

Abstract
We report on an autosomal-recessive variant of Ehlers-Danlos syndrome (EDS) characterized by severe muscle hypotonia at birth, progressive scoliosis, joint hypermobility, hyperelastic skin, myopathy, sensorineural hearing impairment, and normal pyridinoline excretion in urine. Clinically, the disorder shares many features with the kyphoscoliotic type of EDS (EDS VIA) and Ullrich congenital muscular dystrophy. Linkage analysis in a large Tyrolean kindred identified a homozygous frameshift mutation in FKBP14 in two affected individuals. Based on the cardinal clinical characteristics of the disorder, four additional individuals originating from different European countries were identified who carried either homozygous or compound heterozygous mutations in FKBP14. FKBP14 belongs to the family of FK506-binding peptidyl-prolyl cis-trans isomerases (PPIases). ER-resident FKBPs have been suggested to act as folding catalysts by accelerating cis-trans isomerization of peptidyl-prolyl bonds and to act occasionally also as chaperones. We demonstrate that FKBP14 is localized in the endoplasmic reticulum (ER) and that deficiency of FKBP14 leads to enlarged ER cisterns in dermal fibroblasts in vivo. Furthermore, indirect immunofluorescence of FKBP14-deficient fibroblasts indicated an altered assembly of the extracellular matrix in vitro. These findings suggest that a disturbance of protein folding in the ER affecting one or more components of the extracellular matrix might cause the generalized connective tissue involvement in this disorder. FKBP14 mutation analysis should be considered in all individuals with apparent kyphoscoliotic type of EDS and normal urinary pyridinoline excretion, in particular in conjunction with sensorineural hearing impairment.
Introduction
The Ehlers-Danlos syndrome (EDS) comprises a clinically and genetically heterogeneous group of heritable connective tissue disorders that predominantly affect skin, joints, ligaments, blood vessels, and internal organs.1 Clinical hallmarks are skin hyperelasticity, joint hypermobility, and increased tissue fragility. The natural history and mode of inheritance differ among the six major types.1,2 Among them, the kyphoscoliotic type of EDS (formerly named EDS VIA [MIM 225400]) is characterized by severe muscle hypotonia at birth, progressive kyphoscoliosis, marked skin hyperelasticity with widened atrophic scars, and joint hypermobility.1 Additional features are osteopenia without a tendency to fractures, a Marfanoid habitus, microcornea, and occasionally rupture of the arteries and the eye globe. The underlying defect in EDS VIA is a deficiency of the enzyme lysyl hydroxylase 1 (LH1; EC 1.14.11.4; procollagen-lysine, 2-oxoglutarate 5-dioxygenase) caused by mutations in PLOD1 (MIM 153454). This enzyme hydroxylates lysyl residues in -Xaa-Lys-Gly- triplets of collagens and other proteins with collagen-like sequences.3 The resulting hydroxylysyl residues serve as attachment sites for carbohydrate units and are involved in the formation of intra- and intermolecular collagen crosslinks, which provide mechanical stability to collagen fibrils. A deficiency of lysyl hydroxylase results in an abnormal urinary excretion pattern of lysyl pyridinoline (LP) and hydroxylysyl pyridinoline (HP) crosslinks with an increased LP/HP ratio, which is diagnostic for EDS VIA.1,4–6
Because of severe muscle hypotonia and delayed motor development, children with EDS VIA are initially often suspected to have a primary neuromuscular disease.5,7 However, neuromuscular workup in the neonatal period mostly yields normal results. Muscle weakness and fatigability are common symptoms in various types of EDS and have been explained by increased distensibility of tendons, decreased mechanical efficiency resulting from sparse intramuscular connective tissue, and exercise avoidance because of joint instability and/or because of the weak connective tissue surrounding the muscle spindles embedded in the muscles.1 Recent findings8,9 suggest that neuromuscular symptoms in EDS may in part be due to mild to moderate myopathic and/or neuropathic changes that have been attributed to the altered composition of the extracellular matrix in muscle and peripheral nerve. Vice versa, primary myopathies caused by mutations in extracellular matrix components, such as the collagen type VI-related disorders Ullrich congenital muscular dystrophy (UCMD [MIM 254090]) and Bethlem myopathy (MIM 158810), may show substantial connective tissue involvement.10–13,36 UCMD in particular is characterized by kyphoscoliosis, joint hypermobility, joint contractures, and abnormal scar formation in addition to severe muscle weakness.
A subgroup of individuals with EDS type VI has been reported, in whom lysyl hydroxylase activity and urinary LP/HP ratio appear to be normal.1 This condition formerly has been classified as EDS VIB.1,14,15 In some of these individuals, mutations in CHST14 (MIM 608429) coding for carbohydrate (N-acetylgalactosamine 4-O) sulfotransferase 14 (D4ST1) have been reported.16–18 Various names have been coined for disorders associated with mutations in CHST14 such as adducted thumb-club foot syndrome (ATCS),16 musculocontractural EDS (MCEDS [MIM 601776]),17 or EDS Kosho Type (EDSKT).19 Recently, Shimizu et al.19 suggested that ATCS, MCEDS, and EDSKT are a single clinical entity with variable expression and proposed the unifying name “D4ST1-deficient EDS” for the clinical spectrum of manifestations.
Here we report an autosomal-recessive variant of EDS with kyphoscoliosis and normal urinary LP/HP ratio, which we identified in six affected individuals from five unrelated families originating from different European countries. This disorder shares many clinical features with EDS VIA but is also characterized by sensorineural hearing loss. Affected individuals show clear signs of a myopathy as demonstrated by clinical, electrophysiological, imaging, and muscle biopsy findings. We identified causative mutations in FKBP14, which codes for a member of the FK506-binding family of peptidyl-prolyl cis-trans isomerases (PPIases), in all individuals with this disorder.
Subjects and Methods
At 14 years of age, the index person P1 (individual V-2 in family I, Figure 1) was referred to the Department of Paediatrics IV at Innsbruck Medical University (MB) for the evaluation of severe kyphoscoliosis, joint hypermobility, and muscle weakness. He was initially suspected to have EDS VIA, but the urinary LP/HP ratio was within the normal range. Pedigree analysis revealed that the index person's father had a cousin (P2, individual IV-2 in family I, Figure 1) who was affected by the same disorder. The parents of both affected family members were not knowingly consanguineous but originated from neighboring small villages in Tyrol (Austria), and therefore autosomal-recessive inheritance was assumed and linkage analysis was performed in order to identify the candidate gene.
Figure 1.
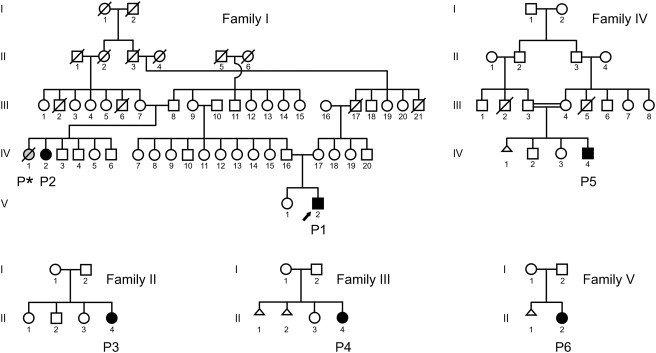
Pedigrees of the Families
The circles indicate females, the squares males, and the triangles spontaneous abortions. Filled symbols indicate affected individuals with proven homozygous or compound heterozygous FKBP14 mutations. The index person P1 in family I (individual V-2) is indicated by an arrow. The deceased, probably affected, member of family I is indicated as P∗ (individual IV-1).
Based on clinical characteristics, four additional individuals affected by the same disorder were identified. Their families originated from Italy, Turkey, France, and Germany, respectively. The pedigrees of the families involved in the study are depicted in Figure 1. Salient clinical and neuromuscular findings are summarized in Tables 1 and 2 and Figures 2 and 3.
Table 1.
Salient Clinical Findings
|
P1 |
P2 |
P∗ |
P3 |
P4 |
P5 |
P6 |
|
|---|---|---|---|---|---|---|---|
|
Current age/gender |
16 y/M |
48 y/F |
12 y/Fa |
11 y/F |
16 y/F |
11 y/M |
3 y/F |
| Origin | Austria | Austria | Austria | Italy | France | Turkey | Germany |
| Skin | |||||||
| hyperelastic | + | (+) | nr | + | – | + | + |
| soft | + | + | nr | + | + | + | + |
| plantar softness | + | – | nr | + | + | + | + |
| follicular hyperkeratosis | + | – | + | + | + | – | + |
| easy bruising | – | + | nr | (+) | – | + | – |
| hypertrophic scars | (+) | – | nr | – | – | – | – |
| atrophic scars | – | – | nr | – | – | – | – |
| Joints | |||||||
| hypermobility of large joints | + | + | + | + | ++ | ++ | + |
| hypermobility of small joints | ++ | + | + | ++ | ++ | ++ | ++ |
| Beighton score | 6/9 | 6/9 | nr | 8/9 | 6/9 | 9/9 | 9/9 |
| recurrent dislocations | – | – | – | – | – | ++ | – |
| joint contractures | – | – | – | – | – | – | – |
| Skeletal | |||||||
| progressive kyphoscoliosis | ++ | ++ (11 y op) | + | ++ (4 y op) | ++ (12 y op) | kyphosis | scoliosis |
| flat feet | + | + | + | + | +, club foot left | + | +, club foot left |
| fractures | – | – | – | – | – | (+) | – |
| Neuromuscular | |||||||
| muscle hypotonia at birth | ++ | ++ | ++ | ++ | ++ | ++ | ++ |
| poor head control in infancy | + | + | + | + | + | + | + |
| weakness improving in infancy | + | + | + | + | + | + | + |
| delayed motor development | + | + | + | ++ | + | ++ | ++ |
| walking independently | 2.5 y | 2.5 y | 2.5 y | 4 y | 2 y | 4 y | 3 y supported |
| muscular atrophy | + | + | (+) | (+) | + | (+) | (+) |
| current MRC muscle score | 3–4 | 3–4 | nr | 4 | 4 | 3–4 | 3–4 |
| Cardiovascular | |||||||
| cardiomyopathy | – | nr | nr | – | – | – | – |
| valvular abnormalities | – | nr | nr | – | tricusp. insuf. I° | mitral and tricusp. insuf. I° | – |
| vascular abnormalities | – | – | aortic rupture | – | – | – | – |
| Eyes and Ears | |||||||
| bluish sclerae | + in infancy | – | nr | – | – | – | – |
| myopia | + | + | + | – | + | – | – |
| microcornea | – | – | nr | – | – | – | – |
| hearing impairment | sensorineural | sensorineural | nr | sensorineural | conductive | sensorineural | sensorineural |
| Miscellaneous | |||||||
| herniae | inguinal | umbilical | – | umbilical | – | umbilical | – |
| bladder diverticulum | + | nr | nr | nr | nr | + | nr |
| cleft soft palate | – | – | – | – | – | + | + |
| retrogenia in infancy | – | – | – | – | – | + | + |
| subdural hygroma | microcephaly, learning difficulties |
||||||
Abbreviations: P∗, probably affected person; y, years; F, female; M, male; (+), mildly present; +, present; ++, explicitly present; –, absent; nr, not recorded; op, operation; MRC, Medical Research Council scale for muscle strength.
