

Abstract
Metallic radionuclides are the mainstay of both diagnostic and therapeutic radiopharmaceuticals. Therapeutic nuclear medicine is less advanced but has tremendous potential if the radionuclide is accurately targeted. Great interest exists in the field of inorganic chemistry for developing target specific radiopharmaceuticals based on radiometals for non-invasive disease detection and cancer radiotherapy. This perspective will focus on the nuclear properties of a few important radiometals and their recent applications to developing radiopharmaceuticals for imaging and therapy. Other topics for discussion will include imaging techniques, radiotherapy, analytical techniques, and radiation safety. The ultimate goal of this perspective is to introduce inorganic chemists to the field of nuclear medicine and radiopharmaceutical development, where many applications of fundamental inorganic chemistry can be found.
Introduction
Radiopharmaceuticals are radioactive drugs. Radiometals are the source of ionizing radiation in radiopharmaceuticals responsible for diagnosing or treating of various diseases. Although several important nonmetallic (organic) radionuclides (18F, 11C, 13N, 15O, 124I, etc.) have been around for quite some time, metallic radionuclides are of particular interest for radiopharmaceutical development, because of their wider range of nuclear properties (half life, decay characteristics etc.), rich coordination chemistry, and easy availability. The huge success1 of historical 99mTc-radioimaging agents such as 99mTc-sestamibi and 99mTc-tetrofosmin for myocardial perfusion has led researchers to explore the potential of other radiometals having different physico-chemical and nuclear properties. Because of this effort, several new radiometal-based drugs have been approved by the Food and Drug Administration (FDA) and many more are in different phases of active clinical trials. In nuclear medicine, radiopharmaceuticals are mainly used in diagnostic radioimaging and radiotherapy.
Diagnostic imaging is a noninvasive method intended to assess the disease or disease states and monitor the effects of treatment. In general, diagnostic radiopharmaceuticals are introduced in to the body at very low concentrations (nano-molar to pico-molar range), and are not intended to have any pharmacological effects. Radiometal selection for diagnostic imaging is mainly based on two radioimaging modalities: single photon emission computed tomography (SPECT) and positron emission tomography (PET). Both are well-established clinical imaging modalities that offer high-quality sensitivity in deep tissue. In SPECT, radiopharmaceuticals labelled with gammaemitting isotopes are injected into the living subject. Gamma emissions directly from the radio isotopes are collected in a gamma camera, which enables us to reconstruct a picture of where the gamma rays originated. From this, we can determine how a particular organ or system is functioning. Radiopharmaceuticals labelled with a positron emitting isotope have been used in PET. As the radio-isotope undergoes decay, it emits a positron that travels for a short distance and interacts with an electron. This encounter produces a pair of 511 keV gamma photons that move in almost the opposite direction (180 degrees) to each other. As soon as they arrive at the circular array of detectors the photons are registered by PET and it is possible to determine their source along a straight line of coincidence. These coincidences are forwarded to the image processing unit to generate PET images via mathematical reconstruction procedures (Fig. 1). In PET, a single positron decay results in two 511 keV gamma photons emitted in opposite directions. Therefore, PET scans offer much better image resolution than SPECT scans. But SPECT scans are comparatively less expensive because of the availability of cost-effective, longer-lived SPECT isotopes (99mTc, 111In, etc.). PET radionuclides (18F, 11C, 13N, 15O) commonly used in research (both clinical and basic) are expensive because they are short-lived and cyclotron-produced.
Fig. 1.

Schematic representation of PET imaging technique.
Therapeutic radiopharmaceuticals deliver cytotoxic non-penetrating radiation (Auger electrons and β− or a particles) doses to diseased sites, resulting in the death of the cancer cells. However, this can lead to DNA mutations in other cells that survive the radiation, which may eventually lead to the development of a second cancer. The main challenges to radiotherapy are the availability of low-cost therapeutic radioisotopes and the techniques to deliver these radioisotopes to diseased tissue (e.g., tumors). Therapeutic radiation dose-delivery techniques that are available include brachytherapy,2 external beam irradiation, and systemic administration. The basic concept of brachytherapy or internal radiotherapy is to insert a radioactive source into a tumor. The radiation source is an implantable ‘seed’ (small radioactive rod), that can be physically placed in the tumor site and will remain there until it is surgically removed. This technique is commonly used to treat cancer of the cervix, prostate, skin, and breast, where the tumor mass can be accessed to implant the ‘seed’.3 In systemic administration, injected radiopharmaceuticals localize to the diseased area to deliver a cytotoxic radiation dose; then, the radiopharmaceuticals clear rapidly from the blood and other organs to minimize the radiation exposure to normal tissues.
When considering the selection of a suitable radionuclide for radiopharmaceutical development, at first one should have two possible aims in mind: either to perform a diagnostic study (i.e., radioimaging) or to destroy the diseased cells (i.e., radiotherapy). The next consideration would be to identify the probable targeting ligands or biomolecules based on biological characteristics (e.g., biological half-life, target, in vivo stability, etc.) and physico-chemical properties. Once the targeting biomolecule is known for a particular application, the radiometal can be identified based on its nuclear properties, which include: physical half-life (should match with the biological half-life of the targeting ligand), decay characteristics, daughter product stability, in vivo red-ox stability, availability, ease of production procedure including purification and isolation, and radiation toxicity. Selected metallic radionuclides are listed in Table 1, along with their half-lives, production method, radiation characteristics, etc.
Table 1.
Characteristics of selected radiometals for imaging and therapya
| Radiometals (symbol) |
Half life (T1/2) |
Decay mode (% branching ratio) |
Production (references) |
Eg/keV (intensity Ig/%) |
Positron Eβ+/KeV (abundance Iβ+/%) |
P-particle Enp/MeV (abundance Inp/%) |
α-particle Eα/MeV (abundance Iα/%) |
Human studies |
|---|---|---|---|---|---|---|---|---|
| Technetium-99m (99mTc) |
6.01 h | it (99.99) |
99Mo/99mTc Generator (6c,7a) |
140(90) | Yes SPECT |
|||
| Indium-111 (111In) |
67.392 h | ec (100) |
111Cd(p, n)lllm,gIn 112Cd(p,2n)111m,gIn Cyclotron (14) |
171(90) 245(94) |
Yes SPECT |
|||
| Gallium-67 (67Ga) |
78.281 h | ec (100) |
natZn(p, x)67Ga 68Zn(p,2n)67Ga Cyclotron (22) |
93(39) 185(21) 300(16) |
Yes SPECT |
|||
| Copper-64 (64Cu) |
12.701 h | ec + β+ (61.5), β+ (17.60), β− (38.5) |
64Ni(p, n)64Cu 67Zn(p,α)64Cu Cyclotron (37b,c) |
511(35) | 278(17) | 0.191(38) | Yes PET |
|
| Copper-62 (62Cu) |
0.16h | ec + β+ (100) β+ (98) |
62Zn/62Cu Generator (37d, 86) |
511(194) | 1316(97) | Yes PET |
||
| Zirconium-89 (89Zr) |
78.41 h | ec + β+ (100), β+ (22.74) |
89Y(p,n)89Zr Cyclotron (60) |
511(45) 909(99) |
395(22) | Yes PET |
||
| Yttrium-86 (86Y) |
14.74 h | ec + β+ (100), β+ (31.9) |
86Sr(p,n)86Y Cyclotron (50, 51) |
511(64) 627(36) 1076(82) |
535(12) | Yes PET |
||
| Gallium-68 (68Ga) |
1.13 h | ec + β+ (100), β+ (89.14) |
68Ge/68Ga Generator (28) |
511(178) | 836(88) | Yes PET |
||
| Rubidium-82 (82Rb) |
0.021 h | β+ (95.43) |
82Sr/82Rb Generator (33) |
511(190) | 1534(81) | Yes PET |
||
| Yttrium-90 (90Y) |
64.00 h | β− (100) |
90Sr/90y Generator (69) |
2.28(100) | Yes β-therapy |
|||
| Lutetium-177 (177Lu) |
6.647 d | β− (100) |
176Lu(n,γ)177Lu 176Yb(n,γ)177Yb→177Lu Nuclear reactor (83) |
208(10) | 0.498(79) 0.177(11) |
Yes β-therapy |
||
| Rhenium-188 (188Re) |
17.00 h | β− (100) |
188w/188Re Generator (71) |
155(15) | 2.12(70) 1.96(26) |
Yes β-therapy |
||
| Samarium-153 (153Sm) |
46.50 h | β− (100) |
152Sm(n, γ)153Sm Nuclear reactor (84) |
103(29) | 0.704(49) 0.634(32) |
Yes β-therapy |
||
| Bismuth-213 (213Bi) |
0.759 h | α (2.20) β− (97.80) |
225Ac/213Bi Generator (85) |
0.435(97) | 5.87(1.94) | Yes α-therapy |
||
| Actinium-225 (225Ac) |
240.0 h | α(100) |
229Th/225Ra/225Ac Generator (85) |
5.83(50) 5.79(18) |
Yes α-therapy |
|||
| Radium-223 (223Ra) |
11.4d | α(100) |
227AC/227Th/223Ra Generator (87) |
5.64(96) | Yes α-therapy |
ec = electron capture; it = isomeric transition; h = hours;d = days; SPECT = Single Photon Emission Computed Tomography; PET = Positron Emission Tomography;”
Some of the data presented in this table were obtained from references (6f, 6i) and http://www.nndc.bnl.gov/nudat2/chartNuc.jsp (Nuclear Structure and Decay Data Searchable Database, National Nuclear Data Center, Brookhaven National Laboratory, USA) accessed on 12/04/2010
Most of the medical radioisotopes are produced in a cyclotron, or generator. Generator-produced radionuclides are generally considered ideal for biomedical applications; because they can be used in places lacking cyclotron access, and high-quality daughter radionuclide products can be obtained throughout the life of the generator. In generator systems, radioactive equilibrium between the parent and daughter nuclides is taken advantage of by eluting the short-lived daughter activity (e.g., 99mTc, 68Ga, 82Rb, etc.) from the longer-lived parent activity (e.g., 99Mo, 68Ge, 82Sr, etc.) using ion-exchange column chromatography or the simple solvent extraction technique. Generators are small and very convenient for transportation, which facilitates in-house radiopharmaceutical production without expensive cyclotrons on site.
The Handbook of Radiopharmaceuticals is an important resource that provides comprehensive information on the production of various nuclides, and an extensive evaluation of the chemistry of conventional radionuclides.4 This perspective covers the nuclear properties of ~17 metallic radionuclides and their potential application in the development of imaging and therapeutic radiopharmaceuticals. It will also briefly discuss the use of analytical tools for quality control and the characterization of radiolabeled molecules. Because some of the radiometals are relatively new in nuclear medicine, radiation safety issues will also be discussed. Since it is impossible to cover all the aspects related to radiopharmaceutical developments and biological applications, the authors would like to apologize to those individuals whose work has not been cited or presented in detail.
Radiopharmaceutical design
Whether designed for diagnosis or therapy, target-specificity of radiopharmaceuticals is essential to minimizing the unnecessary radiation exposure to the body during imaging or radiotherapy. Although some radiometals can target a particular tissue as a simple metal salt or as a simple metal complex, the radio-pharmaceutical chemist is frequently required to coordinate the radiometal in a targeting biomolecule (BM) so the radionuclide will be delivered to the diseased site. These BMs or targeting ligands can be small molecules, peptides, monoclonal antibodies (mAbs), or mAb fragments, and they serve as the vehicle to carry the radionuclide to the target tissue. Ideally, the affinity of a receptor specific ligand for biological targets should be unchanged after radiometal labeling. This rarely happens with the small probes (MW < 1000). Two methods are known to incorporate radiometals to receptor-specific molecules. The most convenient approach to make a stable attachment of a radiometal and a BM, is to use a suitable bifunctional chelate (BFC), which can hold the radiometal tightly and at the same time form a stable conjugation with the active groups of the BM (Fig. 2). Active groups are either naturally present (e.g., −NH2, −SH, etc.) in BMs orare synthetically introduced (e.g., −N3, −C CH, etc.)5a,b into them. In this BFC approach, the BFC is first conjugated to the BM (Fig. 2) and then the radiometal is added to the conjugates. In another approach, chelation of the radiometal is designed in such a way that the resulting complex mimics the shape and size of a target-specific ligand. This is known as an integrated radiolabeling approach. Fig. 3 shows an interesting application of this approach, where an attempt has been made to develop an imaging agent for an estrogen receptor, which is overexpressed on certain tumors. Unfortunately the mimic exhibited very low affinity for the receptor.5c This method is comparatively challenging and examples are rare. For the small probes, in most cases the receptor affinity decreases significantly. This approach may be suitable for high molecular weight species where impact of the size of the metal complex can be minimized.
Fig. 2.

