

Abstract
Acute necrotizing encephalopathy (ANE) is a rapidly progressive encephalopathy that can occur in otherwise healthy children after common viral infections such as influenza and parainfluenza. Most ANE is sporadic and nonrecurrent (isolated ANE). However, we identified a 7 Mb interval containing a susceptibility locus (ANE1) in a family segregating recurrent ANE as an incompletely penetrant, autosomal-dominant trait. We now report that all affected individuals and obligate carriers in this family are heterozygous for a missense mutation (c.1880C→T, p.Thr585Met) in the gene encoding the nuclear pore protein Ran Binding Protein 2 (RANBP2). To determine whether this mutation is the susceptibility allele, we screened controls and other patients with ANE who are unrelated to the index family. Patients from 9 of 15 additional kindreds with familial or recurrent ANE had the identical mutation. It arose de novo in two families and independently in several other families. Two other patients with familial ANE had different RANBP2 missense mutations that altered conserved residues. None of the three RANBP2 missense mutations were found in 19 patients with isolated ANE or in unaffected controls. We conclude that missense mutations in RANBP2 are susceptibility alleles for familial and recurrent cases of ANE.
Introduction
With the proliferation of genetic tools, both hosts and pathogens have been studied for genomic changes that account for variation in disease susceptibility and severity.1,2 Disorders in which hosts respond abnormally to common illnesses provide an additional paradigm for understanding environmentally triggered diseases. In one such host-mediated disorder, acute necrotizing encephalopathy (ANE), previously healthy children develop encephalopathy, seizures, and coma within days after the onset of a common childhood febrile infection, such as influenza A or parainfluenza,3 without evidence of viral infection of the brain or inflammatory cell infiltration.4 Brain T2-weighted MRI reveals characteristic symmetric lesions present in the thalami, pons, and brainstem. Initially reported in Japan and Taiwan, ANE affects children worldwide.5 The fact that different infections can produce the same phenotype suggests that the disorder is mediated by host factors, although some pathogens may be more adept at triggering the host response.2 Increased serum and CSF levels of IL-6 and TNF-α have been reported in children with ANE,6,7 leading to the prevailing hypothesis of a “cytokine storm” as part of the pathogenic mechanism.4 Morbidity and mortality are high.4
We reported a family in which the predisposition to developing ANE segregates as an autosomal-dominant trait, previously abbreviated as ADANE (HGNC: ANE1, OMIM 608033).8 In this family, 16 family members manifested clinical symptoms of ANE and diagnosis was confirmed by pathologic examination in two affected children who had died from their illnesses. Penetrance was estimated at 40% and recurrent episodes occurred in half of the affected individuals. We mapped the ANE1 locus to a 7 million basepair region on chromosome 2q.9 We now report that heterozygous missense mutations in the gene RANBP2, which encodes the nuclear pore component Ran Binding Protein 2, account for genetic susceptibility to ANE in this family and in many other patients with familial or recurrent ANE.
Methods and Materials
Study Subjects
Individuals from 35 unrelated families participated in this study. Nineteen individuals of European (7), Asian (10), or African descent (2) had isolated ANE. The remaining individuals came from 16 families of European descent affected by familial or recurrent ANE. The diagnosis of ANE was based upon the following three criteria: (1) a rapidly progressive encephalopathy beginning 1 to 3 days after the onset of a febrile illness; (2) MRI imaging demonstrating multifocal symmetric lesions of the brain specifically involving the thalamus, but with lesions of the midbrain, pons, and brainstem viewed as supportive; and (3) clinical and laboratory exclusion of meningitis, encephalitis, inborn errors of metabolism, and toxin exposure. Familial or recurrent forms of ANE required at least one of these additional criteria: (1) a relative with confirmed ANE, (2) a relative with a history of unexplained CNS dysfunction occurring in the context of concurrent febrile illness or recent infection, or (3) recurrence of ANE in the proband.
We obtained informed consent, approved by the University Hospitals of Cleveland Institutional Review Board, from all participants. Blood was collected for DNA extraction and for the creation of EBV-transformed lymphoblasts by standard methods.