Patient P∗ died at the age of 12 years.
Table 2.
Neuromuscular Investigations
| P1 | P2 | P∗ | P3 | P4 | P5 | P6 | |
|---|---|---|---|---|---|---|---|
| Current age | 16 y | 48 y | 12 ya | 11 y | 16 y | 11 y | 3 y |
| Laboratory | |||||||
| Creatine kinaseb | normal | slightly elevated (×1.2) | normal | normal | normal | slightly elevated (×1.2) | slightly elevated (×1.3) |
| Urinary crosslinks (LP/HP ratio) | normal | normal | normal | normal | normal | normal | |
| Nerve Conduction | |||||||
| Result | normal | normal | normal | normal | normal | ||
| Electromyography | |||||||
| Age, result | 3 m, normal | 6 y, normal | 6 y, myopathic | 4 m, normal | 1 y, myopathic | ||
| Age, result | 15 y, myopathic | 30 y, myopathic | 2 y, normal | ||||
| Muscle Biopsy | |||||||
| Age, site, result | 2 y, quadriceps, irregular oxidative enzymes; EM: focal myofibrillar rearrangements (Figure 3E) | 2 y, quadriceps, marked fiber atrophy | 6 y, nr, myopathic | 4 m, quadriceps, mildly myopathic | 2 y, quadriceps, mildly myopathic | 1 y, quadriceps, myopathic | 1 y, quadriceps, mildly myopathic with increased variation of fiber diameter (Figures 3A and 3B) |
| Age, site, result | 4 y, nr, marked atrophy, proliferation of fatty tissue | 4 y, paraspinal, areas with fiber atrophy, slightly increased intrafusal fat | 6 y, nr, myopathic | ||||
| Age, site, result | 7 y, anterior tibial, myopathic, proliferation of fatty tissue | 6 y, paraspinal, areas with central activity defects of oxidative enzymes, (Figure 3C); EM: focal myofibrillar rearrangements (Figure 3D) | 12 y, dorsal, myopathic with increased variation of fiber diameter; EM: bifurcation of sarcomeres, small zones of Z-band streaming and some disorganized myofibrils (Figure 3F) | ||||
| Age, site, result | 30 y, paraspinal, mildly myopathic, atrophic fibers | ||||||
| Muscle MRI | |||||||
| Age, result | 15 y, normal in lower limbs | 12 y, marked signal abnormalities in rectus femoris, vastus lateralis and soleus; mild involvement of hip flexors, neck extensors, and shoulder girdle (Figure 3A) | 11 y, marked signal abnormalities in rectus femoris, vastus lateralis, medial head of gastrocnemius, soleus, and paraspinal muscles | ||||
The following symbols and abbreviations are used: P∗, probably affected person; m, months; y, years; EM, electron microscopy.
Patient P∗ died at the age of 12 years.
x-fold upper limit.
Figure 2.
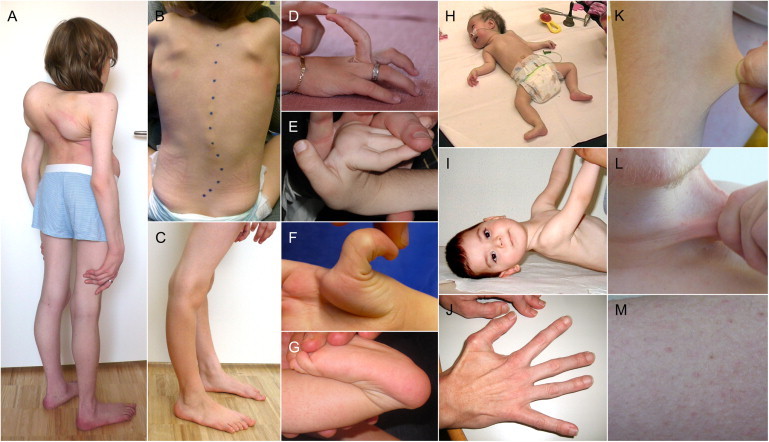
Clinical Phenotype
Clinical findings in individuals with FKBP14-deficient EDS.
(A and B) Severe kyphoscoliosis in P1 (age 15 years) (A) and lumbar scoliosis in P6 (3 years) (B).
(C–G) Genu recurvatum in P3 (C) and distally pronounced joint hypermobility in P3 (D, 10 years), P5 (E, 3 years), and P6 (F and G, 3 years).
(H and I) Muscle hypotonia and weakness in P6 (H, 3 months) and P5 (I, 1.5 years).
(J) Moderate atrophy of intrinsic hand muscles in P2 (47 years).
(K and L) Hyperelastic skin of the forearm of P3 (K, 10 years) and the neck region of P1 (L, 16 years).
(M) Follicular hyperkeratosis in the pretibial region of P3 (10 years).
Figure 3.
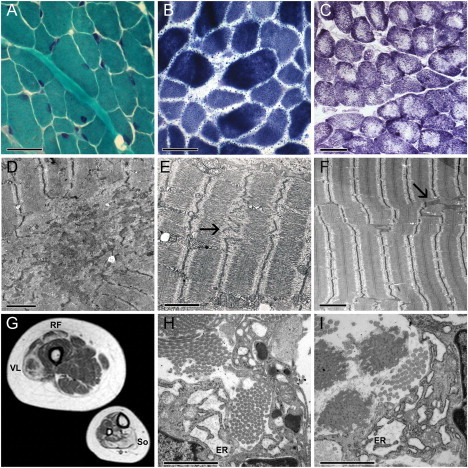
Muscle MRI, Histomorphology, and Ultrastructure of Muscle and Skin
(A and B) Histomorphology of a muscle biopsy taken from the quadriceps muscle of P6 at the age of 1 year showing mild changes with increased variation in muscle fiber diameter: Gomori trichrome (A) and NADH-TR stain (B).
(C and D) Histochemistry (NADH-TR) of a muscle biopsy from the paraspinal muscle of P3 at the age of 6 years shows areas with central activity defects of oxidative enzymes (C), corresponding to focal rearrangements of myofibrils in electron microscopy (D).
(E and F) Transmission electron microscopy of muscle (longitudinal sections) in P1 and P4.
(E) Electron microscopy in P1 shows focal rearrangements of myofibrils with irregular Z lines (arrow) (quadriceps muscle, at the age of 2 years).
(F) Electron microscopy in P4 shows bifurcation of sarcomeres, small zones of Z-band streaming (arrow), and some disorganized myofibrils (dorsal muscle, at the age of 12 years).
(G) Transverse T1-weighted muscle MRI cross sections of the right thigh and lower leg in P4 at the age of 12 years show abnormal signals in the rectus femoris (RF), vastus lateralis (VL), and soleus (So) muscles indicating fatty degeneration of muscle tissue.
(H and I) Two images of transmission electron microscopy of a skin biopsy of P1 show abnormally enlarged ER cisterns filled with a flocculent material; collagen fibrils are normal in shape and diameter.
Scale bars represent 20 μm in light microscopy and 2 μm in electron microscopy.
All individuals participating in the study gave their informed consent. All procedures followed were in accordance with the institutional (Medical University Innsbruck) and national ethical standards of diagnostic and research investigations.
P1
P1 (individual V-2 in family I, Figure 1) was born spontaneously at term after an uneventful pregnancy by vaginal delivery with cephalic presentation. Birth measurements were within the normal range (weight 50th centile, length 10th–25th centile). He presented at birth with severe muscle hypotonia, paucity of antigravity movements, poor sucking, hypermobile joints, and slightly bluish sclerae. Infantile spinal muscular atrophy was excluded. At the age of 2 months, first signs of a spinal deformity developed. In the subsequent months muscle weakness and hypotonia improved. Motor development was delayed, but speech and mental development were normal. He was able to sit without support at 13 months and walked without assistance at 2.5 years. At the age of 2 years, a unilateral inguinal hernia required surgical correction. A chronic subdural hygroma causing increased intracranial pressure was treated with a subdural-peritoneal shunt at the age of 3 years. Bilateral sensorineural hearing impairment affecting predominantly high frequencies (70–80 dB HL at 4–6 kHz) was diagnosed at 6 years and he was provided with a hearing aid. At the age of 9 years, a large bladder diverticulum was surgically removed. Scoliosis progressed despite management with a thoracolumbar orthosis and led to a restrictive ventilation disorder (FVC 34%). Echocardiography and thoraco-abdominal CT scan at the age of 13 years showed no signs of cardiomyopathy and no dilatation of the aorta or other large vessels.