Radiopharmaceutical design: schematic diagram of the bifunc-tional chelator (BFC) approach and different bioconjugation strategies. R is the chelator part of the BFC.
Fig. 3.

Radiopharmaceutical design: an example of an integrated approach.
Each radiometal has a different coordination chemistry and requires BFCs with different donor atoms and chelator frameworks. The choice of an appropriate BFC is largely determined by the oxidation state of the radiometal and its affinity for the chelator’s donor atoms in order to form a kinetically inert and thermodynamically stable complex. In the blood stream, the concentration of radiopharmaceuticals may become so low that the natural chelators (e.g., transferrin or other metal binding enzymes) present in the blood will cause the radiometal to dissociate from its metal–chelate, resulting in the possible accumulation of radioactivity in non-target organs (radiation toxicity). Therefore, a viable BFC should be both kinetically inert and thermodynamically stable to transchelation by these natural chelators. Common bifunctional chelating groups for radiometals are illustrated in Fig. 4. A linker is often used to keep the radiometal chelate away from targeting BM to minimize the overall structural variations (shape and size) of the BM by the metal chelate. A linker is also used to facilitate radiopharmaceutical movement in the body (pharmacokinetics), including distribution and elimination. Coordination chemistry and bioconjugation play important roles in radiopharmaceutical design. Several excellent reviews6 covering these aspects in developing targeted diagnostic or therapeutic radiopharmaceuticals were recently published.
Fig. 4.

Selected BFCs often employed in conjugate labeling strategies.
Radiometals for diagnostic radiopharmaceuticals
Radiometals in diagnostic radiopharmaceuticals are γ or positron emitters. As stated before, the criteria for choosing one particular diagnostic radiometal over another are: imageable gamma or positron emission, little or no emission of particle (α or β−) radiation, stable daughter product, adequate in vivo stability, suitable half-life, well-known chelation chemistry, and ease of production. Two radionuclides commonly used in diagnostic imaging are the non-metallic positron emitter 18F (PET), and the gamma ray emitter 99mTc (SPECT). Their decay characteristics make them ideal for their respective imaging modalities. Although PET has been regarded as a superior imaging modality because of its enhanced sensitivity and image resolution when compared to SPECT, recent invention of multi-pinhole detectors in nano-SPECT imaging has narrowed the spatial resolution gap between the two modalities. The production, chelation chemistry, and radiopharmaceutical applications (preclinical and clinical) of selected SPECT and PET (Table 1) radiometals are described in following section.
Radiometals for SPECT
In Table 1, three low-energy gamma-emitting radiometals (99mTc, 111In, and 67Ga) are listed for potential use in the design of SPECT radiopharmaceuticals. Nearly 80% of all radiopharmaceuticals used in clinical studies are 99mTc-based because of its easy availability (generator produced), optimal nuclear properties, and low cost. According to the recent report from the Society of Nuclear Medicine (SNM),99mTc is currently used in ~70–80% of all radio-diagnostic scans in over 300 000 procedures per week in the U.S.alone.6i Dueto thehigh demand for99mTc and the recent global production shortage (few active reactors) of its parent isotope, 99Mo, the U.S. Congress passed the American Medical Isotopes Production Act (H.R. 3276), which will provide the Department of Energy with $163 million to support U.S.-based production of 99Mo.6i Table 2 lists selected commercially available SPECT and therapeutic agents along with their medical applications.
Table 2.
Selected radiopharmaceuticals for diagnosis or therapy of diseases
| Radiopharmaceuticals | Trade name | Primary use |
|---|---|---|
| Small complexes | ||
| 99mTc–Sestamibi | Cardiolite® | Myocardial perfusion imaging |
| 99mTc–Tetrofosmin | Myoview® | Myocardial perfusion imaging |
| 99mTc–Pentetate (DTPA) | Technescan® | Renal imaging and function studies |
| 99mTc–Bicisate (ECD) | Neurolite® | Cerebral perfusion imaging |
| 99mTc–MDP | Medronate® | Skeletal scintigraphy |
| 99mTc–Teboroxime | Cardiotec® | Myocardial perfusion imaging |
| 111In–Oxyquinoline | Indium-111 oxine® | Leukocyte scintigraphy |
| 111In–Pentetate | Indium-111 DTPA® | Imaging of CSF kinetics |
| 153Sm–EDTMP | Quadramet® | Treatment of bone pain (therapy) |
| 188Re–HEDP | — | Metastatic bone pain (therapy) |
| Peptido- or immuno-conjugates | ||
| 99mTc–Depreotide | Neo Tect® | To evaluate certain lung lesions |
| 99mTc–Arcitumumab | CEA-Scan® | 99mTc-mAb for colorectal cancer imaging |
| 111In–Capromab pendetide | ProstaScint® | Prostate cancer imaging |
| 111In–Pentetreotide | Octreoscan® | Neuroendocrine tumor imaging |
| 111In–Imciromab pentetate | MyoScint® | Imaging of chest pain, suspected to be caused by myocardial infarction |
| 111In–Satumomab pendetide | OncoScint® | Imaging of metastatic disease associated with colorectal and ovarian cancer |
| 90Y–Ibritumomab tiuxetan | Zevalin® | Treatment of non-Hodgkin’s lymphoma (NHL) |
Technetium-99m
Technetium belongs to Group 7 of the periodic table. 99mTc (t1/2 6.01 h) is a metastable γ -emitting (143 keV) radioisotope that decays to highly stable daughter isotope 99Tc. A 6.01 h half-life is long enough for proper radiopharmaceutical preparation and SPECT scanning procedures and is short enough to keep the total patient radiation exposure low. It is a generator-produced daughter product of 99Mo (t1/2 2.78 days), which is a fission product of 235U. In a 99Mo/99mTc generator, 99mTcO4− is formed by the decay of 99MoO4 2−. 99mTcO4− (high specific activity) is eluted from the chromatographic column with saline. The total concentration of technetium in the 99Mo/99mTc generator eluent is in the range of 10−7 to 10−6 M.6c Because of the short half-life, most of the 99mTc-radiopharmaceuticals are simple metal complexes or 99mTc–BFC-conjugated small peptides or proteins.
In 1959, Brookhaven National Laboratory developed the first 99Mo/99mTc generator, which is the historical landmark in the area of nuclear medicine.7 This technology revolutionized radiopharmaceutical chemistry research, and small 99mTc-based radiopharmaceutical research was accelerated to a greater extent.
In the 1980s, radiopharmaceutical research focused mainly on producing 99mTc-based small complexes as perfusion radiotracers for the heart and brain. Extensive research on the coordination chemistry of 99mTc led to the successful development of several first-generation 99mTc-based radiotracers,6c,7 which are known as ‘technetium essential agents’ (Fig. 5). In 1981, Alan Davison, Alun Jones, and Michael Abrams invented one of the most important 99mTc essential agents [99mTc(MIBI)6]+ (MIBI = 2-methoxy-2-methylpropylisonitrile), which showed localization in cardiac tissues after intravenous administration. This is known by the trade name Cardiolite or Sestamibi. This agent can help to assess the amount of blood-perfusing the heart muscle is possible under conditions of rest and stress.1,8 Using this agent, nuclear stress tests can be performed not only on people who are already complaining of heart attack symptoms but also on people who are at risk for a heart attack but have not shown any signs, allowing millions of people to receive better and potentially lifesaving diagnosis and treatment. Like Sestamibi, 99mTc–Tetrofosmin [99mTcO2(tetrofosmin)2]+ (tetrofosmin = 1,2-bis[bis(2-ethoxyethyl)phosphinoethane) is another commercially available myocardial perfusion imaging agent.
Fig. 5.

