

Abstract
In the title molecule, C18H14N2O3, O—H⋯N and N—H⋯O hydrogen bonds influence the molecular conformation; the benzene and naphthalene planes are inclined at a dihedral angle of 11.54 (5)°. In the crystal structure, intermolecular O—H⋯O hydrogen bonds link the molecules into chains running in the [01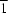 ] direction.
] direction.
Related literature
For useful applications of salicyloyl hydrazide derivatives, see: Sumita et al. (1999 ▶). For the crystal structure of (E)-2-hydroxy-N′-(3-hydroxy-4-methoxybenzylidene)benzohydrazide, see: Luo (2007 ▶).
Experimental
Crystal data
C18H14N2O3
M r = 306.31
Orthorhombic,
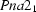
a = 21.124 (2) Å
b = 11.6212 (13) Å
c = 5.9826 (8) Å
V = 1468.6 (3) Å3
Z = 4
Mo Kα radiation
μ = 0.10 mm−1
T = 298 (2) K
0.32 × 0.18 × 0.15 mm
Data collection
Bruker SMART CCD area-detector diffractometer
Absorption correction: multi-scan (SADABS; Sheldrick, 1996 ▶) T min = 0.970, T max = 0.986
6099 measured reflections
1422 independent reflections
896 reflections with I > 2σ(I)
R int = 0.057
Refinement
R[F 2 > 2σ(F 2)] = 0.040
wR(F 2) = 0.076
S = 1.04
1422 reflections
209 parameters
1 restraint
H-atom parameters constrained
Δρmax = 0.15 e Å−3
Δρmin = −0.16 e Å−3
Data collection: SMART (Siemens, 1996 ▶); cell refinement: SAINT (Siemens, 1996 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: SHELXTL (Sheldrick, 2008 ▶); software used to prepare material for publication: SHELXTL.
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536808044073/cv2501sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808044073/cv2501Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O3—H3⋯N2 | 0.82 | 1.90 | 2.623 (5) | 146 |
| N1—H1⋯O2 | 0.86 | 1.92 | 2.620 (4) | 137 |
| O2—H2⋯O1i | 0.82 | 1.81 | 2.573 (4) | 155 |
Symmetry code: (i) 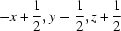 .
.
Acknowledgments
The authors acknowledge the support of the National Natural Science Foundation of Liaocheng University (grant No. X051040).
supplementary crystallographic information
Comment
Salicyloyl hydrazide is an important organic intermediate, it can act as moulding board in inorganic complex (Sumita et al., 1999). In this paper, we present the title compound (I), which was synthesized by the reaction of 2-hydroxyl naphthaldehyde and salicyloyl hydrazide.
In (I) (Fig. 1), the bond lengths and angles are normal and comparable to those observed in the reported compound (Luo, 2007). In the crystal structure, the C8=N2 bond length is 1.279 (5) Å showing the double-bond character. The dihedral angle between the naphthalene ring and C8/N2/N1 is 10.14 (3) Å, the C1/N1/N2 and benzene ring form a dihedral angle of 6.70 (4) Å showing that intramolecular O—H···N and N—H···O hydrogen bonds (Table 1) influence the molecular conformation.
In the crystal, intermolecular O—H···O hydrogen bonds (Table 1) link the molecules into chains running in direction [01–1].
Experimental
Salicyloyl hydrazide (0.5 mmol) and freshly 2-hydroxyl naphthaldehyde (0.5 mmol) were mixed in 50 ml flash. After stirring 30 min at 353 K, the mixture then cooling slowly to room temperature and affording the title compound, then recrystallized from ethanol, affording the title compound as a green crystalline solid. Elemental analysis: calculated for C18H14N2O3: C 70.58, H 4.61, N 9.15%; found: C 70.53, H 4.55, N 9.24%.
Refinement
All H atoms were placed in geometrically idealized positions (N—H 0.86, O—H 0.82 and C—H=0.93 Å) and treated as riding, with Uiso(H) = 1.2 Ueq of the parent atom. In the absence of any significant anomalous scatterers in the molecule, the 1353 Friedel pairs were merged before the final refinement.
Figures
Fig. 1.

ORTEP drawing of the title molecule with atomic numbering scheme and displacement ellipsoids at 30% probability level.
