

Abstract
Sepsis is a major cause of morbidity and mortality, but our understanding of the mechanisms underlying survival or susceptibility is limited. As pathogens often subvert host defence mechanisms, we hypothesised this might influence the outcome of sepsis. Here we used microbiota analysis, faecal microbiota transplant, antibiotic treatment and caecal metabolite analysis to show that gut microbiota-derived tryptophan metabolites including indoles increased host survival during a mouse model of Serratia marcescens sepsis. Infection in macrophage-specific aryl hydrocarbon receptor (AhR) knock-out mice revealed that AhR activation induced transcriptional reprogramming, in macrophages, increased bacterial clearance and host survival. However, culture supernatants from multiple bacterial pathogens inhibited AhR activation in vitro. We showed that the secreted siderophore, enterobactin, also inhibited Ahr activation in vitro, and increased sepsis mortality in vivo. In contrast, oral or systemic tryptophan supplementation increased survival. These findings show that sepsis survival depends upon the interplay between pathogen inhibition and microbiota-derived metabolite-dependent activation of AhR.
Introduction
Despite the many advances in our understanding of the role of the immune system on the pathophysiology of sepsis, the precise factors that govern survival remain poorly understood1,2. Given its definition as a “dysregulated host response to an infection,” survival from sepsis has traditionally been focused on understanding and modulating the immune response following a standardized infectious insult. This immunology centered approach has been difficult to translate to the human condition, as the majority of studies have failed to account for the behavior of the infecting pathogen(s) during the course of infection.
Yet the infecting pathogen (s) may not be the only microbial factors participating in the host response to infection. Recent studies have demonstrated the importance of the role of the gut microbiota and their metabolites on survival following injury and infection3,4. Here we show that the dual, yet distinct, actions of both the microbiota and pathogen-derived metabolites on the aryl hydrocarbon receptor (AhR) activity in macrophages may play a role in survival following intraperitoneal infection. AhR is a cytoplasmic receptor that can interact with microbial ligands and regulate the host immune system.5 In particular loss of AhR sensitizes mice to LPS via its regulation of NFkB6 as well as through microbial tolerance thus limitating the subsequent immunopathology involving IL-107. The ability of AhR to interact with microbial ligands and regulate the innate immune response places it at a unique position to play an essential role in survival from a lethal bacterial infection.
We have previously demonstrated that when the composition and function of the gut microbiota are altered by a combination of diet, antibiotics and surgical stress, mice display an increased susceptibility to severe infection8,9. In particular, increased mortality can be observed following an otherwise recoverable surgical injury in association with decreased gut butyrate5. Consequently, increasing butyrate levels via dietary modulation has been associated with improved survival10. Similarly, when a fecal microbiota transplant (FMT) is administered, gut butyrate concentrations are increased, and survival improves from a an infectious insult9. Although we previously determined that the FMT response occurred in an IRF3 dependent manner9, causality could not be established between the metabolites produced by the gut-microbiota and mortality. Therefore, in the current report we hypothesized that specific gut-microbiota derived metabolites can be identified that are deterministic of “within-group” survival among co-housed, genetically similar, inbred mice subjected to a standardized dose of a monomicrobial or polymicrobial intraperitoneal (IP) inoculum. We further hypothesized that opposing forces from the infecting pathogen itself might overcome this effect and subvert a beneficial effect from gut microbiota-derived metabolites.
In this study, we demonstrated that “within-group mortality” rates following an intraperitoneal LD50 dose of S. marcescens is influenced by the production of indole metabolites by the gut microbiota. Indole metabolites enhanced host survival through activation of AhR on macrophages enhancing bactericidal activity and early resolution of inflammation with increased expression of the M2 phenotype. Finally, we found pathogens actively inhibit AhR activation through the production of enterobactin placing AhR at the center of host-microbiota-pathogen interactions and their influence on survival.
Results
Survival from bacterial sepsis is dependent on the gut microbiota
We began experiments using a monomicrobial model of lethal infection by intraperitoneal delivery of S. marcescens strain MVI (mouse virulent isolate) that was previously demonstrated to cause mortality in several animal models (Fig.1A)8,9. In the present study, to dissociate the effect of systemic illness on mortality and other measures in this model, we used core body temperature to capture a time point at which mice appeared clinically well, but their survival could be accurately predicted prior to becoming systemically ill. 11–14 Both sepsis score (Fig.1B) and core body temperature (Fig.1C) demonstrated distinct alterations over the course infection between surviving and non-surviving mice starting at 8 hours and delineating by 15 hours post-infection. As expected, surviving mice had improved clearance of S. marcescens from the peritoneal cavity at 8- and 15-hours post IP injection compared to the non-survival group (Fig.1D). To confirm the requirement of the gut microbiota on survival from S. marcescens peritonitis in this model, their microbiota was disrupted by administering systemic cefoxitin and oral clindamycin for 5 days that was completed 24 hours prior to IP inoculation of S. marcescens. Mice treated with antibiotics demonstrated a significant increase in mortality (Fig.1E). Further supporting the importance of the microbiota in our model, mice were able to be rescued with an FMT (fecal microbiota transplant) consisting of cecal contents from a healthy littermate control, delivered via enema at the time of infection6 (Fig.1E). In summary, depletion of the microbiota with antibiotics enhanced mortality, whereas microbiota modification with an FMT enhanced survival, supporting a central role of the microbiota on outcome.
Figure 1. Survival from S. marcescens peritonitis is dependent on the gut microbiota and correlates with peritoneal macrophage phenotype.
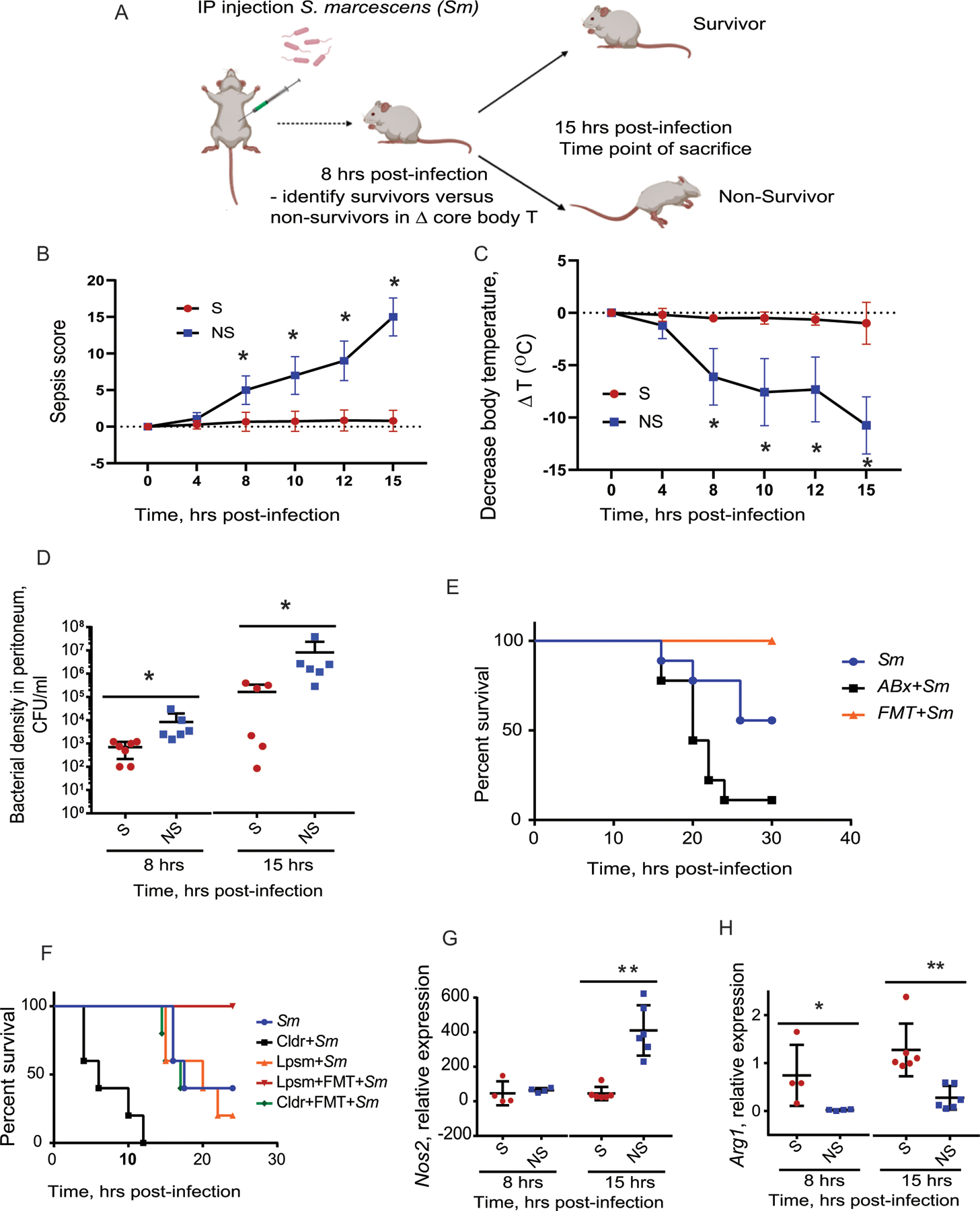
(A), Mouse model of IP Serratia marcescens injection. (B-D), Surviving mice are characterized by lower sepsis score (p = 1.5×10−4 at 8h) (B), higher core body temperature (p = 8.18×10−5 at 8h )(C), and lower S. marcescens density in the peritoneum (D). n=7 mice per group, *p=0.046, Mann-Whitney unpaired t-test. (E), Kaplan-Meyer survival curves demonstrating that disruption of gut microbiota by antibiotics (clindamycin and cefoxitin) increases mortality in infected mice while replenishment of the gut microbiota via FMT protects against mortality. n=10 per group, p<0.0001, Log-rank (Mantel-Cox) test. (F), Kaplan-Meyer survival curves demonstrating that depletion of macrophages by clodronate liposomes increases mortality of mice following intraperitoneal (IP) injection of S. marcescens. n=5 per group, p<0.0001, Log-rank (Mantel-Cox) test. (G,H). pMACs isolated from survivors have lower expression of canonical M1 gene Nos2, *p = **p = 0.0001 at 15h (G) associated with higher expression of the canonical M2 gene Arg1, *p = 0.031 at 8h, **p = 0.00229 at 15h (H). n=4 mice per group for 8 hrs time point; n=6 mice per group for 15 hrs time point. Mann-Whitney unpaired t-test. Sm, S. marcescens; Cldr, clodronate liposomes; Lpsm, liposomes (vehicle control); FMT, fecal microbial transplant. For temperature tracking experiments, n = 52 (Survivors, n = 21; Non-Survivors, n = 31, in 3 independent experiments). Error bars represent standard deviation. All statistical tests were two-sided and BH correction was utilized for multiple hypothesis testing. Created in BioRender. Keskey, R. (2024) BioRender.com/z85h314
To map changes in the microbiota to the response of the immune system, we examined the role of macrophages on survival given their central role in clearing peritoneal infection15. When macrophages were depleted with clodronate liposomes, mortality was significantly increased and FMT was no longer effective at enhancing survival. (Fig.1F). Furthermore, the clearance of S. marcescens was associated with significant differences in canonical M1 and M2 gene expression in peritoneal macrophages (pMACs). On average, pMACs from surviving mice demonstrated lower expression of Nos2, a marker of the M1 phenotype (Fig.1G), and elevated expression of Arg1, a marker of the M2 phenotype, (Fig.1H) when compared to pMACs of non-surviving mice16. Taken together, these data indicate that macrophages are required for both survival and FMT rescue and express a specific phenotype associated with survival.