Ethnically matched controls were taken from 200 control DNA samples, from healthy individuals with self-reported European ancestry, and from 184 European neurologically normal controls (NINDS control panels NDPT009 and NDPT021, Coriell Repositories, Camden, NJ). An additional 1000 controls were taken from the CEPH genome diversity panel.
1297 subjects with multiple sclerosis (MS [OMIM 126200]) and 549 controls were enrolled under a study protocol approved by the Partners Healthcare Institutional Review Board of each institution. Subjects with MS all meet McDonald criteria for MS.10 A summary of the clinical composition of the collection of subjects is given in Table S1 available online.
High-Throughput Sequencing
Genomic DNA from four members of the original ADANE kindred9 was used for high-throughput sequencing; this included two affected individuals with the linked haplotype and two unaffected individuals without the linked haplotype. Exon sequences, including 300 bp of flanking intron, were extracted from the human genome assembly. PCR primers were designed flanking the exons of genes of interest via the program primer311 and purchased from Invitrogen (Carlsbad, CA) in 96-well plates. Sufficient flanking sequence was included to interrogate splice sites as well as coding exons. PCR was performed with 100 ng genomic DNA, 1 μM primer, and Taq Gold Polymerase kits from Applied Biosystems (Foster City, CA) in 10 μl total reaction volume in 384-well plates. Prior to sequencing (Applied Biosystems BigDye v1.1), PCR products were treated with Exonuclease I and shrimp alkaline phosphatase (USB Corporation, Cleveland, OH). Bidirectional sequence data, with the initial PCR primers, was obtained on an Applied Biosystems 3730xl DNA Analyzer. Sequences were assembled with phred and phrap and visualized with the consed software package.
Reverse Transcription PCR of RANBP2
Total RNA was extracted from EBV-transformed lymphoblasts with TRIzol (Invitrogen) according the manufacturer's protocol. First-strand synthesis was performed with Superscript III (Invitrogen) with a RANBP2-specific primer (5′-AGCAAATCTTTAACGAAAGAAC-3′) that is complementary to a unique sequence within the transcript's 3′UTR. The cDNA was then PCR amplified and sequenced (Table S2).
Amplification of Exons 12 and 14 from RANBP2 via Genomic DNA
Primers, PCR conditions, and internal sequencing primers were designed to specifically interrogate RANBP2 and not the related paralogs that are found in segmental duplications (Table S3).
Denaturing High-Performance Liquid Chromatography Screening for the RANBP2 c.1880C→T Variant via the Human Diversity Panel
Members of the original ANE kindred who were either homozygous wild-type or heterozygous for the c.1880C→T variant were used to define the following parameters on the Transgenomic WAVE D7000 (Transgenomic, Omaha, NE) that could identify c.1880C→T heterozgyotes via 5 μl of the exon 12 amplimer (temperature 57°C, initial B% = 55, final B% = 61, gradient 2% B/minute). DNA aliquots from the human genome diversity panel were provided by Jeffrey Murray and Hatem el-Shanti.
Defining Haplotypes that Contain the c.1880C→T Mutation
PCR primers were designed to amplify and sequence contiguous intragenic genomic DNA fragments upstream and downstream of c.1880C→T mutation in exon 12 of RANBP2 to find additional informative SNPs (Table S3). Phase of the SNPs relative to c.1880C→T was determined by evaluating probands, siblings, and parents.
Families that were not informative for intragenic SNPs were analyzed for previously described microsatellite markers (UCSC genome browser) that map from 0.115 to 3.2 Mb from RANBP2. HEX- or FAM-labeled PCR primers in a standard PCR reaction were analyzed on an ABI prism 3700, with the GenoTyper software. Primer sequences for markers of the D2S- designation are publicly available (UCSC genome broswer). Primer sequences for the STR88 marker are (5′HEX-CAGTGGTTGACCTCCTGACC-3′) and (5′-CCAGGATGAAGCAGAGGAA-3′).
