

Abstract
Differentiation and maintenance of recirculating effector memory CD8 T cells (TEM) depends on prolonged cognate antigen stimulation. Whether similar pathways of differentiation exist for recently identified tissue-resident effector memory T cells (TRM), which contribute to rapid local protection upon pathogen re-exposure, is unknown. Memory CD8αβ+ T cells within small intestine epithelium are well-characterized examples of TRM and they maintain a long-lived effector-like phenotype that is highly suggestive of persistent antigen stimulation. This study sought to define the sources and requirements for prolonged Ag-stimulation in programming this differentiation state, including local stimulation via cognate or cross-reactive antigens derived from pathogens, microbial flora, or dietary proteins. Contrary to expectations, we found that prolonged cognate Ag-stimulation was dispensable for intestinal TRM ontogeny. In fact, chronic antigenic stimulation skewed differentiation away from the canonical intestinal T cell phenotype. Resident memory signatures, CD69 and CD103, were expressed in many non-lymphoid tissues including intestine, stomach, kidney, reproductive tract, pancreas, brain, heart, and salivary gland, and could be driven by cytokines. Moreover, TGFβ driven CD103 expression was required for TRM maintenance within intestinal epithelium in vivo. Thus, induction and maintenance of long-lived effector-like intestinal TRM differed from classic models of TEM ontogeny, and were programmed through a novel location-dependent pathway that was required for the persistence of local immunological memory.
Introduction
Effector memory CD8 T cells (CD8 TEM) comprise a heterogenous population that is excluded from lymph node entry through high endothelial venules due to the absence of CD62L or CCR7 expression1,2. Numerous studies have related memory T cell quality and homing potential to protective immunity, thus driving efforts to understand the regulation of memory T cell differentiation state. The regulation of CD8 TEM ontogeny has been extensively studied on recirculating TEM populations isolated from blood, the red pulp of spleen, or non-lymphoid organs such as the liver. This work has revealed a central role for prolonged or potent cognate antigen stimulation in the programming of TEM3. For instance, acute infections that are characterized by repetitive or extended durations of Ag-stimulation favor TEM differentiation4–7. In this context, recirculating TEM may be transient, and either convert to central memory T cells (TCM) or die weeks to months following Ag clearance8–11. In contrast, persistent infections that result in repetitive antigen exposure result in recirculating TEM that may be maintained indefinitely at the population level, although maintenance may be dependent upon Ag persistence10,12.
Recent work has revealed the existence of stable populations of effector memory CD8 T cells that are non-recirculating within intestinal epithelium, the CNS, the skin epidermis, and potentially the salivary gland and a subset of cells within the respiratory mucosa13–21. This subset of TEM has been designated T resident memory (TRM). TRM populations may be very long-lived; for example, mouse and non-human primate studies reported minimal decay over ~1–2y11,22. In addition to lacking CD62L expression, TRM populations are characterized by CD69 expression and typically CD103 expression15,16,18,19,23. CD69 expression is often interpreted as a surrogate for recent TCR engagement and recent studies have proposed that CD103 expression by TRM is induced or maintained by local cognate-Ag15,19. CD8αβ+ TRM that reside within the epithelium of the small intestine (intraepithelial lymphocytes, IELs) have been particularly well characterized and uniformly bear numerous other indications of recent or even ongoing activation, including constitutive retention of lytic function and impaired proliferation potential, and appear similar to CD8 T cells in spleen that are specific for chronic viral infections23–26. This phenotype is compatible with Ag-driven models of TEM differentiation, although the potential source of antigen is unclear27–31. One explanation is that the intestine-specific differentiation state is regulated by continued local stimulation via cognate or cross-reactive antigens derived from pathogens, microbial flora or dietary proteins. For instance, the human gut lumen is colonized with up to 1014 microbes and the average American consumes >30Kg of protein/y; thus intestinal T cells might be exposed to a tremendous diversity of antigenic peptides32,33. Reports indicate that IELs are reduced in number and exhibit a restricted TCRβ repertoire when raised under germ-free conditions34,35 or ectopically transplanted and rendered free from lumenal antigens36. Mice reared on an antigen-minimized diet also exhibit reductions in TCRαβ SI IEL, suggesting that bacterial and food-derived antigens influence the intestinal immune system37. When splenocytes are transferred to SCID mice, CD8 T cells populate the small intestinal epithelium and acquire effector-like properties, which was interpreted to add additional support to the hypothesis that IEL are undergoing a response to lumenal antigens38. Likewise, radiolabeling experiments indicated that blasting, not resting, thoracic duct lymphocytes migrated most efficiently to the intestinal mucosa39. Collectively, these observations are consistent with the hypothesis that recent or constitutive stimulation by antigen may contribute to the activated-like differentiation state of SI IEL.
This study employed a variety of complementary approaches to test the requirement for prolonged Ag-stimulation in programming or maintaining TRM within the intestine. In contrast to expectations, we found that prolonged cognate Ag-stimulation was dispensable for TRM differentiation among intestinal IEL and provide evidence that the characteristic CD103+/CD69+ TRM phenotype was present among a subset of TEM distributed within numerous host tissues, including pancreas, stomach, kidney, and heart. In contrast to classical models of recirculating TEM ontogeny, these findings support the hypothesis that TRM differentiation and maintenance is dependent on tissue microenvironment rather than prolonged Ag-stimulation.
Materials and Methods
Mice and infections
All mice were used in accordance with the Institutional Animal Care and Use Committees guidelines at the University of Minnesota or the University of Pennsylvania. For assessment of LCMV-specific P14 responses, splenocytes containing 9×104 naïve Thy1.1+ transgenic P14 CD8 T cells were isolated from naïve Thy1.1+ P14 transgenic mice52 maintained in our colony and transferred i.v. into C57BL/6J recipients. The following day, recipients were infected with 2×105 pfu LCMV Armstrong i.p. Where indicated, C57Bl/6J mice were infected with 2×105 pfu LCMV Armstrong i.p. or 2×106 pfu LCMV clone 13 i.v. without transfer of P14 CD8 T cells.