Selected diagnostic radiopharmaceuticals based on small metal complexes.
As we march into the 21st century, radiopharmaceutical research has been more focused on the development of radiolabeled receptor specific ligands or target specific radiopharmaceuticals. The application of an integrated approach to the development of 99mTc-based radiopharmaceuticals is very limited. The BFC approach is mostly used to conjugate 99mTc with the receptor-specific molecule. The successful development of 99mTc-based radiotracers largely depends on our understanding of the basic chelation chemistry of Tc. Tc exists between oxidation states −1 and +7, and has complex red-ox chemistry. Stabilizing a metal that can exist in multiple oxidation states is quite challenging. Since there is no effective chemistry that can attach 99mTcO4− directly to the BMs, 99mTc(VII) in 99mTcO4− has to be reduced to a lower oxidation state. Over the decades, several robust 99mTc core structures have been developed to stabilize the lower oxidation state using different chelators and reducing agents. Common core structures for Tc are illustrated in Fig. 6. These stable core structures are primarily used in the development of receptor-specific radiopharmaceuticals. The most extensively studied core is the [Tc(V)O]3+ moiety. The complex of the Tc(V)–oxo core adopted square pyramidal geometry with the oxo group in apical position. The sixth coordinationtrans to the oxo group is uncommon mainly due to the strong trans-influence of the π-bonding oxo group. The core is stabilized by the σ- and π-donating (amino, amido, thiolato, etc.) chelating agents. Another more robust core is the metal–organohydrazino unit, which is readily accessible from the metal–oxo core by a simple condensation reaction with organohydrazine.9 The most effective metal–organohydrazine interaction can be achieved using 6-hydrazinonicotinic acid (HYNIC) because of the chelate effect of HYNIC. A unique feature of the HYNIC system is its simple bioconjugate chemistry, which can be used to attach HYNIC to wide range of receptor-targeting biomolecules.6c Since the HYNIC ligand can act as either a monodentate or a bidentate chelate, the metal–hydrazino core requires a variety of coligands to satisfy the metal coordination requirements. Tricine is one such coligand used mostly to stabilize the metal–hydrazino core in vivo. Because of the solution instability of the coligands and different bonding modalities of the HYNIC, multiple species for binary ligand complexes (99mTc–HYNIC–tricine) may be present in solution.6c,9b Although many efforts have been made to characterize the complexes at the tracer level, the chemistry remains controversial. Because of the increasing regulatory requirements, which include in-depth tracer-level characterization of the products, translating the HYNIC-based radiotracers to regular clinical use will be problematic.6a Alberto and coworkers10a first developed another interesting organometallic Tc-core, [99mTc(H2O)3(CO)3]+ by directly reducing 99mTcO4− with sodium borohydride in aqueous solution in the presence of carbon monoxide. The core has several attractive features, including synthetic ease, water solubility, lipophilicity, and it is organometallic, chemically robust, and highly stable under the harshest conditions. The metal in [Tc(CO)3]+ is d6 low spin, and very inert. Monodentate and bidentate chelators often form complexes with low solution stability, resulting in high protein binding to this core in the blood stream and the complexes are more likely to be retained in the liver and kidneys.10b In contrast, tridentate chelators form [Tc(CO)3]+ complexes with very high stability, which clear rapidly from blood and major organs.10b This difference is because of the presence of an aqua ligand in the monodentate or bidentate complex, which is susceptible to get replaced by proteins in the blood. It is important to note that the tridentate chelator can be easily modified to provide anionic, neutral, or cationic complexes with the [Tc(CO)3]+ core (Fig. 7). Additional functional group can also be introduced to the tridentate ligand for conjugation with the targeting vectors. Overall, the [Tc(CO)3]+ core appears to be an extremely valuable entity for the development of 99mTc-based radiopharmaceuticals.11,12
Fig. 6.

Common technetium core structures (BM is the targeting biomolecule).
Fig. 7.
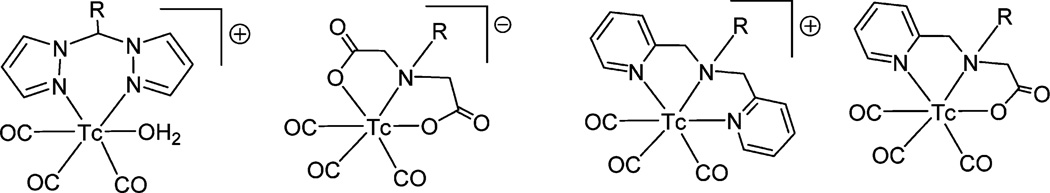
Examples of some ionic and neutral [Tc(CO)3]+ core complexes. The R group may be a biomolecule or a linker attached to the biomolecule.
Research efforts on developing 99mTc-tagged receptor-specific agents resulted in FDA approval of two 99mTc-based antibody agents, 99mTc–LeuTech13a and 99mTc–Arcitumomab13b (CEA-Scan), for infection imaging and colorectal cancer imaging (Table 2). LeuTech is anti-CD 15 antibody, which specifically targets neu-trophils, and 99mTc–LeuTech is used for imaging appendicitis. Arcitumomab recognizes carcinoembryonic antigen (CEA), which is overexpressed in 95% of colorectal cancers.
Indium-111
Indium belongs to Group 13 of the periodic table. The only stable aqueous oxidation state of indium is +3. Due to its large size, the coordination number of In3+ is 7–8. It is a hard acid (pKa 4.0). Therefore, chelators having hard donor atoms should be suitable to form stable metal complexes with In3+. Commonly used chelators are diethylenetriaminepentaacetic acid (DTPA), 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) and 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA) (Fig. 5). 111In is the next most well-known SPECT radiometal after 99mTc. It decays by electron capture, emits gamma photon 173 and 247 keV, and is widely used in gamma scintigraphy. With a 68 h physical half-life, 111In is suitable for developing antibody-based radiopharmaceuticals for radioimmunoimaging (immuno-SPECT). It is a cyclotron-produced radionuclide (Table 1). 111In is usually isolated from Ag or Cd target materials by its co-precipitation with La(OH)3 or Fe(OH)3.14,6c Then 111In is separated from La(III) or Fe(III) by ion-exchange chromatography.
Although some small 111In complexes are commercially available (Table 2) for SPECT imaging, it is mainly used to develop antibody-based SPECT imaging agents. The most common method for attaching 111In to an antibody is via a chelation reaction of a BFC that has previously been conjugated with an antibody molecule. A DTPA derivative with a trans-1,2-diaminocyclohexane backbone, CHX–A″–DTPA, is found to be the most viable chelator for 111In radiometal. 111In-labelled proteins are prepared with high radiochemical yields (>95%) by a simple addition of 111 InCl3 solution to a suitable BFC-conjugated protein solution at room temperature. The specific activity is adjusted to about 4 to 6 mCi per mg of mAb. BFCs are commercially available and can be conjugated with proteins or small peptides. The degree of conjugation (chelate:mAb) should be around 1 to 2, which can be determined by a radiometric metal-binding assay.15
In the past, the majority of radioimmunoimaging studies were done using iodine-131 (a non-metallic radionuclide) as the radioactive source. Comparative studies16 between 131I and 111In have revealed that, 111In-label offers tumor-to-blood ratios at a faster rate, and a higher level. 111In-label also has a longer retention time of radioactivity in the tumor, which results in clearer tumor images. However, there are also some marked disadvantages associated with using 111In. The major disadvantage is the high non-specific uptake of 111In in the liver, kidneys, and spleen of a patient.17 Eliminating this high, non-specific uptake background has been a pressing problem for improving 111In-based immunoimaging. 111In is an iron mimic and can easily be taken up by transferrin, which may cause high liver uptake for 111In-labelled antibodies. Detailed pharmacokinetic studies suggest that the liver uptake may not always be the result of the labeling process, but may be caused by the way the liver handles the antibody (generation of BFC-metabolites).18 Different approaches have been taken to reduce the non-specific bindings, such as changing BFCs, introducing a linker, and administering the chelator.19 111In-labelled small peptides/affibodies are showing promising results (less renal uptake) in SPECT imaging.20 Stability and biodistribution (uptake in tumor, liver, kidneys, etc.) are comparable for all three BFCs (DOTA, NOTA, and DTPA)21 used for 111In labeling.
Recently, three 111In-labelled antibodies (ProstaScint, OncoScint and MyoScint) have been approved by the FDA (Table 2) for use in humans. All three of these approved agents use a DTPA chelator to complex the 111In. The antibody ProstaScint is directed against prostate membrane-specific antigen (PMSA) which is overexpressed in prostate adenocarcinoma cells compared to nonmalignant cells. OncoScint or 111In–satumomab pendetide localizes or binds specifically to a tumor associated glycoprotein (TAG-72), a cell surface antigen, expressedat a high level on nearly all colorectal and ovarian adenocarcinomas. MyoScint is a 111In-labelled mAb (imciromab) that binds the heavy chain of cardiac myosin, which is exposed after loss of the myocyte membrane, as occurs in acute myocardial infarction (AMI) and is detected using immunoscintigraphy.
Gallium-67
Just like indium, the stable oxidation state of gallium is +3. As a hard acidic cation, Ga(III) preferably binds to chelators that have multiple anionic oxygen donor sites (DFO, DOTA, etc.). 67Ga is produced in a cyclotron by proton bombardment of a natZn target. 67Ga uses 15–20 h of cyclotron beam time per production.22. 67Ga(III) is separated with high radionuclide purity from the target material by acid dissolution and chromatography on an organic polymer resin containing no ion-exchange group.22c
67Ga (t1/2 78.3 h) decays to stable 67Zn by electron capture and is extensively used in nuclear medicine.23 Although 67Ga(III)–citrate is not a tumor-specific agent,24 it concentrates in many types of tumors and non-malignant lesions, especially inflammatory ones (e.g., cardiac sarcoidosis).25 Generally, 67Ga(III)–citrate binds to plasma transferrin and is subsequently transported to the tumor tissue. This radioisotope is used extensively to localize a variety of malignant human tumors.26
For 67Ga-immuno-SPECT, mAbs can be efficiently labelled with 67Ga through chelation with desferrioxamine (DFO) or DOTA without losing antigen-binding capabilities. The highest tumor : liver uptake can be obtained by linking a BFC to an antibody with thioether bonds.27
Radiometals for PET
A few positron-emitting radiometals have been selected (Table 1) based on their growing importance to the development of PET pharmaceuticals, as described in many published articles. 18F (t1/2 109 min) and 11C (t1/2 20 min) are very common and widely used as positron-emitting radionuclides for diagnostic imaging. These two radionuclides have relatively short half-lives, which makes them favorable for minimizing the body’s exposure to radiation. 18F and 11C also have decay characteristics that make them optimal for PET imaging. However, their short half-lives and typical labeling condition (high temperature, organic solvent) lower their suitability for use with biomolecules (antibodies and peptides). Alternative or unconventional radionuclides, which have received increasing attention, are the positron emitters 64Cu, 89Zr, 68Ga, 86Y, and 82Rb. Although they have short half-lives, 68Ga and 82Rb can easily be created in-house because they are generator-produced radionuclides. 68Ga can be easily attached to small peptides at low temperatures using a suitable BFC (usually DOTA or NOTA) and 82Rb is used in diagnostic imaging as soon as it is eluted out of the generator.
Gallium-68
68Ga is a generator-produced PET radionuclide which decays to the stable daughter isotope 68Zn. A number of different 68Ge/68Ga generators have been developed based on the composition of stationary phases (inorganic: TiO2, Al2O3, SnO2 etc., or organic polymers).28A typical 68Ge/68Ga generator is usually composed of a column containing a stationary phase, on which a parent isotope, 68Ge (carrier free, t1/2 271 days) is absorbed. The elution of 68Ga is usually completed with 0.05 M ethylenediamine tetraacetate (EDTA) or 0.1 M HCl solution. Generator shelf life is about 1 to 2 years and elution can be completed two to three times per day. The half-life of 68Gaislong enoughtoperform elaborate radiochemical synthesis and purifications. Recent examples demonstrate that small molecules,29a peptides,29b and antibody fragments29c are ideal BMs in the development of 68Ga-based PET pharmaceuticals. 68Ga is often considered an expensive radionuclide for PET pharmaceuticals development30 due to the inefficient production method used for its parent isotope 68Ge.
In aqueous solution, 68Ga(III) ultimately hydrolyses to form the 68Ga(OH)3 precipitate. Transferrin and/or other Fe(III)-binding proteins in physiological fluid can bind to 68Ga(III) with high affinity. Any development of a radiopharmaceutical with 68Ga needs a strong BFC that can resist the exchange of 68Ga(III) from its chelate with transferrin or with OH− ions. Chelators that can form stable complexes with 68Ga(III) are DFO, DOTA, and NOTA.29,31
68Ga labelled DOTA-D-Phe1-Tyr3-octreotide, [68Ga]–DOTA–TOC (Fig. 8) is one of the important 68Ga-based PET tracers. It is the gold standard for 68Ga-based small PET peptide radio-pharmaceuticals and other chelator-based somatostatin analogues for clinical studies.32 Somatostatin receptor proteins are known to be abundant on the surface of several human tumors, including neuroendocrine tumors. Recently, Gabriel et al.32a investigated the potential of [68Ga]–DOTA–TOC for use in diagnosing primary neuroendocrine tumors and related bone metastases. In this clinical study, [68Ga]–DOTA–TOC PET images were compared with conventional radioscintigraphy and CT. In 84 patients, [68Ga]–DOTA–TOC PET was true positive in 69 patients, true negative in 12 patients, false positive in 1 patient, and false negative in 2 patients, indicating a sensitivity of 97%, a specificity of 92%, and an accuracy of 96%.
Fig. 8.

