

Abstract
The recent identification of SATB2 as a candidate gene responsible for the craniofacial dysmorphologies associated with deletions and translocations at 2q32-q33, one of only three regions of the genome for which haploinsufficiency has been significantly associated with isolated cleft palate, led us to investigate the in vivo functions of murine Satb2. We find that, similar to the way in which SATB2 is perceived to act in humans, craniofacial defects due to haploinsufficiency of Satb2, including cleft palate (in ∼25% of cases), phenocopy those seen with 2q32-q33 deletions and translocations in humans. Full functional loss of Satb2 results in amplification of these defects and leads both to increased apoptosis in the craniofacial mesenchyme where Satb2 is usually expressed and to changes in the pattern of expression of three genes implicated in the regulation of craniofacial development in humans and mice: Pax9, Alx4, and Msx1. The Satb2-dosage sensitivity in craniofacial development is conspicuous—along with its control of cell survival, pattern of expression, and reversible functional modification by SUMOylation, it suggests that Satb2/SATB2 function in craniofacial development may prove to be more profound than has been anticipated previously. Because jaw development is Satb2-dosage sensitive, the regulators of Satb2 expression and posttranslational modification become of critical importance both ontogenetically and evolutionarily, especially since such regulators plausibly play undetected roles in jaw and palate development and in the etiology of craniofacial malformations.
The acquisition of jaws, a landmark event in vertebrate evolution, in large part potentiated the diversification and success of the vertebrates. The developmental system that coordinates and patterns the craniofacial primordia that give rise to jaws involves an intricate spatiotemporal series of reciprocal inductive and responsive interactions among the cephalic epithelia (both endodermal and ectodermal) and the cranial neural crest and cephalic mesodermal mesenchyme.1–4 The coordinated regulation of these interactions is critical both to the ontogenetic registration of the jaws, including of the palate, and to the evolutionary elaboration of variable jaw morphologies and designs.5 The importance and sensitivity to perturbation of this developmental system is reflected by the fact that one-third of all human congenital malformations affect the head and face and their developing primordia.6,7 For instance, orofacial clefting, including cleft palate (CP) and/or cleft lip, constitutes one of the most common human birth defects (from 1 in 500 to 1 in 2,000 births, depending on the population) and exhibits a notably complex etiology.8–12 Correlation of the genetic, molecular, and cellular mechanisms underlying the etiology of such malformations lags, however, relative to the importance of the problem.
Cytogenetic evidence, based on interstitial deletions and balanced translocations, has previously implicated human 2q32-q33 (CPI [MIM 119540]) as a craniofacial dysmorphology locus.13–15 Patients with 2q32-q33 deletions are characterized by malformations, including long (occasionally asymmetric) faces with small mouths, high foreheads, cleft (or high-arched) palates, micrognathia, maxillary hypoplasia, and short, broad teeth with large diastemata.15 Similar dysmorphisms, including narrow faces with prominent nasal bridges, small mouths, and CP, are associated with balanced translocations with breakpoints in distal 2q32.14 Notably, 2q32-q33 is one of only three regions of the genome for which haploinsufficiency has been significantly associated with isolated CP.13 Modeling such haploinsufficiency in mice has proven difficult because somatic heterozygous mutations implicated in CP in humans, such as mutations in MSX1 (MIM 142983), PAX9 (MIM 167416), and TBX1 (MIM 602054), are known to lead to CP in only homozygous states in mice.8,11,12,16–22
SATB2 (MIM 608148) has recently been identified as a candidate CP gene at 2q32.10 SATB2 and murine Satb2 are highly conserved (99.6%) members of a small, novel transcription-factor gene family whose members bind to nuclear matrix attachment regions and appear to be involved in the regulation of the tissue-specific organization of chromatin, thereby augmenting the potential for enhancers to act over large distances.10,23,24 Satb2 is, furthermore, a target for SUMOylation, a reversible modification of the protein that modulates its activity as a transcription factor.24 Although exonic polymorphisms have yet to be found in SATB2, a point mutation of SATB2 has recently been identified in a large screen of patients with isolated CP.25 Despite its recently identified association with CP and 2q32-q33 deletions, direct tests of Satb2 function have not been reported elsewhere.
To study the in vivo functions of Satb2, we generated a Satb2 null allele through homologous recombination. In line with the hemizygosity of SATB2 being implicated in CP and the craniofacial malformations linked with 2q32-q33 deletions in humans, we demonstrate similar gene-dosage effects (haploinsufficiency) in the regulation of jaw and palate development exerted by Satb2 in mice. Full functional loss of Satb2, moreover, amplifies the craniofacial deficits observed in Satb2 heterozygotes. Plausibly underlying the mechanistic etiology of these deficits, the loss of Satb2 is associated with increased apoptosis in the discrete, complementary regions of the developing jaw primordia where it is expressed and with the subsequent arrest of regional development, including the down-regulation of the expression of genes associated with craniofacial development, such as Msx1, Alx4 (MIM 605420), and Pax9.16–20,26,27 Coupled with its spatiotemporal expression profile, this marks Satb2 as a potentially key gene coordinating the elaboration of the functional design of jaws, including of the mammalian palate.
