

Abstract
The goal of this study was to apply gastrointestinal simulation technology and integration of physiological parameters to predict biopharmaceutical drug classification. GastroPlus® was used with experimentally determined physicochemical and pharmacokinetic drug properties to simulate the absorption of several weak acid and weak base BCS class II compounds. Simulation of oral drug absorption given physicochemical drug properties and physicochemical parameters will aid justification of biowaivers for selected BCS class II compounds.
Keywords: absorption, bioavailability, bioequivalence, biowaiver, permeability, solubility
INTRODUCTION
A biopharmaceutical drug classification system based on two fundamental parameters, solubility and permeability, originally proposed by Amidon et al. classifies drugs into four different groups: I (high solubility, high permeability), II (low solubility, high permeability), III (high solubility, low permeability) and IV (low solubility, low permeability). The classification of drug solubility is based on the dimensionless dose number D0, defined as the ratio of drug concentration in the administered volume to its saturation solubility in water. The classification of drug permeability is based on a relationship between the intestinal permeability of a drug and its fraction dose absorbed (1). BCS has several goals: identification of rate-limiting steps for biopharmaceutic problem compounds; recommendation of a class of immediate-release solid oral dosage forms for which bioequivalence may be assessed based on in vitro dissolution tests (biowaiver) and thus identification of expendable clinical bioequivalence studies. According to the FDA biowaiver guidance, a waiver for in vivo BE studies can be requested for solid, orally administered immediate-release drug products (>85% release in 30 min) containing highly soluble drugs over the pH range from 1 to 7.5 (dose to solubility ratio <250 ml) which are also highly permeable (fraction absorbed ≥90%) (2). Additionally, the following criteria must be met: the drug must be stable in the gastrointestinal tract, excipients do not affect the rate or extent of absorption, the drug must not have a narrow therapeutic index and the drug product is designed not to be absorbed in the oral cavity. Thus, for BCS class I drug substances, an in vivo bioequivalence study could be replaced by generating suitable in vitro dissolution data. Table I provides a comparison of eligibility for BCS based biowaivers between FDA, CPMP and WHO (2–4).
Table I.
| FDA 2000 | CPMP NFG 2001 | WHO 2005 | |
|---|---|---|---|
| High solubility | pH 1 to 7.5 | pH 1 to 6.8 | pH 1.2 to 6.8 |
| Highest tablet strength | Highest tablet strength | Highest dose | |
| High permeability | fa ≥ 90% | Linear/complete absorption | fa ≥ 85% |
| Dissolution | ≥85% in 30 s | ≥85% in 30 s | ≥85% in 30 s |
| pH 1; 4.5; 6.8 | pH 1; 4.5; 6.8 | pH 1; 4.5; 6.8 | |
| 50 rpm (paddle) | Equipment not specified | 75 rpm (paddle) | |
| 100 rpm (basket) | 100 rpm (basket) |
The WHO modification has widened the eligibility for class I biowaivers of several basic drugs previously considered to be BCS class II members. In the WHO working document additional relaxations for drug products containing BCS class II or class III active pharmaceutical ingredients eligible for biowaivers are presented (4): in vivo BE studies could be waived for very rapidly dissolving products containing BCS class III compounds (≥85% in 15 min in standard media at pH 1.2, 4.5 and 6.8).
Rinaki et al. identified biowaivers among acidic drugs BCS class II (5). Biowaivers could be extended for rapidly dissolving products (≥85% of the labelled amount dissolving within 30 min using USP apparatus I or II in a volume of ≤900 ml of buffer solution) containing BCS class II compounds with weak acidic properties showing high solubility at pH 6.8 but low solubility at pH 4.5 or 1.2. In addition to the modifications proposed by WHO other solubility boundary values for class I drugs have been suggested to include class II compounds with acidic and basic structural elements (6,7). Figure 1 presents BCS class II compounds clustered according to the chemical classes. Furthermore, an intermediate solubility class has been suggested, given the propensity of many acids and bases to be highly soluble at either pH 1.2 or 6.8. Based on relaxation of solubility criteria some bases (e.g. verapamil), categorized as class II compounds due to insufficient solubility at pH 7.5, would be grouped as class I (8). These drugs will show fast and reliable dissolution at physiologically relevant pH values between 4.5 and 6.8 the pH range of the small intestine and will be consistently and completely absorbed.
Fig. 1.

Clustering of BCS Class II Compounds According to Acid/Base Properties
The dissolution profiles of different formulations should be similar as prerequisite for bioequivalence. For weakly acidic and basic class II drugs the pH and composition of the dissolution are the most important parameters. Thus, further extension opportunities for class II drugs to be incorporated in the class I are based on the choice of the dissolution medium simulating a physiologically relevant in vivo situation (9). According to pharmacokinetic simulations performed by Kortejarvi et al., solubility in the stomach, formulation type and gastric emptying were critical factors for Cmax differences of BCS class II drugs (10).
The aim of this study was to use gastrointestinal simulation technology as a tool to recommend extensions of biowaivers for selected BCS class II weak acids and bases. Furthermore, simulations were performed to study how differences in dissolution rates would affect fraction dose absorbed and bioavailability and other PK properties of BCS class II drugs and whether in spite of the difference in dissolution the resulting PK profiles would still be considered bioequivalent, thus justifying a waiver of BE studies also for BCS class II compounds.
MATERIALS AND METHODS
Computer Hardware and Software
GastroPlus® program (version 5.2; Simulations Plus, Inc., CA, USA) was run on a Dell Pentium IV computer. The program enables predictions of rate and extent of drug absorption from the gastrointestinal tract. It also allows input of different dissolution velocities for pharmacokinetic predictions.
Design of the Simulations
The concept of using simulation technology for biowaiver extensions was initially validated to meet BE criteria for BCS class I drugs in IR dosage forms: propranolol hydrochloride (11), metoprolol tartrate (12) and prednisolone (11) were used as model BCS class I compounds. In a second series, simulations with IR dosage forms containing BCS class II compounds were performed.
Input Parameters for Pharmacokinetic Simulations
Characterization of the drugs
Simulations were performed with experimentally determined physicochemical and biopharmaceutic parameters (pKa, log P, and Caco-2 or human jejunal permeability, and solubility). Permeability values for all compounds, except for mefenamic acid (13), ibuprofen (14) and prednisolone (15) (based on Caco-2 cells model), diclofenac, miconazole and terbinafine (16) (estimated using ADMET Predictor®, Simulations Plus, Inc., CA, USA) were based on experimental data (17,18). When no information was available concerning a certain input parameter, default settings proposed by the program were used.
Dosage form
Relevant dosage form properties, used in simulations were: mean precipitation time 900 s, drug particle density 1.2 g/ml and effective particle radius 25 μm. Diffusion coefficient was calculated from the molecular weight of the drug molecule. It was assumed that the dosing volume is 250 ml.
