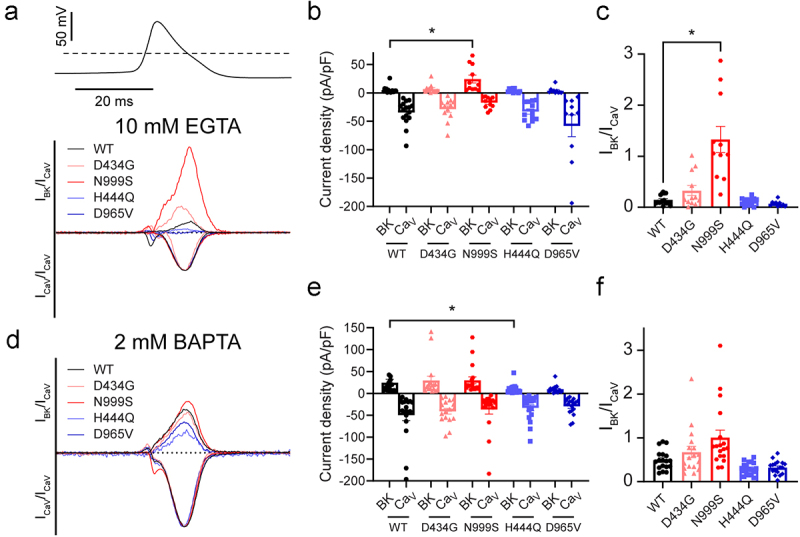
Action potential evoked CaV1.2 and BK channel currents from cells co-expressing BKWT, BKD434G, BKN999S, BKH444Q and BKD965V. (A, D) Representative whole-cell CaV1.2 (inward) and BK (outward) currents from cells co-expressing CaV1.2 (BKWT), CaV1.2 (BKD434G), CaV1.2 (BKN999S), CaV1.2 (BKH444Q), and CaV1.2 (BKD965V) channels in 10 mM EGTA (A) and 2 mM BAPTA (D). Traces are normalized to the absolute value of the peak CaV1.2 channel current. Dotted line represents zero current level. Insets: action potential voltage command. (B, E) BK and CaV1.2 channel currents from the peak of the action potential in 10 mM EGTA (B; N = 10-16 per condition) and 2 mM BAPTA (E; N = 17-18 per condition). BKN999S current was larger than BKWT in EGTA (p = 0.0033). BKH444Q current was smaller than BKWT in BAPTA (p = 0.0422). Expanded y-axis view of BK current levels in panel B shown in supplemental Figure 4. No significant differences in CaV1.2 currents were observed. (C, F) normalized current ratios (IBK/ICav) in 10 mM EGTA (C) and 2 mM BAPTA (F). CaV1.2 (BKN999S) channel current was larger compared to CaV1.2 (BKWT) in EGTA (p = 0.003) but not statistically significant in BAPTA (p = 0.0643).
In 2 mM BAPTA (nanodomain) buffering conditions, CaV1.2 (BKWT) current density averaged −50 ± 12 pA/pF (Figure 4d–e), and there were no statistically significant differences compared to CaV1.2 (BKD434G), CaV1.2 (BKN999S), CaV1.2 (BKH444Q) or CaV1.2 (BKD965V). Outward BKWT current density averaged 25 ± 8 pA/pF, and BKN999S, BKD434G, and BKD965V currents were not statistically different. Thus with BAPTA buffering, the increase in CaV1.2-activated BKN999S current we observed under the two-step voltage protocol was not detectable under an action potential waveform voltage command. BKH444Q showed a decrease in current density compared to BKWT (10 ± 3 pA/pF). This was the only condition where the H444Q variant showed a significant effect on current. However, once the current density was normalized to assess IBK as a function of ICav, no difference was observed between any of the variants (Figure 4f). This result indicates that under certain conditions, such as the rapid BAPTA buffering and millisecond depolarization from a single action potential, even the strong GOF mutant BKN999S channels are unable to produce a detectable difference in CaV1.2-activated BK current.