Material and Methods
Targeted Disruption of Satb2
A Satb2 null allele was generated through homologous recombination by elimination of the second exon in the protein-coding region and replacement with a Cre recombinase–coding sequence and a Neo expression cassette. Before designing the targeting vector, we detected several splice forms of Satb2 message by RT-PCR. To rule out possible exon skipping for the first coding exons, we performed RT-PCR with several sets of primers specific to the first four coding exons (not shown). We were not able to make a firm conclusion regarding skipping of the first coding exon; however, since the second coding exon was present in all RT-PCR products (indicating that it is very unlikely to be the subject of exon skipping), we targeted the second coding exon. The Satb2 construct, along with G418 resistance selection, was electroporated into embryonic stem (ES) cells, from which 150 positive colonies were obtained. Recombinant colonies were identified by Southern-blot analysis of NsiI-digested genomic DNA hybridized with a 3′ external probe designed to detect wild-type and mutant allele fragments of 7.5 kb and 4.6 kb, respectively. Five of the 150 positive ES colonies were selected. To confirm the targeted insertion of the construct into the genomic DNA of the positive colonies, we used PCR amplification with a primer set consisting of an internal primer and a specific 5′ external primer.
Histology
Animals used for histology were fixed in 4% paraformaldehyde (PFA) in PBS, embedded in paraffin or optimum cutting temperature compound, and then were cut into 8–20-μm-thick serial sections. Sections were then prepared with nissel staining.
Differential Bone and Cartilage Staining and Relative Mandibular Measurement
Differential staining of cartilage and bone was achieved through the use of Alcian blue and Alizarin red as per Depew et al.4 Differentially stained wild-type, heterozygous, and homozygous neonatal dentaries were dissected, flat mounted, and photographed en masse at the same magnification. Individual mandibular length was set against the same rule scale (0–110) and was tabulated as the distance from the most proximal tip of the secondary cartilage of the condylar process to the buccal surface of the most distal tip of the ossified dentary proper. The mean length (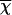 ), SD, and population size were calculated and normalized around an
), SD, and population size were calculated and normalized around an  of 100 for the wild type.
of 100 for the wild type.
Scanning Electron Microscopy
Embryos were taken from timed pregnancies and were fixed at 4°C overnight in 4% PFA in PBS. The embryos were then rinsed three times in PBS and were dehydrated in an increasing series of MeOH. They then were critical-point dried, were sputter coated with gold paladium, and were photographed using an FEI Quanta 200F field-emission scanning electron microscope.
In Situ Hybridization and Satb2 Immunohistochemistry
In situ hybridization was performed as described by Depew et al.4 and Britanova et al.,23 and Satb2 immunoreactivity as described by Britanova et al.23
Apoptosis
Apoptotic cell death was assessed by TUNEL assay on paraffin sections by use of the Apoptag Fluorescein Direct In situ Apoptosis Detection Kit (Chemicon), in accordance with the manufacturer’s instructions.
Results
Expression of Satb2
The identification of SATB2 as a gene associated with CP in humans led us to further clarify Satb2 expression and function during craniofacial development. As anticipated, Satb2 is expressed in the developing jaw primordia during the pharyngula stages of murine development (fig. 1). We did not detect a strong signal by in situ hybridization before embryonic day 10 (E10); however, by E10.5 and E11.5, Satb2 is expressed in a complementary fashion in the mesenchyme at the junction of the maxillary first branchial arch (BA1) and the medial frontonasal process (FNP) and in the distal mandibular BA1 that contributes to the upper and lower jaws, respectively. These Satb2-expression domains demarcate potential developing modules within the overall jaw complex and are strategically placed to act in the coordination of jaw registration. As has been reported elsewhere, we find that expression is regionally maintained during the morphogenetic stages of jaw development (fig. 1).
Figure 1. .

Pharyngeal and early-morphogenetic-stage Satb2 expression. a–c, Satb2 expression detected through in situ hybridization at E10.5 and E11.5 in the mesenchyme of the distal maxillary BA1 (mx), associated FNP (l = lateral; m = medial), and the distal mandibular BA1 (md). Expression is associated with the distal regions but, significantly, is excluded from the actual midlines (arrowheads). op = olfactory pit. d, Radioactive in situ hybridization at E12.5, demonstrating Satb2 expression in the tongue (tg) and in the developing lower (md) and upper (mx) jaws, including in the future palatal shelves (ps).