At the current age of 16 years, he has severe kyphoscoliosis (Figure 2A) and marked hypermobility of the large and small joints (Beighton score 6/9), but no contractures. He has a characteristic handshake with the hands feeling “like a bag of little bones” because the musculoskeletal structure of the hands seems to collapse on pressure. The skin is thin, soft, and hyperelastic (Figure 2L) with follicular hyperkeratosis but no signs of increased fragility or atrophic scars. Over the left deltoid area, where a skin biopsy was taken, two small keloids are visible. There is no skin wrinkling of the palms. He has myopia of 6–7 diopters, and the cornea diameter is within the normal range. Arm span and lower length are within the normal range, but the standing height is reduced because of severe kyphoscoliosis (10 cm below the third centile). He is able to walk up to 1 km and to climb stairs without using the handrail. Muscle strength is between 3− (neck flexion) and 4 on the MRC scale.
P2
P2 (individual IV-2 in family I; Figure 1) is the first cousin of the father of P1. She was born at term after an uneventful pregnancy, but the mother reported feeble fetal movements. Birth measurements were within the normal range (weight 10th–25th centile, length 50th centile). At birth she presented as a floppy infant with severe muscle hypotonia and weakness. Gross motor milestones were delayed. She was able to walk without help at the age of 3 years. At the age of 2 years, a scoliosis was noted, which slowly progressed and required a first surgical correction at the age of 11 years. Moderate bilateral sensorineural hearing impairment affecting predominantly high frequencies was diagnosed at the age of 6 years and she was supplied with a hearing aid. She reported that hearing impairment was not progressive over the years.
In her 40s, she developed progressive weakness of the lower limbs. At the current age of 48 years, she has difficulty climbing stairs and has to rest after a walking distance of 500 m. Toe and heel walking are not possible. Muscle strength is between 3− (hip abduction) and 4 on the MRC scale. Clinical examination shows a moderate degree of muscular atrophy in the legs and intrinsic hand muscles (Figure 2J). She has no sensory deficit. In comparison to the other affected individuals of the present study who are much younger, joint hypermobility is less pronounced, but there is marked instability in the knees and the Beighton score is still 6/9. Her skin is soft and hyperelastic and bruises easily. There is no abnormal scarring. She has myopia of 6 diopters and the cornea diameter is normal. Her weight is at the third centile and her height 3 cm below the third centile.
An older sister of P2 (indicated as P∗ [individual IV-1 in family I] in Figure 1 and Tables 1 and 2) was probably also affected by the same disorder, but clinical information was limited and DNA was not available for molecular confirmation. She was born at 35 weeks of gestation by breech delivery (weight 50th–75th centile) and presented at birth with hypotonia, feeding problems, and a weak cry. Motor milestones were delayed. She was able to walk independently at the age of 2.5 years. At the age of 7.5 years, muscle hypotonia, shoulder girdle weakness, flat feet, and mild but progressive kyphoscoliosis as well as follicular hyperkeratosis were reported. Subsequently, hypotonia and muscle weakness improved over time, and there was no recorded muscle weakness at the age of 10–12 years. There is no information available on the hearing status of P∗. She died unexpectedly of aortic rupture at the age of 12 years.
P3
P3 (individual II-4 in family II, Figure 1) is the youngest of four children of nonconsanguineous parents originating from South Tyrol (Northern Italy). The mother reported feeble fetal movements during pregnancy. The child was born after premature rupture of membranes at 36 weeks + 5 days of gestation by vaginal breech delivery. Birth weight and length were within the normal range (weight 10th–25th centile, length 10th centile). After birth she was noted to have severe hypotonia with few spontaneous movements and distally pronounced joint hypermobility. Her cry was weak and she had feeding problems. Subsequently weakness and hypotonia improved and she achieved head control by the age of 1 year. Unaided sitting was possible at the age of 2 years and she was able to walk independently at the age of 4 years. Since the age of 4 months she developed progressive kyphoscoliosis and underwent spinal surgery at the age of 4 years. At the same time bilateral sensorineural hearing impairment affecting predominantly high frequencies was diagnosed (present status: 10–25 dB HL < 4 kHz and 45–55 dB HL at 6 kHz). Her mental development has been normal.
At the present age of 11 years, her gait is characterized by marked instability in both knees (Figure 2C) and there is also pronounced joint hypermobility of the small joints (Figure 2D) (Beighton score 8/9). After walking more than 200 m, she is exhausted and needs a wheelchair. Muscle strength is between 3− (neck flexion) and 4+ on the MRC scale. Her skin is soft and hyperelastic (Figure 2K) with follicular hyperkeratosis (Figure 2M) but without abnormal scarring. Repeated echocardiographic examinations showed no signs of cardiomyopathy or dilatation of the thoracic aorta. She has no myopia and her cornea diameter is within the normal range. Currently, her weight is between the third and tenth centile and her length is 5 cm below the third centile.
P4
P4 (individual II-4 in family III, Figure 1) is the second child of nonconsanguineous French parents. She presented at birth as a floppy infant with generalized hypotonia and muscle weakness, joint hypermobility, and a left sided club foot. Motor milestones were delayed. She achieved head control at 7 months, was able to sit at the age of 12 months, and walked unaided at the age of 2 years. Early-onset kyphoscoliosis was treated with a brace from the age of 18 months. There was a strong kyphotic component associated with pathological rotation resulting in three curves of the spine. Treatment by serial casts at night stabilized the spinal deformity until the end of growth when she underwent spinal fusion operation. Muscle weakness improved over time; at the current age of 16 years, she can get up from the floor without help, walk up to 1 km, and can climb 20–30 steps of stairs without stopping. The skin on arms and legs shows follicular hyperkeratosis and there are thick and papulous lesions of the skin in the pressure areas of elbows and knees. She has no atrophic scars and doesn't report easy bruising. Her joints are hypermobile (Beighton score 6/9). At the age of 16 years, conductive hearing impairment was diagnosed. Cardiac echography showed a mildly insufficient tricuspid valve. She has myopia of 5 diopters. Her present weight is between the 10th and 25th centile and length is 1 cm below the 3rd centile.
P5
P5 (individual IV-4 in family IV, Figure 1) is the third child of first-degree cousins of Turkish origin. The mother reported reduced fetal movements. She went into spontaneous labor at 36 weeks and a premature rupture of membranes was noted. The baby was in breech presentation and was delivered by emergency caesarean section (weight 90th centile). He was a floppy infant and required nasogastric tube feeding for 9 months because of a cleft of the soft palate and a weak suck. He had micrognathia that later on spontaneously resolved. Hypotonia (Figure 2I), feeding, and motor function slowly improved with age. Sensorineural hearing impairment affecting predominantly high frequencies was diagnosed at the age of 2 years and treated with a hearing aid (present status: 35–55 dB HL ≤ 4 kHz and 90–95 dB HL ≥ 5 kHz). At the present age of 11 years, he has generalized muscle weakness with muscle strength between 3− (axial and neck muscles) and 4 on the MRC scale. He has difficulties with getting up from the floor (Gowers time of 9 s) and with climbing stairs and complains about myalgia and joint pain after walking for more than 15 min. A nonprogressive kyphosis was diagnosed after the first year of life, but so far he has not developed a scoliosis. Hypermobility of the joints (Figure 2E) (Beighton score 9/9) has led to recurrent dislocations of shoulder, elbow, knee, and hip joints. Additionally, dislocations of small finger joints cause significant difficulties with fine motor tasks such as cutting food and dressing. Wound healing is delayed, but he has no atrophic scars or keloids. He has no myopia. In contrast to the other affected individuals of the present study, he has moderate intellectual disability and uses Makaton language to combine spoken words with gestures, signs, and symbols. A brain MRI at the age of 9 years showed mild white matter atrophy. At the age of 11 years during diagnostic work-up for incontinence, a large bladder diverticulum was diagnosed by ultrasound. His present weight is at the 90th centile and length is between the 25th and 50th centile.
P6
P6 (individual II-2 in family V, Figure 1) is the first child of a nonconsanguineous German couple. She was born in the 37th week of gestation after a pregnancy complicated by polyhydramnios. Birth measurements were within the normal range (weight 25th centile, length 10th–25th centile). After birth, she presented as a floppy infant (Figure 2H) with a left sided clubfoot, retrogenia, and a median cleft of the soft palate. She had feeding problems and showed repeatedly inadequate oxygen saturation levels. She was discharged with oxygen support for acute desaturations and a feeding tube. Clubfoot and cleft palate were repaired surgically. At the age of 18 months, first signs of lumbar scoliosis were noted. Hypotonia and muscle weakness gradually improved. However, gross motor milestones continued to be delayed. At the present age of 3 years, she is unable to walk without support and has hypermobile joints (Figures 2F and 2G) (Beighton score 9/9) as well as a lumbar scoliosis (Figure 2B). Moderate sensorineural hearing impairment was diagnosed at the age of 3 years and treated with a hearing aid. The cornea diameter is normal and she has no myopia. Current weight and height measurements are within the normal range (weight between the 3rd and 10th centile, length between the 25th and 50th centile).
Radiological Findings
Original radiographic images were available for individuals P1 and P3 and radiology reports for P2, P∗, and P5. Because all affected individuals have kyphosis and/or scoliosis, the most comprehensive radiological information was available for the vertebral spine. In addition, in individuals P1 and P3, X-ray examinations of the pelvis were performed and information on X-rays of the left hand were available for P1, P2, and P∗.