Survival from bacterial sepsis is associated with indole production
In order to characterize the gut microbiota associated with survival, we compared the gut microbiota of surviving versus non-surviving mice 15 hours following IP S. marcescens infection. Utilizing 16S rRNA sequencing, analysis of the cecal microbiota demonstrated no changes between surviving and non-surviving mice (Fig.2A) as judged by alpha diversity (i.e., Shannon index) (Extended Figure 1A) and beta-diversity (Extended Figure 2A). However, a significant difference in cecal metabolites was observed between survivors and non-survivor mice when normalized to uninfected mice (Fig.2B), including an increase in indole metabolites, products of tryptophan metabolism (Supplemental Table 2). Consistent with this finding was a reduction of cecal tryptophan during S. marcescens infection in both surviving and non-surviving mice, indicating increased tryptophan metabolism (Fig.2C). Yet, by comparison, the concentration of tryptophan was higher in the FMT-treated group (Fig.2C, Table S2). When the total amounts of indole metabolites were accounted for, a significant increase in indoles was observed among surviving mice and mice treated with an FMT compared to non-surviving mice (Fig.2D, Table S2). It is noteworthy that FMT treatment increased cecal tryptophan and indole concentrations to a degree greater than that observed in untreated and uninfected mice (Fig. S1C,D). In addition, surviving mice had a significant increase in tryptophan-derived metabolites despite no alterations in microbiota composition, understanding the limitations of analyzing the microbiota by 16s rRNA alone. We next tested whether tryptophan-derived indoles alter peritoneal macrophages in this model, as others have described17,18. First, we assayed the peritoneal exudate for the presence of indole metabolites and observed a significant increase in several indole metabolites in survivors relative to non-survivors (Fig.2E, Extended Table 2). Similarly, a significant increase in serum concentrations of indole-3 propionic acid was observed (Fig. 2F). Also, consistent with results from our prior study, a significant increase in cecal butyrate levels was observed in surviving mice (Extended Figure 2A–C); however, there were no differences in systemic butyrate between surviving and non-surviving mice (Extended Figure 2D–F). Given the association of gut and peritoneal indole metabolites with survival, we next attempted to determine the mechanism by which indoles production contributed to survival.
Figure 2. Gut microbiota-derived tryptophan metabolites are increased in surviving mice and the aryl hydrocarbon receptor is required for survival in this model.

(A), Relative abundance of cecal microbiota at the phyla level. (B), Gut metabolites abundance of infected mice. (C,D), Tryptophan (C) and relative total indoles (D) in the cecum of mice after 8 hrs of IP injection with S. marcescens (n=6 for S, n = 7 for NS, n = 5 FMT) with IP injection of S. marcescens. S, surviving mice; NS, non-surviving mice; FMT = fecal microbiota transplant. (C): p=0.0049, One-way ANOVA followed by pairwise comparison with Benjamini-Hochberg (BH) correction; ***p = 0.0082, NS vs FMT; *p=0.0504, S vs FMT; p = 0.2401, S vs NS . (D): *p=0.0153, S vs NS; **p= 5.5e-5, NS vs FMT; ***p=0.0055, S vs FMT, pairwise comparison with BH correction. (E), Microbial metabolites of tryptophan in the peritoneum of surviving mice relative to non-surviving mice. (F), Serum levels of indole-3 propionic acid comparing survivor (n = 7) and non-survivor (n = 6) mice. (G), Kaplan-Meyer survival curves demonstrating the abrogation of the FMT rescue effect during AhR inhibition (AhRi). n=5 per group, p<0.0001, Log-rank (Mantel-Cox) test. SM, S. marcescens; AhRi, AhR inhibitor StemRegennin; FMT, fecal microbial transplant. (H), Knockout of AhR within macrophages (LysM-Cre x AhR fl/fl) attenuates survival of mice as demonstrating by Kaplan-Meyer survival curves, p=0.0211, n = 20 mice per group. Error bars represent standard deviation. All statistical tests were two-sided and BH correction was utilized for multiple hypothesis testing.
AhR is required for survival from bacterial sepsis
Indoles are known to activate the aryl hydrocarbon receptor (AhR). The AhR is an intracellular receptor expressed within immune cells that has been demonstrated to impact gene expression and cell phenotype6,19. Therefore, here we hypothesized that AhR activation by indole metabolites is the mechanisms by which the gut microbiota regulate the immune response and drive survival in this model19,20.
To test this, the AhR was first inhibited using the small molecule inhibitor, StemRegenin1 (SR1), which resulted in increased mortality. Furthermore, administration of an FMT in the presence of SR1 no longer prevented mortality (Fig.2F). Additionally, AhR inhibition was associated with a significant increase in S. marcescens in the peritoneum (Extended Figure 3A) and its dissemination in blood (Extended Figure 3B). Given the requirement of macrophages for survival in this model, we next determined if macrophage specific AhR was required for survival. Utilizing a macrophage specific AhR knockout construct (LysM-Cre x AhR fl/fl), we observed a significant increase in mortality (Fig.2G), that correlated with a lower body temperature and higher sepsis score (Extended Figure 3C,D).
AhR signaling in macrophages results in an M2 transcriptional profile
Given that both AhR signaling and macrophages are required for survival following S. marcescens infection, we performed a transcriptional analysis using RNA sequencing of pMACs at 8 hours post infection with S. marcescens across the following groups: surviving mice, non-surviving mice, mice treated with AhRi, mice administered an FMT, and mice treated with AhRi and FMT. pMACs from uninfected mice were used to normalize data to controls. Volcano plots demonstrate differentially expressed genes (logFC >1.5 and FDR < 0.05) between pMACs from surviving mice compared to pMACs from non-surviving mice (Fig.3B) and pMACs from FMT treated mice compared to non-surviving mice (Extended Figure 4A). Further, DEGs were compared between survivors, non-survivors, and FMT treated mice to identify the individual genes associated with survival. There were 826 unique genes associated to survivors, 468 genes unique to non-survivors, and over 2,300 unique to FMT treated mice (Fig.3A). Interestingly, DEGs that were specific to macrophages from surviving mice included genes that have previously been associated with endotoxin tolerance and the regulation of inflammation (Extended Figure 5). For example, Ripk2 limits inflammation related to inflammasome activation21, Dcp1 and Trdmt1 have both been associated with endotoxin tolerance.22,23 In addition, Clu expression was increased in surviving mice which has previously been demonstrated to be reduced in patients who were non-survivors of sepsis24. Furthermore, individual genes that were oppositely expressed between survivors and non-survivors were notable to include: Pik3cd which is an important regulator of NFkB and M1 polarization25, Lpin1 which is an important regulator of M2 macrophages26, Sec141l which is a negative regulator of RIG and M1 polarization27, and Slamf7 which has bene shown to limit inflammation in the setting of polymicrobial sepsis28, Cd47 which negatively regulates NFkb29, and Hif1a which is important for metabolic regulation of macrophages30. In the aggregate, the gene expression data indicate that there is suppression of the immunopathology associated with the septic immune response within macrophages among surviving mice.
Figure 3. Transcriptional analysis of pMACs demonstrates distinct profiles of surviving and non-surviving mice in an AhR dependent manner.

RNA sequencing was performed on pMACs isolated from survivors, non-survivors, n=5 per group. (A) Venn Diagram comparing differentially expressed genes within macrophages of survivors, non-survivors, and FMT treated mice. (B), Volcano plots (logFC >1.5 and FDR < 0.05) between pMACs from surviving mice compared to non-surviving mice. (C), Gene set enrichment (GSE) with g:Profiler comparing survivors to non-survivors demonstrating significant transcription factors. (D), GSE analysis with ImmuneSigDB between pMACs from survivors, non-survivors, and FMT mice. (E), Ingenuity pathway analysis comparing survivors to AhRi (F), Concentration- dependent effect of indoles on AhR activation using a mouse AhR reporter cell line. n=3 biologic replicates for DMSO, n=12 biologic replicates for indoles mix at 0.01 mM concentration and n=3 for indoles mix at 0.1, 1.0 and 10 mM concentrations. p<0.0001, One-way ANOVA; *p=1.5e-9, **p=2e-16, ***p=0.0019, pairwise comparison with BH correction. (G), Relative expression of Cyp1b1 in BMDMs, n=11 p=0.0001, One-way ANOVA test; *p=0.00046 Indoles vs Indoles + AhRi; **p=0.00092 Indoles vs DMSO, pairwise comparison with BH correction. (H), Gentamicin protection assays using BMDMs exposed to 0.01 mM indole mixture with S. marcescens. n= 11 per group, p<0.0001, One-way ANOVA test; *p=2.2e-9 Indole Mix vs Indole Mix + AhRi; p=2.0e-7 Indole Mix vs DMSO; p = 0.046 Indole Mix + AhRi vs DMSO, pairwise comparison with BH correction. (I-J), Relative expression of Arg1 (I) and Il10 (J), in BMDMs after exposure to Sm. I: p<0.0001, One-way ANOVA test; *p<0.0001, DMSO n =12, Indole mix n =14, Indole mix + AhRi n = 9. (J): p=0.0058, One-way ANOVA test; *p=0.00022, pairwise comparison with BH correction. **p=2.3×10−5. (K), IL10 protein by ELISA. p=0.014, One-way ANOVA test; *p=2.0e-7, DMSO vs Indole Mix; p = 2.2×10−9 Indole mix vs Indole mix + AhRi, pairwise comparison completed with BH correction, DMSO n = 6, Indole Mix n = 9, Indole Mix + AhRi n = 6. Error bars represent standard deviation.
Results also indicate a distinct pMAC gene expression pattern associated with survival in this model. Using gene set enrichment analysis of DEGs between the various groups (Fig.3C, Extended Figure 4B), we observed significant differences in predicted transcription factors regulating gene expression, including AhR, among macrophages from surviving and FMT treated mice. Transcription factors known to be important for macrophage polarization including IRF6, IRF4, KLF family of transcription factors, HIF-1a, c-Myc and CREB were found to be significantly altered between surviving and non-surviving mice (Fig.3C) and FMT treated mice compared to non-surviving mice (Extended Figure 4B). Furthermore, when gene set enrichment analysis was examined using ImmuneSigDB, there were significantly altered gene sets unique to each individual group (Fig. 3D). To better understand how AhR-regulated macrophages are characterized among surviving mice, Ingenuity Pathway Analysis (IPA) was applied to the transcriptional profiles of pMACs isolated from survivor compared to AhRi treated mice (Fig.3E) as well as to pMACs isolated from FMT treated mice versus FMT+AhRi (Extended Figure 4C). IPA demonstrated a significant impact of AhR on alternative macrophage activation via IL-10 signaling, toll like signaling, IL-6 and IL-12 production and apoptosis within macrophages in both groups (Fig.3E, Extended Figure 4C). Of note, AhR has been previously demonstrated to regulate NF-kB signaling and IL-10 production31–33. Taken together, these data suggest that AhR’s role in macrophage-mediated clearance of infecting bacteria appears to be critical for survival in this model.
Indole activation of macrophage AhR increase bacteria killing
In order to confirm the ability of the indole ligands to activate AhR in vivo, we tested the activity of the identified indoles in vitro using an AhR-luciferase reporter cell line34. To recapitulate the composition of indoles observed within the peritoneum in vivo, we assembled a multi-component “indole mix” consisting of indole-3-carboxaldehyde, indole-3-acetic acid, indole-3-lactic acid, and tryptophol at various concentrations. We observed an indole-dependent increase in AhR signaling at lower doses between 0.01 mM and 0.1 mM (Fig.3F). Paradoxically, at high concentrations, suppression of AhR signaling was observed beginning at 5 mM (Fig.3F). This pattern of activation and inhibition was also observed when individual metabolites were similarly tested (Extended Figure 5). However, unlike indoles, butyrate was observed to function as a very weak AhR agonist (Extended Figure 7).
To elucidate the impact of the selected indole metabolites and AhR signaling on the macrophage response to S. marcescens, bone marrow derived macrophages (BMDMs) were exposed to S. marcescens in the presence of the indole mix. Activation of AhR in BMDMs in response to the indoles was confirmed by examining the expression of Cyp1b, a canonical AhR-activated downstream gene (Fig.3G). Bactericidal activity was then measured utilizing gentamicin protection assays. When exposed to the indole mix, BMDMs had a significant increase in intracellular killing of S. marcescens, which in turn was abrogated by AhR inhibition (Fig.3H). Next, BMDMs were exposed to lysates of S. marcescens in the presence and absence of the indole mixture. Similarly to the transcriptional alterations observed in vivo, indole metabolites significantly increased the expression of Arg1 and Il10 (Fig.3I,J) without affecting the production of pro-inflammatory genes (Extended Figure 8). Taken together, these results suggest that AhR signaling by indole metabolites can enhance bacterial clearance while potentially limiting any associated immunopathology. AhR has been shown to increase IL-10 production5,35, and this was confirmed in in vitro experiments in which BMDMs were co-cultured with the indole mix and S. marcescens lysates demonstrating increased IL-10 protein production(Fig.3K). These data demonstrate that indole metabolites can modify macrophage responses to S. marcescens.