Crystal data
| C18H14N2O3 | Dx = 1.385 Mg m−3 |
| Mr = 306.31 | Mo Kα radiation, λ = 0.71073 Å |
| Orthorhombic, Pna21 | Cell parameters from 1119 reflections |
| a = 21.124 (2) Å | θ = 2.6–25.4° |
| b = 11.6212 (13) Å | µ = 0.10 mm−1 |
| c = 5.9826 (8) Å | T = 298 K |
| V = 1468.6 (3) Å3 | Block, yellow |
| Z = 4 | 0.32 × 0.18 × 0.15 mm |
| F(000) = 640 |
Data collection
| Bruker SMART CCD area-detector diffractometer | 1422 independent reflections |
| Radiation source: fine-focus sealed tube | 896 reflections with I > 2σ(I) |
| graphite | Rint = 0.057 |
| φ and ω scans | θmax = 25.0°, θmin = 1.9° |
| Absorption correction: multi-scan (SADABS; Sheldrick, 1996) | h = −24→24 |
| Tmin = 0.970, Tmax = 0.986 | k = −13→8 |
| 6099 measured reflections | l = −7→6 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.040 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.076 | H-atom parameters constrained |
| S = 1.04 | w = 1/[σ2(Fo2) + (0.0012P)2 + 0.7621P] where P = (Fo2 + 2Fc2)/3 |
| 1422 reflections | (Δ/σ)max = 0.001 |
| 209 parameters | Δρmax = 0.15 e Å−3 |
| 1 restraint | Δρmin = −0.16 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| N1 | 0.22373 (16) | 0.9395 (3) | 0.7320 (6) | 0.0514 (10) | |
| H1 | 0.2172 | 0.8713 | 0.7828 | 0.062* | |
| N2 | 0.19138 (18) | 0.9753 (3) | 0.5434 (6) | 0.0536 (11) | |
| O1 | 0.27673 (14) | 1.1075 (2) | 0.7748 (6) | 0.0703 (10) | |
| O2 | 0.25278 (12) | 0.7725 (2) | 1.0077 (6) | 0.0574 (9) | |
| H2 | 0.2526 | 0.7107 | 1.0739 | 0.086* | |
| O3 | 0.16012 (13) | 1.1067 (3) | 0.2045 (6) | 0.0664 (10) | |
| H3 | 0.1787 | 1.0892 | 0.3199 | 0.100* | |
| C1 | 0.2649 (2) | 1.0079 (4) | 0.8375 (8) | 0.0504 (12) | |
| C2 | 0.2955 (2) | 0.9611 (3) | 1.0404 (8) | 0.0447 (11) | |
| C3 | 0.2891 (2) | 0.8497 (3) | 1.1229 (7) | 0.0441 (12) | |
| C4 | 0.3185 (2) | 0.8179 (4) | 1.3196 (8) | 0.0544 (13) | |
| H4 | 0.3135 | 0.7435 | 1.3740 | 0.065* | |
| C5 | 0.3548 (2) | 0.8948 (5) | 1.4348 (9) | 0.0672 (15) | |
| H5 | 0.3750 | 0.8718 | 1.5655 | 0.081* | |
| C6 | 0.3618 (2) | 1.0054 (5) | 1.3596 (10) | 0.0718 (16) | |
| H6 | 0.3863 | 1.0578 | 1.4392 | 0.086* | |
| C7 | 0.3321 (2) | 1.0378 (4) | 1.1646 (10) | 0.0614 (14) | |
| H7 | 0.3366 | 1.1130 | 1.1142 | 0.074* | |
| C8 | 0.1536 (2) | 0.8994 (4) | 0.4641 (8) | 0.0546 (13) | |
| H8 | 0.1507 | 0.8288 | 0.5363 | 0.066* | |
| C9 | 0.1152 (2) | 0.9189 (4) | 0.2659 (8) | 0.0493 (12) | |
| C10 | 0.1211 (2) | 1.0193 (4) | 0.1429 (8) | 0.0495 (12) | |
| C11 | 0.0868 (2) | 1.0370 (4) | −0.0549 (8) | 0.0560 (13) | |
| H11 | 0.0914 | 1.1054 | −0.1339 | 0.067* | |
| C12 | 0.0468 (2) | 0.9542 (4) | −0.1312 (9) | 0.0607 (14) | |
| H12 | 0.0255 | 0.9653 | −0.2656 | 0.073* | |
| C13 | 0.0369 (2) | 0.8510 (4) | −0.0089 (9) | 0.0548 (13) | |