Radiographs showed no signs of skeletal dysplasia. Judged by radiology, mild to moderate osteopenia was present in all subjects studied, but there was no increased bone fragility. Bone age corresponded to the chronological age. In individual P1, osteopenia is evidenced by translucent vertebral bodies in comparison to the cortical outline, deformed vertebral bodies, and trabecular prominence in femoral necks (Figure S2 available online).
Neuromuscular Findings
Results of neuromuscular investigations are summarized in Table 2.
In all affected individuals, serum creatine kinase levels were normal or only slightly elevated (range 60–300 IU/l). Nerve conduction studies were normal. Electromyography showed a myopathic pattern in adolescence and adulthood but results obtained in infancy were mostly reported as normal.
In all individuals except P∗, the first muscle biopsy was performed before the age of 2.5 years. These biopsies were taken from the quadriceps muscle. In three affected individuals, repeated biopsies were performed, preferentially during spinal surgery with biopsies from dorsal or paraspinal muscles. Specimens for light microscopy were immediately frozen and processed via standard histological and histochemical techniques. In three affected individuals (P1, P3, and P4), muscle electron microscopy was performed by standard procedures.
Histopathological features ranged from nonspecific mild myopathic changes with increased variation in muscle fiber diameter (Figures 3A and 3B) to more pronounced changes with profound fiber atrophy and proliferation of fatty tissue. Immunofluorescence labeling of type VI collagen in muscle cryosections was performed in P5 and showed normal results. Muscle histochemical studies in P1 and P3 showed irregularities of the oxidative enzymes, including areas with central activity defects in paraspinal muscle from P3 (Figure 3C); the latter changes corresponded to focal rearrangements of myofibrils in electron microscopy (Figure 3D). Likewise, electron microscopy studies of a quadriceps biopsy taken from P1 at the age of 2 years showed focal rearrangements of myofibrils with irregular Z lines (Figure 3E), and electron microscopy of a biopsy from the dorsal muscle of P4 taken at the age of 12 years showed bifurcation of sarcomeres, small zones of Z-band streaming, and some disorganized myofibrils (Figure 3F).
Whole-body muscle MRI performed in P4 at the age of 12 years showed fatty replacement of muscle tissue on T1 weighted images in the rectus femoris, vastus lateralis, and soleus muscles (Figure 3G). There was also mild involvement of neck extensor, shoulder girdle, and hip flexor muscles (not shown).
The muscle MRI of P5 at the age of 11 years showed a similar pattern with fatty infiltration in the paraspinal and leg muscles. In the thighs, the rectus femoris and vastus lateralis were predominantly affected with relative sparing of the adductor compartment. In the calves, the changes were most pronounced in the medial head of the gastrocnemius muscle and in the soleus muscle (not shown). Muscle MRI of the lower limbs in P1 at the age of 15 years was normal.
Biochemical and Ultrastructural Analyses
Total urinary pyridinoline crosslinks, lysyl pyridinoline (LP), and hydroxylysyl pyridinoline (HP) were measured in all affected individuals by HPLC as described.5,6 Dermal fibroblast cultures were established from skin biopsies of P1, P3, and P6 by routine procedures. For the semiquantitative analysis of collagen types I, III, and V, fibroblasts of P1 and P3 were maintained under standard conditions, and radiolabeled collagen samples were prepared by digestion with pepsin, precipitated by ethanol, separated on a 5% SDS-polyacrylamide gel (SDS-PAGE), and visualized by fluorography.20
A skin biopsy of individual P1 taken from the deltoid area was fixed in 3% glutaraldehyde solution for ultrastructural analysis as described.21
Molecular Genetic Studies
Genomic DNA was isolated from peripheral blood leukocytes via standard procedures (BioRobot M48, QIAGEN). A genome-wide scan was performed for 23 subjects (family I in Figure 1) with the HumanCytoSNP-12v2 BeadChip (Illumina, CA) according to the manufacturer's instructions. These arrays contain 294,747 single-nucleotide polymorphism (SNP) markers with a mean intermarker distance of about 10 kb. Raw SNP call data were processed with the Genotyping Analysis Module of GenomeStudio 1.6.3 (Illumina). Parametric linkage analysis was performed with Simwalk2.22 A trait locus mutant allele frequency of 0.0001 was assumed under an autosomal-recessive mode of inheritance with complete penetrance. Marker allele frequencies were determined out of 279 individuals of European descent from our Illumina project. Data management and cleaning was done with ALOHOMORA,23 GRR,24 PedCheck,25 Merlin,26 and Mega2.27 After erasement of Mendelian errors, non-Mendelian errors, and unlikely genotypes, residual markers were selected for a minimal spacing between neighboring markers of 100,000 bp and a minimal minor allele frequency of 0.3. Linkage analysis was calculated with 19,766 markers in marker sets of 100 SNPs. For haplotype analysis and autozygosity mapping in the candidate regions, we used the program Merlin and split the pedigree into two parts, each with a loop and cousin marriage of third order. For those regions we used all markers after normal quality control without minimal spacing.
For mutation analysis of the candidate gene FKBP14 (RefSeq accession number NM_017946.2 [mRNA], NP_060416.1 [protein]), the coding region and the flanking intron-exon boundaries were PCR amplified with primers based on the Ensembl genome browser entry for the genomic DNA (ENSG00000106080) (for primer sequences, please see Supplemental Data). PCR products were purified with ExoSAP-IT (USB Corporation, Cleveland, OH) followed by bidirectional sequencing with M13 forward and reverse primers with BigDye Terminator v3.1 Cycle Sequencing Kit on a fluorescent DNA sequencer (3730s DNA Analyzer, Applied Biosystems).
Relatives as well as 200 European control samples were tested for the detected mutations either by bidirectional sequencing (for the single base pair insertion c.362dup in exon 3) or by PCR amplification and DNA gel electrophoresis (for the 19 base pair deletion c.42_60del in exon 1, QIAXCEL system, QIAGEN) (for primer sequences, please see Supplemental Data).
Gene expression of FKBP14 was analyzed by quantitative RT-PCR (qRT-PCR) in fibroblasts from individuals P1 and P3 and two healthy controls. Total RNA was extracted with RNeasy Mini kit (QIAGEN, Hilden, Germany) according to the manufacturer's protocol. Reverse transcription was performed with 1 μg total RNA and the RevertAid Premium First Strand cDNA Synthesis kit (Fermentas, St. Leon-Rot, Germany). 1/10 volume of this reaction was used to carry out a real-time quantitative PCR amplification via Maxima SYBR Green/ROX qPCR Master Mix (Fermentas) in an ABI Prism 7000 sequence detection system (Applied Biosystems). PCR reaction was carried out by an initial denaturation step 95°C for 10 min followed by 40 cycles of 15 s denaturation at 95°C and 1 min annealing/extension at 60°C and a dissociation analysis (60°C–95°C). The CT values were calculated with sequence-detection system (SDS) software V1.2 (Applied Biosystems) and an automatic setting of base line. The amplification plots and CT values were exported from the exponential phase of PCR directly into a Microsoft Excel worksheet for further analysis.
Quantification of relative gene expression was done by comparative ΔΔCt method with HPRT1 (RefSeq accession number NM_000194.2 [mRNA]) as reference gene (for primer sequences, please see Supplemental Data).28
Western Blot and Indirect Immunofluorescence Microscopy of FKBP14 and Extracellular Matrix Proteins
For western blot analysis, a FKBP14 mouse polyclonal antibody (Abnova) and a mouse anti-α-tubulin monoclonal antibody (1:1000; sc-8035, Santa Cruz Biotechnology, Santa Cruz, CA) were used. Proteins were resolved by electrophoresis in a 12% polyacrylamide gel under denaturing conditions and blotted according to a standard protocol. As secondary antibodies, goat anti-mouse IgG conjugated to horseradish peroxidase were used. The blot was developed with ECL western blotting detection reagents (GE Healthcare, Little Chalfont, Buckinghamshire, UK) according to the manufacturer's instructions.
For immunofluorescence (IF) staining of FKBP14 in fibroblasts of affected individuals and control persons, cells grown in a 8-well chamber slide for 16–20 hr were fixed with 4% paraformaldehyde (PFA), blocked in 10% fetal bovine serum (FBS)/0.1% Triton X-100/phosphate-buffered saline (PBS), incubated for 1.5 hr with FKBP14 rabbit polyclonal antibody 1:200 (ProteinTech 15884-1-AP) and HSP47 mouse monoclonal antibody 1:1000 (Stressgen SPA-470), and subsequently exposed to goat anti-rabbit Alexa Fluor 488 1:500 (Invitrogen A11034) and goat anti-mouse Alexa Fluor 568 1:500 (Invitrogen A11031) for 1 hr. Nuclear counterstaining was done with 4′,6-diamidino-2-phenylindole (DAPI). Images were captured by a Zeiss LSM 510 Confocal imaging system.
For indirect IF microscopy of extracellular matrix proteins, fibroblast cultures of individuals P1 and P3 and two healthy controls were grown in Earle's Modified Eagle Medium (MEM) (Invitrogen-Life Technology, Carlsbad, CA) supplemented with 10% FBS (Invitrogen-Life Technology), 100 IU/ml penicillin, and 100 μg/ml streptomycin (complete MEM) without addition of ascorbic acid at 37°C in a 5% CO2 atmosphere.
Rabbit anti-fibronectin polyclonal antibody, mouse anti-tenascin monoclonal antibody (clone BC-24), recognizing all the tenascins, mouse anti-α5β1 (clone JBS5) and anti-α2β1 (clone BHA.2) integrins and mouse anti-collagen VI (clone 3C4) monoclonal antibodies, and goat anti-collagen type I, anti-collagen type III, and anti-collagen type V antibodies were from Millipore Chemicon Int. Inc. (Billerica, MA). FITC- and TRITC-conjugated goat anti-rabbit and anti-mouse and rabbit anti-goat secondary antibodies were from Calbiochem-Novabiochem INTL. The anti-thrombospondin monoclonal antibody was from NeoMarkers, Lab Vision (Fremont, CA).