Secreted products of pathogens inhibit indole AhR activation
Multiple lines of evidence are accumulating to demonstrate that the success of a pathogen to cause lethality is dependent on its ability to subvert the immune system36. As such, we next determined whether the infecting pathogen, S. marcescens, could itself adversely affect AhR activation by interfering with the effect of indoles on macrophage function. We next determined if the exoproduct of S. marcescens was able to suppress AhR activation in response to indoles. Results indicated that when the AhR reporter cell line was first exposed to S. marcescens supernatant, indole activation of AhR was significantly inhibited in a manner dependent on the growth phase of S. marcescens, with escalating AhR inhibition using supernatant derived from bacteria at late log phase growth versus early log phase growth (Fig.4A,B). When S. marcescens supernatant was fractionated by molecular weight, the < 3kD fraction maintained its ability to inhibit indole activation of AhR whereas fractions > 3kD fraction did not (Fig.4C). These findings suggest that S. marcescens itself can secrete a low molecular weight compound capable of counteracting the AhR- activating indole metabolites. Therefore, it is possible that the extent to which products produced by infecting pathogen inhibit AhR activity, and the extent to which this effect can be mitigated by products from the gut microbiota may determine survival versus mortality. However, further elucidation of the molecular details of this interaction is needed to define a more generalizable principle to explain within-group variability to a standardized infectious challenge36.
Figure 4. S. marcescens exoproduct inhibits indole-mediated AhR activation.

The Impact of S. marcescens on AhR signaling was studied in vitro. Filtered supernatant was collected from S. marcescens at early and late log phase. The AhR reporter cell line was exposed to filtered supernatant in the presence and absence of the indole mixture to determine its impact on AhR signaling. (A) Supernatant from the late log phase resulted in significant repression of AhR activation in the presence of the indole mix at 0.01 mM and 0.1 mM. p<0.0001, One-way ANOVA. *p = 1.6e-13, Indole Mix vs Control; p = 6e-10; p = 3e-7, Indole Mix + Early Log supernatant vs 0.01 Indole Mix; **p<1e-10 for early log, late log, stationary phase vs indole mix, pairwise comparison with BH correction, n = 3 biologic replicates per group. (B) Supernatant from the late log phase resulted in significant repression of AhR activation in the presence of the individual indoles at 0.1 mM. p<0.0001, One-way ANOVA. *p <0.0001, **p=0.0231, unpaired t-test, n = 3 biologic replicates per group. (C), The <3 kDa fraction of the supernatant represses the activation of AhR by the indole mix at 0.01 mM. p<0.0001, One-way ANOVA. *p <0.0001, unpaired t-test, n = 6 biologic replicates per group. (D-G) Supernatants were collected from different clinically relevant pathogens to determine their ability to inhibit indole activation of AhR in vitro, n = 3 biogic replicates per group. (D), The P. aeruginosa supernatants were toxic to cell line and were removed from AhR activation experiment. (E), The AhR reporter cell line (H1L1.1c2) was then exposed to the indole mixture in the presence of supernatant from pathogens that were not cytotoxic to determine their ability to inhibit indole activation of AhR. (F), Enterococcus faecalis and Klebsiella pneumoniae demonstrated an AhR inhibition that was strongly dependent on growth phase. (G), Pathogen supernatants were then fractionated by molecular weight to determine if size (i.e., < 3kD) was a discriminatory factor in the observed inhibition, n = 3 biologic replicates per group. Error bars represent standard deviation. All statistical tests were two-sided and BH correction was utilized for multiple hypothesis testing.
In order to determine if the observed AhR inhibition in response to gut microbiota-derived indoles was generalizable to other pathogens relevant to human sepsis. We utilized our AhR reporter cell line and exposed the cells to indoles in the presence of supernatant isolated from other pathogens, after confirming the supernatant was not cytotoxic (Fig.4D). Of the strains tested, both multidrug resistant S. marcescens (previously designated as ICU1–29) and a virulent strain of S. marcescens MVI inhibited indole activation of AhR. Both Klebsiella oxytoca (previously designated as ICU1–2) and Klebsiella pneumoniae (previously labeled as ICU3–1) also inhibited the activation of AhR in a manner similar to S. marcescens (Fig.4E). AhR inhibition was also seen with gram positive pathogens, Staphylococcus aureus and Enterococcus faecalis, as well as the common fungal pathogen, Candida albicans (Fig.4E). Similar to Serratia marcescens, inhibition of AhR by both Klebsiella species and Enterococcus faecalis was dependent on the phase of growth (Fig.4F). Additionally, supernatants of selected pathogens were fractionated by molecular weight, and similar to our findings seen with S. marcescens, inhibition of AhR activation only occurred within the < 3kD fraction (Fig.4G). The conserved response of AhR inhibition supports an important role by which both gut microbiota-derived metabolites and those of the pathogen, converge on AhR signaling as a potential determinant of their lethality.
Enterobactin inhibits AhR increasing mortality to bacterial sepsis
Given the observation that a soluble factor from pathogens can inhibit indole-mediated AhR activation, experiments were conducted to identify the inhibitory ligand. In silico modeling of AhR interactions with potential microbial ligands was determined via molecular docking. Based on the docking score, we identified several potential microbial candidates capable of interacting with AhR (Extended Figure 9, Table S3) including enterobactin, prodigiosin, tilimycin, tilivalline and melanin, common secondary metabolites of the microorganisms studied. Interestingly, out of the candidate compounds, whole genome sequencing demonstrated that both Klebsiella9 and Serratia species utilized in the above experiments contained the genes encoding enterobactin. Furthermore, when the binding between enterobactin and AhR was evaluated in silico, enterobactin, similar to the AhR antagonist SR1, had more stable binding to AhR than indole-3-acetic acid, further implicating its ability to strongly inhibit AhR (Extended Figure 9, Fig.5A). Mass spectrometry confirmed the ability of both S. marcescens MVI and Klebsiella oxytoca to produce enterobactin (Fig.5B). When tested in vitro, enterobactin significantly inhibited indole activation of AhR (Fig.5C) without being cytotxic (Fig.5D). These findings demonstrate, for the first time, that the virulence factor enterobactin inhibits host AhR signaling. Enterobactin, an iron chelator, has been repeatedly demonstrated to play an important role in iron chelation and the pathogenicity of gram negative bacteria37,38. To determine if iron chelation played a role in enterobactin’s ability to inhibit AhR, we tested enterobactin’s ability to inhibit indole activation of AhR in the presence or absence of iron in vitro (Extended Figure 10A). Iron chelation did not impact enterobactin’s ability to inhibit AhR activation. Furthermore, dihydroxybenzoic acid (DHBA), an enterobactin intermediate that also has the ability to play a role in bacterial virulence through inhibition of host metalloproteinase,39 was tested. When DHBA was tested in vitro, we found that varying concentrations of DHBA could also inhibit AhR activation by indoles (Extended Figure 10B). When enterobactin was delivered at the time of infection with S. marcescens, there was a significant increase in mortality (Fig.5E). Enterobactin delievered at the doses tested did not cause mortality by itself. Taken together, these result indicate that pathogen secretion of enterobactin can subvert the host immune response by suppressing indole-mediated activation of AhR.
Figure 5: Enterobactin can subvert the host immune response and plays a role in the mortality observed following i.p. S. marcescens.

Autodock was utilized to determine potential microbial metabolites capable of interacting with AhR. Three dimensional binding mode (i), surface representation (ii), and two-dimensional illustration of molecular docking between enterobactin and AhR and indole-3-acetic acid and AhR are demonstrated (A). Mass spectrometry demonstrates that both K. oxytoca and S.marcescens are capable of producing enterobactin as both enterobactin and components of enterobactin were detected in <3 KDa fraction of liquid culture, n = 3 biologic replicates per group (B). Enterobactin significantly inhibited indole activation of AhR in vitro (C) without being cytoxic (D) n = 3 biologic replicates per group. When enterobactin was delivered IP, simultaneously with S. marcescens in vivo, there was a significant increase (p=0.0156) in mortality compared to DMSO control, n = 15 per group (E). Error bars represent standard deviation. All statistical tests were two-sided and BH correction was utilized for multiple hypothesis testing.
Oral tryptophan and systemic indoles improve survival from sepsis
Despite testing various doses and dose schedules, oral indole administration did not improve survival in this model. We speculated that this result may be a function of its complex consumption and metabolism by the gut microbiota themselves as it passes from the mouth to the colon. Therefore, we hypothesized that oral supplementation with tryptophan, as others have shown16, would lead to increased indole production by the gut microbiota and improved survival from bacterial peritonitis with S. marcescens, K. oxytoca, and a polymicrobial community (PC: S. marcescens, K. oxytoca, E. faecalis, and C. albicans). K. oxytoca was utilized as a second monomicrobial pathogen that produces enterobactin similarly to S. marcescens. K. oxytoca is also an emerging as a major health threat to humans with sepsis, and the pathogen community consisted of pathogens isolated from critically ill patients with sepsis40–42. We supplemented the drinking water of mice with 1 mM oral tryptophan for 14 days prior to peritoneal infection. Supplementation with oral tryptophan resulted in a significant improvement in survival of mice injected with S. marcescens (Fig.6A), K. oxytoca (Fig.6B), and PC (Fig.6C). In mice that were not supplemented with oral tryptophan, there was a significant reduction in both cecal tryptophan and indoles following infection (Fig.6D,F). Oral tryptophan supplementation maintained gut tryptophan and indoles following infection (Fig.6E,G).
Figure 6. Systemic delivery of the indole mixture or oral supplementation with tryptophan prevents mortality in mice following i.p S. marcescens, Sm; K. oxytoca, Ko; and polymicrobial community, PC consisting of Klebsiella oxytoca, Enterococcus faecalis, Serratia marcescens, and Candida albicans).