Discussion
This study describes the relative BK current levels associated with four representative KCNMA1 channelopathy variants compared to the wild type under CaV1.2 activation. N999S is the most common patient variant and causes more severe neurological disease than the other variants tested in this study [1,4]. In clamped Ca2+ recordings, N999S produces strong GOF changes in multiple aspects of BK channel gating [36,37]. Consistent with this, CaV1.2-activated BKN999S current showed increased steady-state current within both microdomain (EGTA) and nanodomain (BAPTA) Ca2+ buffering conditions (Figures 2,3). Importantly, an increase in CaV1.2-activated BKN999S current was also evident when the BK current was analyzed as a function of the CaV1.2 current magnitude, suggesting that these results reflect the underlying GOF properties of the BKN999S channels and are not due to changes in the Ca2+ current.
While steady-state currents recorded under clamped Ca2+ allow maximal activation of BK channels, we found that the BKN999S GOF effect was still detectable when CaV1.2-activated BK channel current was evoked using an action potential command within Ca2+ microdomain conditions. The activation for CaV1.2 channels ranges from 1–5 ms, depending on membrane potential and auxiliary subunit composition [28,46], and CaV1.2 channels do not achieve maximal Po during short single spikes [28]. The time to peak for the neuronal action potential voltage command used in this study was <0.5 ms [4], and the nanodomain context would further reduce Ca2+-dependent activation. Under this condition, the BKN999S GOF effect, suggesting that the Ca2+ channel openings and BK-CaV coupling context is an important determinant of the GOF current produced by mutant BKN999S channels.
In contrast, the other channelopathy mutations did not show the systematic set of changes across conditions in CaV1.2-activated mutant BK channel current that were observed for N999S. This finding parallels the results from mouse models generated from these mutations. Interestingly, the GOF D434G variant causes similar neurological phenotypes as N999S, but in smaller subset of human patients [38] and with a comparatively milder phenotype in transgenic mice [4]. The LOF H444Q variant is found in a single individual and does not recapitulate the full channelopathy disease phenotype in mice [4]. The D965V variant has also only been found in a single individual so far and has not been tested yet in a transgenic animal model. Thus, finding alterations in CaV1.2-activated BK current for only one of the mutations tested in this study was different from previous work performed in clamped Ca2+ conditions, where all four mutations showed significant differences from the wild type [1]. These results suggest that 1) testing KCNMA1 variants in BK channels recorded in clamped Ca2+ conditions using steady-state voltage protocols may overestimate the potential for pathogenicity, and 2) cellular buffering and dynamic voltage stimuli may differentially affect CaV channel activation of mutant BK channel currents. It remains to be determined whether these contextual elements explain some of the heterogeneity observed in KCNMA1 channelopathy.
Several factors may account for the differences between BK channel activation under steady-state and CaV channel mediated Ca2+ influx, including the type of biophysical alteration produced by the respective channelopathy mutations, the use of paxilline to isolate the BK current, the respective expression levels of BK and CaV1.2 channels, the strength of coupling between BK and CaV channels, and any feedback of BK current on Ca2+ influx. With respect to the biophysical basis of BK channel mutations, the two mutations identified as GOF in clamped Ca2+ conditions differ mechanistically. N999S acts by enhancing voltage sensitivity, and BKN999S channels exhibit a 1.5-fold larger G-V shift and ~3.5-fold faster activation time constant than BKD434G channels [37]. Eliminating calcium-dependent activation in BKN999S channels does not affect the G-V shift [36]. This predominant voltage mechanism could support the GOF activity of BKN999S channels under a wider range of Ca2+ conditions than BKD434G channels. Conversely, BKD434G channels demonstrate increased Ca2+ sensitivity in clamped Ca2+ recording conditions [38–40,42], a finding that could connect their activation more closely to the Ca2+ buffering or BK-CaV coupling context. BKD434G channels also exhibit a more pronounced hyperpolarizing V1/2 shift in the midrange of [Ca2+]i, suggesting that this GOF mutation may behave nonlinearly under dynamic CaV1.2 activation. A single study has probed the activation of BKD434G channels by a CaV channel (CaV2.2; N-type). Under those study conditions, CaV2.2-activated BKD434G channel current showed an acceleration of BK channel activation and reduction in activation lag time compared to BKWT [31]. For LOF mutations characterized in clamped Ca2+ conditions, the biophysical basis for changes in BKH444Q and BKD965V channel gating has not been reported. Both mutations localize to regions of the BK channel gating ring that are involved in Ca2+-dependent activation. The effect of each of these LOF mutations is smaller than the absolute effect of either N999S or D434G [1,4,12], consistent with the data in this study.