Craniofacial Defects Due to Haploinsufficiency of Satb2, Which Phenocopy Those of Human 2q32-33 Deletions
We generated a Satb2 null allele through homologous recombination by eliminating the second exon in the protein-coding region (fig. 2). Heterozygous mice are born and are fertile but, notably, wean at a lower-than-expected frequency (∼35%). In a C57Bl/6 background, the snout of adult heterozygous animals is strongly truncated and occasionally asymmetric. This, coupled with their lower-than-expected frequency, led us to address the early course of craniofacial morphogenesis in Satb2+/− mice. Differential staining of bone and cartilage and histologic analysis of fetal and neonatal Satb2+/− mice demonstrates the early onset of these craniofacial dysmorphologies (fig. 3). With a striking phenotypic similarity to 2q32 deletions and translocations, Satb2+/− neonates exhibit slight microcephaly, small mouths, premaxillary and nasocapsular hypoplasia, micrognathia, and variable incisor hypodontia and/or adontia (fig. 3b, 3e, 3h, and 3t). As an explanation of their lower-than-expected frequency at weaning and in line with SATB2 hemizygosity in humans, approximately one in four perinatal heterozygotes examined have CP (fig. 3b). Significantly, to our knowledge, our targeted mutation of Satb2 provides the first clear murine haploinsufficiency model for the study of a known human haploinsufficient CP genetic locus.
Figure 2. .

Design rationale for the targeted loss of Satb2. a, Schematic representation of the Satb2 locus, the targeting vector, and the targeted locus. Coding exons are shown as unblackened boxes. Restriction sites are SmaI (S), NsiI (N), and PacI (P). Oligonucleotides are indicated by arrows and are designated “a” and “b.” The expected fragments are indicated by the dashed red lines. The 3′ external probe (purple box) identifies a 7.5-kb NsiI fragment in the wild-type allele and a 4.6-kb fragment in the mutant allele. b, Analysis of transfected ES cells (left) and wild-type (WT), heterozygous (+/−), and homozygous (−/−) mice (right) by genomic Southern-blot analysis with a 3′ external probe. Mut = mutant allele. c and d, Immunohistochemical detection of Satb2 protein in E13.5 wild-type (c) and Satb2−/− (d) embryos. Satb2 immunoreactivity (arrowheads) is detected in the forming palatal shelves (ps) of the wild-type embryo, whereas it is not detected in the craniofacial primordia of the Satb2−/− embryo; however, possibly because of exon skipping, some Satb2-positive cells are detected in the spinal cord of the Satb2−/− embryo, just as they are in the wild-type embryo. ns = Nasal septum; sc = spinal cord; tg = tongue.
Figure 3. .

Satb2 gene-dosage sensitivity in craniofacial development. a–c, Differentially stained skulls of wild-type (wt), Satb2+/−, and Satb2−/− neonates demonstrating the effect of gene dosage on upper jaw and palatal (yellow arrowheads) development. White arrows indicate hypoplasia of premaxillae (px) and nasal capsules. Green arrowhead (c) indicates the exacerbated deficiencies of nasal capsules, primary palate, and upper incisors (compare with panel b). Red arrows (b and c) indicate the midline trabecular basal plate. as = Alisphenoid; bs = basisphenoid; mx = maxilla; pg = pterygoid; psm = maxillary palatal shelf; psp = palatine palatal shelf; psx = premaxillary palatal shelf; v = vomer. d–i, Comparative histological sections of nissel-stained wild-type, Satb2+/−, and Satb2−/− postnatal day 0 and E13.5 embryos, highlighting loss of parasagittal hard- and soft-tissue structures. Arrowheads: green = midline nasal septum; peach = upper molar tooth or bud; turquoise = lower molar tooth or bud; red = body of Meckel’s cartilage; blue outlined in yellow = midline of lower jaw and tongue; black outlined in green = developing palatal shelf; black outlined in red = tongue; black outlined in yellow = developing submandibular gland; white = nasopharynx. j and k, Scanning electron micrographs of wild-type and Satb2−/− embryos at E12. Unblackened arrowheads indicate the midline, orange arrowheads highlight relative mandibular hypoplasia, and arrows indicate maxillary and FNP deficits (compare oral aperatures). l and m, Gross anatomy of wild-type and Satb2−/− E16 littermates demonstrating the small mouth, parasagittal hypoplasia, micrognathia, and maintenance of midline structures in mutants. “a–e” indicate relative positions of lateral and mandibular sinus follicles. Arrows are as in panels j and k. n and o, Skulls of the specimen in panels l and m, showing the specific loss of the distal portion of dentaries and distal juxtaposed portion of Meckel’s cartilage (compare orange arrowheads) but the presence of the midline rostral process (unblackened arrowheads). This maintenance of the midline of lower-jaw apparatus is mirrored in the upper jaws by the presence of the nasal septum and associated paraseptal cartilages and vomeronasal organs but the absence of much of the premaxillae and associated nasal capsule and incisors. p and q, Microcephaly, micrognathia (black arrowhead), premaxillary (px) hypoplasia (arrow), and absence of incisors in a neonatal Satb2−/− skull seen in norma lateralis (q) compared with a wild-type skull (p). Note the maintenance of the articular region (purple arrowheads). r, Labeled schema of wild-type neonatal dentary indicating the orientation for measuring relative dentary length. agp = Angular process; 2agp = secondary cartilage of agp; cdp = condylar process; 2cdp = secondary cartilage of cdp; crp = coronoid process; dnt = body of the dentary; ina = incisive alveolus; LI = lower incisor; moa = molar alveolus; mtf = mental foramen; rpMC = rostral process of Meckel’s cartilage. s–u, Measurements of neonatal wild-type, Satb2+/−, and Satb2−/− littermates along with  , SD, and population size (n) of the measured population for which they are samples.