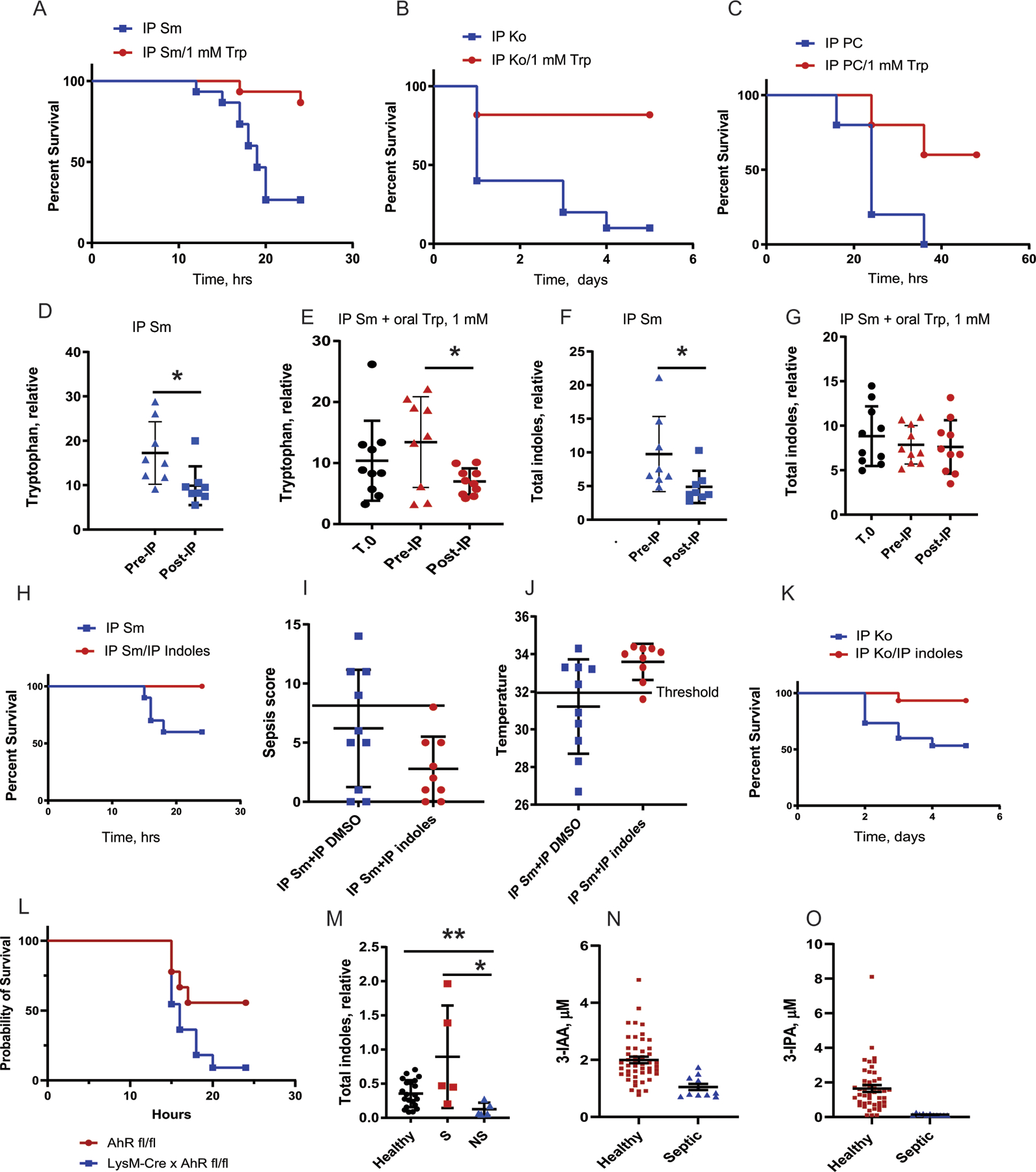
Mice supplemented with oral tryptophan (1mM) in their drinking water for 2 weeks prior to IP infection demonstrated a significant improvement in survival folloiwng IP S. marcecens (A), K. oxytoca (B), and PC (C). (D-G), Metabolomics analysis of the stool demonstrated that that (D), the tryptophan level is decreased after IP injection of Sm. (E), tryptophan supplementation did not significantly increase stool tryptophan prior to infection, however a significant decrease in gut tryptophan was still observed following IP pathogen injection. (F), Similarly,we observed a significant decrease of indoles following i.p pathogen injection that was not observed with Trp supplementation (G). n=10 per group, *p<0.05. (H), Systemic delivery of the indole mixture of 0.01 mM delivered IP at the time of infection with Sm peritonitis demosntrated a significant improvement in survival. Kaplan-Meyer survival curves, n=10 per group, p=0.0291, Log-rank (Mantel-Cox) test (H). (I), Sepsis score. All indole-treated mice sepsis scores were below threshold, sepsis score=8 (n = 10 per group). (J), Core body temperature. All indole-treated mice body temperatures were over the threshold (32°C). (K), Mice were rescued with 0.01 mM indole mixture delivered i.p at the time of infection with Klebsiella oxytoca. (L) LysM Cre x AhR fl/fl were not able to be rescued with i.p injection of indoles compared to AhR fl/fl mice (n= 9 AhR fl/fl, n = 11 LysM-Cre x AhR fl/fl) p = 0.051. (M) Stool indole levels were compared between healthy controls, survivors and non-survivors of sepsis; p = 0.1741, Healthy vs NS; p = 0.0055, Survivor vs Healthy; p = 0.0030, Survivor vs Non-Survivors (Healthy control n = 21, Survivors n = 5, Non-Survivors n= 5). (N) Serum levels of indole-3 acetic acid (I3AA) was compared between septic patients (n = 11, 26 samples) and healthy controls (n =48), p = 0.0003. (O) Serum Indole-3 Propionic Acid (3-IPA) was compared in the serum of the same patients (n = 11, 26 samples), p = 0.0005. Error bars represent standard deviation. All statistical tests were two-sided and BH correction was utilized for multiple hypothesis testing.
We next determined whether systemic administration of indole metabolites would be sufficient to prevent mortality in mice following infection with pathogens capable of AhR inhibition. To test this, we injected the indole mix intraperitoneally simultaneous with a lethal dose of S. marcescens, K. oxytoca, or PC. The indole mix administered into the peritoneum at 0.01 mM significantly increased survival in mice as compared to DMSO vehicle-treated mice after intraperitoneally inoculation with S. marcescens and K. oxytoca (Fig.6H,K). As previously established, surviving mice displayed lower sepsis scores and higher core body temperatures (Fig.6I,J). Reiterative experiments using the human polymicrobial pathogen community did not demonstrate enhanced survival. The indole mix was unable to rescue LysM-Cre x AhR fl mice again demonstrating that the AhR is required for its survival effect (Fig. 6L).
Stool and serum from two separate groups of septic patients were studied to determine if similar trends were seen in indole metabolites in humans. Serum isolated from septic patients demonstrated a significant reduction in indole-3- propionic acid and indole-3- acetic acid (Fig. 6N,6O SupplementalTable 4) when compared to healthy controls. Similarly, stool samples from septic patients admitted to a separate ICU demonstrated a significant increase in stool indoles in patients who survived sepsis compared to those that did not survive (Fig. 6M, Supplemental Table 5). These findings indicate that microbiota production of indole metabolites is involved in the survival of septic patients.
Discussion
Findings from the present study demonstrate that the dual activity of metabolites from the gut microbiota and the infecting pathogen can be deterministic of survival in mice systemically infected with lethal pathogens of relevance to human sepsis. We demonstrate that gut microbiota-derived indole-mediated activation of AhR is a major determinant of survival from a lethal systemic infection. The finding that oral supplementation with tryptophan may preemptively prevent the subversion of AhR activity by the infecting pathogens is important given that these pathogens often contribute to “late-onset sepsis,” now the most common cause of organ failure and mortality during critical illness8. When patients suffer major injury or complications from elective procedures and become critically ill, life-support measures such as ventilator support and dialysis, and the application of polypharmacy such as vasoactive agents, opioids and antibiotics, not only impact the gut microbiome community membership and functional output over time43, but also allow for the proliferation of healthcare-acquired pathogens (HAPs) that are often multidrug resistant and highly virulent. An important mechanism by which these “overgrowth pathogens” are able to survive and reproduce, may underlie their ability to subvert the immune system44. Results from this study suggest that a lack of opposing forces on the AhR by metabolites from the gut microbiota may indeed play a role in the virulence and lethality of HAPs once they predominate. It may be for this reason that the indiscriminate use of antibiotics is associated with adverse outcomes in critically ill patients45.
The gut microbiome is emerging as an important element involved in infectious disease pathogenesis and sepsis physiology. Traditionally, the conceptual framework around the pathobiology of life-threatening infection has consisted of highly polarized investigations focusing on either the pathogen or the host. In general, the microbiologic approach has attempted to hold the host constant while varying the genes or phenotype of the pathogen in an effort to define the determinants of outcome as they relate to the pathogen. Conversely, the immunologic approach often holds the pathogen constant, while varying the host genotype/phenotype to invoke the host response as a key determinant of outcome. Studies that emphasize the host-pathogen interactions as a determinant of virulence and suggest that virulence is neither a property of the pathogen nor that of the host, but rather a property of their interaction46 are needed to advance the field. For example, it is well established that secondary metabolites produced by Pseudomonas aeruginosa can interact with and differentially impact AhR signalling to allow for fine tuning of the host immune response 47,48. Additionally, AhR has been demonstrated to play an important role in the innate immune response to endotoxin controlling both the pro-inflammatory response as well as the development of endotoxin tolerance and guiding resolution of inflammation through production of IL-106,7. It is likely that the anti-inflammatory impact of AhR signaling in our model is occurring via the non-canonical AhR pathway in which AhR is not required to bind to its canonical dioxin response elements to induce an anti-inflammatory response49. However, no work has been done to determine how the microbes within the gut microbiota and the pathogen compete for AhR activation and the extent to which this interaction influences survival.
Considering tryptophan as a unique substrate that can be metabolized by both gut bacteria and host cells to produce various immune-activating metabolites, it may be an important modifiable factor in the battle against sepsis19. Consistent with our findings, others have demonstrated that oral supplementation with tryptophan can increase systemic levels of microbiota-derived indole metabolites with the capacity to alter the host immune response50. It is unclear how metabolites, such as indoles which are produced by the gut microbiota, enrich systemic compartments such as the peritoneum and plasma. Prior studies, have demonstrated that metabolites produced by the gut microbiota are capable of disseminating systemically and can be detected in tissues throughout the body.51 Taken together, these data seem to support our findings of increased indole metabolites in the peritoneal cavity and serum of septic mice in our model.
Our study suggests that there may be a central role for the gut microbiota and AhR activity as a determinant of “within-group” mortality following a systemic infection with a virulent pathogen. Recent studies have now documented that changes in the gut microbiota correlate with variations in core body temperature, a marker of infection-related mortality, following a systemic infection52. To our knowledge, the current study is the first to demonstrate that AhR lies at the center of host-pathogen-microbiota interaction as its activation may be dually influenced by metabolites from both the infecting pathogen and the gut microbiota. The degree and direction by which AhR signaling is affected may play a role in discriminating between those mice that survive versus those that die within a group exposed to a standardized infectious inoculum. The precise fine spatial scale and time-dependent interplay between the concentration of the pathogen produced exoproduct(s) and the concentration and composition of the gut microbiota-derived metabolites, in the background of AhR activation, will require further clarification. Competition for AhR signaling between the host microbiota and S. marcescens, as identified in the current report, adds to the growing examples of the “defense/counter-defense” paradigm and complexity within the host-pathogen interaction44. In this report, we have demonstrated, for the first time, that a pathogen’s secretion of enterobactin, a common pathogen virulence factor, can inhibit indole-mediated AhR signaling in association with impaired survival. While others have demonstrated that interactions between pathogen secreted enterobactin and host lipocalin 2 is important, especially for iron sequestration53–55, here we describe competition between microbiota produced indoles and enterobactin for host AhR activation. The importance of this competition in determining survival from lethal bacterial infection may be significant and will require further confimation.
There are several limitations to this study. As AhR is present on many immune cells beyond macrophages and is considered to be a promiscuous receptor based on its ability to respond to various ligands, other immune cells may be involved in the response herein described. Prior studies have demonstrated the importance of AhR signaling in both the innate and adaptive immune system. In the adaptive immune system, AhR is highly expressed in TH17 and TReg cells influencing their expansion and response to environmental stimuli56,57. Furthermore, AhR has been demonstrated to play an important role in innate lymphoid cells, dendritic cells, and macrophages, typically playing an important role in the anti-inflammatory response of these cells58,59. Therefore, it is likely that many of these cells may also have some degree of influence in our model and were not directly examined. Furthermore, our mouse studies have limited clinical translatability that are typical of animal models of lethal infection. Although our model utilizes a lethal pathogen, it results in rapid mortality within 24 hours which is generally not observed in patients with peritonitis. Mice, unlike humans, were treated in a standard manner with modalities such as antibiotics, fluids, pressors, etc. However, the intent of our study was to understand within group determinants of lethal infection before the clinical condition itself overshadowed survival. It is also important to point out the clinical value of using S. marcescens as it is a lethal pathogen in this model given its ability to subvert the host immune response and its emerging role in intensive care unit infections 60–62. Finally, our compositional analysis of the gut microbiota was limited by 16S sequencing which alone can miss subtle compositional differences that may explain the functional differences seen between survivors and non-survivors in our model. In the future, full metagemonic sequencing may avoid this limitation.
In summary, the aryl hydrocarbon receptor plays a central role in how host-pathogen-microbiota interactions determine survival from lethal sepsis. A conceptual framework may be emerging in which the extent to which a pathogen can subvert the host’s immune clearance mechanisms must be balanced by the extent to which gut microbiota derived metabolites can enhance immunity. Currently, all severe bacterial infections are treated by broad spectrum antibiotics which carry the unintended consequences of eliminating both the offending pathogen as well as multiple components of the gut microbiome, in the context of the present study, considering how to eliminate offending pathogens in a more focused and narrow spectrum may be desired.