Nondimensional biopharmaceutical compound characterization numbers (1)
Dose number (D0) is defined as the ratio of the dose to the amount of drug that will dissolve in 250 ml of test solution at the lowest solubility within the pH range 1 to 8 and was calculated as:
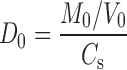 |
1 |
M0—highest dose unit, V0—250 ml, Cs—solubility
Absorption number (An), defined as the ratio of the transit time to the absorption time (1/absorption rate constant), was calculated as:
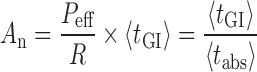 |
2 |
Peff—effective permeability, R—radius of GI, tGI—residence time in GI, tabs—time required for complete absorption
Dissolution number (Dn) is the ratio of the transit time to the dissolution time (1/dissolution rate constant) and was calculated as:
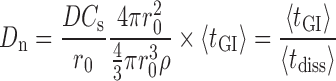 |
3 |
D—diffusivity, Cs—solubility, r0—particle radius, ρ—density, tGI—residence time in GI, tdiss—time required for complete dissolution
Gastrointestinal absorption model
Yu et al. originally developed a Compartmental Absorption and Transit (CAT) model that predicted absorption from the gastrointestinal tract and took into account the flow of the drug substance through the digestive tract, which was divided into a set of seven compartments. The model was further redefined and developed into an Advanced Compartment Absorption and Transit model (ACAT), which accounts for dissolution rate, pH dependence of drug solubility, release rate, absorption in the stomach, the small intestine or the colon, metabolism or degradation in the gut or the liver, or changes in surface area, transporter densities (e.g., efflux protein densities), and other regional factors within the intestinal tract (19). The form of the ACAT model implemented in GastroPlus® is modeled by a system of coupled linear and non-linear rate equations. The equations include the consideration of six states (unreleased, undissolved, dissolved, degraded, metabolized, and absorbed), 18 compartments (stomach, six compartments for the small intestine, two colon and nine enterocyte compartments), three states of excreted material (unreleased, undissolved, and dissolved), and the concentration of drug in physiologically based organ compartments, when tissue partition and flow rate parameters are available.
Human fasted state was the selected setting for all simulations.
Release profiles from immediate-release dosage forms
The dissolution rate is influenced by the physicochemical properties of the substance and the prevailing physiological conditions in the GI tract. The dissolution rate constant (Kd) is given by:
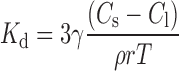 |
4 |
Where γ is the diffusion coefficient, Cs is the solubility at a particular pH, Cl is the lumen concentration, ρ is the density of the substance, r is the particle radius and T is the diffusion layer thickness. The diffusion layer thickness was set equal to the particle radius (25 μm).
Figure 2 summarizes the theoretical dissolution profiles designed for the IR formulations with BCS class I and II compounds used in simulations. RAPID IR was designed to release >90% of the dose within 10 min whereas SLOW IR released 80% in 45 min. For each RAPID and SLOW immediate release formulation, plasma concentrations versus time and absorption curves were predicted.
Fig. 2.

In Silico Dissolution Profiles of Two Immediate Release Drug Products Used as Input Data for Simulations
Disposition parameters for drugs
Values for clearance (CL), volume of central compartment (Vc), transfer rate constants, fraction not bound to proteins (fu), first-pass metabolism used in simulations were taken from the literature. Input body weight for the predictions was 70 kg.
Parameter sensitivity analysis
A parameter sensitivity analysis for particle radius, solubility and precipitation time was performed for BCS class II drug miconazole. Each parameter was varied independently, with all others held constant.
RESULTS AND DISCUSSION
Validation of the Simulation Approach by Using BCS Class I Drugs
Physicochemical properties of propranolol, metoprolol, and prednisolone, used in the simulations, are presented in Table II.
Table II.
Compound Parameters for BCS Class I Compounds Used in the Simulations
| Compound properties | Propranolol | Metoprolol | Prednisolone |
|---|---|---|---|
| Molecular weight (g/mol) | 259.3 | 263.4 | 360.4 |
| Log D | 1.54 (pH 7.4) | −1.72 (pH 4.0) | 2.51 (pH 7.0) |
| pK a | Base 9.3 | Base 9.4 | Acid 12.5 |
| Solubility (mg/ml) | 125 at pH 3.0 | 171 at pH 6.5 | 0.23 at pH 7.0 |
| Human P eff (×10−4 cm/s) | 2.91 | 1.34 | 4.4 |
| Dose (mg) | 40 | 100 | 4, 10, 20 |
| D 0 | 0.0088 | 0.0041 | 0.3478a |
| A n | 5.783 | 2.663 | 8.81a |
| D n | 4.924 × 103 | 6.257 × 103 | 7.398a |
aDose, absorption and dissolution number for prednisolone 20 mg IR tablet
Propranolol is rapidly and completely absorbed from the proximal small intestine. Its bioavailability is highly variable in humans (20–70%), due to high hepatic extraction (54%). The plasma half-life is short-varying from 1.5 to 4 h. It is highly lipophilic and highly bound to plasma proteins (∼90%), with a distribution volume of about 4.2 l/kg and clearance of 1.1 l/h/kg (20–22).
According to the solubility-pH profile of propranolol, solubility will not be a rate-limiting step in its absorption from the GI tract.
Pharmacokinetic profiles of propranolol are not sensitive to the differences in dissolution. Analysis of 14 bioequivalence trials of propranolol IR products (40 and 80 mg) in Germany showed that indeed 11 formulations were bioequivalent with respect to Cmax and AUC. Three formulations were bioinequivalent (two studies failed due to Cmax differences and one study failed due to AUC differences) (23). According to Potthast et al. exemption from in vivo bioequivalence studies is thus feasible for propranolol IR products if the requirement of the WHO Guideline is fulfilled (24). The high pharmacokinetic variability due to considerable first-pass extraction is thus attributed to the drug and not to its dosage form justifying biowaivers based on dissolution similarity of propranolol products.
Metoprolol is also highly soluble in the pH range of the GI tract; it is rapidly and almost completely absorbed from the small intestine, with peak plasma concentrations occurring 1.5 h after oral administration in humans. Due to its high first-pass metabolism (57%) oral systemic availability is as low as 40% to 50%. The degree of binding to plasma proteins is 12%. The total clearance is 30.6 l/h and the volume of distribution is 0.62 l/kg (20,25).
Two metoprolol IR formulations, showing different dissolution profiles as shown in Fig. 2, were simulated and compared. It was found that the poorer in vitro dissolution properties of the SLOW form did not lead to relevant changes in metoprolol pharmacokinetics. Thus, bioequivalence can be assumed due to the excellent solubility and permeability properties of metoprolol. Eight bioequivalence studies on 100 mg metoprolol IR formulations performed in Germany confirmed the results of the simulations, since all drug products were bioequivalent as assessed by AUC and Cmax comparison (23).
The third example from BCS class I comprised prednisolone. Its physicochemical properties are depicted in Table II. Prednisolone is readily absorbed in the GI tract, producing peak concentrations at 1 to 2 h after administration. The oral bioavailability of prednisolone is 70% to 80% in healthy volunteers. First-pass metabolism approximates 13% and plasma half-life is between 2.1 and 3.5 h. Volume of distribution and clearance are dose dependent, most probably due to the concentration-dependent binding to plasma proteins. The following values for volume of distribution and clearance were used in the simulations: 0.2 l/kg and 6.7 l/h for 5 mg prednisolone IR tablets; 0.2 l/kg and 3.66 l/h for 10 mg tablets; 0.3 l/kg and 4.9 l/h for 20 mg tablets, respectively (20,26,27). Although prednisolone has nonlinear pharmacokinetics, the drug is classified as BCS class I. It was reported from human studies, that any nonlinear pharmacokinetics is not relevant for the biowaiver decisions over the full range of tablet strengths (15,28). Exemptions from a bioequivalence study have been granted for example for prednisolone tablets based on comparable dissolution profiles between products at three pH values (1.2; 4.5; 6.8) and permeability information. Four bioequivalence studies of prednisolone immediate-release products 5–20 mg, performed in Germany, indicated the bioequivalence of the prednisolone test preparations as compared to the reference (23,29). This result was also anticipated from the simulations. With respect to Cmax and AUC, prednisolone IR tablets with different dissolution profiles did not show any relevant bioavailability and absorption differences.