| C14 | 0.07102 (19) | 0.8331 (4) | 0.1920 (8) | 0.0511 (12) | |
| C15 | 0.0568 (2) | 0.7320 (4) | 0.3138 (9) | 0.0656 (15) | |
| H15 | 0.0780 | 0.7173 | 0.4470 | 0.079* | |
| C16 | 0.0128 (2) | 0.6564 (5) | 0.2396 (11) | 0.0790 (19) | |
| H16 | 0.0044 | 0.5909 | 0.3239 | 0.095* | |
| C17 | −0.0202 (2) | 0.6734 (5) | 0.0415 (12) | 0.0771 (17) | |
| H17 | −0.0500 | 0.6199 | −0.0067 | 0.093* | |
| C18 | −0.0082 (2) | 0.7700 (4) | −0.0809 (9) | 0.0686 (16) | |
| H18 | −0.0301 | 0.7825 | −0.2136 | 0.082* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| N1 | 0.053 (2) | 0.058 (2) | 0.043 (3) | 0.005 (2) | −0.001 (2) | 0.022 (2) |
| N2 | 0.051 (2) | 0.070 (3) | 0.040 (2) | 0.016 (2) | 0.000 (2) | 0.017 (2) |
| O1 | 0.086 (2) | 0.0454 (18) | 0.079 (3) | 0.0026 (17) | 0.004 (2) | 0.0291 (19) |
| O2 | 0.076 (2) | 0.0427 (16) | 0.053 (2) | −0.0038 (16) | −0.012 (2) | 0.0166 (17) |
| O3 | 0.063 (2) | 0.081 (2) | 0.056 (3) | −0.0053 (18) | −0.0055 (19) | 0.029 (2) |
| C1 | 0.052 (3) | 0.049 (3) | 0.050 (3) | 0.007 (2) | 0.006 (3) | 0.012 (2) |
| C2 | 0.046 (3) | 0.043 (3) | 0.044 (3) | 0.005 (2) | 0.001 (2) | 0.009 (2) |
| C3 | 0.050 (3) | 0.042 (3) | 0.040 (3) | 0.000 (2) | −0.001 (2) | 0.005 (2) |
| C4 | 0.060 (3) | 0.058 (3) | 0.045 (3) | 0.008 (3) | 0.001 (3) | 0.017 (3) |
| C5 | 0.062 (3) | 0.087 (4) | 0.053 (4) | 0.012 (3) | −0.010 (3) | 0.003 (3) |
| C6 | 0.065 (3) | 0.073 (4) | 0.077 (4) | −0.006 (3) | −0.012 (3) | −0.011 (3) |
| C7 | 0.066 (3) | 0.050 (3) | 0.068 (4) | −0.002 (3) | 0.002 (3) | 0.007 (3) |
| C8 | 0.055 (3) | 0.062 (3) | 0.047 (3) | 0.013 (3) | 0.007 (3) | 0.018 (3) |
| C9 | 0.047 (3) | 0.063 (3) | 0.037 (3) | 0.011 (2) | 0.005 (2) | 0.014 (3) |
| C10 | 0.044 (3) | 0.068 (3) | 0.037 (3) | 0.007 (2) | 0.007 (3) | 0.016 (3) |
| C11 | 0.054 (3) | 0.075 (3) | 0.039 (3) | 0.011 (3) | −0.001 (3) | 0.020 (3) |
| C12 | 0.059 (3) | 0.088 (4) | 0.035 (3) | 0.009 (3) | 0.000 (3) | 0.007 (3) |
| C13 | 0.049 (3) | 0.064 (3) | 0.051 (3) | 0.009 (3) | 0.009 (3) | 0.001 (3) |
| C14 | 0.049 (3) | 0.059 (3) | 0.046 (3) | 0.017 (2) | 0.006 (3) | 0.004 (3) |
| C15 | 0.056 (3) | 0.071 (3) | 0.070 (4) | 0.008 (3) | 0.000 (3) | 0.017 (3) |
| C16 | 0.069 (4) | 0.068 (4) | 0.100 (6) | 0.003 (3) | 0.001 (4) | 0.018 (4) |
| C17 | 0.068 (4) | 0.067 (4) | 0.096 (5) | −0.003 (3) | 0.002 (4) | 0.001 (4) |
| C18 | 0.060 (3) | 0.079 (4) | 0.066 (4) | 0.007 (3) | 0.008 (3) | −0.008 (4) |
Geometric parameters (Å, °)
| N1—C1 | 1.336 (5) | C8—C9 | 1.455 (6) |
| N1—N2 | 1.383 (5) | C8—H8 | 0.9300 |
| N1—H1 | 0.8600 | C9—C10 | 1.386 (5) |
| N2—C8 | 1.280 (5) | C9—C14 | 1.435 (5) |
| O1—C1 | 1.242 (5) | C10—C11 | 1.403 (6) |
| O2—C3 | 1.366 (5) | C11—C12 | 1.360 (6) |
| O2—H2 | 0.8200 | C11—H11 | 0.9300 |
| O3—C10 | 1.359 (5) | C12—C13 | 1.420 (6) |