, SD, and population size (n) of the measured population for which they are samples.
Satb2 Dosage Sensitivity in Jaw and Palate Development
Relative to Satb2 heterozygotes, homozygotes are born (and die perinatally) with exacerbated skeletal malformations, including significant micrognathia, increased microcephaly, nasocapsular and premaxillary hypoplasia, and fully penetrant incisor adontia and CP (fig. 3c, 3f, 3i, 3q, and 3u). The jaw deficiencies of the Satb2−/− mutants are focal, with the mesenchymal component of the Satb2-positive jaw module having clearly failed to elaborate local morphogenetic programs. Both the upper and lower jaws are affected in comparable domains, reflective of the symmetrical, complementary regional patterns of Satb2 expression. Notably, the midline structures connected to the upper and lower jaw arcades are patent, as evidenced by the rostral process of Meckel’s cartilage (fig. 3l–3o) in the lower jaws and by the septal cartilages overlying the upper jaws. Parasagittal elements, such as the incisors and their associated alveolar bone, however, fail to form in each jaw quadrant. Elements associated with the region of the articulation of the jaws, such as the proximal dentary and the squamosal, are unaffected (fig. 3p and 3q). Effectively, a lack of Satb2 results in the loss in each jaw quadrant of complementary parasagittal structures, such as the incisors, whose development needs to be coordinated, to keep the jaws functionally in register. As seen in scanning electron micrographs, these focal losses of skeletal structures in the Satb2−/− mutants are clearly presaged by distinct morphological abnormalities by E12 (fig. 3j and 3k).
Additional defects, including shortened limbs, hyoid malformations, and severe microglossia, are evident in the Satb2−/− mutants (fig. 3 and data not shown). Oral epithelial derivatives are, moreover, variably affected—for instance, molar dental buds are enlarged and contain elaborated epithelial folds, and submandibular glands fail to develop (fig. 3).
Early, but Not Late, Molecular Pattern Maintained in Satb2 Null Embryos
To address molecular and cellular mechanistic explanations for the morphologic deficits of the Satb2−/− mutants, we first assayed for changes at E10.5 in regionally expressed genes known to be regulators of jaw development. As anticipated by the apparently relatively late onset of branchial arch Satb2 expression, at E10.5, the expression patterns of Dlx5 (MIM 600028), Msx1, Pitx1 (MIM 602149), Prx2 (MIM 604675), Barx1 (MIM 603260), and dHAND (MIM 602407) are unchanged (fig. 4a and 4b and data not shown), suggesting that fundamental aspects of the initial molecular pattern of the jaw primordia are extant in the Satb2−/− mutant embryos. At later stages of development, however, we detect subregional changes in the expression of genes known to be associated with jaw and palatal development in both humans and mice,16–20,26,27 including that of Pax9 at E11.5 in the BA1 mesenchyme (fig. 4c and 4d) and Alx4 and Msx1 at E13.5 in the base of the forming palate (fig. 4g–4j). Thus, although it is unclear whether these three genes are direct targets of Satb2 protein, it is clear that Satb2 is required for normal expression of genes critical to craniofacial development.
Figure 4. .

Gene expression in Satb2−/− mutants. a and b, In situ hybridization of Msx1 (a) and Dlx5 (b) in wild-type (wt) and Satb2−/− mutant E10.5 embryos indicating the presence of the initial molecular polarity in the developing branchial arches of Satb2−/− mutants. l = Lateral frontonasal process; m = medial frontonasal process; md = mandibular BA1; mx = maxillary BA1; op = olfactory pit. c and d, Marked decrease of Pax9 expression at E11.5 in the mandibular incisor field (if) of the mutant (red arrowhead) presages the loss of the incisor developmental module. The white arrow indicates the midline. di = Diastema; mf = molar field. e and f, Serial histological sections of wild-type (e, g, and i) and Satb2−/− mutant (f, h, and j) embryos at E13.5 highlight the nasal capsular hypoplasia, microglossia, and loss of submandibular glands in the mutant and the subregional loss (compare red arrowheads) of Alx4 (h) and Msx1 (j) expression within the base of the developing palate of the mutant. lm = Lower molar bud; MC = Meckel’s cartilage; nc = nasal capsule; ns = nasal septum; oe = olfactory epithelium; ps = palatal shelf; smg = submandibular gland; tg = tongue; um = upper molar bud.