To assess the role of TGFβ in the adoption of canonical IEL phenotype, 5×104 WT and 5×104 dnTGFβRII OT-I cells 46,53 were co-transferred into naïve C56BL/6J recipients to be infected the following day with 1×106 pfu VSVova i.v. Alternatively, 5×104 WT and 5×104 dnTGFβRII P14 cells were co-transferred into naïve C56BL/6J recipients to be infected the following day with 2×105 pfu LCMV (Armstrong strain) i.p.
To assess the role of CD103 in the generation and maintenance of IEL memory, 5×104 WT and 5×104 CD103-deficient P14 cells were co-transferred into naïve sex-matched C56BL/6J recipients48. The following day, recipients were infected with 2×105 pfu LCMV (Armstrong strain) i.p. For histological analysis, experiments were carried out in a similar fashion, however WT and CD103-deficient P14 cells were transferred into separate recipients.
To assess the role of antigen in expression of the IEL phenotype, we utilized transgenic mice expressing GFP from the gene Nr4a1 (Nur77) locus41. For α-CD3 stimulation, Nur77 mice were given 50μg of α-CD3 antibody i.v. and analyzed 16 hours later. Alternatively, OT-I were transferred into 232–4 transgenic mice, that express ovalbumin under the control of the IFAPB promoter42.
For assessment of phenotype in RAG−/− mice, 1×106 Thy1.1+ P14 or CD45.1+ OT-I were transferred into conventionally housed or germ-free C57Bl/6J mice, as indicated. Experiments involving germ-free mice were performed at the UPENN utilizing mice obtained from the Penn Gnotobiotic Mouse Facility. Germ-free mice were sterilely maintained in plastic isolator units and fed autoclaved LabDiet5021 mouse chow (LabDiet) and autoclaved water.
Isolation and phenotyping of donor CD8 T cells
For isolation of SI IEL, the small intestine was removed, Peyer's Patches were excised, and the intestine was cut longitudinally and then laterally into 1cm pieces. Gut pieces and stomachs were incubated with 15.4mg/ml dithioerythritol in 10% 1xHBSS/Hepes Bicarbonate (30 min at 37°C, stirring at 200rpm) to remove IEL. For LPL isolation, gut and stomach pieces were further treated with 100U/ml Type I Collagenase (Worthington) in 5% RPMI 1640/2mM MgCl2/2mM CaCl2 (45 min at 37°C, stirring at 200rpm). Brains, pancreata, hearts, and kidneys were removed and cut into small pieces prior to treatment with 100U/ml Type I Collagenase (Worthington) in 5% RPMI 1640/2mM MgCl2/2mM CaCl2 (45 min at 37°C, stirring at 200rpm). For isolation of FRT, the uterine horns, cervix, and vaginal tissue were resected and cut into small pieces prior to treatment with 100U/ml Type IV Collagenase (Sigma) 5% RPMI 1640/2mM MgCl2/2mM CaCl2 (45 min at 37°C, stirring at 200rpm). Lymphocytes from intestine, stomach, brain, and FRT were purified on a 44/67% Percoll gradient (800xg at 23°C for 20 min).
In-vitro stimulation assays
The medium used for all cell cultures was RPMI 1640 supplemented with 10% FCS, 2mM L-glutamine, 100U/ml penicillin, 100mg/ml streptomycin, 0.25mg/ml Amphotericin B and 50μ-M 2-ME (RP-10). Single-cell splenic suspensions from intact naïve P14, day 4.5 or memory P14/B6 chimeras were placed in flat-bottom microtiter wells in 200 ml of RP-10 at 7×105 cells/well. Where indicated, cultures were supplemented with the following cytokines: human TGF-β1 at 10ng/mL, mouse IL-23 and IL-33 at 100ng/mL (R&D Systems); murine type I IFN at 5000 U/ml (PBL Biomedical Laboratories); mouse TNFα at 125ng/mL, mouse IL-6 and IL-1β at 100ng/mL, and IFNγ at 2000U/mL (eBioscience). Where indicated, GP33–41 peptide was added at 0.1 mg/mL. Cells were then incubated for 40–60 hours as described and stained with Live/Dead Fixable Aqua per manufacturer's directions (Invitrogen) prior to surface staining to exclude dead cells from the analysis.
Cryopreservation, immunofluorescence and microscopy
Tissues were snap frozen in OCT, cut to 7–8μm thicknesses and fixed for 20 seconds in acetone. Sections were then stained with directly conjugated or antibody pairs purchased from BioLegend, Invitrogen or Acris and counterstained with DAPI. Immunofluorescence microscopy was performed using a Leica DM5500 B microscope. Separate images taken with a 20x objective were collected for each channel and overlaid to obtain a multicolor image. Final image processing was performed using Fiji and Adobe Photoshop software.
In-situ quantification of virus-specific donor CD8 T cells and nucleated cells
P14 cells were identified as Thy1.1+ single positive cell-shaped events of lymphocyte size surrounding a demarcated DAPI positive nucleus. P14s containing nuclei on the luminal side or lamina propria side of the collagen IV+ basement membrane were assigned as IEL or LPL respectively. E-cadherin and collagen IV staining was used to confirm the luminal side of the intestinal basement membrane. Raw TIFF files were imported into Fiji, background was subtracted using a 10μ-m rolling ball radius and DAPI filled nuclei were enumerated using the find maxima function over an average of three threshold settings empirically set to reproduce with best fidelity hand-counted results within a series of spot-checked frames. Cells residing within Peyer's patches were masked out of the analysis.
Results
Phenotype of intestinal resident TEM
To investigate the phenotype of CD8αβ T cells that are first primed against pathogens in secondary lymphoid organs and then disseminate to SI epithelium, monoclonal P14 Thy1.1+ naïve CD8 T cells specific for LCMV were transferred to naïve C57Bl/6J mice. The next day, mice were infected with the Armstrong strain of LCMV and lymphocytes were isolated from spleen and SI epithelium at various time points. Our previous studies indicated that migration of P14 to SI IEL occurs most efficiently during the early stages of the response (D4.5 through D7), after which they become resident with minimal evidence for re-circulation13. As shown in figure 1A–C, virus-specific CD8αβ+ SI IEL contrasted starkly from their clonal counterparts in the spleen because they gradually acquired and maintained an effector-like phenotype and did not upregulate CD62L.