Use of paxilline to isolate the BK current also has a possibility of differentially affecting currents from GOF versus LOF mutations, based on the inverse relationship of paxilline and BK channel open probability. Paxilline inhibition was lower with closed channels compared to maximally open channels, and this relationship is also Ca2+ modulated [47]. Although the patient mutations tested here are not expected to affect paxilline binding based on their locations [48], it is also possible that paxilline affinity differs between mutations. These possibilities await further testing in experiments that specifically control BK channel open probability for each mutation. Such experiments may also be informative for understanding the therapeutic potential of paxilline and related compounds for myotonia [49].
Another factor influencing the functional designations derived from clamped Ca2+ and CaV-mediated BK channel activation conditions is the relative expression level of each channel type, which was not assessed in this study. Differing BK-CaV expression ratios would not be relevant for activating BK channels in clamped Ca2+ but could affect the stoichiometry and coupling strength of CaV1.2-BK channel complexes. The assessment of BK-CaV channel currents in the two buffering conditions tested in this study (EGTA and BAPTA) likely reflects different coupling scenarios, with the EGTA condition including less tightly coupled channels than the BAPTA condition. This is supported by the decrease in CaV-activated BK channel current observed between EGTA and BAPTA. However, the stoichiometry of these coupled channels cannot be assessed in this study and is not resolved in other studies. Some models have suggested a fixed CaV to BK channel stoichoimetry (Prakriya & Lingle, 2000) [20,23], while other investigations only demonstrated a statistical bias for CaV and BK channel proximity with no fixed stoichiometry or geometry within the clusters (Vivas et al., 2017). Additionally, scaffolding proteins could regulate the spatial arrangement of BK-CaV complexes [50], yet it is not known if these proteins function similarly in native and heterologous systems. Interestingly, higher BK channel expression has been shown to compete α2δ away from CaV2.2 channels, reducing the Ca2+ current density [17]. Thus, how the summation of expression affects coupling of BK-CaV channels remains to be determined.
There are two conditions where CaV1.2 currents are altered by co-expression with mutant BK channels in this study: BKN999S (EGTA) and BKD965V (BAPTA). It is possible that recordings for these two variants were easier to obtain from cells that had smaller Ca2+ currents for the GOF mutation and larger Ca2+ currents for the LOF mutation. However, since the Ca2+ current changes were not observed consistently across conditions, whether there is a relevant underlying biological mechanism related to the BK channel mutations or variation in expression related to the mutations remains to be determined. Such a mechanism could involve functional feedback of the BK current on activation of the Ca2+ channel or an altered interaction between those particular mutants and CaV1.2 subunits. Expression, stoichiometry, and coupling are difficult to control in heterologous expression systems using high copy plasmids, and future studies specifically designed to address whether the mutations cause differences in expression and assembly of BK-CaV1.2 complexes will be needed. In addition, patients harbor heterozygous alleles, creating the potential for heterotetrameric BK channels [51]. Nevertheless, this study was able to evaluate the effects of these BK channel mutations by normalizing BK current to the Ca2+ current.
While the central finding of this work was the corroboration of the GOF effect of N999S variant under CaV1.2-mediated BK channel activation, the results for the three other representative channelopathy variants illustrate that the effects of KCNMA1 variants could be highly neuron dependent. Important contextual variations could include different CaV subunits, Ca2+ buffering conditions known to affect coupling strength and type of gating stimuli (waveform and frequency of action potentials). Examining the effects of BK channelopathy mutations in a wider range of contexts, such as within different neuronal types, may yield more realistic predictions about the pathogenicity of novel variants. Improved predictions will provide a more detailed understanding of how dysregulation of BK channel gating may lead to altered neuronal excitability and support stronger genotype-phenotype correlations in KCNMA1-linked disease.
Supplementary Material
Acknowledgments
We thank Ivy Dick and Joerg Striessnig for the Ca2+ channel cDNA constructs and Hans Moldenhauer for data discussions. Figure 1a created with Biorender.com.
Funding Statement
This work was funded by NHLBI R01-HL102758 (ALM) and the NIGMS ASCEND Scholars Program Grant 2RL5GM118972. National Institute of General Medical Sciences [ASCEND Scholars Program Grant 2RL5GM118972]; National Heart Lung and Blood Institute [R01HL102758]
Disclosure statement
No potential conflict of interest was reported by the author(s).
Author contributions
R.L. Dinsdale: research conception and data collection, data analysis, statistical analysis design and execution, and manuscript writing. A.L. Meredith: research conception, data analysis, and manuscript writing. All authors approved the final version of the manuscript.