To analyze the organization of fibronectin, collagen types I, III, and V, and tenascins, 1.0 × 105 cells were grown for 48 hr on glass coverslips in complete MEM, fixed in methanol, and incubated with the specific antibodies, as previously reported.29,30
To analyze the organization of collagen type VI, fibroblasts were cultured for 7 days in the presence of 0.25 mM ascorbic acid, fixed in methanol, and immunoreacted with anti-collagen type VI monoclonal antibody.
The organization of α5β1 and α2β1 integrins was analyzed on 1.0 × 105 cells grown for 48 hr in complete MEM, fixed in 3% paraformaldehyde (PFA) and 60 mM sucrose, and permeabilized in 0.5% Triton X-100. Cells were incubated for 1 hr at room temperature with 1 μg/ml anti-α5β1 and anti-α2β1 integrins monoclonal antibodies. In order to analyze the organization of thrombospondin, the cells were fixed for 10 min in 3% PFA and incubated with 1 μg/ml of the specific monoclonal antibody.
In all IF experiments, the cells were subsequently incubated for 45 min with 10 μg/ml FITC- or TRITC-conjugated anti-rabbit, anti-goat, or anti-mouse IgG. The IF signals were acquired by a CCD black/white TV camera (SensiCam-PCO Computer Optics GmbH, Germany) mounted on a Zeiss fluorescence-Axiovert microscope, and digitalized by Image Pro Plus program (Media Cybernetics, Silver Spring, MD).
Results
Molecular Analysis
Total urinary LP/HP ratios were normal in all affected individuals, thereby excluding EDS VIA.
Because no diagnosis could be established based on clinical findings, linkage analysis was performed in family I. Linkage analysis revealed three candidate loci on chromosomes 7, 11, and 16 with a maximum LOD score of 2.657 (data not shown) spanning 9,106,202 bp (between rs7781656 and rs2709800), 4,997,061 bp (between rs11607726 and rs12786091), and 3,963,702 bp (between rs13332406 and rs657181), respectively. The linkage regions were recalculated with Merlin performing haplotype analysis and autozygosity mapping without marker selection (chr7, 1083 markers; chr11, 617 markers; chr16, 696 markers) to further narrow the linkage regions. Haplotype analysis showed that the affected individuals were homozygous identical by descent (IBD) for only two linkage regions, 7p15.1 and 16q12.2. The size of the homozygous linkage interval on chromosome 7 was narrowed to 670,554 bp (flanking markers rs767430–rs2709800) and the region on chromosome 16 to 724,063 bp (flanking markers rs2110835–rs1486733). Within the linkage regions on chromosomes 7 and 16, there were in total 13 genes/open reading frames: WIPF3 (MIM 612432), SCRN1, FKBP14, PLEKHA8 (MIM 608639), C7orf41, ZNRF2 (MIM 612061), MIRN550-1, DKFZp586I1420, NOD1 (MIM 605980), LOC643911, LOC388279, IRX5 (MIM 606195), and LOC100132339 (annotation according to Build 36.3, which was the genome assembly available at the time of investigation). Considering that the disease affects connective tissue and muscle and based on the putative function and the expression profile, one major candidate gene emerged, FKBP14 (FK506 binding protein 14, 22 kDa, NCBI Gene ID: 55033), which maps to 7p15.1. FKBP14 belongs to the FK506-binding protein (FKBP) class of immunophilins, which have been implicated in catalyzing cis-trans-isomerization of peptidyl-prolyl peptide bonds and are supposed to accelerate protein folding.31
FKBP14 sequence analysis in the affected members of family I revealed a homozygous insertion of one cytosine residue within a 5C-nucleotide repeat in exon 3 (c.362dup), which produces a translational frameshift and a premature stop codon (p.Glu122Argfs∗7). The transcript was predicted to be targeted by nonsense-mediated RNA decay.
We subsequently carried out molecular genetic studies in four unrelated individuals presenting with a similar clinical phenotype (families II–V, Figure 1). Individuals P3, P4, and P5 were found to be homozygous for the c.362dup mutation, whereas P6 was compound heterozygous for c.362dup (on the maternal allele) and a 19 base pair deletion c.42_60del in exon 1 (on the paternal allele), which leads to a premature stop codon (p.Thr15∗). Neither mutation is registered in the available SNP databases, nor was it found in a panel of 200 control persons of European descent, indicating that they are not common polymorphisms. The c.362dup mutation was linked to the same haplotype in all individuals.
FKBP14 Mutation Results In Vitro in Deficiency of FKBP14
In order to determine the subcellular localization of FKBP14 in control cells and the effect of the mutations on the protein level, fibroblasts of affected individuals and healthy controls were analyzed by immunocytochemistry and confocal microscopy as well as by western blot analysis. The results of confocal microscopy are shown in Figure 4B. In control fibroblasts, FKBP14 localizes to the same subcellular compartment as HSP47, an ER-resident collagen-binding glycoprotein, confirming previous reports that FKBP14 localizes to the ER.32 In contrast, FKBP14 was undetectable in fibroblasts of affected individuals whereas HSP47 showed a normal fluorescence pattern. Absence of FKBP14 in fibroblasts of affected individuals was also confirmed by western blot analysis (Figure 4A).
Figure 4.
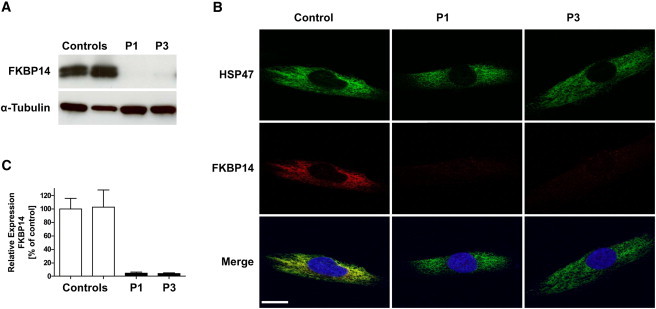
Subcellular Localization of FKBP14, Western Blot Analysis, and Nonsense-Mediated Decay
(A) Immunoblot shows absence of FKBP14 in skin fibroblasts of P1 and P3 in comparison to controls.
(B) Confocal immunofluorescence microscopy shows colocalization of FKBP14 (red) and HSP47 (green) in control fibroblasts. In fibroblasts from P1 and P3, FKBP14 is not detectable. Nuclei are counterstained by DAPI. Scale bar represents 10 μm.
(C) qRT-PCR analysis of FKBP14 in P1, P3, and two control individuals. Relative expression was determined by the ΔΔCt method with HPRT1 as reference gene. Expression of FKBP14 was strongly reduced in patient fibroblasts, indicating nonsense-mediated decay.
To determine whether the transcript harboring the c.362dup mutation undergoes nonsense-mediated RNA decay, FKBP14 transcripts were quantified by qRT-PCR on mRNA extracted from fibroblast cultures of individuals P1 and P3 (both homozygous for c.362dup) in comparison to control fibroblasts. The relative expression ratio of FKBP14 in fibroblasts homozygous for the mutation was significantly reduced (4.7% ± 0.93% in P1 and 3.9% ± 0.73% in P3 [mean ± SD], respectively; Figure 4C), indicating that mutated mRNA is targeted for degradation via nonsense-mediated decay.
FKBP14 Deficiency Leads to Enlarged ER Cisterns In Vivo and Disarray of the Extracellular Matrix In Vitro
To examine the effects of FKBP14 deficiency on collagen biosynthesis and secretion, biochemical analyses of collagens from cultured skin fibroblasts of P1 and P3 were performed. The analyses showed normal proportions and electrophoretic mobility of the α chains of collagen types I, III, and V (Figure S1, Supplemental Data).
In addition, transmission electron microscopy was performed on a skin biopsy sample of P1. The collagen fibrils showed normal contours and diameters; however, the dermal fibroblasts presented with marked enlargement of ER cisterns, which were filled with a flocculent material (Figures 3H and 3I).
In order to understand the pathophysiological consequences of FKBP14 deficiency, the organization of the main components of the extracellular matrix (ECM), i.e., collagen types I, III, V, and VI, fibronectin, tenascins, thrombospondin, and the main receptors of collagens and fibronectin, α2β1 and α5β1 integrins was investigated by indirect immunofluorescence microscopy on cultured skin fibroblasts of individuals P1 and P3 and two controls (Figure 5). Cultured skin fibroblasts of P1 and P3 showed generalized disarray for the majority of the above-mentioned proteins. In comparison to control cells, distinct extracellular networks for collagen types I and III, tenascin, and thrombospondin were missing in FKBP14-deficient cells and those for fibronectin and type VI collagen were reduced and misassembled, the only exception being type V collagen, which was organized in an extracellular network that was similar to control fibroblasts. The main receptors for collagens and fibronectin, α2β1 and α5β1 integrins, were not detectable in FKBP14-deficient cells.
Figure 5.