Figure 1. Virus-specific CD8αβ T cells adopt a recently stimulated phenotype after migration to SI epithelium.

(A) Phenotype of gp33-specific Thy1.1+ P14 CD8 T cells in spleen and SI IEL on the indicated days after LCMV Armstrong infection. All plots are gated on Thy1.1+ lymphocytes. (B) Kinetics of CD62L expression among P14 in spleen and SI IEL after LCMV infection. ***p<0.001, Student's unpaired t test. Error bars indicated SEM. n=5 per time point. (C) Phenotype of gp33-specific P14 CD8 T cells (top row is spleen, bottom row is SI IEL) 37 days after LCMV Armstrong infection. Phenotypes are representative of > 10 mice. (D&E) Mice were infected with the Armstrong or Cl-13 strains of LCMV, and 35 days later, the phenotype of H-2Db/gp33 MHC I tetramer+ splenocytes was compared. (D) Gated on lymphocytes, and (E) gated on CD8+ H-2Db/gp33 MHC I tetramer+ lymphocytes.
To determine whether prolonged Ag-stimulation could induce or maintain effector-like SI IEL phenotypic signatures (Fig. 1A) among CD8 T cells within the spleen, C57Bl/6 mice were infected with the Cl-13 strain of LCMV, which causes a persistent infection. As a control, mice were infected with the Armstrong strain of LCMV, which is cleared from spleen within ~1 week. 35 days later, endogenous LCMV specific CD8 T cells were identified by H-2Db/gp33 tetramer staining (Fig. 1D). In contrast to acute LCMV Armstrong infection, persistent LCMV Cl-13 infection results in a subset of virus specific CD8 T splenocytes that express decreased levels of CD44, low levels of Ly6C, and elevated levels of CD69 and granzyme B (Fig. 1E). These data confirm and extend earlier observations that SI IEL adopt phenotypic signatures indicative of ongoing Ag stimulation in situ, and suggest that many of these markers can be induced outside of the intestinal mucosa in the context of a chronic viral infection23,26.
Role of Ag in the induction and maintenance of IEL phenotype
To interrogate the role of foreign antigen in programming or maintaining phenotypic signatures of SI IEL, we wished to localize Ag-specific CD8 T cells to the SI epithelium in the absence of viral infection. However, neither naïve nor memory CD8 T cells migrate to SI epithelium upon intravenous transfer13. To localize Ag-specific CD8 T cells to the SI epithelium in the absence of infection, we tested whether the `emptiness-driven' proliferation induced among naïve monoclonal CD8 T cells upon transfer to lymphopenic mice would be sufficient to induce CD8 T cell migration to the SI epithelium. 1×106 Thy1.1+ OT-I×RAG−/− CD8 T cells (specific for the SIINFEKL peptide from chicken ovalbumin) were transferred to RAG−/− C57Bl/6 mice (Fig. 2A). It should be noted that mouse chow did not contain chicken ovalbumin. 60 days later, spleens and IELs were harvested and analyzed for the presence of donor OT-I CD8 T cells via staining with MHC I tetramers and confirmed with Thy1.1 staining. Remarkably, homeostatic proliferation in lymphopenic mice was sufficient to induce intestinal homing and OT-I IEL adopted all the cardinal features of effector-like virus-specific IEL (as in Fig. 1). Similar results were observed following transfer of naïve P14 CD8 T cells to RAG−/− mice, suggesting that local induction of an effector-like phenotype was not restricted to ovalbumin-specific CD8 T cells (data not shown). These data ruled out the possibility that occult intestinal infection with LCMV Armstrong was responsible for regulating the SI IEL differentiation state observed in Fig. 1.
Figure 2. Induction and maintenance of activated phenotype is Ag-independent.

(A) Naïve CD45.1+ OT-I × RAG−/− CD8 T cells were transferred to RAG−/− C57Bl/6 mice. 60 days later, OT-I were isolated from spleen and SI IEL and analyzed for the indicated markers (plots gated on CD45.1+ lymphocytes). One representative of 5 mice, experiment was performed 3 times. (B) OT-I were transferred to conventionally housed specific pathogen free (SPF) or germ-free RAG−/− mice. 27 days later, spleen and SI IEL were harvested and analyzed for the indicated markers (plots gated on H-2Kb/SIINFEKL tetramer+ lymphocytes). One representative of 5 mice. (C) PD-1 expression among transferred P14 isolated from spleen and SI IEL 2 months after LCMV Armstrong infection. One representative of 3 mice per experiment, experiment was performed twice. (D) Polyclonal CD8αβ+ lymphocytes were isolated from spleen and SI IEL of nur77-GFP transgenic mice or C57Bl/6 mice were examined for GFP and CD69 expression +/− systemic treatment with anti-CD3 Ab (Plots gated on CD8α+/CD8β+ lymphocytes). One representative of 3 mice per experiment, experiment was performed twice. (A-D) Spleen = solid gray, SI IEL = black line.
However, the lumen of the small intestine is populated with a complex commensal flora which could provide a diverse source of antigens that may have stimulated transferred T cells, resulting in migration to the intestinal epithelium and constitutive maintenance of an activated phenotype. To address this possibility, we transferred OT-I into gnotobiotic RAG−/− mice that lacked microbial exposure. We found no difference compared to conventionally housed mice, suggesting that IEL adopted an activated phenotype independently of the flora (Fig. 2B).
To confirm this interpretation, we assessed markers that serve as surrogates for recent antigenic stimulation. PD-1 is up-regulated upon TCR stimulation and is often maintained on CD8 T cells during chronic infection40. However, P14 IEL isolated from LCMV immune chimeras did not express PD-1 (Fig. 2C). Nur77 is a transcription factor that is transiently expressed upon TCR stimulation41. To assess Nur77 among polyclonal CD8αβ+ IEL, we employed mice that expressed GFP under the Nur77 promoter. In resting mice, IEL were GFP negative, indicating that they did not constitutively transduce a TCR-dependent Nur77-inducing signal (Fig. 2D). As a control, forced stimulation via injection of anti-CD3 promoted GFP expression. Collectively, these results supported the hypothesis that induction and maintenance of the effector-like phenotype among IEL memory CD8 T cells was not dependent on persistent cognate Ag-stimulation.