A comparison of the PK parameters of the three BCS class I drugs obtained by the simulation and those reported from studies in humans are detailed in Table III. Almost identical values for AUC, Cmax and tmax were predicted in comparison with in vivo data validating the simulations approach. Thus, comparative simulations of pharmacokinetics using as input different in vitro dissolution properties of BCS class I drug formulations served as a retrospective justification for biowaivers, implying that dissolution tests may well be used to predict bioequivalence for BCS class I drugs.
Table III.
Simulation Results for Immediate-release Formulations Containing Drugs from BCS Class I
| Dosage form | Propranolol IR tablets (40 mg) | Metoprolol IR tablets (100 mg) | Prednisolone IR tablets | |||||
|---|---|---|---|---|---|---|---|---|
| Rapid | Slow | Rapid | Slow | 5 mg | 10 mg | 20 mg | ||
| Rapid | Slow | |||||||
| Fraction absorbed (%) predicted | 98.8 | 98.7 | 84.7 | 84.3 | 99.7 | 99.6 | 99.7 | 99.8 |
| C max (μg/ml) predicted | 0.031 | 0.032 | 0.153 | 0.153 | 0.15 | 0.37 | 0.61 | 0.64 |
| AUC(0–24 h) (ng h/ml) predicted | 226.8 | 226.3 | 1166.4 | 1159.1 | 651.4 | 2,356.8 | 3,565.9 | 3,567.0 |
| Fraction absorbed (%) observed | >90% | Rapid and complete | Rapid and complete (near 100%) | |||||
| C max (μg/ml) observed | 0.028 ± 0.073 | 0.164 ± 0.057 | 0.144 ± 0.01 | 0.391 ± 0.043 | 0.585 ± 0.11 | n.a. | ||
| AUC(0–48 h) (ng h/ml) observed | 227.8 ± 59.0 | 820.9 ± 70.0 | 713.6 ± 21.0 | 2,354.0 ± 388.0 | 3,278.0 ± 939.0 | n.a. | ||
For comparison, fraction absorbed, C max and AUC (means ± SD) observed in vivo are added from literature (23).
Selection of Candidates for Biowaiver Extension for BCS Class II Using Simulations
For class II drugs, limits are imposed on the absorption by the solubility (D0) or dissolution rate (Dn), either in general or on a regional basis within the GI tract, leading to incomplete absorption, despite the high membrane permeability (An).
Compounds with weak acidic properties
Nonsteroidal anti-inflammatory drug with acetic acid moieties (e.g. diclofenac) and with propionic acid moieties (e.g. ibuprofen, ketoprofen, naproxen) are known for their pH-dependent solubility which increases significantly at the physiological pH range of the intestine thus ensuring complete dissolution at the intestinal pH yet not at typical pH values found in the fasting stomach (13). In the simulations the release characteristics were varied among drug formulations and the fraction absorbed of the API were compared between formulations.
Case study # 1: ibuprofen
Input data for ibuprofen physicochemical properties are shown in Table IV.
Table IV.
Compound Parameters for BCS Class II Weak Acidic Compounds Used in the Simulations
| Compound properties | Ibuprofen | Ketoprofen | Diclofenac | Mefenamic acid | Piroxicam | |
|---|---|---|---|---|---|---|
| Molecular weight (g/mol) | 206.3 | 254.3 | 295.1 | 241.3 | 331.35 | |
| Log P or Log D | 1.2 (pH 7.0) | 0.1 (pH 7.4) | 1.4 (pH 6.8) | 5.12 | 0.72 (pH 6.6) | |
| pK a | 4.6 | 4.39 | 4.2 | 4.2 | 1.86; 5.46 | |
| Solubility (mg/ml) | 3.44 (pH 7.4) | 0.205 (pH 2.0) | 0.001 (pH 1.2) | 0.0002 (pH 1.2) | 0.11 (pH 6.6) | |
| Human P eff (×10−4 cm/s) | 8 | 8.7 | 3.07 | 3.94 | 10.4 | |
| Dose (mg) | 400 | 50, 75 | 50 | 500 | 20 | |
| D 0 | Stomach pH | 42.11 | 0.98 (50 mg) | 2.0 × 102 | 3,330 | 2.31 |
| 1.47 (75 mg) | ||||||
| Intestinal pH | <1a | <1b | <1c | >1d | >1e | |
| A n | 16.02 | 17.29 | 8.91 | 7.89 | 20.67 | |
| D n | Stomach pH | 149.9 | 9.075 | 0.04 | 0.105 | 1.06 |
| Intestinal pH | >1 | >1 | >1 | <1 | <1 | |
aIntestinal pH 6.8; solubility 3.37 mg/ml
bIt was assumed that intestinal D 0 value is <1 due to low stomach D 0
cIntestinal pH 5.0; solubility 2.8 mg/ml
dIntestinal pH 6.8; solubility 0.009 mg/ml
eIntestinal pH 6.6; solubility 0.11 mg/ml
Ibuprofen has limited solubility at acidic pH, but sufficient solubility at neutral pH. Previous studies have shown that the drug is rapidly absorbed from the small intestine with fraction absorbed exceeding 90%, producing peak serum levels in 1 to 2 h with a half-life of about 2 h. Like many other NSAIDs, ibuprofen is highly bound to plasma proteins (>99%) (14,30).
The different in vitro dissolution profiles of ibuprofen IR products, used as input data, are presented in Fig. 3.
Fig. 3.

Dissolution of Ibuprofen from Three IR Products (Two Commercial and One Artificial Out of Specification (OOS) Dissolution Batch), Each Containing 400 mg Ibuprofen. Dissolution Conditions were Phosphate Buffer ph 7.2, Paddle Method, 50 rpm
The corresponding plasma concentrations versus time profiles are depicted in Fig. 4. Whereas the two commercial products were predicted to be bioequivalent (Cmax ratio 1.002; AUC ratio 1.0) the outlier (OOS) did produce differences outside of the BE acceptance criteria (Cmax ratio 1.4; AUC ratio 1.007). Comparison of the simulated PK results is displayed in Table V.
Fig. 4.

Simulated Plasma Concentrations Following Administration of Three Ibuprofen IR Products (400 mg)
Table V.
Simulation Results (Fraction Absorbed, C max, t max and AUC) for Immediate-release Formulations Rapid and Slow Containing Weak Acidic Drugs from BCS Class II
| Dosage form | Ibuprofen IR tablets (400 mg) | Ketoprofen IR tablets (50 mg) | Diclofenac IR tablets (50 mg) | Piroxicam IR tablets (20 mg) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AKTREN SPEZIAL | Gynofug 400 | OOSa | In vivo data (mean ± CV) (23) | Rapid | Slow | Rapid | Slow | Rapid | Slow | |
| Fraction absorbed (%) predicted | 99.9 | 99.9 | 99.3 | >99% | 98.4 | 98.4 | 99.4 | 99.3 | 99.6 | 99.6 |
| C max (μg/ml) predicted | 26.24 | 26.18 | 18.87 | 23.8 ± 21% | 2.23 | 2.22 | 0.57 | 0.56 | 1.75 | 1.73 |
| AUC(0–24 h) (ng h/ml) predicted | 1.14 × 105 | 1.14 × 105 | 1.13 × 105 | 1.02 × 105 ± 17% | 9,755.5 | 9,753.3 | 1,104.1 | 1,103.6 | 3.66 × 104 | 3.65 × 104 |
a OOS Out-of-specification
Similarly, analysis of 25 bioequivalence trials of ibuprofen immediate-release tablets from 200 to 600 mg in Germany showed that 14 studies did not demonstrate bioequivalence, due to Cmax differences by only 4.3% deviation from CI limit on average (23). The extent of exposure as determined by AUC was equivalent.