(A) An example of commercially available 68 Ga-generators (Courtesy of Y. M. Daltorio, E & Z, Germany); (B) Somatostatin analogue 68Ga–DOTA–TOC.
Rubidium-82
In the periodic table rubidium is located in Group 1, just below potassium. Like gallium-68, 82Rb is also a generator-produced PET radionuclide with a very short half-life (75 s). The parent isotope of 82Rb is the longer-lived 82Sr (t1/2 25.55 days), which is produced in a cyclotron by a (p, 4n) reaction induced by >40 MeV protons on a rubidium (85Rb) target. The 82Sr/82Rb generator 33 is composed of a hydrous tin-oxide column on which 100–150 mCi 82Sr is adsorbed. 82Rb is eluted with saline. Since 82Rb has a very short half-life, its elution and infusion into patients is performed by an automated infusion system.34. 82Rb is mainly used in the PET studies of myocardial perfusion. Since 82Rb+ ions mimic the behavior of K+ ions, it is a cost-effective alternative to the use of cyclotron-produced [13N]–NH3 or [15O]–H2O for cardiac perfusion imaging. The ATP-dependent sodium–potassium pump does not differentiate between potassium and rubidium. Like potassium, rubidium is concentrated in the myocardium by the Na/K ATPase pump.35a Furthermore, because of the short half-life of 82Rb, imaging protocols and pharmacokinetics are dominated by the physical characteristics of the nuclide. Myocardial imaging with 82Rb requires rapid acquisition of data, because of the isotope’s short physical half-life, which might make it very attractive for use in rapid repeat studies, such as stress/rest protocols and preintervention/post-intervention studies. 82Rb+ has also been used to provide the useful information on the integrity of the blood–brain barrier, changes in renal perfusion, and myocardial cell membrane viability, etc.6e,35b,c
Copper-64
The predominant oxidation state of the radiocopper in aqueous media is Cu(II), 3d9 system. Cu(II) is a cation of borderline hardness, which has a high affinity for borderline nitrogen donors. Depending on the chelate structure and donor atoms, the coordination number of Cu(II) ranges between 4 and 6, with geometries approximating square planar, square pyramidal, trigonal bipyramidal and octahedral. One or more of its coordinated ligand becomes elongated due to Jahn–Teller distortion of the 3d9 system. Recently, the importance of in vivo bioreduction of Cu(II) has received increasing attention. Cu(II)/Cu(I) bioreduction may be the viable cause of in vivo radiocopper loss in many 64Cu-based radiopharmaceuticals. For in vivo applications, kinetic inertness of Cu(II) complexes is probably more important than their thermodynamic stability.36a
64 Cu (t1/2 12.7 h), produced in a cyclotron is a positron and a beta emitter. These properties make 64Cu a promising radionuclide for both diagnostic PET and ther apeutic radiopharmaceuticals.36 The image quality and spatial resolution are equivalent to 18F and require no major adjustments in image processing and analysis.There have been several methods to produce64Cu.37 Better quality and higher yields of 64Cu can be produced by 12 MeV proton irradiation of enriched 64Ni via 64Ni(p,n)64Cu.37b,c This radionuclide is now commercially available from several sources (MDS Nordion, IBA, Isotrace Tech., etc.).
64 Cu-labelled radiopharmaceuticals can be grouped into three general categories: small molecules, peptides, and antibodies. Among the small molecules, 64Cu–PTSM (pyruvaldehyde bis(N4-dimethylthiosemicarbazone)) (Fig. 9) has been shown to be an agent that measures blood flow in the heart and brain.36d,e Wallhaus and coworkers showed that blood-flow abnormalities identified by 62Cu–PTSM correlated well with angiography in patients with coronary artery disease, and the results were comparable to those obtained with 99mTc–sestamibi and 82Rb.38a Another thiosemicarbazone complex of Cu(II), Cu–ATSM38b (diacetyl-bis(N4-methylthiosemicarbazone)) with a structure similar to 64Cu–PTSM has been shown to be trapped in hypoxic cells. In the presence of hypoxia, Cu(II)–ATSM is reduced to Cu(I)–ATSM. This reduced form is trapped intracellularly, resulting very high tumor uptake in less than 1 h after injection. Clinical studies have shown that when administered in large quantities, 64Cu–ATSMcan predict tumor response to therapy in cervical cancer, non-small cell lung cancer, and to some extent, rectal cancer.38b,c Overall, 64Cu– ATSM has demonstrated potential value in clinical studies as a PET-radiotracer for hypoxia imaging and therapy.
Fig. 9.

Structure of Cu–ATSM and Cu–PTSM (Cu = 62Cu or 64Cu).
To radiolabel peptides and antibodies with 64Cu, a suitable BFC is needed. In biological fluid (human serum), common BFCs based on EDTA or DTPA are rapidly dissociated to a Cu(II) albumin complex. Several BFCs (DOTA, TETA, etc.) have been developed forin vivo studies. Allofthese BFCs and theirderivatives were unsuccessful in preventing metabolic processing of the 64Cu complex, causing elevated liver uptake. Cu-binding endogenous proteins36a (ceruloplasmin and superoxide dismutase) present in the liver compete with BFCs to bind to the 64Cu (transchelation) of 64Cu-radiopharmaceuticals. To overcome the in vivo instability of Cu(II) complexes, cross-bridged tetramine chelators (e.g., CB– TE2A) were developed and evaluated.39 These cross-bridged BFCs provide better geometric coordination and are inert in aqueous solutions.39 Sprauge et al.40 recently performed a comparative PET imaging study between 64Cu–TETA–Y3–TATE and 64Cu– CB–TE2A–Y3–TATE (Fig. 10) on somatostatin receptor positive AR42J tumor-bearing rats. Biodistribution results showed that nonspecific uptake in the blood and liver was lower for 64Cu–CB–TE2A–Y3–TATE. After 4 h blood uptake was ~4-fold lower and liver uptake was 2.4-fold lower than that of 64Cu–TETA–Y3– TATE. In addition to this, tumor uptake was 4.4 times greater with 64Cu–CB–TE2A–Y3–TATE at 4 h post-injection (Fig. 11).
Fig. 10.

Structure of TETA, CB–TE2A conjugates with Tyr3–Octreotate.
Fig. 11.

MicroPET projection images of AR42J tumor bearing rats at 4 h after injection (A); liver, kidney (B); and tumor (C) showing the 64Cu activity uptake at 1, 4, and 24 h post-injection. This image is reprinted with the permission from American Association for Cancer Research: Sprague et al., Clin. Canc. Res., 2004, 10, 8674.
These results have prompted researchers to search for more cross-bridged or cage-like BFCs, which can form extremely stable coordination geometry around 64Cu and can easily be conjugated with different kinds of BMs (small peptides or antibodies) under milder conditions. Smith et al.41 recently developed a BFC, SarAr (Fig. 4), which is a derivative of diamsar, a sarcophagine cage ligand. A pendent aromatic amine of SarAr can be used in a conjugation reaction with carboxylic acid residues of BMs under milder conditions. While more facile in vivo reduction has compromised the utility of other copper radiopharmaceuticals,42 the Cu2+ ion within Sar or SarAr is unusually resistant to reduction (Cu2+/Cu+). A recent evaluation43 of this novel BFC (SarAr) conjugated to anti-GD2 monoclonal murine antibody 14.G2a and corresponding chimeric mAb ch14.8 has demonstrated that using a better chelating agent can greatly improve the tumor to background ratio. MicroPET imaging confirms the significant uptake of radioimmunoconjugate in GD-2-positive tumors, with minimal uptake in non-target tissues of interest such as the liver.43 This study also demonstrates that radiolabeling of SarAr–mAb can be performed in less harsh conditions and the resulting radioimmunoconjugate is stable in the presence of Cu-binding liver proteins.43
More recently Cai and coworkers44 designed a new sarco-phagine-based cage-like BFC named AmBaSar by introducing benzoic acid to diamsar. This ligand can be easily conjugated with the primary amine of peptides and proteins via the well-known 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC)-coupling reaction. A similar kind of modification on diamsar was done by Ma and coworkers,45 where they introduced the aliphatic carboxylic (glutaric) acid group for facile conjugation with the BM (Fig. 12). In a recent preclinical study, compared to the 64Cu-DOTA-RGD peptide, 64Cu–AmBaSar–RGD demonstrated much lower liver accumulation in both biodistribution and microPET imaging of integrin αvβ3 expression.46 Metabolic studies with these two tracers also suggested that 64Cu–AmBaSar–RGD was more stable in vivo and had faster renal clearance than 64Cu-DOTA-RGD.47
Fig. 12.