Persistent antigen alters canonical IEL phenotype
Thus far, we failed to find positive evidence that cognate Ag promoted differentiation and maintenance of the canonical effector memory IEL phenotype. For that reason, we interrogated IEL differentiation state under conditions of purposeful persistent antigenic stimulation. Firstly, we compared endogenous CD8 T cell responses in spleen and SI IEL by staining isolated lymphocytes with gp33/Db MHC I tetramers three weeks after acute LCMV Armstrong and persistent LCMV Cl-13 infections. Within three weeks of acute Armstrong strain infection, most LCMV-specific CD8 T cells in SI IEL had up-regulated CD103. In contrast, persistent Cl-13 strain infection prevented this up-regulation (Fig. 3A&B). To address this further, we exploited a mouse transgenic model of chronic local self-antigen exposure. OT-I were transferred to 232–4 mice that express ovalbumin under the iFABP promoter that targets expression to epithelial cells within the small intestine42. This results in activation of OT-I in associated lymphoid tissues and accumulation of OT-I within the epithelium where they respond to Ag, but does not induce detectable pathology42. As was the case with chronic LCMV Cl-13 infection, persistent self-Ag recognition prevented CD103 up-regulation (measured 150 days after OT-I transfer, Fig. 3C). These observations provide further evidence that maintenance of the canonical SI IEL phenotype, even though it bears markers of recent activation, is not due to persistent Ag-stimulation.
Figure 3. Persistent antigen alters canonical IEL phenotype.

(A) C57Bl/6J mice were infected with the Armstrong (cleared) or Cl-13 (chronic) strains of LCMV. 21 days later, lymphocytes were isolated from spleen and SI IEL (plots gated on CD8α+ lymphocytes). Percent of CD103+ cells among H-2Db/gp33 tetramer+ (upper right) or tetramer- (lower right) is indicated. (B) Mean percent of H-2Db/gp33 tetramer+ IEL that were CD103+, n=5. ***p<0.001, Student's unpaired t test. Error bars indicated SEM. (C) Naïve OT-I were transferred to ovalbumin bearing 232–4 mice and 150 days later isolated SI IEL were examined for expression of CD103 (black histogram gated on OT-I, gray histogram gated on CD8α+ lymphocytes). All plots are representative of at least three mice per experiment and each experiment was performed at least twice.
Cytokine milieu can induce canonical IEL phenotype
Because SI IEL phenotype has indirectly been shown to be regulated by local environment23, and the experiments presented in figures 1–3 excluded a demonstrable role for local Ag, we examined the ability of cytokines to drive the SI IEL differentiation program. TGFβ has been associated with CD103 regulation on regulatory, anti-tumor, and allograft specific T cells43,44. Moreover, TGFβ is constitutively expressed in SI, for instance, as measured by in situ hybridization to detect mRNA in rat45. For this reason we cultured early CD8 T cell effectors, isolated from spleen 4.5 days after infection, for 40–60h with TGFβ. As shown in Fig. 4A, TGFβ induced CD103 up-regulation among effector CD8 T cells. Fully differentiated resting memory CD8 T cells were less likely to undergo TGFβ induced CD103 expression. Consistent with our interpretation of Fig. 3, inclusion of peptide Ag with TGFβ cultured effector cells prevented up-regulation of CD103 (Fig. 4B), further suggesting that CD103+ SI IELs are not chronically antigen stimulated.
Figure 4. Cytokine milieu can induce canonical IEL phenotype.

(A) C57Bl/6J mice were infected with LCMV Armstrong one day after transfer of P14. 4.5 or 30 days after LCMV infection, splenocytes were isolated, cultured with 0.1ng/ml TGFb for 60h, and examined for CD103 expression by flow cytometry. (B) D4.5 splenocytes (as in A) were treated with 0.1ng/ml TGFb for 60h +/− gp33 cognate peptide. (C&D) D4.5 splenocytes were cultured for 40h with the indicated cytokines, then examined for the expression of CD103, Ly6C, and CD69 by flow cytometry. All plots are representative of at least three mice per experiment and each experiment was performed at least three times.
We then tested whether culture with TGFβ and various other cytokines could re-capitulate cardinal phenotypic signatures of SI IEL among splenocytes (Fig. 4C). We found that certain cytokines, particularly TNFα and IL-33, could synergize with TGFβ and resulted in splenocytes that adopted a CD103+/Ly6C-/CD69+ phenotype (Fig. 4D). Because culture experiments were short-term (40–60h) and depended on an effector state of differentiation, granzyme B and other markers that distinguish memory SI IEL from splenocytes were not addressed. Nevertheless, these data demonstrate that the cytokine milieu can induce splenocytes to mimic the SI IEL phenotype.
To test the role of TGFβ in vivo, we bred Tg mice with a dominant negative mutation in TGFβRII46 with Thy1.1+ OT-I mice, resulting in Thy1.1/Thy1.2 heterozygous OT-I mice expressing a single copy of the dominant negative mutation. CD44lo (naïve) OT-I were FACs sorted from these mice, and mixed in a 1:1 ratio with wild type Thy1.1+ OT-I. Cells were co-transferred to wild type C57Bl/6J mice (Fig. 5A). In summary, recipient mice were wild type, half of the transferred OT-I population was wild type, and half of transferred OT-I were refractory to TGFβ mediated signaling. The following day, mice were infected with VSVova, and lymphocytes were isolated from spleen and SI IEL seven days later. As shown in Fig. 5B&C, dnTGFβRII OT-I failed to develop the CD103+/CD69+ subset upon migration to SI epithelium. Similar results were observed when P14s and LCMV were substituted for OT-Is and VSVova in an identical experimental design (Fig. 5D). While this defect in CD103 and CD69 expression was not absolute, it should be noted that effector dnTGFβRII P14 splenocytes up-regulated CD103 in response to TGFβ in vitro (albeit suboptimally, data not shown), consistent with the fact that this dominant negative mutation did not render T cells completely insensitive to TGFβ; likewise, dnTGFβRII P14 that disseminated to IEL upon in vivo activation eventually up-regulated CD103 (data not shown).