Methods
All research methods utilized complied with relevant ethical regulations overseen by the University of Chicago IRB and IACUC standards. No statistical methods were used to predetermine sample size. Sample size was determined on the basis of the minimum number of animals and biological replicates required for good data distribution and statistics. The experiments conducted did not require randomization and the data collection and analysis were not performed blind to the conditions of the experiment.
Mice
Six to eight week old C57/B6 male mice were purchased from Charles River Laboratories. Mice were housed in temperature controlled 12h light/dark cycled rooms at University of Chicago Animal Facility. AhR fl/fl mice were purchased from Jackson Laboratory (Stock No: 006203, RRID: IMSR_JAX:006203) and bred with LysM Cre (Stock No: 004781, RRID: IMSR_JAX:004781) to obtain macrophage specific knockout of AhR. All mice were co-housed in groups of 5 prior to experiments to allow for a shared microbiome and maintained on a standard rodent chow (Envigo Teklad NC1434463).
Bacteria
Serratia marcescens MVI, a known virulent strain, was isolated from the spleen of septic, western diet-fed mouse who was moribund following a surgical stress8. This strain was used in all experiments. For additional experiments to test the role of AhR and indole in survival from human pathogens, pathogens were obtained from the stool of critically ill patients in the ICU and injected intraperitoneally to induce lethal peritonitis, as previously described9,40. The human pathogens used included Klebsiella oxytoca ICU1–29 and a polymicrobial community (referred to as the pathogen community consisting of four microbes: Serratia marcescens ICU1–2, Klebsiella oxytoca ICU1–2, Enterocccus faecalis ICU1–2, and Candida albicans ICU1–2, as previously described9.
Mouse peritonitis model
This model was performed under the protocol IACUC71744. Six to eight week old male mice were used in all experiments and co-housed in temperature controlled 12h light/dark cycled rooms at University of Chicago. For infection, mice were inoculated via intraperitoneal injection (IP) with a single dose of S. marcescens MVI. S. marcescens was grown on MacConkey agar for 32 hrs and resuspended in 10% glycerol to an OD 0.1 and was further diluted 1:600 to achieve a final concentration of 5× 105CFU/mL. Mice were injected intraperitoneally with 1cc of bacteria and their core body temperature measured hourly during the course of infection. The validated MSS scoring system (Supplemental Table 1) was used to track the course of infection 11,14. Mice were deemed to be “non-surviving” when a drop in their temperature was greater than 1 degree Celsius at 8 hours post injection as determined by our time course studies. We used temperature drop as a marker of non-survival, as validated by others, to disassociate derangement of the septic state from parameters of interest11. At 15 hours post injection, mice with a total sepsis score greater than 8 and a core body temperature less than 31 degrees Celsius were determined to be a non-survivors. FMT treatment was conducted by sacrificing a healthy littermate and resuspending their cecal contents in sterile normal saline for a final concentration of approximately 50mg/mL. Approximately 1cc of FMT was delivered to each mouse via enema at the time of IP pathogen injection as previously described6. For macrophage depletion experiments, Clodronate liposomes or control liposomes (FormuMax Scientific Inc, California) were administered intraperitoneally 24 hrs prior to intraperitoneal (IP) injection with S. marcescens. Indole mixture i.p treatment was conducted by delivering 100μL of the indole mixture simultaneous with the S. marcescens injection. For AhR inhibition, 100μL of 1 mM StemRegennin (Selleck, Houston TX) was administered to each mouse i.p at the time of infection. Oral tryptophan was administered in the drinking water a concentration of 1mM for 14 days prior to injection with bacterial pathogens. For enterobactin experiments, commercially available Enterobactin (Sigma-aldrich) was injected i.p at a dose of 100ug /mouse at the time of infection and compared to vehicle controls (i.e. dimethyl sulfoxide-DMSO). The dose of enterobactin utilized in these experiments was not lethal on its own.
16S rRNA gene amplicon sequencing analysis of microbiota in cecal contents
Cecal contents were collected in a sterile manner. DNA was extracted using the power-fecal DNA extraction kit. For library preparation, DNA was amplified using the barcoded 12-bp Golay primer set designed for the Earth Microbiome Project (EMP). PCR was performed according to the manufacturer’s protocol using the EMP primers, mPNA, AccuStart II PCR ToughMix, and the extracted DNA (Quntabio). After amplification, the PCR products were quantified by using a PicoGreen dsDNA quantitation assay (Invitrogen). The results of the quantification were used to normalize the amount of DNA from the PCR product used for sequencing to ensure that each amplicon was represented evenly during sequencing. Finally, an aliquot of the final pool was taken, and the DNA purified by using an Agencourt AMPure XP PCR* purification system (Beckman-Coulter). The samples were then run on an Illumina MiSeq at Argonne National Laboratory (150 bp × 2). Qiime263 was utilized for16S rRNA gene sequence analysis and demux emp-paired-end command was used to demultiplex and join the paired-end reads. Quality filtering was performed using Deblur. Taxonomy and OTUs were assigned using a Greengenes classifier. Sequences were further analyzed utilizing the Phyloseq64 package within R. Samples were rarefied to a depth of 10,000 reads per sample. Raw sequences uploaded to NCBI SRA (Reference number: PRJNA838642).
Metabolomic analysis
Metabolite Extraction:
Upon sacrifice, cecal samples were flash frozen and stored at −80°C before the analysis. Metabolite extraction from cecal samples was performed with 80% methanol spiked with deuterated internal standards, at a ratio of 100 mg of material/mL of extraction solvent, in beadruptor tubes (Fisherbrand, cat# 15-340-154). Samples were homogenized at 4°C on a Bead Mill 24 Homogenizer (Fisher; 15-340-163), set at 1.6 m/s with 6 thirty-second cycles, 5 seconds off per cycle. Samples were then centrifuged at −10°C, 20,000 x g for 15 min and the supernatant was used for subsequent metabolomic analysis.
Metabolite Detection with PFBBr Derivatization and GC-nCI-MS Analysis:
The metabolite extract (100 μL) was added to 100 μL of 100 mM borate buffer (pH 10) (Thermo Fisher, 28341), 400 μL of 100 mM pentafluorobenzyl bromide (Millipore Sigma; 90257) in acetonitrile (Fisher; A955-4), and 400 μL of n-hexane (Acros Organics; 160780010) in a capped mass spectrometry autosampler vial (Microliter; 09-1200). Samples were heated in a thermomixer C (Eppendorf) to 65°C for 1 hour while shaking at 1300 rpm. After cooling to room temperature, samples were centrifuged at 4°C, 2000 x g for 5 min, allowing phase separation. The hexanes phase (100 μL) (top layer) was transferred to an autosampler vial containing a glass insert and the vial was sealed. Another 100 μL of the hexanes phase was diluted with 900 μL of n-hexane in an autosampler vial. Concentrated and dilute samples were separated and analyzed using a gas chromatography – mass spectrometer (GC-MS) (Agilent 7890A GC system, Agilent 5975C MS detector) operating in negative chemical ionization (nCI) mode, using a HP-5MSUI column (30 m x 0.25 mm, 0.25 μm; Agilent Technologies 19091S-433UI), methane as the reagent gas (99.999% pure) and 1 μL split injection (1:10 split ratio). Oven ramp parameters: 1 min hold at 60°C, 25°C per min up to 300°C with a 2.5 min hold at 300°C. Inlet temperature was 280°C and transfer line was 310°C. A 10-point calibration curve was prepared with acetate (100 mM), propionate (25 mM), butyrate (12.5 mM), and succinate (50 mM), with 9 subsequent 2x serial dilutions. Data analysis was performed using MassHunter Quantitative Analysis software (version B.10, Agilent Technologies) and endogenous compound identifies were confirmed by comparison to retention time, m/z, and fragmentation pattern of authentic standards. Normalized peak areas were calculated by dividing raw peak areas of targeted analytes by averaged raw peak areas of internal standards.
Indole/Tryptophan Detection with UPLC-(+)QQQ-MS Analysis:
Tryptophan metabolites and indoles were separated and analyzed by ultra performance liquid chromatography coupled to a triple quadrupole (UPLC-QQQ) mass spectrometer. The metabolite extract (400 μL) was added to pre-labeled microcentrifuge tubes. Samples were dried down completely using a Genevac EZ-2 Elite. Samples were resuspended in 100 μL of 50:50 water:methanol and added to a thermomixer C (Eppendorf) C at 4°C, 1000 rpm for 15 min to resuspend analytes. Samples were then centrifuged at 4°C, 20,000 x g for 15 min to remove insoluble debris. The supernatant (80 μL) was transferred to a fresh, prelabeled MS vial with inserts or 96 deep-well plate (Agilent 5065–4402). Samples were analyzed on an Agilent 1290 infinity II liquid chromatography system coupled to an Agilent 6470 triple quadrupole mass spectrometer, operating in positive mode, equipped with an Agilent Jet Stream Electrospray Ionization source. Each sample (2 μL) was injected into a Acquity UPLC HSS PFP column, 1.8 μm, 2.1 × 100 mm (Waters; 186005967) equipped with a Acquity UPLC HSS PFP VanGuard Precolumn, 100Å, 1.8 μm, 2.1 mm X 5 mm (Waters; 186005974) at 45°C. Mobile phase A was 0.3% formic acid in water and mobile phase B was 0.3% formic acid in 95:5 acetonitrile:water. The flow rate was set to 0.5 mL/min starting at 0% B held constant for 3 min, then linearly increased to 50% over 5 min, then linearly increased to 95% B over 1 min, and held at 100% B for 3 min. Mobile phase B was decreased to 0% over 0.5 min and held at 0% for reequilibration for 2.5 min. The QQQ electrospray conditions were set with capillary voltage at 4 kV, nozzle voltage at 500 V, and Dynamic MRM with cycle time of 500 ms. Transitions were monitored in positive ion mode for 46 analytes. An 11-point calibration curve (ranging from 0.88 nM to 909 μM) was prepared for tryptophan, tyrosine, phenylalanine, serotonin, 5-HIAA, melatonin, tryptamine, kynurenine, kynurenic acid, anthranilic acid, and niacin. Data analysis was performed using MassHunter Quant software (version B.10, Agilent Technologies) and endogenous compound identifies were confirmed by comparison to retention time, m/z, and fragmentation pattern of comparison with authentic standards. Normalized peak areas were calculated by dividing raw peak areas of targeted analytes by averaged raw peak areas of internal standards.
Metabolite Detection with TMS-MOX Derivatization and Electron Impact GC-MS Analysis:
Metabolites were analyzed using GC-MS with electron impact ionization. The metabolite extract (100 µL) was transferred to mass spectrometer autosampler vials (Microliter; 09-1200) and dried down completely under a nitrogen stream at 30 L/min (top) 1 L/min (bottom) at 30°C (Biotage SPE Dry 96 Dual; 3579M). To dried samples, 50 µL of freshly prepared 20 mg/mL methoxyamine (Sigma; 226904) in pyridine (Sigma; 270970) was added and incubated in a thermomixer C (Eppendorf) for 90 min at 30°C and 1400 rpm. After samples are cooled to room temperature, 80 µL of derivatizing reagent (BSTFA + 1% TMCS; Sigma; B-023) and 70 µL of ethyl acetate (Sigma; 439169) were added and samples were incubated in a thermomixer at 70°C for 1 hour and 1400 rpm. Samples were cooled to room temperature and 400 µL of ethyl acetate was added to dilute samples. Turbid samples were transferred to microcentrifuge tubes and centrifuged at 4°C, 20,000 x g for 15 min. Supernatants were then added to mass spec vials for GC-MS analysis (Agilent 7890A GC system, Agilent 5975C MS detector) operating in electron impact (EI) ionization mode, using a HP-5MSUI column (30 m x 0.25 mm, 0.25 µm; Agilent Technologies 19091S-433UI) and 1 µL injection. Oven ramp parameters: 1 min hold at 60°C, 16°C per min up to 300°C with a 7 min hold at 300°C. Inlet temperature was 280°C and transfer line was 300°C. Data analysis was performed using MassHunter Quantitative Analysis software (version B.10, Agilent Technologies) and endogenous compound identifies were confirmed by comparison to retention time, m/z, and fragmentation pattern of to authentic standards. Normalized peak areas were calculated by dividing raw peak areas of targeted analytes by averaged raw peak areas of internal standards. Heatmaps were developed utilizing ggplot in R65.