High ibuprofen intestinal absorption (>99%) implies that ibuprofen dissolves completely in the intestine. Bioinequivalent products are likely to differ in rate (Cmax) rather than extent (AUC) of drug input. Simulations predicted these changes of Cmax for products with insufficient dissolution rates and showed that the behaviour of ibuprofen can be estimated based on physicochemical properties in conjunction with dissolution profiles.
Generic ibuprofen products have been approved by the MPA (Sweden) based on comparative dissolution data without request of an in vivo bioequivalence study (31). It has further been pointed out that for ibuprofen drug products therapeutic risks in case of bioinequivalence can be considered as being irrelevant (risk analysis).
Ibuprofen, which has been categorized as class II compound at the high dose (800 mg), will meet the criteria of high solubility at the low dose (200 mg), based on its pH 7.4 solubility value. BfArM classified ibuprofen as BCS class I, since the drug has a wide therapeutic index and non critical therapeutic use (32–35).
Case study # 2: ketoprofen
Ketoprofen is a potent nonsteroidal anti-inflammatory drug (NSAID) of the 2-arylpropionic acid class. Ketoprofen would be grouped into BCS Class II due to its low solubility and high permeability characteristics. Table IV summarizes physicochemical characteristics of ketoprofen. Ketoprofen solubility increases dramatically at pH values representative for the small intestine.
Absorption of ketoprofen is rapid and almost complete when given orally (≥95%). It binds extensively to plasma albumin (99%), has a short half-life (2–4 h), has negligible first-pass effect, and a broad therapeutic window. The volume of distribution varies from 7–14 l (0.15 l/kg) and the clearance is 5.04 l/h (36–38).
Fraction dose absorbed of ketoprofen that results from simulations with IR formulations, which differed in dissolution profiles are very similar. For ketoprofen, such differences in dissolution profiles should not lead to pharmacokinetic differences based on comparison of BE parameter point estimates (Table V).
Ketoprofen would have minimal solubility/dissolution in stomach, yet it is completely ionized and highly soluble at intestinal pH, leading to 100% dissolution of the dose. Similar to the simulations and animal experiments demonstrate high dissolution of ketoprofen in the intestine going along with complete oral absorption in humans (39,40). Ketoprofen is thus a candidate for BCS class II biowaiver.
Case study # 3: diclofenac
Physicochemical properties for the phenylacetic acid derivative diclofenac are given in Table IV. After oral administration diclofenac is rapidly absorbed (>90%) with extensive hepatic metabolism (40%). The peak plasma concentration was achieved after approximately 1 h. The compound exhibits a volume of distribution of 0.12 l/kg, clearance of 27 l/h and 99.5% plasma protein binding (20).
The dissolution behaviour of the two hypothetical diclofenac formulations resulted in similar predicted pharmacokinetic parameters as can be seen in Table V.
Despite high Dose number (>1) and low Dissolution number (<1) at stomach pH diclofenac solubility and dissolution in the small intestine are satisfactory and result in complete absorption. The pharmacokinetic results predicted by the modelling approach are in accordance with data from in vivo human studies (23,41).
From the 23 comparative bioequivalence trials with 25 and 50 mg diclofenac products in Germany it is known that only five test products failed BE criteria in terms of Cmax rather than AUC. Several trials showed that Cmax differences were caused by the occurrence of double peaks which may be related to physiological events and first-pass metabolism (13). Furthermore, differences in the kinetics of release of the formulations, effect of excipients on the precipitation of the neutral form in the stomach or diclofenac salt selection may have contributed as well. This is pointed out by O'Connor and Corrigan where they show that diclofenac salt selection has also an influence on solubility and intrinsic dissolution rate (42).
Weak acid diclofenac can be defined as “highly soluble” compound at pH > 6.8, but also satisfies solubility requirements at lower intestinal pH values (intestinal D0 < 1). Considering its high permeability it can be concluded that diclofenac exhibits absorption similar to that of a BCS class I compound (43).
Case study # 4: mefenamic acid
The physicochemical properties of the anthranilic acid mefenamic acid, along with the recommended dose and absorption in humans are presented in Table IV.
Mefenamic acid is rapidly but incompletely absorbed after oral administration. The bound fraction of mefenamic acid to albumin exceeds 90% (20,44–46). Due to insufficient solubility in the physiological pH range the dissolution of mefenamic acid drug product is highly formulation dependent and will be incomplete in physiological dissolution media. Thus a fundamental principle for the conversion of a BCS class II drug to obtain BCS class I biowaiver—complete dissolution and absorption in the GI tract—is violated. Simulation results clearly demonstrate that small differences in mefenamic acid formulations may affect systemic availability. Predicted influence of particle size on absorption and plasma-concentration versus time profiles of mefenamic acid products were compared to in vivo human data published by Hummel et al. This study of influence of particle size on dissolution and PK parameters of mefenamic acid tablets confirmed in silico predictions in that micronization resulted in increase of absorption and bioavailability (47).
The simulation also predicted solubility- and dissolution-limited absorption of mefenamic acid in the small intestine. The lipophilic drug has low solubility in acidic and neutral media. Its solubility increases only far from physiologic pH values. In order to establish a sufficient solubility of the API for the dissolution test of mefenamic acid drug products, unphysiological dissolution media with pH values of 8 were utilized. By comparison of in vitro release profiles and in vivo bioequivalence parameters it was found that in vitro dissolution was not predictive for the biopharmaceutic behaviour of the dosage form (48). Thus one fundamental principle of granting a biowaiver for mefenamic acid, the establishment of a predictive dissolution method, is violated. A second violation is to be seen in the fact, that for mefenamic acid the dose will not be dissolved completely within the gastrointestinal fluid.
Case study # 5: piroxicam
The physicochemical parameters of piroxicam are given in Table IV. Solubility of piroxicam largely depends on the pH value of the medium. Under acidic conditions piroxicam shows low solubility and dissolution rate.
Piroxicam exhibits excellent absorption (fa ∼ 100%) and negligible first-pass metabolism. Its systemic clearance is 0.147 l/h and the volume of distribution approaches 0.15 l/kg. The drug is extensively bound to plasma proteins (99%) (20,49).
Although piroxicam shows low aqueous solubility at acidic pH, the pharmacokinetic data show that its absorption is not impaired, even at the highest dose of 20 mg. Thus, in spite of their apparent dissolution differences, RAPID and SLOW IR tablets resulted in similar pharmacokinetic parameters (Table V).
Differences in dissolution of piroxicam formulations have been observed resulting from variations in particle size (50). A parameter sensitivity analysis conducted for particle size, predicts that large variation in particle size e.g. from 2.5 to >100 μm would be necessary to affect bioavailability by 20% or more.
Validation of the computer predictions was performed again by noting the probability of failed bioequivalence trials respectively. Piroxicam product BE trials published in Germany demonstrated bioequivalence for all products (23,51). It was furthermore noted that dissolution tests were able to discriminate between formulations that contained different piroxicam polymorphic forms, yet the differences did not result in significant BE differences, in accordance with simulation results. According to Yazdanian et al. piroxicam met the “high solubility” criteria in all tested pH conditions (1.2–7.4) and would be classified as class I drug (13). Predicted high bioavailability suggested that piroxicam absorption kinetics is not limited by drug solubility and release in the intestinal pH, thus piroxicam could be a candidate for a BCS class II biowaiver.