Figure 5. TGFβ induces CD103+/CD69+ SI IEL population in vivo.

(A–B) Thy1.1+ OT-I and Thy1.1/Thy1.2 dnTGFbRII OT-I were mixed in a 1:1 ratio and co-transferred to C57Bl/6J mice. The following day, recipients were infected with VSVova, and lymphocytes were isolated from spleen and SI IEL 7 days later, and analyzed for CD103 and CD69 expression by flow cytometry. Plots gated on WT OT-I (black) or dnTGFbRII OT-I, as indicated. (C) Number of recovered WT (black) and dnTGFβRII OT-I (gray) SI IEL among the indicated subsets. (D) Identical experimental design as in A–C, however WT and mutant P14 were substituted for OT-I and mice were infected with LCMV Armstrong. **p<0.01, Student's unpaired t test. Error bars indicated SEM. Flow plots are representative of five mice per experiment. Each experiment was performed at least three times.
CD103 expression is required for maintenance of virus-specific memory IEL
CD103 binds E-cadherin, expressed by SI epithelial cells, and has been reported to play a role in retention in several contexts47,48. Although maintenance of virus-specific memory CD8αβ+ SI IEL has been reported to be CD103 independent49, the TGFβ dependent loss of CD69+/CD103+ SI IEL (Fig. 5B–D) suggested there may be an underlying survival defect. Thus, we redressed this issue. CD103−/−mice were bred to P14 TCR Tg mice. Similar to the experimental design in Fig. 5, wild type and CD103−/− P14 were co-transferred to wild type C57Bl/6J mice, and mice were infected with LCMV Armstrong. On the indicated days post infection, lymphocytes were harvested from various tissues and the ratio of WT:CD103−/−gp33-specific CD8 T cells were determined by flow cytometry (Fig. 6A). Early during the response, WT and CD103−/− cells were distributed equivalently in spleen and intestinal epithelium. However, CD103−/− P14 were specifically underrepresented within the small intestine by day 30 post-infection. Together with previous work defining the kinetics of P14 migration to SI IEL following LCMV infection13, these data suggest that there is no requirement for CD103 in migration to SI, but a defect in maintenance or survival. As this result differed from the previous study analyzing the requirement of CD103 in SI IEL maintenance following an acute viral infection, we confirmed our analysis histologically. C57Bl/6J mice received either Thy1.1+ WT or CD103−/− P14, and were then infected with LCMV Armstrong. 30 days later, the entire length of the SI was frozen in pinwheels, which allowed a single region (indicated in Fig. 6B) to capture portions along the entire length of the SI. IEL (Fig. 6C) and LPL (Fig. 6D) could be discriminated on the basis of orientation to the basement membrane (defined by collagen IV staining). Thy1.1+ IEL and LPL were counted throughout the SI region of mice that received WT or CD103−/− P14, and enumerated as the number of each subset per 106 SI nuclei (Fig. 6E), or as a ratio of SI IEL to SI LPL (Fig. 6F). These data confirmed that induction of CD103 is required for the maintenance of resident pathogen-specific memory CD8 T cells within small intestine epithelium.
Figure 6. TGFβ and CD103 expression is required for maintenance of IEL memory.
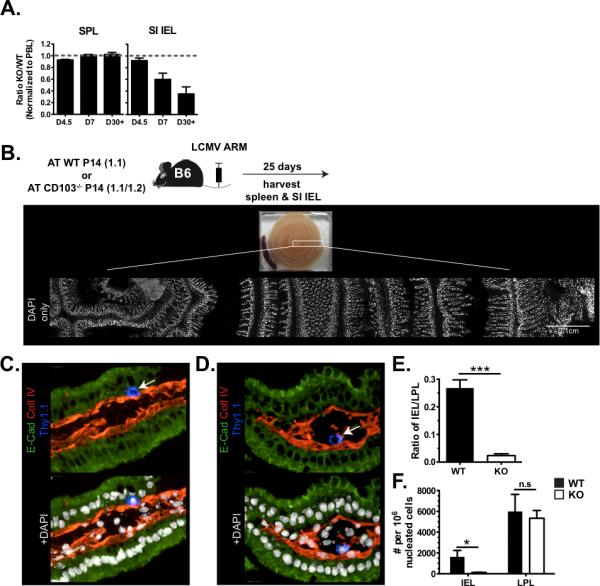
(A) C57Bl/6J mice were infected with LCMV Armstrong one day after transfer of a 1:1 ratio of WT P14 and CD103−/− P14. 4.5, 7, and 30 days later, the ratio of each population was assessed in the indicated tissues. (B–F) C57Bl/6J mice that received either WT Thy1.1+ P14 or CD103−/− Thy1.1+ P14 and infected with LCMV Armstrong were analyzed by immunohistochemistry 30 days later. (B) The indicated region of frozen small intestine `pinwheels' were assessed for SI IEL (C) and SI LPL (D, representative images, DAPI indicates nuclei, Thy1.1 indicates P14). (E) Number of WT or CD103−/− P14 counted in epithelium or lamina propria per 106 nucleated cells (approximately 1.5×105 nuclei were counted per mouse within the indicated region). n=3. (F) The ratio of SI IEL vs. LPL P14 that were counted in B–E. ***p<0.001, *p<0.05, Student's unpaired t test. Error bars indicated SEM.