Genotype Screening in a Multiple Sclerosis Population
RANBP2 was genotyped for one susceptibility allele, the T or C at c.1880, via the hME Sequenom MassARRAY platform (Sequenom, San Diego, CA). These data generated met standards for high-quality data (>95% genotype call rate, HWE p value > 0.001) in healthy control subjects. The RANBP2 assay consisted of two extension primers: 5′-AGCGGATAACTTTTAGACCTGGAAACGTAGCA-3′ and 5′-AGCGGATAACAAGGCTGACCATCTACACCTC-3′ for an amplicon length of 522 bp. The detection primer had the following sequence: 5′-CAGAATGCCTTCAGAAAA-3′. The PCR parameters were as follows: 5 min at 95°C, 39 cycles of 1 min at 94°C, 50 s at 63°C, and 50 s at 72°C, followed by 10 min at 72°C, then hold at 10°C.
Results
Thirty-five unrelated patients or families affected by ANE participated in this study. Sixteen probands had familial or recurrent ANE (Figure 1) and 19 had isolated ANE.
Figure 1.

Pedigrees of 16 Families with Familial or Recurrent ANE
Pedigrees with mutations in RANBP2 are given in (A) whereas those without mutation are shown in (B). Family numbers are given at top. Symbol notations for (A) as follows: filled, mutation carrier, affected with ANE; filled with white lower right quandrant, mutation carrier, recurrent ANE; vertical bar, mutation carrier; horizontal bar, encephalopathy, unconfirmed as ANE; underlined symbol, DNA studied; circled symbol, molecularly proven de novo mutation carrier. For simplicity, not all siblings and spouses are shown in pedigree 586. Symbol notations for (B) are equivalent to (A) except without mutation.
At the initiation of this study, only one family with a clear genetic susceptibility to ANE had been identified. DNA from this family had been used to define a 7 Mb candidate interval.9 Exons of all known genes, predicted genes, and spliced ESTs within the interval were sequenced with DNA from two affected and two unaffected family members. No disease-causing mutation was identified. However, 117 out of 363 exons failed sequencing because they are part of large segmental duplications. These segmental duplications (Figure 2A) contain sequence derived from RANBP2 and a trans-Golgi component, Grip coiled-coil domain-containing protein 2 (GCC2). RANBP2 and GCC2 underwent a partial duplication and genomic rearrangement to form a novel gene encoding “RANBP2-like and GRIP Domain-containing protein” (RGPD). RGPD subsequently underwent further genomic duplication in the human and chimpanzee progenitor to produce a total of eight RGPDs (RGPD1-8).12,13 The strong conservation of genomic organization (Figure 2B) and sequence between RANBP2 and RGPDs 1-8 initially limited the ability to reliably interrogate these genes with genomic DNA.
Figure 2.
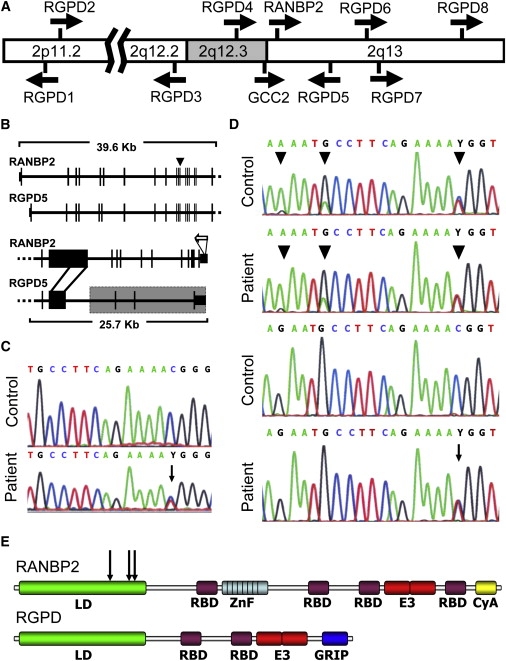
Mutation Identification in RANBP2
(A) The locations and orientations of RANBP2, GCC2, and the RGPDs are shown on a partial ideogram of chromosome 2.
(B) A comparison of the genomic regions of RANBP2 and RGPD5 indicating strong structural and sequence conservation for their exons 1-18 (upper lines) and the divergence of their 3′ regions (lower lines). RGPD5 is deleted for a portion of exon 20 and has its final three exons derived from GCC2 (shaded box). This enables RANBP2-specific cDNA to be synthesized with a primer (hollow arrow) in the gene's 3′UTR. Also shown is the location of the c.1880C→T mutation in exon 12 of RANBP2 (arrowhead).
(C) Sequencing electropherograms of RANBP2-specific cDNA reveals a c.1880C→T transition in the patient but not the control.