Necessity of Satb2 for Cell Survival in Its Expression Domains
The notable loss of gene expression at E11.5 and loss of structure by E12, coupled with the maintenance of initial molecular pattern at E10.5, strongly suggested that differential cell survival and/or proliferation between these time points underlay the phenotypic defects observed. We therefore assayed for changes in the levels of apoptosis in the developing jaw primordia at E11.5. A marked increase in the number of apoptotic cells in the mandibular BA1, maxillary BA1, and FNP in the regions normally expressing Satb2 was observed in Satb2−/− mutant embryos relative to wild-type embryos (fig. 5), thus providing a partial mechanistic explanation for the subsequent morphogenetic deficits found in the Satb2−/− mutants. In line with the more variable severity of the deficits (fig. 3r–3u), we detected a more variable increase in apoptotic cells in the Satb2+/– embryos (data not shown).
Figure 5. .

Increased cell death at E11.5 in Satb2−/− mutants. Parasagittal (a and b) and coronal (c and d) sections of E11.5 wild-type (a and c) and Satb2−/− (b and d) embryos assayed for apoptosis (TUNEL) demonstrate focal increases in cell death in the mandibular (md [red arrowheads]), maxillary (mx [yellow arrowheads]), and frontonasal (fnp [yellow arrowheads]) mesenchyme. White arrowheads indicate the midline, green arrowheads indicate the lambdoidal junction, and purple arrowheads point to typical apoptosis found at the maxillo-mandibular junction. fb = Forebrain; hrt = heart; hy = hyoid arch; op = olfactory pit.
Discussion
The identification of SATB2 as a candidate gene responsible for the craniofacial dysmorphologies associated with 2q32-q33 deletions and translocations led us to investigate the in vivo functions of Satb2. By generating a Satb2 null allele in mice through homologous recombination, we have demonstrated that Satb2 acts in a dosage-dependant manner, similar to the way in which SATB2 is perceived to act in humans, as an essential regulator of murine jaw and palate development and morphogenesis. Moreover, its absence leads both to increased apoptosis in a highly localized fashion in craniofacial mesenchyme and to changes in the pattern of expression of three genes known to regulate craniofacial development in humans and mice—Pax9, Alx4, and Msx1.
While this manuscript was under review, another loss-of-function investigation of Satb2 was published.28 In many respects, such as the early maintenance of expression of a number of genes known to be essential for proper craniofacial development, our analysis of the consequences of the loss of Satb2 for craniofacial development coincides with the analysis of Dobreva et al.28; however, perhaps because of variance in targeting strategies, differences exist in several critical respects. For instance, although the report of Dobreva et al.28 states that heterozygous Satb2+/− mice are phenotypically normal and fertile, we find significant phenotypic defects due to Satb2 haploinsufficiency. This is notable with regard to human pathogenicity, since 2q32-q33 is a locus significantly linked with craniofacial malformations and, importantly, is one of only three regions of the genome for which haploinsufficiency has been significantly associated with isolated CP.13 Our Satb2+/− mice exhibit phenotypic similarities corresponding to those seen with 2q32 deletions and translocations in humans, including CP (in ∼25% of cases), microcephaly, reduced oral aperture, hypoplasia of the premaxillae and nasal capsules, lower jaw micrognathia, and variable incisor hypodontia and/or adontia. Uniquely, our targeted mutation of Satb2 provides the first clear murine haploinsufficiency model for the study of a known human haploinsufficient CP genetic locus. Previously, modeling such haploinsufficiency in mice proved difficult because somatic heterozygous mutations implicated in CP in humans (e.g., MSX1, TBX1, and PAX9) are known to lead to CP in only homozygous states in mice.16–22
Notably, the cleft or arched palate and microglossia associated with 2q32-q33 deletions may be correlated with Pierre Robin sequence,29,30 a clinical phenotype characterized by micrognathia and CP with glossoptosis and often considered to result from a series of causal events. Although no single nosologic etiology represents Pierre Robin sequence, Satb2 haploinsufficiency results in variable cleft or arched palate, micrognathia, and microglossia. Since cell death is regionally associated with the primodia of the palate (in the maxillary arch) and that portion of the mandibular first arch that is associated with the tongue, in this instance the phenotypic correlation of Satb2 loss and Pierre Robin sequence is likely due to independent growth deficiencies.
A second significant point of difference between our analysis and that of Dobreva et al.28 involves the apparent coordination of defects between the jaw primordia. Dobreva et al. present important data regarding a role for Satb2 in osteogenesis. This includes data indicating an interaction during calvarial osteogenesis between Satb2 and Hoxa2 (MIM 604685), a gene that is not normally expressed in the jaw forming BA1 and whose loss in mice affects the caudal, non–jaw-forming branchial arches. This, together with the fact that branchial arch expression of Hoxa2, as reported by Dobreva et al., is relatively normal in Satb2−/− embryos, suggests that explanations for the coordinated, focal jaw-related defects of the Satb2−/− mutants must extend beyond an interaction with Hoxa2 during osteogenesis.