Compounds with weak basic properties
Weak bases with low pH-dependent water solubility and high intestinal permeability may exhibit distinct absorption profiles based on different physiologic conditions. They usually dissolve during their residence time in the stomach, due to low gastric pH. Once in solution bases from BCS class II readily permeate membranes easily resulting in ≥90% absorption. World Health Organization redefined the solubility boundary values for the highest single dose administered to <250 ml of buffer solution over a pH range of 1.2 to 6.8. This modification has widened the eligibility of several basic drugs previously considered to be class II members for class I biowaiver.
Case study # 1: verapamil
Table VI encloses the physicochemical properties of verapamil. After administration of IR verapamil tablets, the drug will be dissolved and absorbed in the upper part of the GI tract (pH < 6.8) such that the low solubility at high pH should not be limiting verapamil absorption.
Table VI.
Compound Parameters for BCS Class II Weak Bases Used in the Simulations
| Compound properties | Verapamil | Miconazole | Terbinafine | |
|---|---|---|---|---|
| Molecular weight (g/mol) | 455 | 416.14 | 291.44 | |
| Log P or Log D | 4.47 | 4.5 (pH 6.8) | 5.5 (pH 6.8) | |
| pK a | 8.6 | 6.7 | 7.82 | |
| Solubility (mg/ml) | 0.44 (pH 7.32) | 0.0029 (pH 6.8) | 0.0009 (pH 8.05) | |
| Human P eff (×10−4 cm/s) | 6.7 | 12.0 | 12.0 | |
| Dose (mg) | 120 | 250 | 250 | |
| D 0 | Stomach pH | <1a | 8.33b | 0.37c |
| Intestinal pH | 2.85a | 2.78 × 102b | 1.18 × 103c | |
| A n | 13.41 | 24.03 | 24.03 | |
| D n | 40.9 | 0.089 | 0.21 | |
aStomach pH 2.32; solubility 82 mg/ml; intestinal pH 7.32; solubility 0.44 mg/ml
bStomach pH 1.0; solubility 0.12 mg/ml; intestinal pH 6.8; solubility 0.003 mg/ml
cStomach pH 2.0; solubility 2.7 mg/ml; intestinal pH 8.05; solubility 0.0009 mg/ml
Verapamil undergoes extensive first-pass elimination and is commonly regarded as highly variable drug. Following oral absorption more than 90% of labelled 14C-verapamil is absorbed. The protein bound fraction of verapamil is moderate (90%) (20,52).
Simulations show that absorption of verapamil from an IR dosage form is complete in the upper small intestine; therefore low solubility in the distal GI regions has little impact on rate and extent of its absorption. Simulated cumulative absorption versus time profiles for rapid and slow dissolving formulations of verapamil demonstrated similar contour curves. Comparison of the simulated pharmacokinetic parameters of the two verapamil IR products RAPID and SLOW is displayed in Table VII.
Table VII.
Simulation Results (Fraction Absorbed, C max, t max and AUC) for Immediate-release Formulations Rapid and Slow Containing Weak Bases from BCS Class II
| Dosage form | Verapamil IR tablets (120 mg) | Terbinafine IR tablets (250 mg) | |||
|---|---|---|---|---|---|
| Rapid | Slow | Rapid | Slow | In vivo data (68) | |
| Fraction absorbed (%) predicted | 99.97 | 99.96 | 99.68 | 99.62 | – |
| C max (μg/ml) predicted | 0.088 | 0.088 | 0.69 | 0.67 | 0.92 ± 0.43 |
| AUC(0–24 h) (ng h/ml) predicted | 416.7 | 416.65 | 2,959.5 | 2,951.9 | 4,598 ± 1,811a |
a
Analysis of 30 bioequivalence trials with 40 to 120 mg verapamil IR tablets in Germany showed that 20 studies did not demonstrate bioequivalence: seven failed due to AUC, seven failed due to Cmax and six failed due to AUC and Cmax differences (23). Bioinequivalence of products with highly variable drugs is harder to be predicted from solubility, permeability and dissolution data alone. The observed differences in rate and extent of systemic drug input can be also related to high variability in verapamil pharmacokinetics, and not just due to formulation differences (53). Verapamil undergoes first-pass extraction (80%) by CYP 3A4 and 1A2 in the intestine and liver, therefore additional information on the variations in population physiology need to be integrated in the model to possibly predict bioinequivalence. A virtual trial was performed to estimate the effect of distributions of values for systemic clearance and first-pass effect. Stochastic simulations based on the means and the log normal distributions were used with the following coefficients of variations: systemic clearance = 40% and first-pass effect = 20%. Table VIII shows both the variability associated with the observed clinical data as well as the variability that would be expected from the simulations given our best assumptions for the coefficients of variations for two metabolism-related physiological parameters.
Table VIII.
Stochastic Virtual Trial Simulation with 120 mg Verapamil Immediate Release Tablet
| Rapid IR product | Slow IR product | In vivo data (53) | |
|---|---|---|---|
| Mean (CV) | Mean (CV) | Mean (CVrange) | |
| C max (ng/ml) | 123.6 (49.3%) | 127.6 (49.7%) | 121.9 (42–66%) |
| t max (h) | 1.61 | 1.90 | 1.67 |
| AUC (ng h/ml) | 581.9 (49.5%) | 597 (49.5%) | 445.6 (38–41%) |
Simulated results are presented as the mean (CV%) compared with observed data (53) presented as the mean (CV% and range %).
Verapamil hydrochloride is a BCS Class II substance on the borderline, classified in a provisional classification of the WHO Essential Drugs as BCS Class I drug, and can be considered as a candidate for granting a biowaiver when the IR tablets show rapid in vitro dissolution, and meet the dissolution profile comparison criteria.
Case study # 2: miconazole
Miconazole, a synthetic imidazole derivate, is suitable for the oral therapy of systemic candidiasis and other systemic fungal infections. This antifungal agent is also used for local treatment of vaginal, skin and nail infections. Physicochemical properties of miconazole are given in Table VI. Since miconazole is a weak base, it can be solubilised only under very acidic conditions.
Miconazole is poorly absorbed from the gastrointestinal tract (fa ∼ 20%). The pharmacokinetics of miconazole is characterized by a clearance of 70 l/h and volume of distribution of 20 l/kg. Between 91% and 93% of miconazole is bound to plasma proteins and the drug shows no first-pass metabolism (20,54).
Miconazole exhibits a high Dose number D0 and a low Dissolution number Dn, indicating the insufficient solubilisation capacity of the intestinal fluid for the entire dose of miconazole. Fraction absorbed can be improved by increasing drug solubility. It is shown that miconazole fraction absorbed depends also on the particle size, since Dn < 1 at particle size of 25 μm (Fig. 5).
Fig. 5.

Parameter Sensitivity Analyses for 250 mg Miconazole Peroral Administration. The y-Axis Is the Simulated Fraction Absorbed. The x-Axis Scale Shows the Direction and Magnitude of the Perturbation from the Initial Parameter Value
In Fig. 6 the predicted miconazole absorption and dissolution profiles are presented. From the unusual shape of the amount dissolved vs. time profile it can be concluded that a fraction of the dose goes into solution in the acidic environment of the stomach and there is a partial precipitation of the weak base in intestine. It might be expected due to the high permeability of the drug, that fraction absorbed will be higher. The simulation results however show that under neutral pH conditions in the small intestine the drug does not re-dissolve sufficiently. In the case of miconazole dose to solubility ratio is high and the oral absorption of this drug is limited by dissolution rate and by the saturated solubility. Parameter sensitivity analysis presented in Fig. 5 verifies that the drug precipitates in the intestine, but precipitation time has not strongly affected fraction absorbed.