Copper complex of sarcophagine-based cage-like BFC and an ORTEP representation shows a distorted octahedral structure with axial Jahn–Teller elongation (Ma et al. Chem. Commun. 2009, 22, 3237; reproduced by the permission of the Royal Society of Chemistry).
All these studies clearly show that in vivo stability of radiocopper largely depends on the structure of the BFCs. The possible stability order can be DOTA ~ TETA < CB-TE2A 〈〈 SarAr ~ AmBaSar.
Although excellent results have been obtained while imaging intact antibodies with 64Cu, based on 64Cu’s physical half-life, it is a more suitable radiometal for imaging when applied to antibody fragments, peptides, and other engineered target vectors, which have faster localization and clearance.48 In spite of efforts to reduce the non-specific uptake by developing better BFCs and linkers, liver accumulation of 64Cu remains a concern.
Several clinical studies have been published on the application of Cu-ATSM (small molecule) in diagnosis and therapy.49a–c To date, there has been only one clinical study that utilized 64Cu-mAb as diagnostic immuno-PET agent. This clinical study (Phase I/II) was completed by Philpott and coworkers49d using 64Cu-TETA-1A3 (1A3 is an anti-colorectal carcinoma mAb) to evaluate 36 patients with suspected primary or advanced colorectal cancer. All patients had CT and 18F-FDG PET scans 4 to 36 h after being injected with 64Cu–TETA–1A3 specific to lipid antigen present in human colon carcinoma. This study demonstrated that 64Cu–TETA–1A3 was more specific for detecting colorectal cancer than 18F–FDG and 64Cu-immuno-PET may have better applications in clinical oncology.
Yttrium-86
Highly specific activity 86Y can be produced in a medical cyclotron by irradiating SrCO3 or SrO applying 86Sr(p,n)86Y nuclear reaction.50 A simple, semiautomated method has been developed by Germastani et al., to purify 86 Y.51 The main purpose of studying the viability of 86Y as a diagnostic PET pharmaceutical was to use it as an imaging surrogate for the therapeutic radionuclide 90 Y (t1/2 64 h), which is a purely β− emitter. The half-life of 86Y (t1/2 14.2 h) is long enough to monitor 90Y-therapy over several days. The high positron energy of 3.14 MeV (34% abundance) and additional γ-emission of 1.08 MeV (83% abundance) significantly affects the spatial resolution and image quality, respectively.
Chelation chemistry with Y(iii) is well-established. Generally, the derivatives DOTA and DTPA have been used to develop im-munoconjugates for 86Y-immuno-PET.52 Therapeutic studies with 90Y–DTPA–immunoconjugate in patients demonstrated that 90 Y was released and deposited in bones, causing radiation toxicity.53 In order to improve the in vivo stability of the radioimmunoconju-gate, several backbone-substituted DTPA have been evaluated.54 Among all derivatives, CHX–A“–DTPA, (enantiomeric form of CHX–DTPA) (Fig. 4) has demonstrated excellent stability in vitro and in vivo.55 Stability was improved further using DOTA derivatives, such as SCN–Bz–DOTA.56 Numerous preclinical and clinical studies have been published that report the use of DTPA- and DOTA-based chelating agents for 90Y immunotherapy and, thus, for 86Y immunoimaging.57 86Y–CHX–A“–DTPA– trastuzumab and 86Y–CHX–A”–DTPA–cetuximab are the most popular 86Y-radiopharmaceuticals frequently used for diagnostic imaging of different cancer xenografts in mice.58 A recent clinical study demonstrated the successful use of 86Y–DOTA–Phe1–Tyr3– octreotide (SMT-487) for the dosimetry of 90Y–SMT-487 radiotherapy (Fig. 15)59 in patients with metastatic carcinoid tumors.
Fig. 15.

Structures of some therapeutic radiopharmaceuticals. A: 153Sm–EDTMP (Quadramet); B: 177Lu and 90Y-labelled DOTATOC, a somatostatin analog useful for tumor imaging and therapy.
Zirconium-89
Zirconium belongs to Group 4 of the periodic table. Based on Pearson’s soft-hard-acid-base (SHAB) theory, Zr(IV) is an extremely acidic cation, which is the main reason of its strong preference for polyatomic hard donor chelators. Only limited ther-modynamic, kinetic, and structural data are available for Zr(IV)-chelator complexes compared to other common positron emitting metals (e.g., 64Cu, 68Ga, 86Y, etc.). This indicates that Zr(IV) is inert and has a strong preference for a particular type of chelators. In recent years, 89 Zr has emerged as a promising positron-emitting radionuclide for diagnostic immuno-PET imaging because of its longer half -life (78.4 h), matching with the biological half-life of an intact mAb; positron yield (22.7%), which is comparable to 64Cu and 86Y; and inertness in biological system. Additionally, based on clinical studies in Europe, 89Zr-labelled mAbs are safe for use in humans.
89Zr is a cyclotron-produced nuclide and a promising longer-lived (t1/2 78.4 h) positron emitter for labeling mAbs for use in immuno-PET imaging. 89Zr decays to an ultra short-lived daughter, 89mY (t1/2 16 s), with 23% positron emission and 77% electron capture. 89Zr can be produced through a 89Y(p,n)89Zr reaction upon irradiation of natural yttrium foil with a 14– 14.5 MeV proton beam; the target material is separated by ion-exchange chromatography or solvent extraction.60
The most convenient BFC for 89Zr labeling to proteins and antibodies is DFO.62 Verel et al.63a developed a succinimidyl ester derivative of DFO (N-SucDFO) to make a stable conjugate with antibodies. In this method, the chelating site of N-SucDFO was temporarily blocked by the Fe(III) ion. The N-SucDFO–Fe complex was then esterified and conjugated with antibodies. After that, Fe(III) was removed by reduction to Fe(II) and transchelation to EDTA. Finally, the mAb–N-SucDFO conjugate was radiola-beled with 89Zr at pH 7.2–7.4 after a 30 min incubation period at room temperature.63 Recently, this multistep conjugation and radiolabeling method has been further simplified (Perk et al.),64 using a commercially available (Macrocyclics, Inc.) amine reactive BFC, p-isothiocyanatobenzyldesferrioxamine B (DFO–Bz–NCS) (Fig. 4). The DFO–Bz–NCS–mAb conjugates was created by adding a 3-fold molar excess of DFO–Bz–NCS into the antibody at pH 9.0 and incubating the reaction mixture for 30 min at 37 °C. DFO–Bz–NCS–mAb was then purified using size-exclusion chromatography (PD10, GE-Healthcare). Radiolabeling with 89Zr was performed at pH 7.2–7.4 by incubating the conjugate with 89Zr(IV)–oxalate at room temperature for 60 min. Finally, 89Zr–DFO–Bz–NCS–mAb (Fig. 13) can be purified by size-exclusion chromatography. Although some other methods are reported65 in literature, these two methods are primarily used to develop 89Zr-immunoconjugates.
Fig. 13.

Structure of 89Zr labelled desferrioxamine conjugate with mAb.
86Y is receiving clinical attention as a PET surrogate for 90Y-immunotherapy because of their similar chelation chemistry. However, the half-life of 86Y is relatively short for optimal imaging and dosimetry with an intact antibody which usually requires 2– 4 days to distribute the entire body. Because the physical half-life of 89Zr (t1/2 78.4 h) is almost 5 times greater than that of 86Y (t1/2 14.2 h), 89Zr-labelled antibodies are currently being evaluated as PET surrogates for both 90Y and 177Lu therapy studies. In 2006, Perk et al.,66 developed 89Zr–Zevalin and showed that PET with 89Zr–Zevalin was very safe and attractive for quantitative prediction of pharmacokinetics, biodistribution, and dosimetry of 90Y–Zevalin radioimmunotherapy (RIT) in patients with CD20+ B-cell non-Hodgkin’s lymphoma (NHL). The same year, Börjesson et al.67 completed the first clinical trial of 89Zr-labelled U36, a chimeric anti-CD44v6 mAb used to detect squamous cell carcinoma of the head and neck (HNSCC). In this study, 20 HNSCC patients received 75 MBq of 89Zr–U36 along with 10 mg of antibody. Preoperative findings obtained with 89Zr-immuno-PET were compared with the histopathologic findings, where 25 tumors and 121 operated levels were involved. 89Zr-immuno-PET detected tumors in 18 of 25 positive levels (72% sensitivity) and the interpretation was correct in 112 of 121 operated levels (93% accuracy). Immuno-PET scans were acquired (Fig. 14) up to 144 h post-injection and compared with diagnostic results obtained using 18F-FDG PET, CT, and MRI in the same patients. Further clinical studies with 89Zr-immuno-PET are underway in both the United States and Europe.
Fig. 14.