Data availability statement
The data are available from the corresponding author, ALM, upon reasonable request.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19336950.2024.2396346
References
- [1].Meredith AL. BK channelopathies and KCNMA1-linked disease models. Annu Rev Physiol. 2024 [Feb 12];86(1):277–13. doi: 10.1146/annurev-physiol-030323-042845 [DOI] [PubMed] [Google Scholar]
- [2].Bailey CS, Moldenhauer HJ, Park SM, et al. KCNMA1-linked channelopathy. J Gen Physiol. 2019 [Oct 7];151(10):1173–1189. doi: 10.1085/jgp.201912457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Miller JP, Moldenhauer HJ, Keros S, et al. An emerging spectrum of variants and clinical features in KCNMA1-linked channelopathy. Channels (Austin). 2021. [Dec;15(1):447–464. doi: 10.1080/19336950.2021.1938852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Park SM, Roache CE, Iffland PH.. 2nd, BK channel properties correlate with neurobehavioral severity in three KCNMA1-linked channelopathy mouse models. Elife. 2022 [Jul 12];11:e77953. doi: 10.7554/eLife.77953 et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dong P, Zhang Y, Hunanyan AS, et al. Neuronal mechanism of a BK channelopathy in absence epilepsy and dyskinesia. Proc Natl Acad Sci. 2022 [Mar 22];119(12):e2200140119. doi: 10.1073/pnas.2200140119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dinsdale RL, Roache CE, Meredith AL. Disease-associated KCNMA1 variants decrease circadian clock robustness in channelopathy mouse models. J Gener Physiol. 2023;155(11). doi: 10.1085/jgp.202313357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sun AX, Yuan Q, Fukuda M, et al. Potassium channel dysfunction in human neuronal models of Angelman syndrome. Science. 2019 [Dec 20];366(6472):1486–1492. doi: 10.1126/science.aav5386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhu Y, Zhang S, Feng Y, et al. The Yin and Yang of BK channels in epilepsy. CNS Neurol Disord Drug Targets. 2018;17(4):272–279. doi: 10.2174/1871527317666180213142403 [DOI] [PubMed] [Google Scholar]
- [9].Perche O, Lesne F, Patat A, et al. Large-conductance calcium-activated potassium channel haploinsufficiency leads to sensory deficits in the visual system: a case report. J Med Case Rep. 2022 [May 5];16(1):180. doi: 10.1186/s13256-022-03387-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Du X, Carvalho-de-Souza JL, Wei C, et al. Loss-of-function BK channel mutation causes impaired mitochondria and progressive cerebellar ataxia. Proc Natl Acad Sci USA. 2020 [Mar 4];117(11):6023–6034. doi: 10.1073/pnas.1920008117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Deng PY, Klyachko VA. Genetic upregulation of BK channel activity normalizes multiple synaptic and circuit defects in a mouse model of fragile X syndrome. J Physiol. 2016 [Jan 1];594(1):83–97. doi: 10.1113/JP271031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Moldenhauer HJ, Tammen K, Meredith AL. Structural mapping of patient-associated KCNMA1 gene variants. Biophys J. 2024 [Jul 16];123(14):1984–2000. doi: 10.1016/j.bpj.2023.11.3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tao X, MacKinnon R. Molecular structures of the human Slo1 K+ channel in complex with beta4. Elife. 2019 [Dec 9];8. doi: 10.7554/eLife.51409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shah KR, Guan X, Yan J. Structural and functional coupling of calcium-activated BK channels and calcium-permeable channels within nanodomain signaling complexes. Front Physiol. 2022;12:12. doi: 10.3389/fphys.2021.796540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Raimondo JV, Burman RJ, Katz AA, et al. Ion dynamics during seizures [review]. Front Cell Neurosci. 2015 [Oct 21];9. doi: 10.3389/fncel.2015.00419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fakler B, Adelman JP. Control of KCa channels by calcium nano/microdomains [review]. Neuron. [2008 Sep 25];59(6):873–881. doi: 10.1016/j.neuron.2008.09.001 [DOI] [PubMed] [Google Scholar]