Fig. 6.

Prediction of Dissolution and Absorption Profiles of the 250 mg Miconazole RAPID IR Product (Human Fasted Condition—Stomach pH 1.3 and Stomach pH 5.0)
Figure 6 shows the effect of gastric pH on the plasma concentration versus time profile of miconazole. By an increase of the stomach pH to 5 as compared to pH 1.3, the absorption of miconazole is predicted to be reduced from 29.4% to 23.8%. This may occur following meal intake, administration of gastric acid blockers or by patients with gastric hypoacidity or achlorhydria. Influence of gastric pH on absorption has been clearly illustrated in human clinical trials with weak bases e.g. ketoconazole, gefitinib, cinnarizine and dipyridamole (55–58). Biowaiver for miconazole products can thus not be recommended since the fraction absorbed is very low due to miconazole low solubility and the fact that dissolution of a highly dosed base should elevate the intragastric pH (57) resulting probably in the precipitation of the drug.
Case study # 3: terbinafine
Terbinafine is a topically and orally active synthetic antifungal agent of the allylamine class, which is indicated for the treatment of fungal infections of the skin and nails. The physicochemical parameters of terbinafine are summarized in Table VI. As weak base, it shows pH-dependent solubility with maximum values at acidic pH.
Terbinafine is rapidly absorbed, reaching peak concentrations approximately 1 to 2 h postdosing. It binds strongly to plasma proteins (99%). The volume of distribution of terbinafine in healthy volunteers is high and the drug is extensively distributed into tissues after oral doses (20,59,60).
Terbinafine absorption and release profile are depicted in Fig. 7. Weak bases such as ketoconazole precipitate rapidly from their acidic solution as the pH of the dissolution medium exceeds 5.5, pH values that are encountered in the upper small intestine (61). For terbinafine, likewise, a fraction of the dose dissolves and precipitates in the stomach and at the pH of the small intestine. High permeability of the drug and long intestinal transit time contribute that re-dissolved drug will be completely absorbed.
Fig. 7.

Prediction of Dissolution and Absorption Profiles of the 250 mg Terbinafine RAPID IR (Human Fasted Condition—Stomach pH 1.3 and Stomach pH 5.0)
Figure 7 shows that at gastric pH 5.0 a lower fraction of the dose is dissolved in the stomach leading to a reduction in the precipitation material. From the simulations it is apparent that the solubility of the drug in the stomach has impact on overall fraction absorbed since the rate and extent of absorption were reduced after gastric pH was elevated from 94.7% to 77.7% and affected strongly the pharmacokinetics of terbinafine (Cmax(pH 1.3)/Cmax(pH 5.0): 1.7; AUC(pH 1.3)/AUC(5.0): 1.265). The simulations are also suitable to demonstrate the influence of variation in gastric transit time on terbinafine dissolution/absorption. Figure 8 shows the predicted amounts of terbinafine as a function of time and physicochemical state (dissolved/undissolved) in the duodenum and jejunum at a stomach residence time of 0.25 versus 3 h. It can be seen that gastric transit time influenced the fraction of dose that dissolved in the stomach and remained undissolved in upper GIT, but variation of stomach transit time did not strongly affected terbinafine fraction absorbed (fa = 94.7% in case of 0.25 h stomach transit time versus 99.6% for 3 h stomach transit time). Variation of stomach transit time was reflected by Cmax differences (Cmax(0.25 h)/Cmax(2 h): 1.23), while at the same time practically no difference in AUC was observed (AUC(0.25 h)/AUC(2 h): 0.98).
Fig. 8.

Predicted Terbinafine Amount Remained Undissolved in Duodenum and in Upper Jejunum (Stomach Transit Time 0.25 Versus 3 h)
Further simulations aimed at predicting in vivo performance of terbinafine SLOW IR product (Fig. 9) in comparison to the RAPID IR product. Virtually no differences in fraction absorbed (99.68% versus 99.62% for RAPID and SLOW IR products respectively) among terbinafine tablets were predicted (Table VII).
Fig. 9.

Prediction of Absorption and Dissolution Profile of 250 mg Terbinafine SLOW IR Product at the Human Fasted Condition
Terbinafine might be a candidate for granting a biowaiver provided the existence of a discriminating and predictive in vitro dissolution test, since the dissolution of two IR products did not affect the plasma concentration of the drug and most of the administered powder could dissolve in the GI tract producing a high fraction absorbed. Quick absorption of slowly dissolved drug that slow arrived in intestine created “sink” conditions, so it was less like that precipitation of the base occurred.
Routine bioequivalence studies with 250 mg immediate release terbinafine tablets in Germany confirmed no significant differences in the rate and extent of absorption between the generic products and the test product (62–66). In one study, published by Koytchev et al., the bioequivalence acceptance range for Cmax value was expanded to 0.7–1.43 since the rate of absorption was considered as not relevant for terbinafine clinical efficacy (67).
CONCLUSIONS
In silico models are useful to identify BCS class II biowaiver candidate drugs. The risk of bioinequivalence in terms of Cmax has shown to be higher than for AUC. Class II weak acids and bases in immediate release dosage forms may be eligible for biowaivers provided that the dose dissolves completely before reaching middle jejunum. Biowaivers for some class II drugs also necessitate the availability of a discriminative and in vivo predictive in vitro dissolution method. Thus they should be complemented by prospective in vitro/in vivo correlation studies to validate the proper selection of biowaiver candidate drugs.
References
- 1.Amidon G. L., Lennernäs H., Shah V. P., Crison J. R. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995;12(3):413–420. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
- 2.F. D. A. Guidance. Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system. US Food and Drug Administration Center for Drug Evaluation and Research (2000).
- 3.CPMC (EU). CPMP Note for Guidance on the Investigation of Bioavailability and Bioequivalence (CPMP/EWP/QWP/1401/98). (2001).