Immuno-PET images with 89Zr–cmAb U36 of a head and neck cancer patient with a tumor in left tonsil (large arrow) and lymph node metastases (small arrows). Images were obtained 72 h post-injection. A, sagittal image; B, axial image; and C, coronal image. This image is reprinted with the permission from American Association for Cancer Research: Bo¨rjesson et al., Clin. Canc. Res., 2006, 12, 2133.
Radiometals for therapeutic radiopharmaceuticals
The use of high-energy radiation from X-rays, gamma rays, neutrons, protons, and other sources to kill cancer cells and shrink tumors is called radiotherapy. Radionuclides that predominantly emit Auger electrons and β− or α particles are usually used to develop therapeutic radiopharmaceuticals. Radionuclides selected for therapy use should have complementary properties to the radionuclides used in diagnoses. The radiation emission of these radionuclides should mainly contain damaging and non-penetrating particulate radiation with little to no accompanying gamma emission so that a highly-localized dose can be delivered without inducing any unnecessary radiation damage to normal tissues. However, it should be noted that 64 Cu decays via the b− particle (38.5%) and its therapeutic potential has been investigated.68 Diagnostic radiotracers could also be used to monitor the efficacy of the radiotherapy treatment. The use of radionuclides in therapy is less advanced than in diagnosis because destructive doses require much more accurate targeting than tracer doses. Therapeutic radiopharmaceuticals should have high tumor uptake, a high tumor to background ratio, long residence time, and fast renal clearance. A few β− and a particle-emitting radiometals for potential use in the design of therapeutic radiopharmaceuticals are listed in Table 1.
β−-Emitting radionuclides
90Y (t1/2 64 h) is an important generator-produced69 radiometal that has been thoroughly studied for use in β-therapy. It is a pure β−-emitter without any γ-emission for imaging. The coordination chemistry of 90Y (like 86Y) is well established. Yttrium and lanthanide (Sm, Lu, etc.) metal ions favor a +3 oxidation state and are coordinated by a number of water molecules in aqueous solution. Due to their large sizes, the coordination numbers of yttrium and lanthanide metal ions are typically between 7 and 10. Thus, CHX–A”–DTPA, SCN–Bz–DOTA, and DOTA– NHS-ester are the common BFCs for developing 90Y and other lanthanide radiometal-based therapeutic agents (Fig. 15).6h,21b,70
188Re (t1/2 16.8 h) is a generator-produced71 daughter radionuclide of 188W (t1/2 60 days). Design of 188W/188Re generator is similar to the commonly used 99mTc generator. 188Re is eluted with saline solution as Na[188ReO4]. A generator can last 2 to 6 months. 188Re (rhenium-188) is an excellent candidate for β-therapy. 188Re emits β− particles with longer tissue range (10–11 mm), which are suitable for large tumor masses.72. 188Re’s low-energy γ-ray emission (0.155 MeV) can be exploited for dosimetric purposes and to observe biological distribution during therapy.73 In the periodic table Re is located just below the Tc, Group 7 congener. The coordination chemistry of 188Re is very similar to 99mTc.
In 2002, 90Y−Zevalin was approved for clinical use in RIT-treatment of B-cell lymphoma.70a,74 Several clinical trials are ongoing to assess the therapeutic efficacy of 90Y-labelled antibodies in the treatment of NHL, colorectal, glioblastoma, prostate and renal cancers.75. 90Y–DOTA–TOC and 177Lu–DOTA–TATE are peptide based therapeutic agents recently being used to treat the patients with somatostatin receptor-positive neuroendocrine tumors in advanced stages.76 The Phase I/II data of 177Lu–rituximab (anti-CD20) for the treatment of relapsed lymphoma have demonstrated that this regimen is quite effective.77 Researchers at Cornell University in New York City are currently conducting a Phase II clinical trial to assess the therapeutic efficacy of 177Lu-labelled J591, an anti-PSMA antibody, in patients with prostate cancer (Clinical Trial #NCT00859781).6i
153Sm–ethylenediamine tetramethylene phosphonic acid (153Sm– EDTMP, Fig. 15) commercially known as Quadramet®, is under a Phase 2.5 study (2007–2012), sponsored by the National Cancer Institute (NCT00450619) to treat advanced prostate cancer. It is administered through a vein, where it can directly target bone tumors and relieve the pain caused by bone lesions. Radiation also increases the level of certain proteins inside the tumor, making it easier for the immune system to find and kill the tumor cells. This study is currently recruiting participants.
188Re-hydroxyethylidinediphosphonate (188Re-HEDP), an analogue of 99mTc-MDP, has shown therapeutic efficacy in the treatment of metastatic bone pain associated with prostate, breast, and colorectal cancers.78
α-Emitting radionuclides
Most of the radiometals that emit α-particles are heavy-metal elements. There is considerable research involving α-emitters, where the shorter particle range and high-energy deposition can be used to destroy DNA directly. α-Emitting radiopharmaceuticals offer less radiotoxicity and more favorable dosimetry for background tissues. Clinical trials are in progress to determine the efficacy of the α-RIT as potential therapeutic radiopharmaceuticals. For example, researchers of the Memorial Sloan-Kettering Cancer Center and the National Cancer Institute are conducting Phase I/II clinical trials to assess the treatment efficacy of 213Bi (t1/2 45.59 min)-labelled and 225Ac (t1/2 10 days)-labelled HuM 195, a humanized anti-CD33 antibody used to treat patients with leukemia or myelodysplastic syndrome.6i
Radium-223 (t1/2 11.4 d) is one of the well known a-emitting radionuclides used in large scale clinical trials.79 Cationic 223Ra in the form of a solution containing 223RaCl2 (Alpharadin™) used in clinical trial can selectively accumulate in skeletal metastases resulting from primary tumors of prostate and breast. The clinical results show an impressive anti-tumor activity and improved overall survival in prostate cancer patients with bone metastases. Because of the unique chemical properties of Ra, it is challenging to find a suitable BFC to develop 223Ra-based immunotherapeutic agents.
Heavy-halogen astatine-211, which resembles iodineinsolution, is another well known α-emitting radionuclide. Targeted radiotherapy is possible with astatine-211. Just like radioactive iodine, it can easily form stable chemical bond with BMs.80a Zalutsky et al.80b evaluated the feasibility, safety, and efficacy of administration of chimeric anti-tenascin mAb (ch81C6) labelled with 211At (211At–ch81C6) into the surgically created resection cavity (SCRC) of patients with recurrent malignant brain tumors. Monoclonal antibody ch81C6 is an IgG2b that reacts with tenascin, an extracellular matrix glycoprotein expressed in high-grade gliomas but not in normal brain tissue. Results showed that the regional administration of 211At–ch81C6 is feasible, safe, and associated with a promising antitumor benefit in patients with malignant central nervous system tumors.
The potential therapeutic value of many radiometals has not yet been properly explored because of some scientific and regulatory barriers.74 The main limiting factor in using therapeutic radiopharmaceuticals is the potential for radiation toxicity to the kidneys, bone marrow, and other internal organs. Antibody-based therapeutic radiopharmaceuticals (long biological half-life) may cause severe damage as they pass slowly through various organs. When considering radiation exposure from radiation therapy treatment, the benefits generally outweigh the risks. Much effort is still required to overcome the challenges (scientific and regulatory) that currently inhibit the development of therapeutic radiopharmaceuticals for the clinic. Finally, treating cancer with drugs is a trial and error process. Some patients may respond better to one drug than another and tolerable limits vary from patient to patient. All of these factors allow clinicians to try new therapeutic drugs with the hope that they will produce positive responses.
Techniques for quality control (QC) of radiopharmaceuticals
Common analytical techniques (NMR, IR, UV-Vis, X-ray, etc.) are not useful for characterizing radiopharmaceuticals at their tracer levels. Few analytical tools and techniques (ITLC, HPLC, LC–MS, etc.) are routinely used in the radiopharmaceutical laboratory. In both preclinical and clinical set-up, these techniques are essential for quality control of radiolabeled drugs.
Instant thin layer chromatography (ITLC)
ITLC is one of the easiest and quickest analytical techniques used to determine the radiochemical yield (i.e. the percentage of bound radiometals) in radiopharmaceuticals. The whole process takes 5–15 min. In ITLC, a paper strip or plate is used to separate free radiometals and radiometal–BFC–BM conjugates. For radioimmuno conjugates (e.g., 111In–CHXA”–DTPA–trastuzumab), the mobile phase (aqueous buffers) is often selected in such a way that free radio-metals migrate to the solvent front and the conjugated radiometals remain in the origin. Forsmall radiolabeled molecules, organic solvent is normally used as the mobile phase where radiopharmaceuticals migrate to the solvent front and unbound radioisotopes remain in the origin. The ITLC strip is then cut into three parts (top, middle, and bottom) and the amount of radioactivity in each part is measured in a well counter (e.g., gamma well counter) to determine the radiochemical yield.
High pressure liquid chromatography (HPLC)
HPLC is frequently used to determine the radiochemical purity of radiometal-labelled bioconjugates. Typically, a highly sensitive radiation detector is attached in the liquid line just after the UV detector so the radiation peak appears along with the UV peak. Radio-HPLC is essential not only for determining radiochemical purity, but also for characterizing and isolating the radiolabeled compounds after radiosynthesis, which may generate multiple side products. HPLC can also be used to determine the specific activity (radioactivity mole−1) of a radiopharmaceutical. It is a very powerful tool to separate epimers and diastereomers. The choice of the HPLC column depends on the nature of the biomolecules to which the radiometal is attached. For large protein molecules or antibodies, the size exclusion column is used; whereas, the C-18 column is ideal for small peptides or small molecules. Radio-HPLC also has its limitations. It may not be possible to assess the amount of free radiometal present (e.g., size exclusion chromatography) in the reaction mixture. Therefore, an ITLC method is needed in combination with radio-HPLC to determine both radiochemical yield and the purity of the radiolabeled BMs.
Liquid chromatography–mass spectrometry (LC–MS)
LC– MS is primarily used to study radiopharmaceutical metabolism. It can also help to determine in vitro stability and the probable decomposition pathway including ligand exchange or transchelation. It can provide the molecular weight of all species present in a sample along with their liquid chromatograms. This information is very useful to determine the possible structure, oxidation state, charge of the species, and probable pathway of radiotracer decomposition. Currently LC–MS is regarded as a powerful technique for the in-depth characterization of small-molecule and peptide-based radiotracers. However, the high cost and service charge of this instrument is the biggest obstacle to its widespread use in radiopharmaceutical laboratories.
Gas chromatography (GC)
GC is primarily used to determine the volatile solvent impurities in radiopharmaceuticals. It is commonly used in the clinical laboratory to analyzethe percentage of organic solvents (acetonitrile, ethanol, etc.) present in a human dose. Usually only a few mL of radiotracer are injected in the column at a high temperature (100–250 °C). The percentage of solvent impurities is determined from the area of the characteristic peak after comparing the impurities to the standard calibration curve for that particular solvent. The drug will be released after the acceptable level of volatile solvent impurities has been confirmed. This technique is useful to small molecule-based radiopharmaceuticals that require organic or toxic solvent at the time of synthesis.
To meet USP compliance (USP 823) and to satisfy FDA requirements on human dose, more QC analysis needs to be performed on radiopharmaceuticals, including a toxicity test, a radionuclide purity test (using a multi channel analyzer), a sterility test, and others. Unlike the sterility test, which takes several days, all other tests are quick and easy, and should be performed the same day of production before releasing the drug to the clinic.
Radiation safety
Radiation safety is another important part of the radiopharmaceutical development process. Because of the recent advancement of cyclotron targetry, radiochemistry, and bioconjugation methods, more new radionuclides with different decay characteristics are entering biomedical research and clinical trials. Therefore, there is a clear need to reconsider the radiation safety aspects in the laboratory or the radiopharmaceutical production facility. Most of the nuclear medicine facilities around the world are designed to manipulate 99mTc and/or 18F. The shielding specifications and efficiencies for the other radiometals (89Zr, 64Cu, 225Ac, 68Ga, 82Rb etc.) are not very well researched or properly implemented. As per the recent statement of the American Association of Physicists in Medicine (AAPM), it is clear that PET facilities designed for 18F may not have appropriate shielding for the longer-lived radionuclides that have positron or high-energy gamma emissions.81 Holland et al.6i recently described their experience with unexpected radiation exposure while manipulating some unconventional radionuclides. Adequate shielding is essential in all cyclotron and nuclear medicine facilities to reduce the risk of radiation exposure to workers. The purpose of the shielding is to attenuate radiation by scattering, which protects radiation workers by reducing their exposure. The important shielding parameters in lead, which include a specific gamma ray dose constant (C constant) and a tenth-value layer (TVL), were found to vary widely from one radionuclide to another. These parameters are available in different sources, including the Handbook of Health Physics and Radiological Health. The design of proper shielding requires the use of accurate C constant and TVL (cmpb) numbers. In summary, the amount of radiation from nuclear medicine procedures should be kept within a safe limit following the “ALARA” (As Low As Reasonably Achievable) principle. Based on the type of radionuclide,the design and radiation safety features of current facilities need to be adjusted appropriately.
Conclusions
It is clear from the examples cited in this perspective that efforts to develop new radiopharmaceuticals for use in clinical trials are continuing. Many academic institutions (medical schools) have started setting up their own “Nuclear Medicine Centers” to develop imaging agents, using PET or SPECT, and to practice “individual nuclear medicine.”
Radiometals are now frequently being used to develop target-specific radiopharmaceuticals because of their longer half-lives, which make them ideal for developing various target-specific protein- and peptide-based radiopharmaceuticals. Identifying the biological target and BMs are the two critical aspects for radiopharmaceutical development. It is not an easy task to attach a radiometal to a BM through a suitable BFC without affecting the target specificity of the BM. The metal chelate’s impact on the biological properties of radiopharmaceuticals is significant. Little modification in a BFC may result in huge variations in in vivo data (biodistribution, metal–chelate stability, pharmacokinetics). Therefore, coordination chemistry continues to play a pivotal role in developing new radiopharmaceuticals.
The clinical success of a new radiopharmaceutical largely depends on the purpose of its use, the overall cost associated with its production, and the process of validation through clinical trials, which is gradually increasing. For example, if an agent needs a rare and expensive radionuclide or a BM, and the application of this agent is too specific, then this expensive drug will be unable to reach a large population of patients and will not be successful from a commercial and medical point of view. Because of their wide applications and low production cost, 18F–FDG and 99mTc-labelled compounds will remain dominant in the near future. However, a few other agents based on 64Cu, 89Zr, and 225Ac are showing promising results and active clinical trials are underway. Because of the continuous research on technology advancement and radiochemistry techniques, the production cost of these agents is gradually decreasing. These radionuclides are becoming available at reasonable prices.
The process of converting a promising radiochemical to a radiopharmaceutical for the clinic is rife with scientific and regulatory obstacles.6f,74 Tremendous effort is needed to overcome these barriers before realizing the true potential of the targeted radiopharmaceuticals. We would like to end our discussion with a statement written by Professor Susan Lever in her review:82“Regardless of the beauty of the science involved in the development of the radiotracer, the ultimate goal is not the science, but the ability to improve the quality of life.”
Acknowledgements
This article is dedicated to Professor Animesh Chakravorty in celebration of his 75th birthday.88 The authors are grateful to all the talented researchers cited amongst the references for their significant contributions in this field of research. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U. S. Government.
Biographies