In search of mechanistic explanations for the observed defects, both investigations assessed apoptotic cell death in the branchial arches. Notably, Dobreva et al.28 found increased cell death at E10.5 in the region of the maxillo-mandibular junction, a region in which we failed to detect Satb2 transcript at this same stage (fig. 1b). A day later (E11.5), however, we observed a marked increase in the number of apoptotic cells in Satb2−/− mutant embryos in the mandibular and maxillary BA1 and FNP in those parasagittal regions where we also detected expression of Satb2 (figs. 1 and 5). Notably and in line with the observed maintenance of midline structures, such as the rostral process of Meckel’s cartilage, the midlines are relatively spared (fig. 3). Thus, we find that a partial explanation of the jaw defects resides in the increased, coordinated, and localized cell death that results from a loss of Satb2. This is coupled, directly or indirectly, with changes in gene expression of Alx4, Msx1, and Pax9, three genes known to be critical to craniofacial development.
Moreover, the full functional loss of Satb2 results in an amplification of the defects found in Satb2+/− mice, and this revelation of Satb2-dosage sensitivity for craniofacial development is conspicuous. Coupled with its control of cell survival, its pattern of expression, and its reversible functional posttranslational modification by SUMOylation, this suggests that Satb2/SATB2 function in craniofacial development may prove to be more profound than has been anticipated previously, and its potentially crucial significance for understanding the complex etiology of various craniofacial dysmorphologies, such as CP, specifically and of craniofacial development and evolution in general should not be overlooked.
Jaws are fundamental, functional cranial units that were patent factors in the diversification and success of the vertebrates. As mechanisms, jaws are composed of two articulated, or hinged, appositional units for which developmental polarity and modularity are inherent characteristics5 (fig. 6). Functionality demands that the developmental system that generates jaws be coordinated, to keep these appositional units in register while elaborating the morphology necessary to meet the specific demands of an organism—for instance, it must result in the appropriately matched occlusion of the teeth in the upper and lower jaws (fig. 6b). This system is known to involve a highly regulated series of inductive and responsive interactions between the BA1 and FNP epithelia and subjacent ectomesenchyme.1–4 The functional registration of jaws is thought to be manifested through the integration of positional information stemming from distinct signaling centers located at the junction of maxillary and mandibular BA1 (including the pharyngeal plate) with that from the most-distal BA1 midline and the lambdoidal junction where the maxillary BA1 meets the FNP. These centers thus generate a system of polarity and modularity along the axes of the developing craniofacial primordia (fig. 6c–6e).
Figure 6. .

Modeling the significance of Satb2 for coordination and elaboration of jaw development. a and b, Hemisected human (a) and canine (b) skulls showing the nature of jaws as hinged, appositional mechanisms of which polarity and potential modularity are characteristics. The translucent circle around the jaw articulation represents the hinge as a region, whereas the blue arrows highlight the polarity generated by the position of the hinge. The potential for modularity inherent in polarized structures is depicted by the relative purple shading of the jaws. c, Functional demands, such as for the matched occlusion of the homodont dentition of the jaws of the Ganges River dolphin (Platanista gangetica), dictate that jaw registration must be ontogenetically coordinated. The lower jaw is lavender to better differentiate the jaws. d and e, Diagrams of E10.5 murine embryos, highlighting the integration of some of the signaling centers believed to act in setting pattern, polarity, and modularity in the developing jaw primordia. The translucent circle represents the presumptive hinge region, whereas the blue arrows indicate positional information, such as Fgf expression, emanating from the first pharyngeal plate and the oral ectoderm at the junction of the maxillary (mxBA1) and mandibular (mdBA1) first arch in this region. This positional information is integrated with midline signaling sources coming from the distal midline mdBA1 and the lambdoidal junction (λ), where mxBA1 meets the medial (mFNP) and lateral (lFNP) frontonasal processes. The relative purple shading signifies the polarity and potential modularity of the primorida set up by this integration. The orange arrowhead indicates the mdBA1 midline, whereas the blue discs indicate the relative positions of Satb2 expression in the left-side upper- and lower-jaw primordia. Modified from Depew et al.4 f, Diagram integrating pattern, polarity, and modularity between embryonic and adult jaws. The disc represents the articulation of the upper and lower jaws, whereas the arrows represent the integrated signaling from the hinge region (blue) and distal ends (red). Polarity and modularity are marked by the toothed modules differentially shaded in purple. g–i, Three (of many) hypothetical scenarios for the modification of jaw modules through the differential regulation of Satb2 activity. Although it is assumed that the default state lies with the equivalence of each complementary module, the functional demands of any particular organism may require the modification of one or more complementary modules. This can occur by altering Satb2 activity at multiple levels, including both transcriptionally and posttranscriptionally. g, Scenario one: simultaneous down-regulation (red bars) of Satb2 activity in the blue modules of both the upper and lower jaws may result in either the diminution of growth of these modules (top arrow) or the loss of these modules (bottom arrow), a scenario essentially examined in our targeted loss of Satb2. h, Scenario two: specific down-regulation (red bars) of Satb2 activity in the upper-jaw blue module, its up-regulation (green arrow) in the lower jaw, or both may result in relative lengthening of the lower jaw, such as seen in the mastodon, Phiomia. Modified from Romer after Andrews.31 i, Scenario three: specific up-regulation (green arrow) of Satb2 activity in the upper-jaw blue module, its down-regulation (red bars) in the lower jaw, or both may result in relative lengthening of the upper jaw, such as seen in the thecodont, Chasmatosaurus. Modified from Romer.32
We have identified Satb2 as a key regulator of jaw and palate development, where it controls cell survival in the developmental modules in which it is expressed. With respect to the polarity and potential modularity of the jaws, Satb2 expression demarcates complementary domains that are within the early-developing upper and lower jaw primordia distad to the future jaw articulation but that are significantly excluded from the actual midlines (figs. 1 and 6d). These domains are strategically placed to act in the coordination of jaw registration by coordinating the relative survival, within symmetrical modules, of cell populations of fundamental importance to the elaboration, registration, and coordination of the upper and lower jaws. Because jaw development is Satb2-dosage sensitive, the role of regulators of Satb2 expression and posttranslational modification reaches paramount importance both ontogenetically and evolutionarily (fig. 6f and 6g). Modification of the Satb2/SATB2 regulatory cascade leading to changes in cell survival can conceivably play significant yet underappreciated and possibly undetected roles in jaw and palate development. We suggest that Satb2 acts as an integral component of complementary developmental domains that will potentially be under their own selective pressures.