- [17].Zhang FX, Gadotti VM, Souza IA, et al. BK potassium channels suppress Cavalpha2delta subunit function to reduce inflammatory and neuropathic pain. Cell Rep. 2018 [Feb 20];22(8):1956–1964. doi: 10.1016/j.celrep.2018.01.073 [DOI] [PubMed] [Google Scholar]
- [18].Rehak R, Bartoletti TM, Engbers JD, et al. Low voltage activation of KCa1.1 current by Cav3-KCa1.1 complexes. PLoS One. 2013;8(4):e61844. doi: 10.1371/journal.pone.0061844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Augustine GJ, Santamaria F, Tanaka K. Local calcium signaling in neurons. Neuron. 2003 [Oct 9];40(2):331–346. doi: 10.1016/S0896-6273(03)00639-1 [DOI] [PubMed] [Google Scholar]
- [20].Fakler B, Adelman JP. Control of K(Ca) channels by calcium nano/microdomains. Neuron. 2008 [Sep 25];59(6):873–881. doi: 10.1016/j.neuron.2008.09.001 [DOI] [PubMed] [Google Scholar]
- [21].Naraghi M, Neher E. Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of [Ca2+] at the mouth of a calcium channel. J Neurosci. 1997 [Sep 15];17(18):6961–6973. doi: 10.1523/JNEUROSCI.17-18-06961.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Vivas O, Moreno CM, Santana LF, et al. Proximal clustering between BK and CaV1.3 channels promotes functional coupling and BK channel activation at low voltage. Elife. 2017 [Jun 30]:e28029 doi: 10.7554/eLife.28029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cox DH. Modeling a Ca2+ channel/BKCa channel complex at the single-complex level. Biophys J. 2014 [Dec 16];107(12):2797–2814. doi: 10.1016/j.bpj.2014.10.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Berkefeld H, Sailer CA, Bildl W, et al. BKCa-CaV channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science. 2006 [Oct 27];314(5799):615–620. doi: 10.1126/science.1132915 [DOI] [PubMed] [Google Scholar]
- [25].Engbers JD, Zamponi GW, Turner RW. Modeling interactions between voltage-gated Ca2+ channels and KCa1.1 channels. Channels (Austin). 2013. [Nov-Dec];7(6):524–529. doi: 10.4161/chan.25867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Whitt JP, Ba M, Meredith AL. Differential contribution of Ca2+ sources to day and night BK current activation in the circadian clock. J Gen Physiol. 2018 [Feb 5];150(2):259–275. doi: 10.1085/jgp.201711945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Plante AE, Whitt JP, Meredith AL. BK channel activation by L-type Ca2+ channels CaV.2 and CaV.3 during the subthreshold phase of an action potential. J Neurophysiol. 2021 [Aug 1];126(2):427–439. doi: 10.1152/jn.00089.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Berkefeld H, Fakler B. Repolarizing responses of BKCa-Cav complexes are distinctly shaped by their cav subunits. J Neurosci. 2008 [Aug 13];28(33):8238–8245. doi: 10.1523/JNEUROSCI.2274-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Blömer LA, Giacalone E, Abbas F, et al. Kinetics and functional consequences of BK channels activation by N-type Ca2+ channels in the dendrite of mouse neocortical layer-5 pyramidal neurons. Front Cell Neurosci. 2024. 2024-Feb 14;18:18. doi: 10.3389/fncel.2024.1353895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Loane DJ, Lima PA, Marrion NV. Co-assembly of N-type Ca2+ and BK channels underlies functional coupling in rat brain. J Cell Sci. 2007 [Mar 15];120(Pt 6):985–995. doi: 10.1242/jcs.03399 [DOI] [PubMed] [Google Scholar]
- [31].Berkefeld H, Fakler B. Ligand-gating by Ca2+ is rate limiting for physiological operation of BKCa channels. J Neurosci. 2013 [Apr 24];33(17):7358–7367. doi: 10.1523/JNEUROSCI.5443-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Catterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol. 2011. Aug;3(8):a003947. doi: 10.1101/cshperspect.a003947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Grunnet M, Kaufmann WA. Coassembly of big conductance Ca2+-activated K+ channels and L-type voltage-gated Ca2+ channels in rat brain. J Biol Chem. 2004 [Aug 27];279(35):36445–36453. doi: 10.1074/jbc.M402254200 [DOI] [PubMed] [Google Scholar]
- [34].Herold KG, Hussey JW, Dick IE. CACNA1C-Related channelopathies. Handb Exp Pharmacol. 2023;279:159–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Liang L, Liu H, Bartholdi D, et al. Identification and functional analysis of two new de novo KCNMA1 variants associated with Liang–Wang syndrome. Acta Physiol. 2022. Feb;13(1):e13800. doi: 10.1111/apha.13800 [DOI] [PubMed] [Google Scholar]