Sibaprasad Bhattacharyya
Sibaprasad Bhattacharyya received his Ph.D. under Professor Animesh Chakravorty from the Indian Association for the Cultivation of Science in 2001. A subsequent postdoctoral position at chemistry department of Indiana University, Blooming-ton with Professor Jeffrey M. Zaleski focused on metal ion induced enediyne cyclization. Dr Bhattacharyya then investigated photoacoustic and nuclear imaging (PET and SPECT) probes at Indiana University and Harvard Medical School with Professor Timothy R. DeGrado. In 2009, he started a new radiochemistry program at NCI-Frederick to support the Cancer Imaging Program of NCI with variety of imaging probes for preclinical and clinical studies. Currently, Dr Bhattacharyya is a senior scientist and the head of the radiopharmaceutical chemistry laboratory.

Manish Dixit
Manish Dixit received his Ph. D. degree in Medicinal Chemistry under Dr A. Goel at the Central Drug Research Institute Lucknow, India in 2007. Dr Dixit then joined the University of Arizona as a postdoctoral fellow with Professor Laurence Hurley, where he worked on the design and synthesis of G-Quadruplex DNA binding agents for anti-cancer activity. He joined Dr Bhattacharyya’s group in 2009, and is now investigating radio-tracers for PET/SPECT imaging
Footnotes
Dedicated to Professor Animesh Chakravorty on the occasion of his 75th birthday
References
- 1.(a) Bucerius J, Ahmadzadehfar H, Biersack H. 99mTc-Sestamibi Clinical applications. 1st edn. Germany: Springer; 2011. (ISBN: 978-3-642-04232-4) [Google Scholar]; (b) Sheikine Y, Berman D, Di Carli MF. Clin. Cardiol. 2010;33:E39–E45. doi: 10.1002/clc.20519. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Fleming RM, Harrington GM, Baqir R, Jay S, Sridevi C, Avery K, Jim G. Metho. DeBakey Cardio. J. 2009;5(3):42–48. doi: 10.14797/mdcj-5-3-42. [DOI] [PubMed] [Google Scholar]
- 2.Gupta VK. J. Med. Phys. 1995;20:31. [Google Scholar]
- 3.Hoskin P, Coyle C. Radiotherapy in Practice: Brachytherapy. NY: Oxford University Press; 2005. [Google Scholar]
- 4.Welch MJ, Redvanly CS. Handbook of radiopharmaceuticals: radiochemistry and applications. New York: Wiley; 2003. [Google Scholar]
- 5.(a) Mindt TL, Muller C, Stuker F, Salazar JF, Hohn A, Mueggler T, Rubin M, Schibli R. Bioconjugate Chem. 2008;20:1940. doi: 10.1021/bc900276b. [DOI] [PubMed] [Google Scholar]; (b) Mindt TL, Muller C, Melis M, de ong M, Schibli R. Bioconjugate Chem. 2008;19:1689. doi: 10.1021/bc800183r. [DOI] [PubMed] [Google Scholar]; (c) Hom RK, Katzenellenbogen JA. J. Org. Chem. 1997;62:6290. doi: 10.1021/jo951995k. [DOI] [PubMed] [Google Scholar]
- 6.(a) Bartholoma D, Louie AS, Valliant JF, Zubieta J. Chem. Rev. 2010;110:2903. doi: 10.1021/cr1000755. [DOI] [PubMed] [Google Scholar]; (b) Liu S. Adv. Drug Delivery Rev. 2008;60:1347. doi: 10.1016/j.addr.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu S. Chem. Soc. Rev. 2004;33:445. doi: 10.1039/b309961j. [DOI] [PubMed] [Google Scholar]; (d) Dilworth JR, Parrott SJ. Chem. Soc. Rev. 1998;27:43. [Google Scholar]; (e) Anderson CJ, Welch MJ. Chem. Rev. 1999;99:2219. doi: 10.1021/cr980451q. [DOI] [PubMed] [Google Scholar]; (f) Nayak TK, Brechbiel MW. Bioconjugate Chem. 2009;20:825. doi: 10.1021/bc800299f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Blower P. Dalton Trans. 2006:1705. doi: 10.1039/b516860k. [DOI] [PubMed] [Google Scholar]; (h) Liu S, Edwards DS. Bioconjugate Chem. 2001;12:7. doi: 10.1021/bc000070v. [DOI] [PubMed] [Google Scholar]; (i) Holland JP, Williamson MJ, Lewis JS. Mol. Imaging. 2010;9:1. [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Harper PV, Beck R, Charleston D, Lathrop KA. Nucleonics. 1964;22:1137. [Google Scholar]; (b) Banerjee S, Pillai MRA, Ramamoorthy N. Semin. Nucl. Med. 2001;31:260. doi: 10.1053/snuc.2001.26205. [DOI] [PubMed] [Google Scholar]; (c) Jain D. Semin. Nucl. Med. 1999;29:221. doi: 10.1016/s0001-2998(99)80012-9. [DOI] [PubMed] [Google Scholar]
- 8.Society of Nuclear Medicine press release. SNM Honors Originators of Imaging Agent for Nuclear Heart Stress Test. 2009 Jun;12 [Google Scholar]
- 9.(a) Schwartz DA, Abrams MJ, Hauser MM, Gaul FE, Larsen SK, Rauh D, Zubieta J. Bioconjugate Chem. 1991;2:333. doi: 10.1021/bc00011a007. [DOI] [PubMed] [Google Scholar]; (b) Liu S, Edwards DS. Chem. Rev. 1999;99:2235. doi: 10.1021/cr980436l. [DOI] [PubMed] [Google Scholar]
- 10.(a) Alberto R, Schibli R, Egli A, Schubiger AP. J. Am. Chem. Soc. 1998;120:7987. doi: 10.1021/ja003932b. [DOI] [PubMed] [Google Scholar]; (b) Schibli R, Schubiger PG. Eur. J. Nucl. Med. Mol. Imaging. 2002;29:1529. doi: 10.1007/s00259-002-0900-8. [DOI] [PubMed] [Google Scholar]
- 11.(a) Alberto R, Braband H, N’Dongo HWP. Curr. Radiopharm. 2009;2:254. [Google Scholar]; (b) Banerjee SR, Maresca KP, Francesconi L, Valliant J, Babich JW, Zubieta J. Nucl. Med. Biol. 2005;32:1. doi: 10.1016/j.nucmedbio.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 12.(a) Alberto R, Ortner K, Wheatley N, Schibli R, Schubiger AP. J. Am. Chem. Soc. 2001;123:3135. doi: 10.1021/ja003932b. [DOI] [PubMed] [Google Scholar]; (b) Torres R, de Rosales M, Finucane C, Mather SJ, Blower PJ. Chem. Commun. 2009:4847. doi: 10.1039/b908652h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Banerjee SR, Foss CA, Castanares M, Mease RC, Byun Y, Fox JJ, Hilton J, Lupold SE, Kozikowski AP, Pomper MG. J. Med. Chem. 2008;51:4504. doi: 10.1021/jm800111u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Kipper SL, Rypins EB, Evans DG, Thakur ML, Smith TD, Rhodes B. J. Nucl. Med. 2000;41:449. [PubMed] [Google Scholar]; (b) Hughes K. Proc. Amer. Soc. Clin. Oncol. 1995;14:544. [Google Scholar]
- 14.(a) Tárkányi F, Szelecsényi F, Kopecký P, Molnár T, Andó L, Mikecz P, Tóth Gy, Rydl A. Appl. Radiat. Isot. 1994;45:239. doi: 10.1016/0969-8043(94)90018-3. [DOI] [PubMed] [Google Scholar]; (b) Filossofov DV, Lebedev NA, Novgorodov AF, Bontchevabd GD, Starodub GY. Appl. Radiat. Isot. 2001;55:293. doi: 10.1016/s0969-8043(00)00376-6. [DOI] [PubMed] [Google Scholar]
- 15.(a) Meares CF, McCall MJ, Reardan DT, Goodwin DA, Diamanti CI, McTigue M. Anal. Biochem. 1984;142:68–78. doi: 10.1016/0003-2697(84)90517-7. [DOI] [PubMed] [Google Scholar]; (b) Bhattacharyya S, Cheal SM, Hill GC, Griffiths GL. Am. Chem. Soc. Meeting. Washington DC: 2009. abstract. [Google Scholar]; (c) Bhattacharyya S, Wei L, Dixit M, Hill GC. J. Nuc. Med. 2010;51(suppl. 2):315 pp. [Google Scholar]
- 16.(a) Armitage NC, Perkins AC, Pimm MV, et al. Nucl. Med. Commun. 1985;6:623. doi: 10.1097/00006231-198510000-00003. [DOI] [PubMed] [Google Scholar]; (b) Pimm MV, Perkins AC, Baldwin RW. Eur. J. Nucl. Med. Mol. Imaging. 1985;11:300. doi: 10.1007/BF00252341. [DOI] [PubMed] [Google Scholar]; (c) Perkins AC, Pimm MV. Eur. J. Nucl. Med. Mol. Imaging. 1985;11:295. doi: 10.1007/BF00252340. [DOI] [PubMed] [Google Scholar]
- 17.(a) Halpern SE. Nucl. Med. Biol. 1988;13:195. [Google Scholar]; (b) Larson SM, Carrasquillo JA. Nucl. Med. Biol. 1988;15:231. doi: 10.1016/0883-2897(88)90100-6. [DOI] [PubMed] [Google Scholar]