Acknowledgments
This study was supported in part by the Royal Society (to M.J.D.), by the Max Planck Society, by DFG through the DFG Research Center for Molecular Physiology of the Brain, and by Volkswagenstiftung grant 78577 (to V.T.). O.B. was a recipient of a Humboldt Fellowship, and I.M. was supported by the Medical Research Council.
Web Resource
The URL for data presented herein is as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for CPI, MSX1, PAS9, TBX1, SATB2, Alx4, Dlx5, Pitx1, Prx2, Barx1, dHand, and Hoxa2)
References
- 1.Depew MJ, Tucker AS, Sharpe PT (2002) Craniofacial development. In: Rossant J, Tam PPL (eds) Mouse development: patterning, morphogenesis, and organogenesis. Academic Press, San Diego, pp 421–498 [Google Scholar]
- 2.Santagati F, Rijli FM (2003) Cranial neural crest and the building of the vertebrate head. Nat Rev Neurosci 4:806–818 10.1038/nrn1221 [DOI] [PubMed] [Google Scholar]
- 3.Tucker A, Sharpe P (2004) The cutting-edge of mammalian development: how the embryo makes teeth. Nat Rev Genet 5:499–508 10.1038/nrg1380 [DOI] [PubMed] [Google Scholar]
- 4.Depew MJ, Lufkin T, Rubenstein JL (2002) Specification of jaw subdivisions by Dlx genes. Science 298:381–385 10.1126/science.1075703 [DOI] [PubMed] [Google Scholar]
- 5.Depew MJ, Simpson CA (2006) 21st Century neontology and the comparative development of the vertebrate skull. Dev Dyn 235:1256–1291 10.1002/dvdy.20796 [DOI] [PubMed] [Google Scholar]
- 6.Gorlin RJ, Cohen MM Jr, Hennekan RM (2001) Syndromes of the head and neck. Oxford University Press, New York [Google Scholar]
- 7.Thorogood P (1997) Embryos, genes and birth defects. Wiley, New York [Google Scholar]
- 8.Rice DP (2005) Craniofacial anomalies: from development to molecular pathogenesis. Curr Mol Med 5:699–722 10.2174/156652405774641043 [DOI] [PubMed] [Google Scholar]
- 9.Marazita ML, Murray JC, Lidral AC, Arcos-Burgos M, Cooper ME, Goldstein T, Maher BS, et al (2004) Meta-analysis of 13 genome scans reveals multiple cleft lip/palate genes with novel loci on 9q21 and 2q32-35. Am J Hum Genet 75:161–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.FitzPatrick DR, Carr IM, McLaren L, Leek JP, Wightman P, Williamson K, Gautier P, McGill N, Hayward C, Firth H, Markham AF, Fantes JA, Bonthron DT (2003) Identification of SATB2 as the cleft palate gene on 2q32-q33. Hum Mol Genet 12:2491–2501 10.1093/hmg/ddg248 [DOI] [PubMed] [Google Scholar]
- 11.Murray JC, Schutte BC (2004) Cleft palate: players, pathways, and pursuits. J Clin Invest 113:1676–1678 10.1172/JCI200422154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jugessur A, Murray JC (2005) Orofacial clefting: recent insights into a complex trait. Curr Opin Genet Dev 15:270–278 10.1016/j.gde.2005.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brewer C, Holloway S, Zawalnyski P, Schinzel A, FitzPatrick D (1998) A chromosomal deletion map of human malformations. Am J Hum Genet 63:1153–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brewer CM, Leek JP, Green AJ, Holloway S, Bonthron DT, Markham AF, FitzPatrick DR (1999) A locus for isolated cleft palate, located on human chromosome 2q32. Am J Hum Genet 65:387–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Buggenhout G, Van Ravenswaaij-Arts C, Mc Maas N, Thoelen R, Vogels A, Smeets D, Salden I, Matthijs G, Fryns JP, Vermeesch JR (2005) The del(2)(q32.2q33) deletion syndrome defined by clinical and molecular characterization of four patients. Eur J Med Genet 48:276–289 10.1016/j.ejmg.2005.05.005 [DOI] [PubMed] [Google Scholar]
- 16.van den Boogaard MJ, Dorland M, Beemer FA, van Amstel HK (2000) MSX1 mutation is associated with orofacial clefting and tooth agenesis in humans. Nat Genet 24:342–343 10.1038/74155 [DOI] [PubMed] [Google Scholar]