(D) Intronic primers that amplify exon 12 of RANBP2 and the RGPD paralogs (upper two electropherograms) demonstrate multiple paralog-derived signal alterations (arrowheads) in the patient and the control. In contrast, sequence specific to exon 12 of RANBP2 (lower electropherograms) shows the absence of paralog-derived signals in the patient and control, but persistence of the c.1880C→T mutation (arrow) in the patient.
(E) Schematic diagram of RANBP2 protein indicating the “leucine-rich” domain (LD), four Ran binding domains (RBD), eight zinc finger repeats (ZnF), a cyclophilin A domain (CyA), and a SUMO E3 ligase domain (E3). In the partial deletion of the 20th exon, the RGPDs lose the ZnF and second RBD domain. The derived exons from GCC2 provide a new C-terminal GRIP domain which targets the RGPDs to the Golgi. The sites of the p.Thr585Met, p.Thr653Ile, and p.Ile656Val LD domain substitutions associated with ANE1 are indicated by arrows.
To sequence RANBP2, we therefore used gene-specific primers and performed RT-PCR with mRNA derived from patient lymphoblasts (Figure 2B). This led us to identify a heterozygous C→T transition at position 1880 of the cDNA (c.1880C→T), which is predicted to cause an amino acid substitution (p.Thr585Met) (Figures 2C and 2D). All affected family members and obligate carriers are heterozygous for this mutation. We did not observe the mutation in 384 control DNAs from individuals with self-reported European ancestry or in 1000 DNAs from the human genome diversity panel.
We then screened RANBP2 in 15 additional families suspected of having familial or recurrent ANE and found 9 with the same heterozygous c.1880C→T mutation. In 2 of these 9 families (pedigrees 100 and 121, Figure 1), the mutation arose de novo. Six other families did not share a common linked haplotype (Figure 3), indicating additional independent origins for this mutation. Two families (pedigrees 671 and 613, Figure 1) did not have the c.1880C→T mutation, but were heterozygous for other missense mutations in RANBP2 (either c.2085C→T; p.Thr653Ile or c.2094A→G; p.Ile656Val). These mutations also altered highly conserved residues and were not observed in 200 controls. Finding de novo and independently arising missense mutations in affected families, and not in controls, clearly implicates RANBP2 as a susceptibility allele for familial or recurrent ANE.
Figure 3.

Comparison of Mutation-Bearing Haplotypes in Seven Families with c.1880C→T Mutation
The haplotypes of seven families show differences in intragenic and extragenic polymorphic markers, suggesting independent origins for each family of the linked c.1880C→T mutation. Alleles are shown in their contiguous arrangement and represented as STR PCR fragment size (e.g., D2S2229), nucleotide allele (e.g., rs826559), or repeat count (ss76880141). Marker distances are given relative to the c.1880C→T mutation site (0 Kb), with orientation along the plus strand of the chromosome. RANBP2 intragenic markers are delineated between horizontal lines at −4.1 Kb and +25.8 Kb. To highlight the potential for, or lack of, ancestral relationship, matching alleles are given the same unique color. When noninformative, both alleles are shown (e.g., family 102, D2S293).
RANBP2 mutations were not found in 4 of the 15 additional families. Linkage to RANBP2 could be excluded by haplotype analysis in one family (108, Figure 1). In another, parental consanguinity, multiple affected children, and lack of family history suggests recessive inheritance (611, Figure 1). Although RANBP2 missense mutations are not the sole susceptibility alleles for familial or recurrent ANE, they accounted for 75% (12 of 16) of the familial or recurrent cases in this series. In light of the potential locus heterogeneity, we propose using the term “ANE1” when disease is associated with a RANBP2 missense mutation.