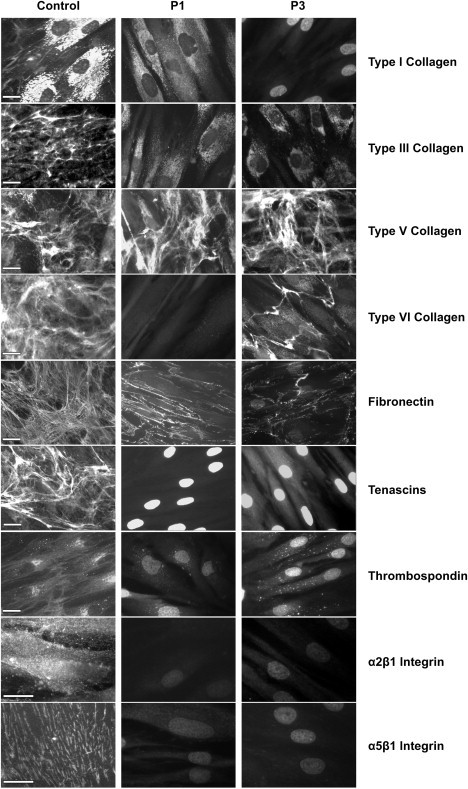
Immunofluorescence of Extracellular Matrix Proteins in Cultured Skin Fibroblasts from Individuals with FKBP14 Deficiency
Disarray of collagen types I, III, VI, fibronectin, tenascins, thrombospondin, and the integrin receptors α2β1 and α5β1 is demonstrated in FKBP14-deficient fibroblasts of P1 and P3 in comparison to control fibroblasts (one of two control cultures is shown; the scale bars represent 10 μm). In control cells, collagen type I was synthesized and mainly detected in the cytoplasm and only a few fibrils were assembled in the extracellular compartment. In FKBP14-deficient cells, collagen type I was reduced or not detectable in the cytoplasm and absent in the ECM. Collagen types III, V, and VI, fibronectin, and tenascins were assembled by control fibroblasts in distinct extracellular networks whereas FKBP14-deficient cells organized an extracellular network only for collagen type V and did not assemble a network for collagen type III and tenascins. The extracellular networks for collagen type VI and fibronectin were almost completely disorganized in FKBP14-deficient cells. In control cells, thrombospondin was detected either in the extracellular space or in the cytoplasm in the perinuclear ER; in contrast, FKBP14-deficient cells did not organize the protein in an extracellular network and only a few intracellular deposits were detected. The main receptors for collagens and fibronectin, α2β1 and α5β1 integrins, were not detectable in FKBP14-deficient cells.
Discussion
We describe an autosomal-recessive connective tissue disorder caused by mutations in FKBP14. Clinically, FKBP14-deficient EDS is characterized by the following features: (1) severe generalized hypotonia at birth with marked muscle weakness that improve in infancy; (2) early-onset progressive (kypho)scoliosis; (3) joint hypermobility without contractures; (4) hyperelastic skin with follicular hyperkeratosis, easy bruising, and occasional abnormal scarring; (5) myopathy as confirmed by muscle MRI, histology, and electron microscopy; (6) hearing impairment, which is predominantly sensorineural; and (7) a normal ratio of lysyl pyridinoline to hydroxylysyl pyridinoline (LP/HP) in urine. Occasional features that underline systemic connective tissue involvement are aortic rupture (in P∗, the probably affected sister of P2), subdural hygroma (potentially resulting from subdural bleeding or spontaneous intracranial hypotension),33 insufficiency of cardiac valves, bluish sclerae, bladder diverticula, inguinal or umbilical herniae, and premature rupture of membranes.
The course of the disease is characterized by delayed motor development with gradual improvement of hypotonia and muscle weakness during childhood. Ages of unassisted walk ranged from 2.5 years to 4 years. All affected individuals of the present study have reduced muscle strength and report difficulties in walking long distances and easy fatigability. Primary muscle disease was confirmed on muscle biopsies, which showed mild to moderate histological abnormalities by light microscopy and focal myofibrillar rearrangements and Z line anomalies by electron microscopy, albeit without a specific recognizable pattern. In addition, muscle MRI indicated fatty degeneration of muscle tissue, an observation that is typically found in myopathies and muscular dystrophies.34,35 Whether the decline in motor function that started in the only adult person of the present study in her forties is a consistent feature in the natural course of FKBP14-deficient EDS remains to be clarified.
This disorder shares many clinical features with EDS VIA and D4ST1-deficient EDS (EDS VIB) on the one hand and the collagen VI-related congenital myopathies Ullrich congenital muscular dystrophy (UCMD) and Bethlem myopathy on the other hand; however, there are also clear differences (Table 3).
Table 3.
Differential Diagnosis Based on Clinical Characteristics
| Disease | FKBP14-Deficient EDS | LH1-Deficient EDS (VIA) | D4ST1-Deficient EDS (VIB) | Collagen VI-Related Ullrich CMD |
|---|---|---|---|---|
| Skin | ||||
| Hyperelastic skin | + | + | + | – |
| Follicular hyperkeratosis | + | (+) | – | + |
| Striae distensae | – | – | – | (+) |
| Easy bruising | + | + | + | – |
| Fragile skin | (+) | + | + | (+) |
| Hypertrophic scars | ∗ | (+) | – | + |
| Atrophic scars | – | + | + | + |
| Joints/skeleton | ||||
| Joint hypermobility | + | + | + | + |
| Joint dislocations | (+) | + | + | (+) |
| Joint contractures | – | – | + | + |
| Clubfoot deformities | (+) | (+) | + | (+) |
| Progressive kyphoscoliosis | + | + | (+) | + |
| Neuromuscular | ||||
| Muscle hypotonia at birth | + | + | (+) | + |
| Delayed motor development | + | + | + | + |
| Weakness | + | + | + | ++ |
| Respiratory muscle failure | – | – | – | + |
| Vascular | ||||
| Rupture of large arteries | ∗ | § | – | – |
| Eyes | ||||
| Bluish sclerae | ∗ | (+) | + | (+) |
| Microcornea | – | (+) | (+) | – |
| Retinal detachment | – | (+) | (+) | – |
| Rupture of eye globe | – | § | – | – |
| Ears | ||||
| Hearing impairment | + | – | (+) | – |
| Stature | ||||
| Marfanoid habitus | – | + | + | – |
| Miscellaneous | ||||
| Herniae, diverticulae | + | (+) | (+) | – |
| Cleft palate/cleft soft palate | (+) | – | (+) | – |
| Inheritance | AR | AR | AR | AD/AR |
The following symbols and abbreviations are used: +, present; (+), occasionally present; §, less common but characteristic if present; –, absent; ++, progressive weakness; ∗, yet uncertain criterion in this entity; AR, autosomal-recessive; AD, autosomal-dominant.
Individuals with UCMD as well as those with FKBP14-deficient EDS present with generalized hypotonia, muscle weakness, and joint hypermobility at birth. In addition, both disorders show signs of myopathy and most individuals with UCMD also develop (kypho)scolisis.13,36 But in contrast to UCMD, FKBP14-deficient EDS does not seem to be associated with progressive contractures of large joints in particular hips, knees, and elbows13,36 whereas skin hyperextensibility, one of the cardinal features of FKBP14-deficient EDS, is not found in UCMD. A clear difference is also observed in the natural course of both disorders. In contrast to UCMD, motor function in individuals with FKPB14 deficiency improves during childhood and there is no marked decline in respiratory function.
Features that are common to EDS VIA, D4ST1-deficient EDS, and FKBP14-deficient EDS are severe hypotonia at birth with delayed motor development, joint hypermobility, progressive (kypho)scoliosis, and hyperelastic skin that bruises easily. High-frequency sensorineural hearing loss was found in five of six of the individuals with FKBP14-deficient EDS and has also been reported in D4ST1-deficient EDS.17,19,37 Talipes deformities, one of the hallmarks in D4ST1-deficient EDS,19 may occasionally also occur in FKBP14-deficient EDS. However, congenital contractures, particularly adduction-flexion contractures of thumbs, which constitute another cardinal feature of D4ST1-deficient EDS, as well as facial dysmorphisms, arachnodactyly, and wrinkled skin over the palms and fingers were not found in our cohort of individuals with FKBP14-deficient EDS.
The most significant clinical overlap exists between FKBP14-deficient EDS and EDS VIA. In addition to the above-mentioned similarities, clinical features that are common to both disorders are the potential risk for catastrophic vascular incidents and premature rupture of membranes. However, FKBP14-deficient EDS differs from EDS VIA first by the hearing impairment, which developed in most individuals of the present study during early childhood (and which to the best of our knowledge has not been reported so far in EDS VIA) and second by the absence of an abnormal LP/HP ratio in urine, which is diagnostic for EDS VIA. A third crucial point is muscle involvement in individuals with FKBP14-deficient EDS in comparison to EDS VIA. So far, studies focusing on the natural course of muscle function in EDS VIA are scarce. In one recently reported individual with EDS VIA, muscle biopsy in early infancy was unremarkable but a follow-up study at the age of 16 years showed myopathic changes and polyneuropathy.8 In a current study Rohrbach et al.38 comment on the ability to walk of 15 newly diagnosed individuals with EDS VIA, and point out that one of them (aged 16 years) had severe scoliosis, hip dysplasia, and intellectual disability and never learned to walk. It remains open whether some degree of myopathy contributed to the inability to walk in this person.
Considering the relatively small number of affected individuals with FKBP14-deficient EDS in the present study and the limited data on muscle involvement in EDS VIA, it is too early to claim that the myopathic aspect in FKBP14-deficient EDS is more pronounced than in EDS VIA. Therefore the most important distinctive features of this disorder in comparison to EDS VIA are sensorineural hearing impairment and a normal urinary LP/HP ratio. Because hearing impairment may not manifest in the first months of life, the only reliable distinctive criterion in the neonatal period is normal pyridinoline excretion.