- 17.Jezewski PA, Vieira AR, Nishimura C, Ludwig B, Johnson M, O’Brien SE, Daack-Hirsch S, Schultz RE, Weber A, Nepomucena B, Romitti PA, Christensen K, Orioli IM, Castilla EE, Machida J, Natsume N, Murray JC (2003) Complete sequencing shows a role for MSX1 in non-syndromic cleft lip and palate. J Med Genet 40:399–407 10.1136/jmg.40.6.399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Satokata I, Maas R (1994) Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat Genet 6:348–356 10.1038/ng0494-348 [DOI] [PubMed] [Google Scholar]
- 19.Schuffenhauer S, Leifheit HJ, Lichtner P, Peters H, Murken J, Emmerich P (1999) De novo deletion (14)(q11.2q13) including PAX9: clinical and molecular findings. J Med Genet 36:233–236 [PMC free article] [PubMed] [Google Scholar]
- 20.Peters H, Neubuser A, Kratochwil K, Balling R (1998) Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev 12:2735–2747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, Kamatani N, Momma K, Takao A, Nakazawa M, Shimizu N, Matsuoka R (2003) Role of TBX1 in human del22q11.2 syndrome. Lancet 362:1366–1373 10.1016/S0140-6736(03)14632-6 [DOI] [PubMed] [Google Scholar]
- 22.Jerome LA, Papaioannou VE (2001) DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet 27:286–291 10.1038/85845 [DOI] [PubMed] [Google Scholar]
- 23.Britanova O, Akopov S, Lukyanov S, Gruss P, Tarabykin V (2005) Novel transcription factor Satb2 interacts with matrix attachment region DNA elements in a tissue-specific manner and demonstrates cell-type-dependent expression in the developing mouse CNS. Eur J Neurosci 21:658–668 10.1111/j.1460-9568.2005.03897.x [DOI] [PubMed] [Google Scholar]
- 24.Dobreva G, Dambacher J, Grosschedl R (2003) SUMO modification of a novel MAR-binding protein, SATB2, modulates immunoglobulin μ gene expression. Genes Dev 17:3048–3061 10.1101/gad.1153003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vieira AR, Avila JR, Daack-Hirsch S, Dragan E, Felix TM, Rahimov F, Harrington J, Schultz RR, Watanabe Y, Johnson M, Fang J, O’Brien SE, Orioli IM, Castilla EE, Fitzpatrick DR, Jiang R, Marazita ML, Murray JC (2005) Medical sequencing of candidate genes for nonsyndromic cleft lip and palate. PLoS Genet 1:e64 10.1371/journal.pgen.0010064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park JW, Cai J, McIntosh I, Jabs EW, Fallin MD, Ingersoll R, Hetmanski JB, Vekemans M, Attie-Bitach T, Michael L, Scott AF, Beaty TH (2006) High throughput SNP and expression analyses of candidate genes for non-syndromic oral clefts. J Med Genet 43:598–608 10.1136/jmg.2005.040162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beverdam A, Brouwer A, Reijnen M, Korving J, Meijlink F (2001) Severe nasal clefting and abnormal embryonic apoptosis in Alx3/Alx4 double mutant mice. Development 128:3975–3986 [DOI] [PubMed] [Google Scholar]
- 28.Dobreva G, Chahrour M, Dautzenberg M, Chirivella L, Kanzler B, Farinas I, Karsenty G, Grosschedl R (2006) SATB2 is a multifunctional determinant of craniofacial patterning and osteoblast differentiation. Cell 125:971–986 10.1016/j.cell.2006.05.012 [DOI] [PubMed] [Google Scholar]
- 29.Cohen MM Jr (1999) Robin sequences and complexes: causal heterogeneity and pathogenetic/phenotypic variability. Am J Med Genet 84:311–315 [DOI] [PubMed] [Google Scholar]
- 30.Jakobsen LP, Knudsen MA, Lespinasse J, Garcia Ayuso C, Ramos C, Fryns J-P, Bugge M, Tommerup N (2006) The genetic basis of Pierre Robin sequence. Cleft Palate Craniofac J 43:155–159 10.1597/05-008.1 [DOI] [PubMed] [Google Scholar]
- 31.Romer AS (1947) Man and the vertebrates. University of Chicago Press, Chicago [Google Scholar]
- 32.Romer AS (1956) Osteology of the reptiles. University of Chicago Press, Chicago [Google Scholar]