We did not find RANBP2 mutations in the 19 patients who had isolated ANE, indicating that ANE1 is etiologically distinct. We initially relied on family history or disease recurrence to differentiate familial from isolated ANE. Having found a mutation, we could determine whether patients with ANE1 had other features that may distinguish them from patients with isolated ANE. As with isolated ANE, infection was the antecedent of neurologic deterioration in ANE1. Specific infectious agents implicated in 10 of 28 ANE1 episodes include influenza A (7 episodes), influenza B (1 episode), parainfluenza II (1 episode), and Mycoplasma pneumoniae (1 episode). Similar to isolated ANE, patients with ANE1 had a high incidence of seizure (59%), coma (100%), and CSF protein elevation (85%). Unlike patients with isolated ANE, 82% of whom have been reported to have elevations of serum transaminases,3 no elevation was observed in patients with ANE1. Also unusual for isolated ANE, which affects young children, three patients with ANE1 had disease onset during adolescence or adulthood (ages 12, 14, and 37 years).
Our inclusion criteria for ANE required MRI showing symmetric alterations of the thalami, with lesions of the brainstem and pons viewed as supportive for the diagnosis (Figure 4). Although each ANE1 patient demonstrated this pattern at least once, recurrent episodes revealed variability in presentation, with the thalami being affected in 16 of 17 initial and 7 of 9 recurrent episodes (17 patients). The pons was affected frequently (23 of 26 total episodes) and lesions of the medulla were less common (8 of 26 episodes). Patients with ANE1 also had involvement of additional CNS structures, including the external capsule and claustrum (19 of 26 episodes), medial temporal lobes and limbic structures including amygdalae, hippocampi, or medial temporal lobes (19 of 26), and spinal cord (3 of 26 episodes). These additional sites have not been previously reported in patients with isolated ANE.
Figure 4.

Brain MRI Findings in a Child (690, II:1) with Familial and Recurrent Acute Necrotizing Encephalopathy
T2-weighted images obtained during the acute encephalopathy (top) are shown in comparison to resolved images obtained 6 months later (middle) and images from a recurrent episode 3 years later (bottom). During acute events, abnormal signal is visible in the pons (white arrow), midbrain (thin white arrow), amygdala (black arrow), thalamus (black arrowhead), and external capsule (white arrowhead).
Two families in our study reported a positive family history for demyelinating disease suggestive of multiple sclerosis, of whom one was a carrier of the c.1880C→T mutation. However, the carrier of the c.1880C→T mutation had a brain MRI that was more suggestive for ANE (G.D., unpublished data). This indicated that some adults with manifestations of ANE1, especially if recurrent, could be misdiagnosed with MS. We tested a well-characterized cohort of 1297 MS patients and 547 accompanying controls for the c.1880C→T mutation to determine whether rare patients diagnosed with MS might have actually had adult-onset ANE1. We did not find the c.1880C→T mutation in any MS cases or controls. Thus, according to stringent diagnostic criteria for MS,14 c.1880C→T does not contribute to this disease. It remains to be determined whether genetic risk factors that predispose to MS alter the penetrance of ANE1.
Discussion
We have identified a missense mutation in RANBP2 as a genetic risk factor for an environmentally triggered neurologic disease, ANE1. The c.1880C→T allele occurred de novo in two patients and arose independently in several families. Two other missense alleles, which were found in single families, require further study to demonstrate their role in ANE1 susceptibility. RANBP2 is not the sole susceptibility gene for ANE, as indicated by the fact that we did not find mutations in 4 of 16 kindreds with familial or recurrent disease or in patients who had isolated ANE. Nevertheless, the unexpected finding that a missense mutation in a component of the nuclear pore predisposes individuals to infection-triggered neurologic disease opens new avenues of research.
RANBP2 is a 358 kD protein located on the cytoplasmic surface of the nuclear pore.15 It has numerous roles throughout the cell cycle. During interphase, its nuclear envelope-associated functions include facilitation of protein import and export and sumoylation of protein cargoes.16 However, in certain cell types including neurons, it is detected associated with microtubules17 and/or mitochondria,18,19 suggesting roles in intracellular trafficking or energy maintenance. During mitosis, RANBP2 is important for nuclear envelope breakdown20 and it localizes to the kinetochore,21 where it facilitates sister chromatid resolution.22
We do not know whether the RANBP2 mutations predispose to disease by disrupting one of the protein's many endogenous functions or by creating a novel biologic activity. Support for the latter possibility derives from a comparison between the RANBP2 protein sequence, which is part of the nuclear pore complex, and the eight RGPD proteins, which localize to the trans-Golgi apparatus.13 The mutated amino acid residues p.Thr585, p.Thr653, and p.Ile656 are conserved in all mammalian RANBP2 proteins. In contrast, the p.Thr585Met amino acid substitution is present as a stable variant in four of the eight RGPDs, which have undergone further positive selection for sequence divergence.13 These trans-Golgi localized proteins likely have altered biologic properties in their new context, and amino acid changes that confer selective advantage in the RGPDs may be deleterious to the original RANBP2. The fact that the p.Thr585Met RANBP2 substitution occurred de novo twice and was found in two thirds of familial or recurrent ANE cases, and not in 3230 controls or non-ANE subjects, supports this notion.