- 4.W. Preparations, G. W. H. Organization. WHO Expert Committee on Specifications for Pharmaceutical Preparations: World Health Organization (2006). [PubMed]
- 5.Rinaki E., Dokoumetzidis A., Valsami G., Macheras P. Identification of biowaivers among class II drugs: theoretical justification and practical examples. Pharm. Res. 2004;21(9):1567–1572. doi: 10.1023/B:PHAM.0000041450.25106.c8. [DOI] [PubMed] [Google Scholar]
- 6.Dressman J., Butler J., Hempenstall J., Reppas C. The BCS: where do we go from here. Pharm. Tech. 2001;25(7):68–76. [Google Scholar]
- 7.Yu L., Amidon G., Polli J., et al. Biopharmaceutics classification system: the scientific basis for biowaiver extensions. Pharm. Res. 2002;19(7):921–925. doi: 10.1023/A:1016473601633. [DOI] [PubMed] [Google Scholar]
- 8.Vogelpoel H., Welink J., Amidon G., et al. Biowaiver monographs for immediate release solid oral dosage forms based on biopharmaceutics classification system (BCS) literature data: verapamil hydrochloride, propranolol hydrochloride, and atenolol. J. Pharm. Sci. 2004;93(8):1945–1956. doi: 10.1002/jps.20131. [DOI] [PubMed] [Google Scholar]
- 9.Galia E., Nicolaides E., Hörter D., Löbenberg R., Reppas C., Dressman J. Evaluation of various dissolution media for predicting in vivo performance of class I and II drugs. Pharm. Res. 1998;15(5):698–705. doi: 10.1023/A:1011910801212. [DOI] [PubMed] [Google Scholar]
- 10.Kortejarvi H., Urtti A., Yliperttula M. Pharmacokinetic simulation of biowaiver criteria: the effects of gastric emptying, dissolution, absorption and elimination rates. Eur. J. Pharm. Sci. 2007;30(2):155–166. doi: 10.1016/j.ejps.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 11.Kasim N., Whitehouse M., Ramachandran C., et al. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol. Pharm. 2004;1(1):85–96. doi: 10.1021/mp034006h. [DOI] [PubMed] [Google Scholar]
- 12.Yu L. X., Straughn A. B., Faustino P. J., et al. The effect of food on the relative bioavailability of rapidly dissolving immediate-release solid oral products containing highly soluble drugs. Mol. Pharmaceutics. 2004;1(5):357–362. doi: 10.1021/mp0499407. [DOI] [PubMed] [Google Scholar]
- 13.Yazdanian M., Briggs K., Jankovsky C., Hawi A. The “High Solubility” definition of the current FDA guidance on biopharmaceutical classification system may be too strict for acidic drugs. Pharm. Res. 2004;21(2):293–299. doi: 10.1023/B:PHAM.0000016242.48642.71. [DOI] [PubMed] [Google Scholar]
- 14.Potthast H., Dressman J., Junginger H., et al. Biowaiver monographs for immediate release solid oral dosage forms: ibuprofen. J. Pharm. Sci. 2005;94(10):2121–2131. doi: 10.1002/jps.20444. [DOI] [PubMed] [Google Scholar]
- 15.Vogt M., Derendorf H., Krämer J., et al. Biowaiver monographs for immediate release solid oral dosage forms: prednisolone. J. Pharm. Sci. 2007;96(1):27–37. doi: 10.1002/jps.20768. [DOI] [PubMed] [Google Scholar]
- 16.Hendriksen B., Felix M., Bolger M. The composite solubility versus pH profile and its role in intestinal absorption prediction. AAPS PharmSci. 2003;5(1):E4. doi: 10.1208/ps050104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Available at: http://trslinc.com/services/bcs/search.cfm. Accessed August 12 (2007).
- 18.Kim J-S., Mitchell S., Kijek P., Tsume Y., Hilfinger J., Amidon G. L. The suitability of an in situ perfusion model for permeability determinations: utility for BCS class I biowaiver requests. Mol. Pharmaceutics. 2006;3(6):686–694. doi: 10.1021/mp060042f. [DOI] [PubMed] [Google Scholar]
- 19.Agoram B., Woltosz W., Bolger M. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv. Drug Deliv. Rev. 2001;50(Supplement 1):S41–S67. doi: 10.1016/S0169-409X(01)00179-X. [DOI] [PubMed] [Google Scholar]
- 20.Dollery L. Therapeutic Drugs. Edinburgh: Churchill Livingstone; 1999. [Google Scholar]
- 21.Routledge P., Shand D. Clinical pharmacokinetics of propranolol. Clin. Pharmacokinet. 1979;4(2):73–90. doi: 10.2165/00003088-197904020-00001. [DOI] [PubMed] [Google Scholar]
- 22.Silber B., Holford N., Riegelman S. Dose-dependent elimination of propranolol and its major metabolites in humans. J. Pharm. Sci. 1983;72(7):725–732. doi: 10.1002/jps.2600720703. [DOI] [PubMed] [Google Scholar]
- 23.H. Blume, and E. Mutschler. Bioäquivalenz, Qualitätsbewertung wirkstoffgleicher Fertigarzneimittel, Teil I/II, Isosorbiddinitrat 6. Ergänzungslieferung, Govi-Verlag Pharmazeutischer Verlag, Frankfurt/Main-Eschborn (1996).
- 24.Potthast H., Mircioiu I., Möller H. Propranolol Präparate Biopharmazeutische Charakterisierung. Pharm. Ztg. 2001;146:574. [Google Scholar]
- 25.Rekhi G., Eddington N., Fossler M., Schwartz P., Lesko L., Augsburger L. Evaluation of in vitro release rate and in vivo absorption characteristics of four metoprolol tartrate immediate-release tablet formulations. Pharm. Dev. Technol. 1997;2(1):11–24. doi: 10.3109/10837459709022605. [DOI] [PubMed] [Google Scholar]
- 26.Bergrem H., Grøttum P., Rugstad H. Pharmacokinetics and protein binding of prednisolone after oral and intravenous administration. Eur. J. Clin. Pharmacol. 1983;24(3):415–419. doi: 10.1007/BF00610064. [DOI] [PubMed] [Google Scholar]
- 27.Luippold G., Schneider S., Marto M., Benohr P., Muhlbauer B. Pharmacokinetics of two oral prednisolone tablet formulations in healthy volunteers. Arzneimittelforschung. 2001;51(11):911–915. doi: 10.1055/s-0031-1300136. [DOI] [PubMed] [Google Scholar]
- 28.Rose J., Yurchak A., Jusko W. Dose dependent pharmacokinetics of prednisone and prednisolone in man. J. Pharmacokinet. Pharmacodyn. 1981;9(4):389–417. doi: 10.1007/BF01060885. [DOI] [PubMed] [Google Scholar]
- 29.Luippold G., Benohr P., Schneider S., Marto M., Muhlbauer B. Bioequivalence of different prednisolone tablet formulations. Arzneimittelforschung. 2001;51(8):638–642. doi: 10.1055/s-0031-1300094. [DOI] [PubMed] [Google Scholar]
- 30.Davies N. Clinical pharmacokinetics of ibuprofen. Clin Pharmacokinet. 1998;34(2):101–154. doi: 10.2165/00003088-199834020-00002. [DOI] [PubMed] [Google Scholar]
- 31.C. Graffner. Nfg on bioavailability and bioequivalence (CPMP/EWP/QWP/1401/98): exemption from in vivo bioequivalence studies. Practical applications of MPA (Sweden) 2006; Lisboa.