Collection of peritoneal samples.
At fifteen hours post injection with S. marcescens, mice were sacrificed and grouped as survivors and non-survivors as described above. A small incision was made over the inferior portion of the abdomen and the peritoneum was carefully opened. A sterile sponge was inserted into the peritoneal cavity to absorb peritoneal fluid. The sponge was then placed into a sterile 10cc syringe housed in a 15cc sterile tube which was centrifuged for 15 min at 15,000 x g to allow for removal of the concentrated peritoneal exudate from the sponge. The peritoneal exudate was collected in this manner to avoid dilution with peritoneal lavage and allow detection of metabolites. Samples were incubated at −80°C for at least one hour, or up to overnight. Extraction solvent (4 volumes of 100% methanol spiked with internal standards and stored at −80°C) was added to the liquid sample (1 volume) in a microcentrifuge tube. Tubes were then centrifuged at −10°C, 20,000 x g for 15 min and supernatant was used for subsequent metabolomic analysis. Extraction solvent (4 volumes of 100% methanol spiked with internal standards and stored at −80°C) was added to the liquid sample (1 volume) in a microcentrifuge tube. Tubes were then centrifuged at −10°C, 20,000 x g for 15 min and supernatant was used for subsequent metabolomic analysis. Peritoneal exudate metabolites were analyzed using GC-MS with electron impact ionization. The metabolite extract (100 µL) was transferred to mass spectrometer autosampler vials (Microliter; 09-1200) and dried down completely under a nitrogen stream at 30 L/min (top) 1 L/min (bottom) at 30°C (Biotage SPE Dry 96 Dual; 3579M). To dried samples, 50 µL of freshly prepared 20 mg/mL methoxyamine (Sigma; 226904) in pyridine (Sigma; 270970) was added and incubated in a thermomixer C (Eppendorf) for 90 min at 30°C and 1400 rpm. After samples are cooled to room temperature, 80 µL of derivatizing reagent (BSTFA + 1% TMCS; Sigma; B-023) and 70 µL of ethyl acetate (Sigma; 439169) were added and samples were incubated in a thermomixer at 70°C for 1 hour and 1400 rpm. Samples were cooled to room temperature and 400 µL of ethyl acetate was added to dilute samples. Turbid samples were transferred to microcentrifuge tubes and centrifuged at 4°C, 20,000 x g for 15 min. Supernatants were then added to mass spec vials for GC-MS analysis (Agilent 7890A GC system, Agilent 5975C MS detector) operating in electron impact (EI) ionization mode, using a HP-5MSUI column (30 m x 0.25 mm, 0.25 µm; Agilent Technologies 19091S-433UI) and 1 µL injection. Oven ramp parameters: 1 min hold at 60°C, 16°C per min up to 300°C with a 7 min hold at 300°C. Inlet temperature was 280°C and transfer line was 300°C. Data analysis was performed using MassHunter Quantitative Analysis software (version B.10, Agilent Technologies) and endogenous compound identifies were confirmed by comparison to retention time, m/z, and fragmentation pattern of to authentic standards. Normalized peak areas were calculated by dividing raw peak areas of targeted analytes by averaged raw peak areas of internal standards. Given the lack of concentrated peritoneal exudate in uninfected mice, a relative amount of metabolites were calculated by dividing the survival values by the average of the non-survivors.
Peritoneal macrophage isolation and RNA sequencing
Peritoneal macrophages were isolated utilizing the macrophage isolation kit (Miltenyi Biotec, 130-110-434). Briefly, the mouse peritoneal cavity was flushed with 5cc of ice cold of macrophage buffer (PBS, pH 7.2, 0.5% bovine serum albumin, 2mM EDTA). Peritoneal exudate was then blocked utilizing the manufacturer Fc blocking regatent, and stained with Biotin- Antibody cocktail from the peritoneal macrophage isolation kit according to the manufacturer’s protocol (Miltenyi Biotec) and run over an LS magnetic bead column to remove non-targeted cells. Once macrophages were isolated via bead isolation, RNA was extracted using the Qiagen RNeasy extraction kit. RNA quality control was conducted utilizing the Agilent 2100 Bioanalyzer (Santa Clara, CA). A RNA integrity number (RIN) of 8 was required to proceed with RNA sequencing. RNA sequencing was conducted by Igenbio Inc. Library preparation was conducted using the NEBNext Ultra II with poly-A selection kit according to manufacturer’s protocol. QC was performed on the libraries and they were in turn sequenced on the Illumina NovaSeq S4 machine yielding 38M to 138M 2× 150 bp paired end reads per sample. Quality analysis was performed with ERGOs Read QC workflow with mean base sequences with a PHRED score > 35. Transcript abundances were obtained using Kallisto(v 0.46.1) which utilizes pseudo-alignment to determine the compatibility of read with target mRNA. The reference assembly and annotation of Mus musculus version GRCm39 was used for quantification. The abundances were imported into R using tximport (v 1.14.2) and counts were obtained using DESeqDAtaSetFromTxImport from DESeq2 (v1.26.0). PCA analysis was conducted using plot PCA function of DESeq2. Significantly differentially expressed genes, log2 > 1.5 or < −1.5 and FDR < 0.05, were obtained utilized DeSeq2 comparing experimental groups to uninfected mice. Gene set enrichment analysis was performed utilizing DEGs using g:Profiler. DEGs were then uploaded into Ingenuity Pathway Analysis for further comparisons of the group. Raw RNA sequences uploaded to NCBI SRA (Reference number: PRJNA850672).
AhR activation assay using hepatoma luciferase reporter cell lines
Mouse hepatoma AhR luciferase cells (H1L1.1c2 cells) were obtained from Michael Denison PhD from UC-Davis66. As previously described, Mouse hepatoma cells were grown in alpha MEM media. For Luciferase assay, the cells were split into 96 well plates and seeded at 75,000 cells per well and allowed to grow overnight. The cells were then exposed to the metabolite of interest. After four hours of exposure to the metabolite, the hepatoma cells were washed and lysed utilizing the Promega luciferase assay kit. Luminescence was measured using SpectraMax i3x (Molecular devices), and AhR activity was determined based on relative luminescence to DMSO treated cells. For experiments studying the suppressive effects of S. marcescens, S. marcesens was grown in aMEM media at 37°C on a rotary shaker, and the optical density measured over the course of 32 hours to determine the growth curve. Supernatant from early and late log phase of the growth was isolated by centrifuging the pelleted bacterial cells and the supernatant obtained by filtering using a 0.2 micron filter. Further fractionation of the filtered supernatant was performed using 3kD molecular weight filters (Centrifugal filter units, Millipore) that allowed for separation of a < 3kD and > 3kD fractions. Both fractions were restored to original volume using sterile aMEM cell culture media. The filtered 0.22 µm fractions were then placed on the AhR cell line with or without the indole mixture to determine their impact on AhR signaling.
Cytotoxicity assay
The ATP bioluminescence assay was performed to quantify cell cytotoxicity following vendor direction (ATP Cell Viability Luciferase Assay, Sigma-Aldrigh).
BMDM experiments
Bone marrow was isolated from WT mice as previously described67. Macrophages were derived utilizing L-conditioned media over 7 days of incubation. Following conditioning, macrophages were exposed to the experimental conditions at the same time as S. marcescens lysate. Briefly, S. marcescens was grown over night on MacConkey agar and diluted in liquid RPMI media to an MOI of 5. S. marcescens was then lysed utilizing the mBio tissue homogenizer. Supernatant was added to the BMDM cultures with or without experimental conditions including an indole mixture at a concentration of 0.01 mM and/or AhR inhibitor. Following incubation for six hours, supernatant was collected for protein expression and cells were lysed. RNA was extracted utilizing RNeasy extraction kits following the manufacturer protocol. Furthermore, cDNA was reverse transcribed from RNA utilizing the Biorad reverse transcription kit. The cDNA was then utilized for RT-PCR (QuantStudio 3 Real Time PCR System, ThermoFisher) utilizing the primers of interests and fold change was calculated utilizing the delta-delta CT method.
Phagocytic activity (Gentamicin protection assay).
Gentamicin protection assays were performed after deriving macrophages as previously described68. The BMDMs were exposed to Serratia marcescens at an MOI of 5. After exposure to S. marcescens, the cell plates were spun for 30 min at 900xG and underwent an additional 30 min of incubation at 37°C. Following completion of the incubation, the cells were incubated for an hour with 100µg/mL of gentamicin. The macrophage supernatant was replaced with 25µg/mL gentamicin and incubated for a total of 4 hours to eliminate extracellular bacteria. Macrophages were lysed at 0, 2, and 4 hours and lysates were plated on MacConkey agar to assess for intracellular S. marcescens.
S. marcescens inhibition of Aryl hydrocarbon receptor
S. marcescens was grown in liquid MacConkey media at 37°C and the optical density monitored over 24 hours to determine its growth curve. Once the growth curve was determined, supernatant was collected at the early log, mid log, and late log phase. The supernatant was filtered with a 0.2 micron filter. The supernatant was combined with the indole mix at a concentration of 0.01 mM and added to the AhR reporter cell line; AhR signaling was measured as described above.
Molecular docking
A molecular docking analysis was conducted to determine the degree of AhR binding between the AhR and small molecules produced by the microorganisms used in this model (S. marcescens, K. oxytoca, E. faecalis, C. albicans) as previously described 69–71. The crystal structure of AhR was downloaded from PDB database. Then the organics, solvents, and ions were removed by PyMOL software, and hydrogens were removed by AutoDockTools-1.5.7 software. Finally, the AhR crystal structure was saved in ‘pdbqt’ by AutoDockTools-1.5.7 software. The 2D structures of enterobactin, prodigiosin, and melanin were obtained from PubChem (https://pubchem.ncbi.nlm.nih.gov/), which were then imported into ChemBio3D 14.0 software to minimize free energy and obtain 3D structures. These structures were further entered into AutoDockTools-1.5.7 and transferred into “pdbqt” format. Finally, AutoDock Vina software was used for molecular docking. The 3D and 2D results with the highest negative binding of free energy (kcal/mol) were visualized by PyMOL and LigPlot+ software. Potential candidates for AhR suppression were chosen based on the docking score in the range of AhR antagonist stemreginin.
Patient samples
Two separate cohorts of patient samples were included in this study to demonstrate the role of indoles in septic patients. Informed consent was obtained determined by the individual IRB. The first study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Federal Research and Clinical Center of Intensive Care Medicine and Rehabilitology, Moscow, RF (Protocol code № 5/21/2 from 23 December 2021).
Control blood samples were taken from healthy donors, n = 48: these were 19 women and 29 men aged 20 to 67 years. The donors showed no signs of an acute cold or inflammation. Cases of chronic liver, kidney, etc. diseases were excluded.
Patients’ blood samples (n=26) were taken from 11 patients admitted in January-February 2024 to the intensive care unit (ICU) specializing in the treatment of acute pancreatitis and other severe abdominal infections at risk of sepsis. All patients included in the study had procalcitonin(PCT, a common lab marker of a bacterial infection) levels above the reference PCT value of 0.05 μg/L, which confirms the presence of bacterial-mediated inflammation. Clinical information of the patients can be found in supplemental Table S3.
All blood samples were collected from a peripheral vein into anticoagulant-free test tubes. Serum samples were obtained via blood centrifugation at 1500× g for 10 min on the same day. Serum aliquots were poured into disposable Eppendorf tubes, frozen, and stored at −80 °C. UPLC-MS/MS analysis was carried out in the Labs Bioanalytical Laboratory, as previously described72.
In a second cohort of patient samples, stool samples were obtained from a prior study in Covid -19 patients that were hospitalized in the medical intensive care unit under University of Chicago under IRB protocol (20–1102)1. From this corhort of samples, a subset of septic patients were selected that met Sepsis-3 criteria (qSOFA score ≥ 2 and a known infection other than or in addition to also having Covid-19) and their fecal metabolome was compared based on survival.