Mutations in RANBP2 predispose to ANE1, but by themselves are insufficient to make the phenotype fully penetrant. Therefore, additional genetic and environmental factors are required. Infection precedes episodes of neurologic deterioration. However, neither the infectious agent nor the individual's genetic background fully explains the incomplete penetrance or episodic nature of this disease. For example, the neurologic responses during the same viral illness were often discordant in a pair of identical twins (pedigree 586) with the c.1880C→T mutation. Environmental factors such as differences in viral inoculum, route of viral exposure (oral versus intranasal),23 and nutritional state could play a role. If the amino acid substitutions result in temperature sensitivity, resulting in protein misfolding and alteration of function, variations in the severity and duration of fever could contribute to the reduced penetrance. Alternatively, the somatic recombinations and mutations that create the diversity present in immunoglobulins and T cell receptors may also provide a source of variation for the initiation of ANE1.24
At present, the complex roles of RANBP2 make it difficult to speculate on the mechanism of the mutational effect. The three mutations identified cluster in the “leucine-rich domain” (LD) that has been described as a domain necessary for localization to the both the nucleus and microtubules.17 However, the p.Thr585Met mutation occurs outside of the subregion of the LD associated with nuclear association,17 suggesting that the mutational effect could impact its interaction with microtubules. The lesions of necrotizing encephalopathy suggested the involvement of energy metabolism, because it involves the same constellation of pathological findings described in Wernicke encephalopathy (associated with thiamine deficiency; [OMIM 277730]) and Leigh syndrome (which can be due to various defects of mitochondrial ATP production; [OMIM 256000]).3 We previously reported a coupling defect of oxidative phosphorylation in one of the ANE1 cases.8 RANBP2 has recently been shown to contribute to the subcellular distribution of mitochondria through its kinesin-binding domain.18 Mice haploinsufficient for RanBP2 have reduced amounts of brain hexokinase19 and altered lipid metabolism.25 Age-related defects in free fatty acid uptake lead to impaired weight gain and resistance of retinal cells to apoptosis.25 Thus, intracellular trafficking of mitochondria, energy production, and lipid peroxidation are potential targets for RANBP2 mutational effects. However, other processes that could be affected by RANBP2 mutations include viral entry, antigen presentation, cytokine signaling, immune responsiveness, and blood-brain barrier maintenance. Studies at the cell-biologic and whole-organism levels will be required to delineate the effects of RANBP2 mutations.
Our study was not designed to prospectively ascertain all patients who have been diagnosed with ANE. Therefore, we cannot yet estimate the frequency of ANE1 among unselected individuals with ANE. Clinical features in ANE1 substantially overlap those of isolated ANE, although lesions occurring outside of the brainstem and thalamus and normal serum transaminase levels may help clinically differentiate the two. At present we suggest that ANE1 be considered in the differential diagnosis of any patient with findings consistent with ANE and (1) family history of neurologic symptoms occurring in the context of infection, (2) recurrent disease, or (3) MRI changes of ANE that affect the external capsule, claustrum, limbic structures, or temporal lobes. In order to prevent or lessen the neurologic consequences of ANE1, it remains important to determine how missense mutations in RANBP2 predispose to CNS dysfunction.
Supplemental Data
Supplemental Data include three tables and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Phred, phrap, and consed, http://www.phrap.org
Primer3, http://frodo.wi.mit.edu/
UCSC Genome Browser, http://genome.ucsc.edu
Acknowledgments
The authors thank the research subjects and families for their participation. We thank Masaya Kubota, David Michelson, Nancy Leslie, Lazlo Sztriha, Timothy Chin, Muge Calikoglu, Yoshihiro Maegaki, Yusuke Goto, Tomohide Goto, Vivian Wong, Dragana Josifova, Peter van Hasselt, and Hitoshi Osaka for patient referrals. We thank Jeffrey Murray and Hatem el-Shanti for their generous assistance in testing control samples. This work was supported by NIH grant NINDS K08NS050331, by a Clinician Scientist Award in Translational Research to M.L.W. from the Burroughs Wellcome Fund, and by the Howard Hughes Medical Institute. The authors report no conflicts of interest.