Induction of TRM phenotypic signatures outside of intestine
We next tested the distribution of TRM phenotypic signatures among subsets of T cells present outside of the intestinal mucosa, as well as the role of persistent cognate Ag stimulation in their induction and maintenance. P14 CD8 T cells were transferred to naïve mice prior to infection with LCMV Armstrong. Three months later, lymphocytes were harvested from spleen, SI IEL, SI LP, female reproductive tract, and brain. As shown in Figure 7A&B, each of these non-lymphoid tissues contained a subset of CD103+ memory CD8 T cells which expressed higher levels of granzyme B compared to splenocytes, and was invariably CD69+. We then further expanded our analysis to include the kidney, pancreas, stomach, and heart, all of which had a subset of CD62L−/CD69+/CD103+ memory CD8 T cells (Fig. 7B). These data provide further evidence that intestine-specific sources of persistent antigen, such as proximity to the commensal flora or dietary antigens, were not responsible for the maintenance of the stereotypic TRM phenotype embodied by SI IEL.
Figure 7. Signature IEL markers are expressed in other non-lymphoid tissues.

(A–B) C57Bl/6J mice were infected with LCMV Armstrong one day after receiving naïve Thy1.1+ P14. Three months later, lymphocytes were isolated from the indicated tissues and analyzed by flow cytometry for expression of CD103, (A) granzyme B, or (B) CD69 and CD62L. Plots gated on CD8+ Thy1.1+ lymphocytes. (C) Naïve CD45.1+ OT-I × RAG−/− CD8 T cells were transferred to RAG−/− C57Bl/6 mice. 60 days later, OT-I were isolated from the indicated tissues and analyzed for CD103, CD69, and CD62L expression. Plots gated on CD8+ CD45.1+ lymphocytes.
To exclude a role for local or prolonged stimulation with cognate viral-derived Ag in the induction and maintenance of TRM in these non-lymphoid tissues, we localized Ag-specific CD8 T cells to the SI epithelium in the absence of infection, by transferring naïve monoclonal CD8 T cells to lymphopenic mice. 1×106 CD45.1+ OT-I × RAG−/− CD8 T cells were transferred to RAG−/− C57Bl/6 mice. We found that “emptiness driven proliferation,” was sufficient to drive dissemination of OT-I into all non-lymphoid tissues tested. Furthermore, the proportion of CD62L−/CD103+/CD69+ CD8 T cells was similar between models of LCMV infection vs. “emptiness induced proliferation” induced migration and differentiation. It should be noted that brain was an exception; in the absence of infection bearing cognate Ag, CD8 T cells that migrated to brain exhibited a paucity of CD103+ CD8 T cells, consistent with a proposed role for local Ag in inducing this TRM phenotype within this tissue. Nevertheless, these data demonstrate that the TRM phenotypic signature was broadly distributed in many non-lymphoid tissues, and was largely independent of persistent or local cognate-Ag interaction.
Discussion
This study clearly demonstrates that the maintenance of the resident TEM differentiation state that typifies SI IEL memory CD8αβ T cells is regulated independently of antigen, and also provides evidence that cytokines induce location-specific T cell properties required for the maintenance of local immunity. These findings precipitate consideration of the relationship between SI IEL, other tissue-resident TRM subsets, and re-circulating TEM pools. Implications for protective immunity and existing models of T cell differentiation are discussed.
SI IEL were rapidly incorporated into the emerging TCM/TEM classification scheme because constitutive maintenance of cytotoxic potential was originally a defining feature of TEM22,50. In mice, the definition of TEM has since broadened to include all T cells that lack expression of CD62L and/or CCR7. Given the growing evidence of a relationship between memory CD8 T cell subsets and protective immunity, there is considerable interest in understanding the regulation of commitment to TEM lineage(s)1. Existing models largely focus on the role of antigen, and indicate that high levels of antigen during priming, repetitive stimulation events, or chronic infections, all favor TEM development1. Compared to TCM, various studies provide evidence that recirculating TEM may be relatively short-lived in the absence of continued antigen-stimulation8,9. However, it has remained unclear whether these conclusions should be conflated with memory CD8 T regulation within the intestinal mucosa; particularly because SI IEL show little evidence of re-circulation13. Given the potential exposure of the intestinal mucosa to numerous foreign peptides, we examined the mechanisms regulating SI IEL phenotype.
This study provides several complementary lines of evidence indicating that foreign antigen-dependent signals are not required for the induction or maintenance of “effector-like” CD8αβ SI IEL. Homeostatic proliferation induced among naïve transgenic CD8 T cells upon transfer into lymphopenic recipients was sufficient to induce migration to the intestinal mucosa and acquisition of SI IEL effector-like phenotypic signatures (Fig 2A). This experiment demonstrated that “programming” fully differentiated SI IEL was not LCMV-infection dependent, and it ruled out a role of persistent stimulation by chronic occult LCMV Armstrong infection. This result was re-capitulated in germ-free RAG−/− mice, indicating that cross-reactivity with microbial antigens was also not required (Fig. 2B). Key features of the SI IEL phenotype were also displayed by CD103+ memory CD8αβ T cells isolated from the female reproductive tract, kidney, brain, etc., suggesting that a “resting effector” phenotype might extend to other tissue-resident memory CD8 T cell populations and further ruling out a requirement for both intestinal flora and also dietary antigens (Fig. 7). Absence of PD-1 or Nur77 expression further supported the hypothesis that SI IEL were not persistently stimulated with cognate antigen (Fig. 2C&D). Indeed, when a source of constitutive antigen was provided via infection with a chronic strain of LCMV or by expression of cognate antigen by intestinal epithelial cells, SI IEL deviated from their conventional phenotype and failed to acquire normal levels of CD103 expression (Fig. 3).
Interestingly, ex vivo culture of effector CD8 T splenocytes with combinations of TGFβ and IL-33 or TNFα promoted acquisition of SI IEL phenotypic signatures, as measured by CD103, CD69, and Ly6C expression. The presence of cognate antigen prevented the ability of TGFβ to induce CD103 expression among effector splenocytes. In this light, it will be interesting to determine whether CD103 expression may serve as a useful marker to distinguish resting vs. recently antigen-stimulated CD8αβ T cells in certain nonlymphoid compartments.