- 32.Gleiter C., Klotz U., Kuhlmann J., et al. When are bioavailability studies required? A German proposal. J. Clin. Pharmacol. 1998;38(10):904–911. doi: 10.1002/j.1552-4604.1998.tb04385.x. [DOI] [PubMed] [Google Scholar]
- 33.Lenhard G., Kieferndorf U., Berner G., Engels B., Vogtle-Junkert U. Pharmakokinetik und bioaqiovalenz von zwei ibuprofen-formulierungen. Arzneimittel-Forschung. 1990;40(12):1358–1362. [PubMed] [Google Scholar]
- 34.Potthast H., Winter S., Bastian B., Möller H. Ibuprofen-Präparate im Vergleich. Pharm. Ztg. 2002;147(19):26–30. [Google Scholar]
- 35.Wagener H., Vogtle-Junkert U. Intrasubject variability in bioequivalence studies illustrated by the example of ibuprofen. Int. J. Clin. Pharmacol. Ther. Toxicol. 1996;34(1):21–31. [PubMed] [Google Scholar]
- 36.Foster R., Jamali F., Russell A., Alballa S. Pharmacokinetics of ketoprofen enantiomers in healthy subjects following single and multiple doses. J. Pharm. Sci. 1988;77(1):70–73. doi: 10.1002/jps.2600770113. [DOI] [PubMed] [Google Scholar]
- 37.Jamali F., Brocks D. Clinical pharmacokinetics of ketoprofen and its enantiomers. Clin. Pharmacokinet. 1990;19(3):197–217. doi: 10.2165/00003088-199019030-00004. [DOI] [PubMed] [Google Scholar]
- 38.Kantor T. Ketoprofen: a review of its pharmacologic and clinical properties. Pharmacotherapy. 1986;6(3):93–103. doi: 10.1002/j.1875-9114.1986.tb03459.x. [DOI] [PubMed] [Google Scholar]
- 39.Granero G., Ramachandran C., Amidon G. Rapid in vivo dissolution of ketoprofen: implications on the biopharmaceutics classification system. Pharmazie. 2006;61(8):673–676. [PubMed] [Google Scholar]
- 40.Sheng J., Kasim N., Chandrasekharan R., Amidon G. Solubilization and dissolution of insoluble weak acid, ketoprofen: effects of pH combined with surfactant. Eur. J. Pharm. Sci. 2006;29:306–314. doi: 10.1016/j.ejps.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 41.Hasan M., Najib N., Rawashdeh N., Sallam E., Shubair M., Alawneh Y. Comparative bioavailability of two tablet formulations of diclofenac sodium in normal subjects. Int. J. Clin. Pharmacol. Ther. Toxicol. 1991;29(5):178–183. [PubMed] [Google Scholar]
- 42.O'Connor K., Corrigan O. Preparation and characterisation of a range of diclofenac salts. Int. J. Pharm. 2001;226(1–2):163–179. doi: 10.1016/S0378-5173(01)00800-6. [DOI] [PubMed] [Google Scholar]
- 43.Potthast H., Mircioiu I., Bastian B., Möller H. Biopharmazeutische Characterisierung von Diclofenac-Präparaten. Pharm. Zeit. 2001;146(45):1–10. [Google Scholar]
- 44.Glomme A., Maerz J., Dressman J. Comparison of a miniaturized shake-flask solubility method with automated potentiometric acid/base titrations and calculated solubilities. J. Pharm. Sci. 2005;94(1):1–16. doi: 10.1002/jps.20212. [DOI] [PubMed] [Google Scholar]
- 45.TenHoor C., Bakatselou V., Dressman J. Solubility of mefenamic acid under simulated fed-and fasted-state conditions. Pharm. Res. 1991;8(9):1203–1205. doi: 10.1023/A:1015874906665. [DOI] [PubMed] [Google Scholar]
- 46.Winder C. V., Wax J., Scotti L., Scherrer R. A., Jones E. M., Short F. W. Anti-inflammatory, antipyretic and antinociceptive properties of mefenamic acid. J. Pharmacol. Exp. Ther. 1962;138(3):405–413. [PubMed] [Google Scholar]
- 47.Hummel D., Buchmann S. Einfluss der Teilchengrösse von Mefenaminsäure auf Dissolution und Bioverfügbarkeit von Tabletten. Pharm. Ind. 2000;62(6):452–456. [Google Scholar]
- 48.Ngo S. When do differences in dissolution profiles predict clinical problems? J. Clin. Pharm. Ther. 2007;32(2):111–112. doi: 10.1111/j.1365-2710.2007.00807.x. [DOI] [PubMed] [Google Scholar]
- 49.Brogden R., Heel R., Speight T., Avery G. Piroxicam. A reappraisal of its pharmacology and therapeutic efficacy. Drugs. 1984;28(4):292–323. doi: 10.2165/00003495-198428040-00002. [DOI] [PubMed] [Google Scholar]
- 50.Swanepoel E., Liebenberg W., de Villiers M., Dekker T. Dissolution properties of piroxicam powders and capsules as a function of particle size and the agglomeration of powders. Drug Dev. Ind. Pharm. 2000;26(10):1067–1076. doi: 10.1081/DDC-100100270. [DOI] [PubMed] [Google Scholar]
- 51.Michotte Y., van Klaveren H., Detaevernier M., Gusdorf C., Vanhaelst L. Bioequivalence f two formulations of piroxicam. Arzneimittel-Forschung. 1991;41(3):244–246. [PubMed] [Google Scholar]
- 52.Kroemer H., Gautier J., Beaune P., Henderson C., Roland Wolf C., Eichelbaum M. Identification of P450 enzymes involved in metabolism of verapamil in humans. Naunyn-Schmiedeberg's Arch. Pharmacol. 1993;348(3):332–337. doi: 10.1007/BF00169164. [DOI] [PubMed] [Google Scholar]
- 53.Tsang Y., Pop R., Gordon P., Hems J., Spino M. High variability in drug pharmacokinetics complicates determination of bioequivalence: experience with verapamil. Pharm. Res. 1996;13(6):846–850. doi: 10.1023/A:1016040825844. [DOI] [PubMed] [Google Scholar]
- 54.Sawyer P., Brogden R., Pinder R., Speight T., Avery G. Miconazole: a review of its antifungal activity and therapeutic efficacy. Drugs. 1975;9(6):406–423. doi: 10.2165/00003495-197509060-00002. [DOI] [PubMed] [Google Scholar]
- 55.Chin T., Loeb M., Fong I. Effects of an acidic beverage (Coca-Cola) on absorption of ketoconazole. Antimicrob. Agents Chemother. 1995;39:1671–1675. doi: 10.1128/aac.39.8.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hurwitz A., Ruhl C., Kimler B., Topp E., Mayo M. Gastric function in the elderly: effects on absorption of ketoconazole. J. Clin. Pharmacol. 2003;43(9):996–1002. doi: 10.1177/0091270003255645. [DOI] [PubMed] [Google Scholar]
- 57.Vertzoni M., Pastelli E., Psachoulias D., Kalantzi L., Reppas C. Estimation of intragastric solubility of drugs: in what medium? Pharm. Res. 2007;24(5):909–917. doi: 10.1007/s11095-006-9209-9. [DOI] [PubMed] [Google Scholar]
- 58.Zhou R., Moench P., Heran C., et al. pH-Dependent dissolution in vitro and absorption in vivo of weakly basic drugs: development of a canine model. Pharm. Res. 2005;22(2):188–192. doi: 10.1007/s11095-004-1185-3. [DOI] [PubMed] [Google Scholar]
- 59.Jensen J. C. Clinical pharmacokinetics of terbinafine (Lamisil) Clin. Exp. Dermatol. 1989;14(2):110–113. doi: 10.1111/j.1365-2230.1989.tb00904.x. [DOI] [PubMed] [Google Scholar]
- 60.Ryders N. The mechanism of action of terbinafine. Clin. Exp. Dermatol. 1989;14(2):98–100. doi: 10.1111/j.1365-2230.1989.tb00900.x. [DOI] [PubMed] [Google Scholar]
- 61.Carlson J., Mann H., Canafax D. Effect of pH on disintegration and dissolution of ketoconazole tablets. Am. J. Health-syst. Pharm. 1983;40(8):1334–1336. [PubMed] [Google Scholar]
- 62.Intendis Dermatologie. Fachinformation “Amiada®”. (2005).
- 63.Merck Dura. Product Information “Terbinafin dura 250 mg Tabletten”. (2006).
- 64.Pharma Novartis. Product Information “Lamisil®”. (2006).
- 65.Pharmaceuticals Sandoz. Product Information “Verapamil Sandoz®”. (2005).
- 66.Aventis Sanofi. Product Information “Octosan 250 mg Tabletten”. (2006).
- 67.Koytchev R., Erenmemisoglu A., van der Meer M., Alpan S. Clinical relevance of bioequivalence acceptance criteria. The example of terbinafine. Arzneimittelforschung. 2003;53(4):289–293. doi: 10.1055/s-0031-1297110. [DOI] [PubMed] [Google Scholar]
- 68.Medicines Evaluation Board, Netherlands. Terbinafine 250A, tablets 250 mg Apothecon/NL. Public assessment report. (2006).