References
- 1.Liu R., Paxton W.A., Choe S., Ceradini D., Martin S.R., Horuk R., MacDonald M.E., Stuhlmann H., Koup R.A., Landau N.R. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86:367–377. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- 2.Mori S.I., Nagashima M., Sasaki Y., Mori K., Tabei Y., Yoshida Y., Yamazaki K., Hirata I., Sekine H., Ito T. A novel amino acid substitution at the receptor-binding site on the hemagglutinin of H3N2 influenza A viruses isolated from 6 cases with acute encephalopathy during the 1997–1998 season in Tokyo. Arch. Virol. 1999;144:147–155. doi: 10.1007/s007050050491. [DOI] [PubMed] [Google Scholar]
- 3.Mizuguchi M. Acute necrotizing encephalopathy of childhood: a novel form of acute encephalopathy prevalent in Japan and Taiwan. Brain Dev. 1997;19:81–92. doi: 10.1016/s0387-7604(96)00063-0. [DOI] [PubMed] [Google Scholar]
- 4.Mizuguchi M., Yamanouchi H., Ichiyama T., Shiomi M. Acute encephalopathy associated with influenza and other viral infections. Acta Neurol. Scand. 2007;115:45–56. doi: 10.1111/j.1600-0404.2007.00809.x. [DOI] [PubMed] [Google Scholar]
- 5.Mastroyianni S.D., Gionnis D., Voudris K., Skardoutsou A., Mizuguchi M. Acute necrotizing encephalopathy of childhood in non-Asian patients: report of three cases and literature review. J. Child Neurol. 2006;21:872–879. doi: 10.1177/08830738060210101401. [DOI] [PubMed] [Google Scholar]
- 6.Ichiyama T., Endo S., Kaneko M., Isumi H., Matsubara T., Furukawa S. Serum cytokine concentrations of influenza-associated acute necrotizing encephalopathy. Pediatr. Int. 2003;45:734–736. doi: 10.1111/j.1442-200x.2003.01822.x. [DOI] [PubMed] [Google Scholar]
- 7.Ichiyama T., Isumi H., Ozawa H., Matsubara T., Morishima T., Furukawa S. Cerebrospinal fluid and serum levels of cytokines and soluble tumor necrosis factor receptor in influenza virus-associated encephalopathy. Scand. J. Infect. Dis. 2003;35:59–61. doi: 10.1080/0036554021000026986. [DOI] [PubMed] [Google Scholar]
- 8.Neilson D.E., Eiben R.M., Waniewski S., Hoppel C.L., Varnes M.E., Bangert B.A., Wiznitzer M., Warman M.L., Kerr D.S. Autosomal dominant acute necrotizing encephalopathy. Neurology. 2003;61:226–230. doi: 10.1212/01.wnl.0000073544.28775.1a. [DOI] [PubMed] [Google Scholar]
- 9.Neilson D.E., Feiler H.S., Wilhelmsen K.C., Lynn A., Eiben R.M., Kerr D.S., Warman M.L. Autosomal dominant acute necrotizing encephalopathy maps to 2q12.1–2q13. Ann. Neurol. 2004;55:291–294. doi: 10.1002/ana.10849. [DOI] [PubMed] [Google Scholar]
- 10.McDonald W.I., Compston A., Edan G., Goodkin D., Hartung H.P., Lublin F.D., McFarland H.F., Paty D.W., Polman C.H., Reingold S.C. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann. Neurol. 2001;50:121–127. doi: 10.1002/ana.1032. [DOI] [PubMed] [Google Scholar]
- 11.Rozen S., Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 12.Hillier L.W., Graves T.A., Fulton R.S., Fulton L.A., Pepin K.H., Minx P., Wagner-McPherson C., Layman D., Wylie K., Sekhon M. Generation and annotation of the DNA sequences of human chromosomes 2 and 4. Nature. 2005;434:724–731. doi: 10.1038/nature03466. [DOI] [PubMed] [Google Scholar]