The results of this study do not diminish the central role of Ag in regulating T cell differentiation. However, our data do indicate that anatomic location, independently of antigen-stimulation history, also significantly and directly affects CD8 T cell differentiation state. This suggests that TEM phenotype is not invariably a reliable indicator of stimulation history. It also suggests that effector-like phenotypes may be acquired and maintained in certain tissues after vaccination without requiring a continuous source of antigen stimulation, and cytokine milieu may play a central role in regulating this process. Of course, vaccines that do establish persistent sources of Ag may still modify CD8 T cell differentiation state, abundance, anatomic distribution, and function in ways that could favor protective immunity51. Nevertheless, from a teleological perspective, tissue directed regulation of memory CD8 T cell development may be essential for ensuring that T cells adapt to the unique survival and functional requirements of their particular environment. In support of this concept, TGFβ-dependent induction of CD103 following migration to the SI epithelium was required for maintenance of memory CD8 T cells in this compartment (Fig. 5&6). In the future, it will be interesting to determine whether CD103 expression among memory CD8 T cells in other locations is under the direction of local TGF-β and whether it marks resident populations that do not re-circulate under homeostatic conditions. Regardless, localization of T cells to the SI epithelium without cognate antigen stimulation (Fig. 2) was sufficient to induce maintenance of granzyme B expression, suggesting that cells positioned on the front lines of this common portal of infection are poised for rapid cytotoxicity regardless of “programming” cues during priming in lymphoid tissue.
Taken together, these results suggest that tissues provide instructional cues that play a major role in defining T cell differentiation state and optimize the maintenance of host protective immunity at common portals of pathogen entry.
Acknowledgements
The authors thank Marion Pepper, Boton Igyarto, Stephen Jameson, and Michael Gerner for helpful discussions.
References
- 1.Jameson SC, Masopust D. Diversity in T cell memory: an embarrassment of riches. Immunity. 2009;31:859–71. doi: 10.1016/j.immuni.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lefrancois L, Marzo AL. The descent of memory T-cell subsets. Nat Rev Immunol. 2006;6:618–23. doi: 10.1038/nri1866. [DOI] [PubMed] [Google Scholar]
- 3.Lanzavecchia A, Sallusto F. Understanding the generation and function of memory T cell subsets. Curr Opin Immunol. 2005;17:326–32. doi: 10.1016/j.coi.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 4.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–63. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 5.van Faassen H, et al. Reducing the stimulation of CD8+ T cells during infection with intracellular bacteria promotes differentiation primarily into a central (CD62LhighCD44high) subset. J Immunol. 2005;174:5341–50. doi: 10.4049/jimmunol.174.9.5341. [DOI] [PubMed] [Google Scholar]
- 6.Masopust D, Ha SJ, Vezys V, Ahmed R. Stimulation history dictates memory CD8 T cell phenotype: implications for prime-boost vaccination. J Immunol. 2006;177:831–9. doi: 10.4049/jimmunol.177.2.831. [DOI] [PubMed] [Google Scholar]
- 7.Sarkar S, et al. Strength of stimulus and clonal competition impact the rate of memory CD8 T cell differentiation. J Immunol. 2007;179:6704–14. doi: 10.4049/jimmunol.179.10.6704. [DOI] [PubMed] [Google Scholar]
- 8.Wherry EJ, et al. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol. 2003;4:225–34. doi: 10.1038/ni889. [DOI] [PubMed] [Google Scholar]
- 9.Marzo AL, et al. Initial T cell frequency dictates memory CD8+ T cell lineage commitment. Nat Immunol. 2005;6:793–9. doi: 10.1038/ni1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaech SM, Wherry EJ. Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity. 2007;27:393–405. doi: 10.1016/j.immuni.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, et al. Durable mucosal simian immunodeficiency virus-specific effector memory T lymphocyte responses elicited by recombinant adenovirus vectors in rhesus monkeys. J Virol. 2011;85:11007–15. doi: 10.1128/JVI.05346-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shin H, Blackburn SD, Blattman JN, Wherry EJ. Viral antigen and extensive division maintain virus-specific CD8 T cells during chronic infection. J Exp Med. 2007;204:941–9. doi: 10.1084/jem.20061937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Masopust D, et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med. 2010;207:553–64. doi: 10.1084/jem.20090858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klonowski KD, et al. Dynamics of blood-borne CD8 memory T cell migration in vivo. Immunity. 2004;20:551–62. doi: 10.1016/s1074-7613(04)00103-7. [DOI] [PubMed] [Google Scholar]
- 15.Wakim LM, Woodward-Davis A, Bevan MJ. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc Natl Acad Sci U S A. 2010;107:17872–9. doi: 10.1073/pnas.1010201107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gebhardt T, et al. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol. 2009;10:524–30. doi: 10.1038/ni.1718. [DOI] [PubMed] [Google Scholar]
- 17.Gebhardt T, et al. Different patterns of peripheral migration by memory CD4(+) and CD8(+) T cells. Nature. 2011 doi: 10.1038/nature10339. [DOI] [PubMed] [Google Scholar]
- 18.Hofmann M, Pircher H. E-cadherin promotes accumulation of a unique memory CD8 T-cell population in murine salivary glands. Proc Natl Acad Sci U S A. 2011;108:16741–6. doi: 10.1073/pnas.1107200108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee YT, et al. Environmental and antigen receptor-derived signals support sustained surveillance of the lungs by pathogen-specific cytotoxic T lymphocytes. J Virol. 2011;85:4085–94. doi: 10.1128/JVI.02493-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clark RA, et al. The vast majority of CLA+ T cells are resident in normal skin. J Immunol. 2006;176:4431–9. doi: 10.4049/jimmunol.176.7.4431. [DOI] [PubMed] [Google Scholar]
- 21.Purwar R, et al. Resident memory T cells (T(RM)) are abundant in human lung: diversity, function, and antigen specificity. PLoS One. 2011;6:e16245. doi: 10.1371/journal.pone.0016245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masopust D, Vezys V, Marzo AL, Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 2001;291:2413–7. doi: 10.1126/science.1058867. [DOI] [PubMed] [Google Scholar]