| O3—H3 | 0.8200 | C12—H12 | 0.9300 |
| C1—C2 | 1.479 (6) | C13—C18 | 1.407 (6) |
| C2—C3 | 1.392 (5) | C13—C14 | 1.417 (6) |
| C2—C7 | 1.395 (6) | C14—C15 | 1.415 (6) |
| C3—C4 | 1.381 (6) | C15—C16 | 1.354 (6) |
| C4—C5 | 1.364 (6) | C15—H15 | 0.9300 |
| C4—H4 | 0.9300 | C16—C17 | 1.389 (7) |
| C5—C6 | 1.370 (7) | C16—H16 | 0.9300 |
| C5—H5 | 0.9300 | C17—C18 | 1.364 (6) |
| C6—C7 | 1.377 (7) | C17—H17 | 0.9300 |
| C6—H6 | 0.9300 | C18—H18 | 0.9300 |
| C7—H7 | 0.9300 | ||
| C1—N1—N2 | 121.8 (4) | C10—C9—C14 | 118.7 (4) |
| C1—N1—H1 | 119.1 | C10—C9—C8 | 120.9 (4) |
| N2—N1—H1 | 119.1 | C14—C9—C8 | 120.4 (4) |
| C8—N2—N1 | 113.8 (4) | O3—C10—C9 | 122.7 (4) |
| C3—O2—H2 | 109.5 | O3—C10—C11 | 115.7 (4) |
| C10—O3—H3 | 109.5 | C9—C10—C11 | 121.6 (5) |
| O1—C1—N1 | 122.9 (4) | C12—C11—C10 | 120.1 (5) |
| O1—C1—C2 | 120.2 (5) | C12—C11—H11 | 120.0 |
| N1—C1—C2 | 116.9 (4) | C10—C11—H11 | 120.0 |
| C3—C2—C7 | 117.3 (4) | C11—C12—C13 | 121.1 (5) |
| C3—C2—C1 | 126.2 (4) | C11—C12—H12 | 119.5 |
| C7—C2—C1 | 116.4 (4) | C13—C12—H12 | 119.5 |
| O2—C3—C4 | 120.5 (4) | C18—C13—C14 | 120.4 (5) |
| O2—C3—C2 | 119.1 (4) | C18—C13—C12 | 120.4 (5) |
| C4—C3—C2 | 120.5 (4) | C14—C13—C12 | 119.1 (5) |
| C5—C4—C3 | 120.6 (5) | C15—C14—C13 | 116.8 (5) |
| C5—C4—H4 | 119.7 | C15—C14—C9 | 123.8 (5) |
| C3—C4—H4 | 119.7 | C13—C14—C9 | 119.4 (4) |
| C4—C5—C6 | 120.6 (5) | C16—C15—C14 | 121.1 (5) |
| C4—C5—H5 | 119.7 | C16—C15—H15 | 119.5 |
| C6—C5—H5 | 119.7 | C14—C15—H15 | 119.5 |
| C5—C6—C7 | 119.0 (5) | C15—C16—C17 | 122.1 (6) |
| C5—C6—H6 | 120.5 | C15—C16—H16 | 118.9 |
| C7—C6—H6 | 120.5 | C17—C16—H16 | 118.9 |
| C6—C7—C2 | 122.0 (5) | C18—C17—C16 | 118.8 (6) |
| C6—C7—H7 | 119.0 | C18—C17—H17 | 120.6 |
| C2—C7—H7 | 119.0 | C16—C17—H17 | 120.6 |
| N2—C8—C9 | 122.9 (4) | C17—C18—C13 | 120.8 (6) |
| N2—C8—H8 | 118.6 | C17—C18—H18 | 119.6 |
| C9—C8—H8 | 118.6 | C13—C18—H18 | 119.6 |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O3—H3···N2 | 0.82 | 1.90 | 2.623 (5) | 146 |
| N1—H1···O2 | 0.86 | 1.92 | 2.620 (4) | 137 |
| O2—H2···O1i | 0.82 | 1.81 | 2.573 (4) | 155 |
Symmetry codes: (i) −x+1/2, y−1/2, z+1/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: CV2501).
References
- Luo, Z.-G. (2007). Acta Cryst. E63, o3672.
- Sheldrick, G. M. (1996). SADABS University of Göttingen, Germany.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Siemens (1996). SMART and SAINT Siemens Analytical X-ray Instruments Inc., Madison, Wisconsin, USA.
- Sumita, N. R., Munshi, K. N., Nageswara, R. N., Bhadbhade, M. M. & Suresh, E. (1999). Polyhedron, 18, 2491–2497.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536808044073/cv2501sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536808044073/cv2501Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report