FKBP14 encodes for a 22 kDa protein that belongs to the FK506-binding protein (FKBP)-family of peptidyl-prolyl cis-trans isomerases (PPIases). cis-trans isomerization in peptidyl-prolyl bonds is considered to be a rate-limiting step in protein folding, and especially in folding of procollagens because of their abundance of prolyl-residues.39–42 This process is accelerated by PPIases, which act as folding catalysts and sometimes also have chaperone function.43 So far, available information on the function of FKBP14 has been scarce. According to electronic annotation, FKBP14 is predicted to have a PPIase FKBP-type domain and to carry two EF-hand motifs and an ER-retention signal (UniProtKB/SwissProt).44 In fact, ER localization of FKBP14 has been demonstrated by Raykhel et al.32 and was also confirmed by our experiments in human control fibroblasts (Figure 4B). The mutations in FKBP14 found in the present study lead to FKBP14 deficiency in the ER. Electron microscopy of a skin biopsy of P1 showed normal shape and diameter of the collagen fibrils, and biochemical analysis of collagens showed normal distribution and electrophoretic migration of the α chains of collagen types I, III, and V, respectively (Figure S1, Supplemental Data). Moreover, the relative amount of collagen types I, III, and V in the medium and the cell layers of FKBP14-deficient fibroblasts was comparable to that of the controls. However, on electron microscopy the ER cisterns of skin fibroblasts were dilated and filled with flocculent material. An influence of FKBP14 on extracellular matrix components is suggested by immunofluorescence experiments on FKBP14-deficient skin fibroblasts, which showed disturbed distribution and assembly of several ECM components. In particular, a disarray of the extracellular network of collagens type I and type III and fibronectin as well as their receptors was observed. This is a common trait of in-vitro-cultured fibroblasts derived from individuals with different types of EDS, irrespective of the gene mutated.29,30 Indeed, mutations in the genes for collagen types I, III, and V as well as PLOD1, which is involved in the maturation of collagen molecules, affect the assembly of various components of the ECM in EDS skin fibroblasts. Matrix assembly is considered to be a step-wise process that is usually initiated by ECM glycoproteins binding to cell surface receptors such as fibronectin dimers binding to integrin receptors. Once assembled, the fibronectin matrix contributes to the assembly of other ECM components.45 Hence, impaired assembly of one ECM component may entail disturbed assembly of others. Based on these considerations, FKBP14 might act on one or several components of the ECM.
FKBP10 (MIM 607063), another ER-resident member of the FKBP family of PPIases, has recently been shown to interact with the collagen triple helix and to act as a chaperone.43 Loss-of-function mutations in FKBP10 affect secretion of type I collagen and lead to an osteogenesis imperfecta and Bruck syndrome phenotype (MIM 610968).46–48 It is tempting to speculate that FKBP14 might also act as a folding catalyst or chaperone for one or more components of the extracellular matrix and that ER stress in response to the accumulation of misfolded proteins might play a role in the pathophysiology of FKBP14-deficient EDS. Biochemical analysis in FKBP14-deficient cells showed normal proportions and electrophoretic mobility of the α chains of collagen types I, III, and V. However, altered secretion of other extracellular matrix components such as fibronectin, collagen type VI, and integrin subunits cannot be excluded. More functional data are needed to unravel the exact pathogenetic mechanisms of FKBP14 deficiency.
Remarkably, a 1 base pair insertion of a cytosine residue within a 5C-nucleotide stretch in exon 3 was found in all affected individuals on at least one allele. Analysis of flanking SNP markers showed that the 1 base pair insertion is linked to the same haplotype in all mutation carriers. Despite the geographically diverse origin of the individuals involved in this study, it is possible that this mutation traces back to the same founder event. However, a recurrent mutation resulting from replication slippage in the polycytidine tract cannot be excluded.
In conclusion, we report an autosomal-recessive variant of EDS caused by mutations in FKBP14. The wide spectrum of clinical features in this condition may be due to a disturbance of protein folding in the ER affecting one or more components of the extracellular matrix in various body parts and organs such as joints, ligaments, skin, vessels, ear, eye, and muscle. FKBP14 mutation analysis should be considered in all individuals with apparent kyphoscoliotic type of EDS and normal pyridinoline excretion, in particular in conjunction with sensorineural hearing impairment.
Acknowledgments
We express our gratitude to all families who participated in this study. We thank Marius Kraenzlin for urinary pyridinoline analysis, Ingrid Hausser-Siller and Patricie Paesold-Burda for helpful discussions, Angelika Schwarze for skilful technical assistance, Rudolf Funke for providing photographs of individual P6, Beril Talim, Haluk Topaloglu, and Julia Wanschitz for providing muscle biopsy data, and Dan Lipsker for skin examination results in individual P4. This work was supported by a grant from the Gottfried und Julia Bangerter-Rhyner Stiftung to C.G. and M.R. The support of the Muscular Dystrophy Campaign Centre grant to the Dubowitz Neuromuscular Centre is gratefully acknowledged. F.M. is supported by the Great Ormond Street Hospital Children's Charity.
Contributor Information
Matthias Baumann, Email: matthias.baumann@uki.at.
Christine Fauth, Email: christine.fauth@i-med.ac.at.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl, http://www.ensembl.org/index.html
Entrez Gene, http://www.ncbi.nlm.nih.gov/sites/entrez
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Steinmann B., Royce P.M., Superti-Furga A. The Ehlers-Danlos syndrome. In: Royce P.M., Steinmann B., editors. Connective Tissue and Its Heritable Disorders. 2nd ed. Wiley-Liss; New York: 2002. pp. 431–523. [Google Scholar]
- 2.Beighton P., De Paepe A., Steinmann B., Tsipouras P., Wenstrup R.J., Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK) Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Am. J. Med. Genet. 1998;77:31–37. doi: 10.1002/(sici)1096-8628(19980428)77:1<31::aid-ajmg8>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 3.Kivirikko K.I., Pihlajaniemi T. Collagen hydroxylases and the protein disulfide isomerase subunit of prolyl 4-hydroxylases. Adv. Enzymol. Relat. Areas Mol. Biol. 1998;72:325–398. doi: 10.1002/9780470123188.ch9. [DOI] [PubMed] [Google Scholar]
- 4.Steinmann B., Eyre D.R., Shao P. Urinary pyridinoline cross-links in Ehlers-Danlos syndrome type VI. Am. J. Hum. Genet. 1995;57:1505–1508. [PMC free article] [PubMed] [Google Scholar]
- 5.Giunta C., Randolph A., Al-Gazali L.I., Brunner H.G., Kraenzlin M.E., Steinmann B. Nevo syndrome is allelic to the kyphoscoliotic type of the Ehlers-Danlos syndrome (EDS VIA) Am. J. Med. Genet. A. 2005;133A:158–164. doi: 10.1002/ajmg.a.30529. [DOI] [PubMed] [Google Scholar]
- 6.Kraenzlin M.E., Kraenzlin C.A., Meier C., Giunta C., Steinmann B. Automated HPLC assay for urinary collagen cross-links: effect of age, menopause, and metabolic bone diseases. Clin. Chem. 2008;54:1546–1553. doi: 10.1373/clinchem.2008.105262. [DOI] [PubMed] [Google Scholar]
- 7.Yiş U., Dirik E., Chambaz C., Steinmann B., Giunta C. Differential diagnosis of muscular hypotonia in infants: the kyphoscoliotic type of Ehlers-Danlos syndrome (EDS VI) Neuromuscul. Disord. 2008;18:210–214. doi: 10.1016/j.nmd.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 8.Voermans N.C., Bönnemann C.G., Lammens M., van Engelen B.G., Hamel B.C. Myopathy and polyneuropathy in an adolescent with the kyphoscoliotic type of Ehlers-Danlos syndrome. Am. J. Med. Genet. A. 2009;149A:2311–2316. doi: 10.1002/ajmg.a.32997. [DOI] [PubMed] [Google Scholar]
- 9.Voermans N.C., van Alfen N., Pillen S., Lammens M., Schalkwijk J., Zwarts M.J., van Rooij I.A., Hamel B.C., van Engelen B.G. Neuromuscular involvement in various types of Ehlers-Danlos syndrome. Ann. Neurol. 2009;65:687–697. doi: 10.1002/ana.21643. [DOI] [PubMed] [Google Scholar]
- 10.Lampe A.K., Bushby K.M. Collagen VI related muscle disorders. J. Med. Genet. 2005;42:673–685. doi: 10.1136/jmg.2002.002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirschner J., Hausser I., Zou Y., Schreiber G., Christen H.J., Brown S.C., Anton-Lamprecht I., Muntoni F., Hanefeld F., Bönnemann C.G. Ullrich congenital muscular dystrophy: connective tissue abnormalities in the skin support overlap with Ehlers-Danlos syndromes. Am. J. Med. Genet. A. 2005;132A:296–301. doi: 10.1002/ajmg.a.30443. [DOI] [PubMed] [Google Scholar]