- 23.Masopust D, Vezys V, Wherry EJ, Barber DL, Ahmed R. Cutting edge: gut microenvironment promotes differentiation of a unique memory CD8 T cell population. J Immunol. 2006;176:2079–83. doi: 10.4049/jimmunol.176.4.2079. [DOI] [PubMed] [Google Scholar]
- 24.Ibraghimov AR, Lynch RG. Heterogeneity and biased T cell receptor alpha/beta repertoire of mucosal CD8+ cells from murine large intestine: implications for functional state. J Exp Med. 1994;180:433–44. doi: 10.1084/jem.180.2.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rocha B, Guy-Grand D, Vassalli P. Extrathymic T cell differentiation. Curr Opin Immunol. 1995;7:235–42. doi: 10.1016/0952-7915(95)80008-5. [DOI] [PubMed] [Google Scholar]
- 26.Wherry EJ, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–84. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 27.Goodman T, Lefrancois L. Intraepithelial lymphocytes. Anatomical site, not T cell receptor form, dictates phenotype and function. J Exp Med. 1989;170:1569–81. doi: 10.1084/jem.170.5.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huleatt JW, Lefrancois L. Antigen-driven induction of CD11c on intestinal intraepithelial lymphocytes and CD8+ T cells in vivo. J Immunol. 1995;154:5684–93. [PubMed] [Google Scholar]
- 29.Arstila T, et al. Identical T cell clones are located within the mouse gut epithelium and lamina propia and circulate in the thoracic duct lymph. J Exp Med. 2000;191:823–34. doi: 10.1084/jem.191.5.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shires J, Theodoridis E, Hayday AC. Biological insights into TCRgammadelta+ and TCRalphabeta+ intraepithelial lymphocytes provided by serial analysis of gene expression (SAGE) Immunity. 2001;15:419–34. doi: 10.1016/s1074-7613(01)00192-3. [DOI] [PubMed] [Google Scholar]
- 31.Montufar-Solis D, Garza T, Klein JR. T-cell activation in the intestinal mucosa. Immunol Rev. 2007;215:189–201. doi: 10.1111/j.1600-065X.2006.00471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grigg D. The Pattern of World Protein Consumption. Geoforum. 1995;26:1–17. [Google Scholar]
- 33.Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Hostbacterial mutualism in the human intestine. Science. 2005;307:1915–20. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 34.Helgeland L, Vaage JT, Rolstad B, Midtvedt T, Brandtzaeg P. Microbial colonization influences composition and T-cell receptor V beta repertoire of intraepithelial lymphocytes in rat intestine. Immunology. 1996;89:494–501. doi: 10.1046/j.1365-2567.1996.d01-783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Helgeland L, et al. Microbial colonization induces oligoclonal expansions of intraepithelial CD8 T cells in the gut. Eur J Immunol. 2004;34:3389–400. doi: 10.1002/eji.200425122. [DOI] [PubMed] [Google Scholar]
- 36.Ferguson A, Parrott DM. The effect of antigen deprivation on thymus-dependent and thymus-independent lymphocytes in the small intestine of the mouse. Clin Exp Immunol. 1972;12:477–88. [PMC free article] [PubMed] [Google Scholar]
- 37.Kawaguchi-Miyashita M, et al. Development and cytolytic function of intestinal intraepithelial T lymphocytes in antigen-minimized mice. Immunology. 1996;89:268–73. doi: 10.1046/j.1365-2567.1996.d01-740.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Camerini V, et al. Generation of intestinal mucosal lymphocytes in SCID mice reconstituted with mature, thymus-derived T cells. J Immunol. 1998;160:2608–18. [PubMed] [Google Scholar]
- 39.Ferguson A. Intraepithelial lymphocytes of the small intestine. Gut. 1977;18:921–37. doi: 10.1136/gut.18.11.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007;8:239–45. doi: 10.1038/ni1443. [DOI] [PubMed] [Google Scholar]
- 41.Moran AE, et al. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp Med. 2011;208:1279–89. doi: 10.1084/jem.20110308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vezys V, Olson S, Lefrancois L. Expression of intestine-specific antigen reveals novel pathways of CD8 T cell tolerance induction. Immunity. 2000;12:505–14. doi: 10.1016/s1074-7613(00)80202-2. [DOI] [PubMed] [Google Scholar]
- 43.Kilshaw PJ, Murant SJ. A new surface antigen on intraepithelial lymphocytes in the intestine. Eur J Immunol. 1990;20:2201–7. doi: 10.1002/eji.1830201008. [DOI] [PubMed] [Google Scholar]
- 44.El-Asady R, et al. TGF-{beta}-dependent CD103 expression by CD8(+) T cells promotes selective destruction of the host intestinal epithelium during graft-versus-host disease. J Exp Med. 2005;201:1647–57. doi: 10.1084/jem.20041044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koyama SY, Podolsky DK. Differential expression of transforming growth factors alpha and beta in rat intestinal epithelial cells. J Clin Invest. 1989;83:1768–73. doi: 10.1172/JCI114080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lucas PJ, Kim SJ, Melby SJ, Gress RE. Disruption of T cell homeostasis in mice expressing a T cell-specific dominant negative transforming growth factor beta II receptor. J Exp Med. 2000;191:1187–96. doi: 10.1084/jem.191.7.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Denucci CC, Mitchell JS, Shimizu Y. Integrin function in T-cell homing to lymphoid and nonlymphoid sites: getting there and staying there. Crit Rev Immunol. 2009;29:87–109. doi: 10.1615/critrevimmunol.v29.i2.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schon MP, et al. Mucosal T lymphocyte numbers are selectively reduced in integrin alpha E (CD103)-deficient mice. J Immunol. 1999;162:6641–9. [PubMed] [Google Scholar]
- 49.Lefrancois L, et al. The role of beta7 integrins in CD8 T cell trafficking during an antiviral immune response. J Exp Med. 1999;189:1631–8. doi: 10.1084/jem.189.10.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–12. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 51.Hansen SG, et al. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature. 2011;473:523–7. doi: 10.1038/nature10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pircher H, Rohrer UH, Moskophidis D, Zinkernagel RM, Hengartner H. Lower receptor avidity required for thymic clonal deletion than for effector T-cell function. Nature. 1991;351:482–5. doi: 10.1038/351482a0. [DOI] [PubMed] [Google Scholar]
- 53.Hogquist KA, et al. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]