- [36].Li X, Poschmann S, Chen Q, et al. De Novo BK channel variant causes epilepsy by affecting voltage gating but not Ca2+ sensitivity. Eur J Hum Genet. 2018. Feb;26(2):220–229. doi: 10.1038/s41431-017-0073-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Moldenhauer HJ, Matychak KK, Meredith AL. Comparative gain-of-function effects of the KCNMA1-N999S mutation on human BK channel properties. J Neurophysiol. 2020 [Feb 1];123(2):560–570. doi: 10.1152/jn.00626.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Du W, Bautista JF, Yang H, et al. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet. 2005. Jul;37(7):733–738. doi: 10.1038/ng1585 [DOI] [PubMed] [Google Scholar]
- [39].Wang B, Rothberg BS, Brenner R. Mechanism of increased BK channel activation from a channel mutation that causes epilepsy. J Gen Physiol. 2009. Mar;133(3):283–294. doi: 10.1085/jgp.200810141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yang J, Krishnamoorthy G, Saxena A, et al. An epilepsy/dyskinesia-associated mutation enhances BK channel activation by potentiating Ca2+ sensing. Neuron. 2010 [Jun 24];66(6):871–883. doi: 10.1016/j.neuron.2010.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Plante AE, Lai MH, Lu J, et al. Effects of single nucleotide polymorphisms in human KCNMA1 on BK current properties. Front Mol Neurosci. 2019;12:285. doi: 10.3389/fnmol.2019.00285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Diez-Sampedro A, Silverman WR, Bautista JF, et al. Mechanism of increased open probability by a mutation of the BK channel. J Neurophysiol. 2006. Sep;96(3):1507–1516. doi: 10.1152/jn.00461.2006 [DOI] [PubMed] [Google Scholar]
- [43].Moldenhauer H, Park SM, Meredith AL. Characterization of new human KCNMA1 loss-of-function mutations. Biophys J. 2020;118(3):114a. doi: 10.1016/j.bpj.2019.11.767 [DOI] [Google Scholar]
- [44].Dolphin AC. The alpha2delta subunits of voltage-gated calcium channels. Biochim Biophys Acta. 2013. Jul;1828(7):1541–1549. doi: 10.1016/j.bbamem.2012.11.019 [DOI] [PubMed] [Google Scholar]
- [45].Ferrandiz-Huertas C, Gil-Minguez M, Lujan R. Regional expression and subcellular localization of the voltage-gated calcium channel beta subunits in the developing mouse brain. J Neurochem. 2012. Sep;122(6):1095–1107. doi: 10.1111/j.1471-4159.2012.07853.x [DOI] [PubMed] [Google Scholar]
- [46].Helton TD, Xu W, Lipscombe D. Neuronal L-type calcium channels open quickly and are inhibited slowly. J Neurosci. 2005 [Nov 2];25(44):10247–10251. doi: 10.1523/JNEUROSCI.1089-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhou Y, Lingle CJ. Paxilline inhibits BK channels by an almost exclusively closed-channel block mechanism. J Gen Physiol. 2014. Nov;144(5):415–440. doi: 10.1085/jgp.201411259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhou Y, Xia XM, Lingle CJ. The functionally relevant site for paxilline inhibition of BK channels. Proc Natl Acad Sci. 2020 [Jan 14];117(2):1021–1026. doi: 10.1073/pnas.1912623117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hoppe K, Sartorius T, Chaiklieng S, et al. Paxilline prevents the onset of myotonic stiffness in pharmacologically induced myotonia: a preclinical investigation. Front Physiol. 2020;11:533946. doi: 10.3389/fphys.2020.533946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Sclip A, Acuna C, Luo F, et al. Rim-binding proteins recruit bk-channels to presynaptic release sites adjacent to voltage-gated Ca2+-channels. Embo J. 2018 [Aug 15];37(16). doi: 10.15252/embj.201798637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Geng Y, Li P, Butler A, et al. BK channels of five different subunit combinations underlie the de novo KCNMA1 G375R channelopathy. J Gen Physiol. 2023 [May 1];155(5):e202213302. doi: 10.1085/jgp.202213302 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data are available from the corresponding author, ALM, upon reasonable request.