- 13.Ciccarelli F.D., von Mering C., Suyama M., Harrington E.D., Izaurralde E., Bork P. Complex genomic rearrangements lead to novel primate gene function. Genome Res. 2005;15:343–351. doi: 10.1101/gr.3266405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Polman C.H., Reingold S.C., Edan G., Filippi M., Hartung H.P., Kappos L., Lublin F.D., Metz L.M., McFarland H.F., O'Connor P.W. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann. Neurol. 2005;58:840–846. doi: 10.1002/ana.20703. [DOI] [PubMed] [Google Scholar]
- 15.Yokoyama N., Hayashi N., Seki T., Pante N., Ohba T., Nishii K., Kuma K., Hayashida T., Miyata T., Aebi U. A giant nucleopore protein that binds Ran/TC4. Nature. 1995;376:184–188. doi: 10.1038/376184a0. [DOI] [PubMed] [Google Scholar]
- 16.Pichler A., Gast A., Seeler J.S., Dejean A., Melchior F. The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell. 2002;108:109–120. doi: 10.1016/s0092-8674(01)00633-x. [DOI] [PubMed] [Google Scholar]
- 17.Joseph J., Dasso M. The nucleoporin Nup358 associates with and regulates interphase microtubules. FEBS Lett. 2008;582:190–196. doi: 10.1016/j.febslet.2007.11.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cho K.I., Cai Y., Yi H., Yeh A., Aslanukov A., Ferreira P.A. Association of the kinesin-binding domain of RanBP2 to KIF5B and KIF5C determines mitochondria localization and function. Traffic. 2007;8:1722–1735. doi: 10.1111/j.1600-0854.2007.00647.x. [DOI] [PubMed] [Google Scholar]
- 19.Aslanukov A., Bhowmick R., Guruju M., Oswald J., Raz D., Bush R.A., Sieving P.A., Lu X., Bock C.B., Ferreira P.A. RanBP2 modulates Cox11 and hexokinase I activities and haploinsufficiency of RanBP2 causes deficits in glucose metabolism. PLoS Genet. 2006;2:e177. doi: 10.1371/journal.pgen.0020177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prunuske A.J., Liu J., Elgort S., Joseph J., Dasso M., Ullman K.S. Nuclear envelope breakdown is coordinated by both Nup358/RanBP2 and Nup153, two nucleoporins with zinc finger modules. Mol. Biol. Cell. 2006;17:760–769. doi: 10.1091/mbc.E05-06-0485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joseph J., Liu S.T., Jablonski S.A., Yen T.J., Dasso M. The RanGAP1-RanBP2 complex is essential for microtubule-kinetochore interactions in vivo. Curr. Biol. 2004;14:611–617. doi: 10.1016/j.cub.2004.03.031. [DOI] [PubMed] [Google Scholar]
- 22.Dawlaty M.M., Malureanu L., Jeganathan K.B., Kao E., Sustmann C., Tahk S., Shuai K., Grosschedl R., van Deursen J.M. Resolution of sister centromeres requires RanBP2-mediated SUMOylation of topoisomerase IIalpha. Cell. 2008;133:103–115. doi: 10.1016/j.cell.2008.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mori I., Kimura Y. Neuropathogenesis of influenza virus infection in mice. Microbes Infect. 2001;3:475–479. doi: 10.1016/s1286-4579(01)01403-4. [DOI] [PubMed] [Google Scholar]
- 24.Schwartz R.S. Shattuck lecture: diversity of the immune repertoire and immunoregulation. N. Engl. J. Med. 2003;348:1017–1026. doi: 10.1056/NEJMsa022766. [DOI] [PubMed] [Google Scholar]
- 25.Cho K.I., Yi H., Yeh A., Tserentsoodol N., Cuadrado L., Searle K., Hao Y., Ferreira P.A. Haploinsufficiency of RanBP2 is neuroprotective against light-elicited and age-dependent degeneration of photoreceptor neurons. Cell Death Differ. 2008 doi: 10.1038/cdd.2008.153. in press. Published online October 24, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.