Extended Data
Extended Figure 1: Comparison of alpha and beta diversity between gut microbiota of Survivors, Non-Survivors and FMT treated mice.

16S sequencing was completed of the cecal microbiota obtained from survivors (n =7), non-survivors (n=8), and FMT (n=5). The Shannon alpha diversity was compared between groups (A) and the Beta diversity, weighted unifrac (B). Boxplots – lower and upper lines represent first and third quartiles, middle line represents the median, whiskers are 1.5xIQR.
Extended Figure 2: Short chain fatty acids (SCFA) were compared between survivors and non-survivors in the cecum and peritoneum.

Cecal concentrations of acetate (A), propionate (B), and butyrate (C) were compared between survivors and non-survivors (n = 5). Levels of acetate (D), propionate (E). and butyrate (F) in the peritoneum of survivors and non-survivors (n = 3 per group).
Extended Figure 3: Bacterial dissemination and clinical severity of AhR inhibition in S. marcescens peritonitis.

Bacterial culture of the peritoneum (A) and blood (B) comparing survivors (n = 6), non-survivors (n =6), AhR inhibitor with SR1 (n = 5), FMT (n =5), and FMT + AhRi ( n =5). One way ANOVA followed by pairwise comparison with BH correction. p values, * < 0.05, ** < 0.01, *** <0.001
Extended Figure 4: Transcriptional profiles of peritoneal macropahges from FMT treated mice.

RNA sequencing was performed on peritoneal macrophages from FMT treated mice (n =5) compared to non-surviving mice (n = 5). Differentially expressed genes comparing FMT to non-surviving mice are demonstrated in the volcano plot in A. Gene set enrichment demonstrated significant transcription factors between FMT and non-survivors (B). Ingenuity pathway analysis was completed comparing differentially expressed genes between peritoneal macrophages isolated from FMT and FMT+AhRi demonstrating significantly altered pathways (C).
Extended Figure 5: Uniquely differentially expressed genes in peritoneal macrophages from surviving mice.

RNA sequencing was performed on peritoneal macrophages isolated from survivors (n = 5), non-survivors (n =5), and FMT (n =5). Differentially expressed genes (absolute log FC > 1.5 and FDR < 0.05) that were either unique to survivors or oppositely expressed between survivors and non-survivors are listed in the panels.
Extended Figure 6: AhR activation in vitro by individual indole metabolites.

A murine hepatoma AhR reporter cell line was utilized to determine the dose response of AhR activation by individual indole metabolites (indole-3 acetic acid, indole-3 carboxaldehyde, indole-3 lactic acid, and tryptophol). Each metabolite was tested at concentrations ranging from 0.01 mM to 5 mM, n = 3 bioligic replicates per concentration for each metabolite. Boxplots – lower and upper lines represent first and third quartiles, middle line represents the median, whiskers are 1.5xIQR.
Extended Figure 7: AhR activation in vitro by short chain fatty acids.
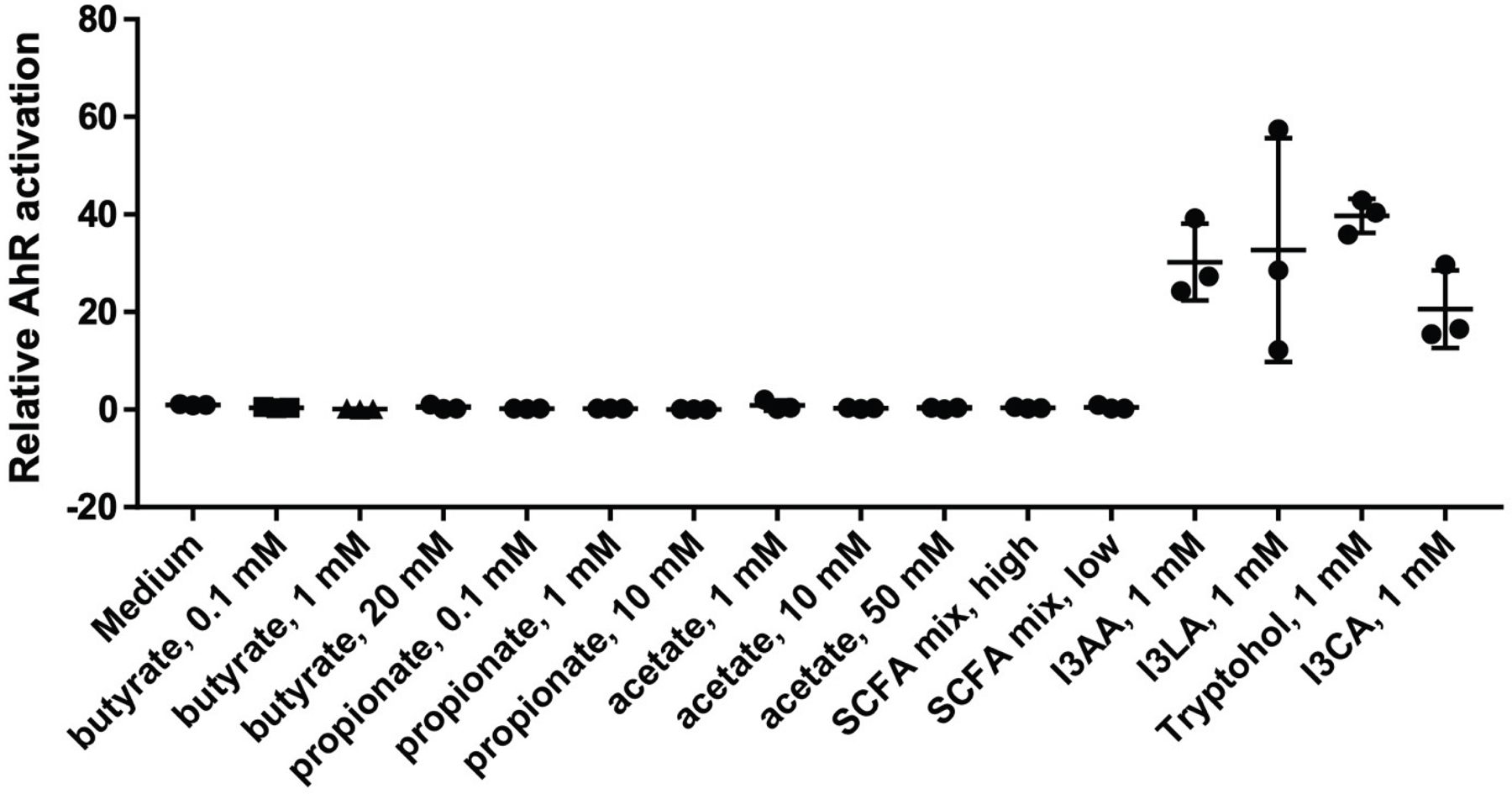
A murine hepatoma AhR reporter cell line was utilized to determine the dose response of AhR activation by individual short chain fatty acids (SCFAs: acetate, propionate, and butyrate) and a mixture of SCFAs (high = 10mM, low = 1mM). The AhR activation was compared to indole metabolites at 1mM (indole-3 acetic acid, indole-3 lactic acid, indole-3 carboxaldehyde, and tryptophol). n = 3 biologic replicates per concentration for each metabolite.
Extended Figure 8: Expression of pro-inflammatory cytokines by macrophages in response to S. marcescens and indole metabolites in vitro.

Bone marrow derived macrophages (BMDMs) were stimulated in vitro in the presence of S. marcesens, S. marcescens with 0.01 mM indole metabolite mixture (indole-3 lactic acid, indole-3 acetic acid, indole-3 carboxaldehyde, and tryptophol), and S. marcescens in the presence of indole mixture with AhR inhibitor (SR1). Gene expression of Nos2, Il6, and TNFa was compared between groups (DMSO n =6, Indole Mix n = 9, and Indole Mix + AhRi n = 6). Boxplots – lower and upper lines represent first and third quartiles, middle line represents the median, whiskers are 1.5xIQR.
Extended Figure 9: Molecular docking was utilized in silico to determine potential pathogen secreted exoproducts capable of inhibiting AhR.

Prodiogosin and enterobactin are two secondary metabolites common to pathogens that are predicted to be AhR Antagonists. Three dimensional binding mode (i), surface representation (ii), and two-dimensional illustration of molecular docking between enterobactin and AhR and prodiogosin and AhR are demonstrated. The binding energies and docking scores are demonstrated in extended table 3.
Extended Figure 10: Iron chelation does not impact enterobactin inhibition of AhR and enterobactin intermediate dihydroxybenzoic acid can inhibit AhR in vitro.

Murine hepatoma AhR reporter cell line was utilized to determine the ability of iron to impact enterobactin inhibition of AhR activation by indole metabolites (A). Furthermore, enterobactin intermediate dihydroxybenzoic acid (DHBA) was utilized to determine if it could inhibit indole activation of AhR (B). n = 3 biologic replicates per group. Boxplots – lower and upper lines represent first and third quartiles, middle line represents the median, whiskers are 1.5xIQR.
Supplementary Material
Supplemental Table 1: Murine sepsis score. Clinical sepsis score utilized to characterize mice following bacterial infection
Supplemental Table 2: Cecal and peritoneal metabolites. Raw data of cecal and peritoneal metabolites determined by mass spec.
Supplemental Table 3: Autodock binding energies and docking scores of pathogen exoproducts predicted to inhibit AhR. Molecular docking was utilized to determine pathogen associated exoproducts capable of inhibiting AhR. Docking score and binding energy (B.E.) are displayed in the table.
Supplemental Table 4: Clinical description of septic ICU patients utilized determine serum indole metabolite levels. PCT = procalcitonin, qSOFA = quick sequential organ failure assessment, APACHE-II = Acute Physiology and Chronic Health Evaluation II
Supplemental Table 5: Clinical description of septic ICU patients utilized to determine levels of stool indole metabolites. qSOFA = quick sequential organ failure assessment
Acknowledgments
We acknowledge the Duchossois Family Institute Host-Microbe Metabolomics Facility for running the 16S RNA amplicon sequencing and analysis and for the metabolomics analyses; Dr. Michael Denison and his lab (University of Califorina Davis) for providing the hepatoma AhR reporter cell line; and Dr. Lev Becker for helpful discussions and advise on the macrophage-related experiments. This work was supported by NIH grant R01GMO62344–22 (JCA).
Footnotes
Competing Interests: None of the authors have any competing interests
Data Availability
Sequence availability for 16S sequencing is available at NCBI SRA (Reference number: PRJNA83864) and RNA sequencing data from peritoneal macropahges is available at NCBI SRA (Reference number: PRJNA850672). Raw data from metabolomics is available in extended table 2.