- 12.Voermans N.C., Bönnemann C.G., Huijing P.A., Hamel B.C., van Kuppevelt T.H., de Haan A., Schalkwijk J., van Engelen B.G., Jenniskens G.J. Clinical and molecular overlap between myopathies and inherited connective tissue diseases. Neuromuscul. Disord. 2008;18:843–856. doi: 10.1016/j.nmd.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 13.Nadeau A., Kinali M., Main M., Jimenez-Mallebrera C., Aloysius A., Clement E., North B., Manzur A.Y., Robb S.A., Mercuri E., Muntoni F. Natural history of Ullrich congenital muscular dystrophy. Neurology. 2009;73:25–31. doi: 10.1212/WNL.0b013e3181aae851. [DOI] [PubMed] [Google Scholar]
- 14.Cadle R., Hames B., Waziri M. Phenotypic Ehlers-Danlos type VI with normal lysyl hydroxylase activity and macrocephaly. Am. J. Hum. Genet. 1985;37:A48. [Google Scholar]
- 15.Royce P.M., Moser U., Steinmann B. Ehlers-Danlos syndrome type VI with normal lysyl hydroxylase activity cannot be explained by a defect in cellular uptake of ascorbic acid. Matrix. 1989;9:147–149. doi: 10.1016/s0934-8832(89)80033-2. [DOI] [PubMed] [Google Scholar]
- 16.Dündar M., Müller T., Zhang Q., Pan J., Steinmann B., Vodopiutz J., Gruber R., Sonoda T., Krabichler B., Utermann G. Loss of dermatan-4-sulfotransferase 1 function results in adducted thumb-clubfoot syndrome. Am. J. Hum. Genet. 2009;85:873–882. doi: 10.1016/j.ajhg.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malfait F., Syx D., Vlummens P., Symoens S., Nampoothiri S., Hermanns-Lê T., Van Laer L., De Paepe A. Musculocontractural Ehlers-Danlos Syndrome (former EDS type VIB) and adducted thumb clubfoot syndrome (ATCS) represent a single clinical entity caused by mutations in the dermatan-4-sulfotransferase 1 encoding CHST14 gene. Hum. Mutat. 2010;31:1233–1239. doi: 10.1002/humu.21355. [DOI] [PubMed] [Google Scholar]
- 18.Miyake N., Kosho T., Mizumoto S., Furuichi T., Hatamochi A., Nagashima Y., Arai E., Takahashi K., Kawamura R., Wakui K. Loss-of-function mutations of CHST14 in a new type of Ehlers-Danlos syndrome. Hum. Mutat. 2010;31:966–974. doi: 10.1002/humu.21300. [DOI] [PubMed] [Google Scholar]
- 19.Shimizu K., Okamoto N., Miyake N., Taira K., Sato Y., Matsuda K., Akimaru N., Ohashi H., Wakui K., Fukushima Y. Delineation of dermatan 4-O-sulfotransferase 1 deficient Ehlers-Danlos syndrome: observation of two additional patients and comprehensive review of 20 reported patients. Am. J. Med. Genet. A. 2011;155A:1949–1958. doi: 10.1002/ajmg.a.34115. [DOI] [PubMed] [Google Scholar]
- 20.Steinmann B., Rao V.H., Vogel A., Bruckner P., Gitzelmann R., Byers P.H. Cysteine in the triple-helical domain of one allelic product of the alpha 1(I) gene of type I collagen produces a lethal form of osteogenesis imperfecta. J. Biol. Chem. 1984;259:11129–11138. [PubMed] [Google Scholar]
- 21.Vogel A., Holbrook K.A., Steinmann B., Gitzelmann R., Byers P.H. Abnormal collagen fibril structure in the gravis form (type I) of Ehlers-Danlos syndrome. Lab. Invest. 1979;40:201–206. [PubMed] [Google Scholar]
- 22.Sobel E., Lange K. Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am. J. Hum. Genet. 1996;58:1323–1337. [PMC free article] [PubMed] [Google Scholar]
- 23.Rüschendorf F., Nürnberg P. ALOHOMORA: a tool for linkage analysis using 10K SNP array data. Bioinformatics. 2005;21:2123–2125. doi: 10.1093/bioinformatics/bti264. [DOI] [PubMed] [Google Scholar]
- 24.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. GRR: graphical representation of relationship errors. Bioinformatics. 2001;17:742–743. doi: 10.1093/bioinformatics/17.8.742. [DOI] [PubMed] [Google Scholar]
- 25.O'Connell J.R., Weeks D.E. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abecasis G.R., Cherny S.S., Cookson W.O.C., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 27.Mukhopadhyay N., Almasy L., Schroeder M., Mulvihill W.P., Weeks D.E. Mega2: data-handling for facilitating genetic linkage and association analyses. Bioinformatics. 2005;21:2556–2557. doi: 10.1093/bioinformatics/bti364. [DOI] [PubMed] [Google Scholar]
- 28.Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zoppi N., Gardella R., De Paepe A., Barlati S., Colombi M. Human fibroblasts with mutations in COL5A1 and COL3A1 genes do not organize collagens and fibronectin in the extracellular matrix, down-regulate α2β1 integrin, and recruit αvβ3 instead of α5β1 integrin. J. Biol. Chem. 2004;279:18157–18168. doi: 10.1074/jbc.M312609200. [DOI] [PubMed] [Google Scholar]
- 30.Zoppi N., Barlati S., Colombi M. FAK-independent αvβ3 integrin-paxillin-EGFR complexes rescue from anoikis matrix-defective fibroblasts. Biochim. Biophys. Acta. Mol. Cell Res. 2008;1783:1177–1188. doi: 10.1016/j.bbamcr.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 31.Galat A. Peptidylprolyl cis/trans isomerases (immunophilins): biological diversity—targets—functions. Curr. Top. Med. Chem. 2003;3:1315–1347. doi: 10.2174/1568026033451862. [DOI] [PubMed] [Google Scholar]
- 32.Raykhel I., Alanen H., Salo K., Jurvansuu J., Nguyen V.D., Latva-Ranta M., Ruddock L. A molecular specificity code for the three mammalian KDEL receptors. J. Cell Biol. 2007;179:1193–1204. doi: 10.1083/jcb.200705180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schievink W.I., Gordon O.K., Tourje J. Connective tissue disorders with spontaneous spinal cerebrospinal fluid leaks and intracranial hypotension: a prospective study. Neurosurgery. 2004;54:65–70. doi: 10.1227/01.neu.0000097200.18478.7b. discussion 70–71. [DOI] [PubMed] [Google Scholar]
- 34.Mercuri E., Jungbluth H., Muntoni F. Muscle imaging in clinical practice: diagnostic value of muscle magnetic resonance imaging in inherited neuromuscular disorders. Curr. Opin. Neurol. 2005;18:526–537. doi: 10.1097/01.wco.0000183947.01362.fe. [DOI] [PubMed] [Google Scholar]
- 35.Wattjes M.P., Kley R.A., Fischer D. Neuromuscular imaging in inherited muscle diseases. Eur. Radiol. 2010;20:2447–2460. doi: 10.1007/s00330-010-1799-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bönnemann C.G. The collagen VI-related myopathies: muscle meets its matrix. Nat. Rev. Neurol. 2011;7:379–390. doi: 10.1038/nrneurol.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kosho T., Takahashi J., Ohashi H., Nishimura G., Kato H., Fukushima Y. Ehlers-Danlos syndrome type VIB with characteristic facies, decreased curvatures of the spinal column, and joint contractures in two unrelated girls. Am. J. Med. Genet. A. 2005;138A:282–287. doi: 10.1002/ajmg.a.30965. [DOI] [PubMed] [Google Scholar]
- 38.Rohrbach M., Vandersteen A., Yiş U., Serdaroglu G., Ataman E., Chopra M., Garcia S., Jones K., Kariminejad A., Kraenzlin M. Phenotypic variability of the kyphoscoliotic type of Ehlers-Danlos syndrome (EDS VIA): clinical, molecular and biochemical delineation. Orphanet J. Rare Dis. 2011;6:46. doi: 10.1186/1750-1172-6-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brandts J.F., Halvorson H.R., Brennan M. Consideration of the possibility that the slow step in protein denaturation reactions is due to cis-trans isomerism of proline residues. Biochemistry. 1975;14:4953–4963. doi: 10.1021/bi00693a026. [DOI] [PubMed] [Google Scholar]
- 40.Schmid F.X., Baldwin R.L. Acid catalysis of the formation of the slow-folding species of RNase A: evidence that the reaction is proline isomerization. Proc. Natl. Acad. Sci. USA. 1978;75:4764–4768. doi: 10.1073/pnas.75.10.4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steinmann B., Bruckner P., Superti-Furga A. Cyclosporin A slows collagen triple-helix formation in vivo: indirect evidence for a physiologic role of peptidyl-prolyl cis-trans-isomerase. J. Biol. Chem. 1991;266:1299–1303. [PubMed] [Google Scholar]
- 42.Schmid F.X., Mayr L.M., Mücke M., Schönbrunner E.R. Prolyl isomerases: role in protein folding. Adv. Protein Chem. 1993;44:25–66. doi: 10.1016/s0065-3233(08)60563-x. [DOI] [PubMed] [Google Scholar]
- 43.Ishikawa Y., Vranka J., Wirz J., Nagata K., Bächinger H.P. The rough endoplasmic reticulum-resident FK506-binding protein FKBP65 is a molecular chaperone that interacts with collagens. J. Biol. Chem. 2008;283:31584–31590. doi: 10.1074/jbc.M802535200. [DOI] [PubMed] [Google Scholar]
- 44.Schiene-Fischer C., Aumüller T., Fischer G. Peptide bond cis/trans isomerases: a biocatalysis perspective of conformational dynamics in proteins. Top. Curr. Chem. 2011 doi: 10.1007/128_2011_151. in press. Published online May 20, 2011. 10.1007/128_2011_151. [DOI] [PubMed] [Google Scholar]
- 45.Singh P., Carraher C., Schwarzbauer J.E. Assembly of fibronectin extracellular matrix. Annu. Rev. Cell Dev. Biol. 2010;26:397–419. doi: 10.1146/annurev-cellbio-100109-104020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alanay Y., Avaygan H., Camacho N., Utine G.E., Boduroglu K., Aktas D., Alikasifoglu M., Tuncbilek E., Orhan D., Bakar F.T. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010;86:551–559. doi: 10.1016/j.ajhg.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shaheen R., Al-Owain M., Sakati N., Alzayed Z.S., Alkuraya F.S. FKBP10 and Bruck syndrome: phenotypic heterogeneity or call for reclassification? Am. J. Hum. Genet. 2010;87:306–307. doi: 10.1016/j.ajhg.2010.05.020. author reply 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kelley B.P., Malfait F., Bonafe L., Baldridge D., Homan E., Symoens S., Willaert A., Elcioglu N., Van Maldergem L., Verellen-Dumoulin C. Mutations in FKBP10 cause recessive osteogenesis imperfecta and Bruck syndrome. J. Bone Miner. Res. 2011;26:666–672. doi: 10.1002/jbmr.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.