References
- 1.Leligdowicz A & Matthay MA Heterogeneity in sepsis: new biological evidence with clinical applications. Crit Care 23, 80 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marshall JC Why have clinical trials in sepsis failed? Trends in Molecular Medicine 20, 195–203 (2014). [DOI] [PubMed] [Google Scholar]
- 3.McCarville JL, Chen GY, Cuevas VD, Troha K & Ayres JS Microbiota Metabolites in Health and Disease. Annu. Rev. Immunol 38, 147–170 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Casadevall A & Pirofski L Host-Pathogen Interactions: Basic Concepts of Microbial Commensalism, Colonization, Infection, and Disease. Infect Immun 68, 6511–6518 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gutiérrez-Vázquez C & Quintana FJ Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 48, 19–33 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y & Kishimoto T Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proceedings of the National Academy of Sciences 105, 9721–9726 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bessede A et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature 511, 184–190 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hyoju SK et al. Mice Fed an Obesogenic Western Diet, Administered Antibiotics, and Subjected to a Sterile Surgical Procedure Develop Lethal Septicemia with Multidrug-Resistant Pathobionts. mBio 10, e00903–19, /mbio/10/4/mBio.00903-19.atom (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim SM et al. Fecal microbiota transplant rescues mice from human pathogen mediated sepsis by restoring systemic immunity. Nat Commun 11, 2354 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keskey R et al. Defining Microbiome Readiness for Surgery: Dietary Prehabilitation and Stool Biomarkers as Predictive Tools to Improve Outcome. Annals of Surgery Publish Ahead of Print, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mai SHC et al. Body temperature and mouse scoring systems as surrogate markers of death in cecal ligation and puncture sepsis. ICMx 6, 20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mei J et al. Body temperature measurement in mice during acute illness: implantable temperature transponder versus surface infrared thermometry. Sci Rep 8, 3526 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stolarski AE, Kim J, Nudel J, Gunn S & Remick DG Defining Sepsis Phenotypes—Two Murine Models of Sepsis and Machine Learning. Shock 57, 268–273 (2022). [DOI] [PubMed] [Google Scholar]
- 14.Bhavani SV et al. Identifying Novel Sepsis Subphenotypes Using Temperature Trajectories. Am J Respir Crit Care Med 200, 327–335 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bain CC & Jenkins SJ The biology of serous cavity macrophages. Cellular Immunology 330, 126–135 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Murray PJ et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 41, 14–20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shinde R et al. Apoptotic cell–induced AhR activity is required for immunological tolerance and suppression of systemic lupus erythematosus in mice and humans. Nat Immunol 19, 571–582 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hezaveh K et al. Tryptophan-derived microbial metabolites activate the aryl hydrocarbon receptor in tumor-associated macrophages to suppress anti-tumor immunity. Immunity 55, 324–340.e8 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stockinger B, Meglio PD, Gialitakis M & Duarte JH The Aryl Hydrocarbon Receptor: Multitasking in the Immune System. Annu. Rev. Immunol 32, 403–432 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Han H, Safe S, Jayaraman A & Chapkin RS Diet–Host–Microbiota Interactions Shape Aryl Hydrocarbon Receptor Ligand Production to Modulate Intestinal Homeostasis. Annu. Rev. Nutr 41, 455–478 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu Z et al. Card9 protects sepsis by regulating Ripk2-mediated activation of NLRP3 inflammasome in macrophages. Cell Death Dis 13, 502 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McClure C, Brudecki L, Yao ZQ, McCall CE & El Gazzar M Processing Body Formation Limits Proinflammatory Cytokine Synthesis in Endotoxin-Tolerant Monocytes and Murine Septic Macrophages. J Innate Immun 7, 572–583 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Z et al. TRDMT1 exhibited protective effects against LPS -induced inflammation in rats through TLR4-NF-κB / MAPK-TNF -α pathway. Anim Models and Exp Med 5, 172–182 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Augusto J-F et al. Clusterin Neutralizes the Inflammatory and Cytotoxic Properties of Extracellular Histones in Sepsis. Am J Respir Crit Care Med 208, 176–187 (2023). [DOI] [PubMed] [Google Scholar]
- 25.Wang J et al. FAM76B regulates PI3K/Akt/NF-κB-mediated M1 macrophage polarization by influencing the stability of PIK3CD mRNA. Cell. Mol. Life Sci 81, 107 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chandran S et al. Lipin-1 Contributes to IL-4 Mediated Macrophage Polarization. Front. Immunol 11, 787 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quicke KM, Diamond MS & Suthar MS Negative regulators of the RIG-I-like receptor signaling pathway. Eur J Immunol 47, 615–628 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu Y et al. SLAMF7 regulates the inflammatory response in macrophages during polymicrobial sepsis. Journal of Clinical Investigation 133, e150224 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stein EV, Miller TW, Ivins-O’Keefe K, Kaur S & Roberts DD Secreted Thrombospondin-1 Regulates Macrophage Interleukin-1β Production and Activation through CD47. Sci Rep 6, 19684 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McGettrick AF & O’Neill LAJ The Role of HIF in Immunity and Inflammation. Cell Metabolism 32, 524–536 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Wen AY, Sakamoto KM & Miller LS The Role of the Transcription Factor CREB in Immune Function. J.I 185, 6413–6419 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sekine H et al. Hypersensitivity of Aryl Hydrocarbon Receptor-Deficient Mice to Lipopolysaccharide-Induced Septic Shock. Mol Cell Biol 29, 6391–6400 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park JM et al. Signaling Pathways and Genes that Inhibit Pathogen-Induced Macrophage Apoptosis— CREB and NF-κB as Key Regulators. Immunity 23, 319–329 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Denison MS & Heath-Pagliuso S The Ah Receptor: A Regulator of the Biochemical and Toxicological Actions of Structurally Diverse Chemicals. Bull. Environ. Contam. Toxicol 61, 557–568 (1998). [DOI] [PubMed] [Google Scholar]
- 35.Zhu J et al. Aryl Hydrocarbon Receptor Promotes IL-10 Expression in Inflammatory Macrophages Through Src-STAT3 Signaling Pathway. Front. Immunol 9, 2033 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nizet V Bacteria and Phagocytes: Mortal Enemies. J Innate Immun 2, 505–507 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Hantke K, Nicholson G, Rabsch W & Winkelmann G Salmochelins, siderophores of Salmonella enterica and uropathogenic Escherichia coli strains, are recognized by the outer membrane receptor IroN. Proc. Natl. Acad. Sci. U.S.A 100, 3677–3682 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raymond KN, Dertz EA & Kim SS Enterobactin: An archetype for microbial iron transport. Proc. Natl. Acad. Sci. U.S.A 100, 3584–3588 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh V et al. Interplay between enterobactin, myeloperoxidase and lipocalin 2 regulates E. coli survival in the inflamed gut. Nat Commun 6, 7113 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zaborin A et al. Membership and Behavior of Ultra-Low-Diversity Pathogen Communities Present in the Gut of Humans during Prolonged Critical Illness. mBio 5, e01361–14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Collias D et al. Systematically attenuating DNA targeting enables CRISPR-driven editing in bacteria. Nat Commun 14, 680 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Campos-Madueno EI et al. Evaluation of Phenotypic Tests to Detect Extended-Spectrum β-Lactamase (ESBL)-Producing Klebsiella oxytoca Complex Strains. J Clin Microbiol e01706–22 (2023) doi: 10.1128/jcm.01706-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller WD, Keskey R & Alverdy JC Sepsis and the Microbiome: A Vicious Cycle. The Journal of Infectious Diseases 223, S264–S269 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baxt LA, Garza-Mayers AC & Goldberg MB Bacterial Subversion of Host Innate Immune Pathways. Science 340, 697–701 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Chanderraj R et al. In critically ill patients, anti-anaerobic antibiotics increase risk of adverse clinical outcomes. Eur Respir J 2200910 (2022) doi: 10.1183/13993003.00910-2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Casadevall A Expanding the Pathogenic Potential Concept To Incorporate Fulminancy, Time, and Virulence Factors. mSphere 7, e01021–21 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moura-Alves P et al. AhR sensing of bacterial pigments regulates antibacterial defence. Nature 512, 387–392 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Moura-Alves P et al. Host monitoring of quorum sensing during Pseudomonas aeruginosa infection. Science 366, eaaw1629 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Murray IA et al. Evidence for Ligand-Mediated Selective Modulation of Aryl Hydrocarbon Receptor Activity. Mol Pharmacol 77, 247–254 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tintelnot J et al. Microbiota-derived 3-IAA influences chemotherapy efficacy in pancreatic cancer. Nature 615, 168–174 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Uchimura Y et al. Antibodies Set Boundaries Limiting Microbial Metabolite Penetration and the Resultant Mammalian Host Response. Immunity 49, 545–559.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bongers KS et al. The Gut Microbiome Modulates Body Temperature Both in Sepsis and Health. Am J Respir Crit Care Med 207, 1030–1041 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen GY & Ayres JS Beyond tug-of-war: Iron metabolism in cooperative host–microbe interactions. PLoS Pathog 16, e1008698 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Raffatellu M et al. Lipocalin-2 Resistance Confers an Advantage to Salmonella enterica Serotype Typhimurium for Growth and Survival in the Inflamed Intestine. Cell Host & Microbe 5, 476–486 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hughes ER & Winter SE Enterococcus faecalis: E. coli ‘s Siderophore-Inducing Sidekick. Cell Host & Microbe 20, 411–412 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quintana FJ et al. Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature 453, 65–71 (2008). [DOI] [PubMed] [Google Scholar]
- 57.Veldhoen M et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453, 106–109 (2008). [DOI] [PubMed] [Google Scholar]
- 58.Goudot C et al. Aryl Hydrocarbon Receptor Controls Monocyte Differentiation into Dendritic Cells versus Macrophages. Immunity 47, 582–596.e6 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Qiu J et al. The Aryl Hydrocarbon Receptor Regulates Gut Immunity through Modulation of Innate Lymphoid Cells. Immunity 36, 92–104 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.González-Juarbe N et al. Requirement for Serratia marcescens Cytolysin in a Murine Model of Hemorrhagic Pneumonia. Infect Immun 83, 614–624 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ishii K et al. Serratia marcescens Induces Apoptotic Cell Death in Host Immune Cells via a Lipopolysaccharide- and Flagella-dependent Mechanism. Journal of Biological Chemistry 287, 36582–36592 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krzymińska S, Ochocka K & Kaznowski A Apoptosis of Epithelial Cells and Macrophages due to Nonpigmented Serratia marcescens Strains. The Scientific World Journal 2012, 1–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bolyen E et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37, 852–857 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McMurdie PJ & Holmes S phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 8, e61217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wickham H Ggplot2: Elegant Graphics for Data Analysis. (Springer-Verlag New York, 2016). [Google Scholar]
- 66.Garrison P Species-Specific Recombinant Cell Lines as Bioassay Systems for the Detection of 2,3,7,8-Tetrachlorodibenzo-p-dioxin-like Chemicals. Fundamental and Applied Toxicology 30, 194–203 (1996). [DOI] [PubMed] [Google Scholar]
- 67.Kratz M et al. Metabolic Dysfunction Drives a Mechanistically Distinct Proinflammatory Phenotype in Adipose Tissue Macrophages. Cell Metabolism 20, 614–625 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang D et al. Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gaurav I et al. Synthesis, In-Vitro and In-Silico Evaluation of Silver Nanoparticles with Root Extract of Withania somnifera for Antibacterial Activity via Binding of Penicillin-Binding Protein-4. CPB 21, 1674–1687 (2020). [DOI] [PubMed] [Google Scholar]
- 70.Gaurav I et al. Delivery of Apoplastic Extracellular Vesicles Encapsulating Green-Synthesized Silver Nanoparticles to Treat Citrus Canker. Nanomaterials 13, 1306 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hu X et al. Cortex Mori extracts induce apoptosis and inhibit tumor invasion via blockage of the PI3K/AKT signaling in melanoma cells. Front. Pharmacol 13, 1007279 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sobolev PD, Burnakova NA, Beloborodova NV, Revelsky AI & Pautova AK Analysis of 4-Hydroxyphenyllactic Acid and Other Diagnostically Important Metabolites of α-Amino Acids in Human Blood Serum Using a Validated and Sensitive Ultra-High-Pressure Liquid Chromatography-Tandem Mass Spectrometry Method. Metabolites 13, 1128 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1: Murine sepsis score. Clinical sepsis score utilized to characterize mice following bacterial infection
Supplemental Table 2: Cecal and peritoneal metabolites. Raw data of cecal and peritoneal metabolites determined by mass spec.
Supplemental Table 3: Autodock binding energies and docking scores of pathogen exoproducts predicted to inhibit AhR. Molecular docking was utilized to determine pathogen associated exoproducts capable of inhibiting AhR. Docking score and binding energy (B.E.) are displayed in the table.
Supplemental Table 4: Clinical description of septic ICU patients utilized determine serum indole metabolite levels. PCT = procalcitonin, qSOFA = quick sequential organ failure assessment, APACHE-II = Acute Physiology and Chronic Health Evaluation II
Supplemental Table 5: Clinical description of septic ICU patients utilized to determine levels of stool indole metabolites. qSOFA = quick sequential organ failure assessment
Data Availability Statement
Sequence availability for 16S sequencing is available at NCBI SRA (Reference number: PRJNA83864) and RNA sequencing data from peritoneal macropahges is available at NCBI SRA (Reference number: PRJNA850672). Raw data from metabolomics is available in extended table 2.