- 18.(a) Arano Y, Mukai T, Akizawa H, Uezono T, Motonari H, Wakisaka K, Kairiyama C, Yokoyama A. Nucl. Med. Biol. 1995;22:555. doi: 10.1016/0969-8051(95)00009-m. [DOI] [PubMed] [Google Scholar]; (b) Farrano FN, Edwards WB, Welch MJ, Duncun JR. Nucl. Med. Biol. 1994;21:1023. doi: 10.1016/0969-8051(94)90174-0. [DOI] [PubMed] [Google Scholar]
- 19.(a) Haseman MK, Goodwin DA, Meares CF, et al. Eur. J. Nuc. Med. 1986;12:455. doi: 10.1007/BF00254750. [DOI] [PubMed] [Google Scholar]; (b) Meares CF, McCall MJ, Deshpande SV, et al. Int. J. Cancer. 1988;41:99. doi: 10.1002/ijc.2910410723. [DOI] [PubMed] [Google Scholar]; (c) Quadri SM, Paik CH, Yokoyama K, Reba RC. J. Label. Comp. Radiopharm. 1986;23:1291. [Google Scholar]; (d) Arano Y. Q. J. Nucl. Med. 1998;42:262. [PubMed] [Google Scholar]; (e) Arano Y, Fujioka Y, et al. Cancer Res. 1999;59:128. [PubMed] [Google Scholar]
- 20.(a) Tsai SW, Li L, Anderson AL, Raubitschek AA, Shively JE. Bioconjugate Chem. 2001;12:264. doi: 10.1021/bc0000987. [DOI] [PubMed] [Google Scholar]; (b) Guo H, Yang J, Gallazzi F, Prostinz ER, Sklar LA, Miao Y. Bioconjugate Chem. 2009;20:2162. doi: 10.1021/bc9003475. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ke CY, Mathias CJ, Green MA. J. A m. Chem. Soc. 2005;127:7421. doi: 10.1021/ja043006n. [DOI] [PubMed] [Google Scholar]
- 21.(a) Lee J, Garmestani K, Wu C, Brechbiel MW, Chang HK, Choi CW, Gansow OA, Carrasquillo JA, Paik CH. Nucl. Med. Biol. 1997;24:225. doi: 10.1016/s0969-8051(97)00056-5. [DOI] [PubMed] [Google Scholar]; (b) Mohsin H, Fitzsimmons J, Shelton T, Hoffman TJ, Cutler CS, Lewis MR, Athey PS, Gulyas G, Kiefer GE, Frank RK, Simone J, Lever SZ, Jurrison SJ. Nucl. Med. Biol. 2007;34:493. doi: 10.1016/j.nucmedbio.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 22.(a) Szelecsenyi F, Tarkanyi F, Kovacs Z. Acta Radiol. Suppl. 1991;376:62. [PubMed] [Google Scholar]; (b) Nayak D, Lahiri S. Appl. Radiat. Isot. 2001;54:189. doi: 10.1016/s0969-8043(00)00194-9. [DOI] [PubMed] [Google Scholar]; (c) Naidoo C, van der Walt TN. Appl. Radiat. Isot. 2001;54:915. doi: 10.1016/s0969-8043(00)00360-2. [DOI] [PubMed] [Google Scholar]
- 23.Green MA, Welch MJ. J. Nucl. Med. Biol. 1989;16:435. doi: 10.1016/0883-2897(89)90053-6. [DOI] [PubMed] [Google Scholar]
- 24.Vallabhajosula SR, Harwig JF, Wolf W. Int. J. Nucl. Med. Biol. 1981;8:363. doi: 10.1016/0047-0740(81)90044-9. [DOI] [PubMed] [Google Scholar]
- 25.Nakazawa A, Ikeda K, Ito Y, Iwase M, Sato K, Ueda R, Dohi Y. Chest. 2004;126:1372. doi: 10.1378/chest.126.4.1372. [DOI] [PubMed] [Google Scholar]
- 26.Freeman LM. Nuclear Medicine Annual. Philadelphia, PA: Lippincott Williams and Wilkens Publishers; 1999. p. 165. [Google Scholar]
- 27.(a) Koizumi M, Endo K, Kunimatsu M, Sakahara H, Nakashima T, Kawamura Y, Watanabe Y, Saga T, Konishi J, Yamamuro T, Hosoi S, Toyania S, Arano Y, Yokoyama A. Cancer Res. 1988;48:1189. [PubMed] [Google Scholar]; (b) Antunes P, Ginj M, Zhang H, Waser B, Baum RP, Reubi JC, Maecke H. Eur. J. Nucl. Med. Mol. Imaging. 2007;34:982. doi: 10.1007/s00259-006-0317-x. [DOI] [PubMed] [Google Scholar]
- 28.(a) Fani M, Andre JP, Maecke HR. Contrast Media Mol. Imaging. 2008;3:53. doi: 10.1002/cmmi.232. [DOI] [PubMed] [Google Scholar]; (b) Ehrhardt GJ, Welch MJ. J. Nucl. Med. 2007;19:925. [PubMed] [Google Scholar]; (c) Zhernosekov KP, Filosofov DV, Baum RP. J. Nucl. Med. 2007;48:1741. doi: 10.2967/jnumed.107.040378. [DOI] [PubMed] [Google Scholar]
- 29.(a) Mathias CJ, Lewis MR, Reichert DE. Nucl. Med. Biol. 2003;30:725. doi: 10.1016/s0969-8051(03)00080-5. [DOI] [PubMed] [Google Scholar]; (b) Smith-Jones PM, Stolz B, Bruns C. J. Nucl. Med. 1994;35:317. [PubMed] [Google Scholar]; (c) Smith-Jones PM, Solit DB, Akhurst T, Afroze F, Rosen N, M Larson S. Nat. Biotechnol. 2004;22:701. doi: 10.1038/nbt968. [DOI] [PubMed] [Google Scholar]
- 30.Welch M, McCarthy TJ. J. Nucl. Med. 2000;41:315. [PubMed] [Google Scholar]
- 31.Maecke HR, Hoffman M, Haberkorn U. J. Nuc. Med. 2005;46:172S. [PubMed] [Google Scholar]
- 32.(a) Gabriel M, Decristoforo C, Kendler D. J. Nucl. Med. 2007;48:508. doi: 10.2967/jnumed.106.035667. [DOI] [PubMed] [Google Scholar]; (b) Gabriel M, Oberauer A, Dobrozemsky G. J. Nucl. Med. 2009;50:1427. doi: 10.2967/jnumed.108.053421. [DOI] [PubMed] [Google Scholar]; (c) Putzer D, Gabriel M, Henninger B. J. Nucl. Med. 2009;50:1214. doi: 10.2967/jnumed.108.060236. [DOI] [PubMed] [Google Scholar]
- 33.(a) Yano Y, Chu P, Budinger TF. J. Nucl. Med. 1977;18:46. [PubMed] [Google Scholar]; (b) Horlock PL, Clark JC, Goodier IW. J. Radioanal. Chem. 1981;64:263. [Google Scholar]; (c) Bilewicz A, Bartos B, Misiak R, Petelenz B. J. Radioanal. Nucl. Chem. 2006;268:485. [Google Scholar]
- 34.Saha GB, Go RT, Macinetyre WJ, Marwick TH, Beachler A, king JL, Neumann DR. Nucl. Med. Biol. 1990;17:763. doi: 10.1016/0883-2897(90)90023-t. [DOI] [PubMed] [Google Scholar]
- 35.(a) Selwyn AP, Allan RM, L’Abbate A. Am. J. Cardiol. 1982;50:112. doi: 10.1016/0002-9149(82)90016-9. [DOI] [PubMed] [Google Scholar]; (b) Prakash R, DeKemp RA, Ruddy TD. J. Nucl. Cardiol. 2004;11:440. doi: 10.1016/j.nuclcard.2004.04.005. [DOI] [PubMed] [Google Scholar]; (c) Epstein NJ, Benelfassi A, Beanlands RSB, DeKemp RA. Appl. Radiat. Isot. 2004;60:921. doi: 10.1016/j.apradiso.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 36.(a) Bass LA, Wang M, Welch MJ, Anderson CJ. Bioconjugate Chem. 2000;11:527. doi: 10.1021/bc990167l. [DOI] [PubMed] [Google Scholar]; (b) Connett JM, Buettner TL, Anderson CJ. Clin. Cancer Res. 1999;5:3207s. [PubMed] [Google Scholar]; (c) Blower PJ, Lewis JS, Zweit J. Nucl. Med. Biol. 1996;23:957. doi: 10.1016/s0969-8051(96)00130-8. [DOI] [PubMed] [Google Scholar]; (d) Green MA, Klippenstein DL, Tennison JR. J. Nucl. Med. 1988;29:1549. [PubMed] [Google Scholar]; (e) Green MA, Mathias CJ, Willis LR, Handa RK, Lacy JL, Miller MA, Hutchins GD. Nucl. Med. Biol. 2007;34:247. doi: 10.1016/j.nucmedbio.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 37.(a) McCarthy DW, Shefer RE, Klinkowstein RE, Bass LA, Margeneau WH, Cutler CS, Anderson CJ, Welch MJ. Nucl. Med. Biol. 1997;24:35. doi: 10.1016/s0969-8051(96)00157-6. [DOI] [PubMed] [Google Scholar]; (b) Obata A, Kasamatsu S, McCarthy DW, Welch MJ, Saji H, Yonekura Y, Fujibayashi Y. Nucl. Med. Biol. 2003;30:535. doi: 10.1016/s0969-8051(03)00024-6. [DOI] [PubMed] [Google Scholar]; (c) Avila Rodriguez MA, Nye JA, Nickles RJ. Appl. Radiat. Isot. 2007;65:1115. doi: 10.1016/j.apradiso.2007.05.012. [DOI] [PubMed] [Google Scholar]; (d) Wadas TJ, Wong EH, Weisman GH, Anderson CJ. Chem. Rev. 2010;110:2858. doi: 10.1021/cr900325h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.(a) Wallhaus TR, Lacy J, Stewart R, Bianco J, Green MA, Nayak N, Stone CK. J. Nucl. Cardiol. 2001;8:67. doi: 10.1067/mnc.2001.109929. [DOI] [PubMed] [Google Scholar]; (b) A, Vave re L, Lewis JS. Dalton Trans. 2007:4893. doi: 10.1039/b705989b. [DOI] [PubMed] [Google Scholar]; (c) Chitneni SK, Palmer GM, Zalutsky MR, Dewhirst MW. J. Nucl. Med. 2011;52:165. doi: 10.2967/jnumed.110.075663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anderson CJ, Wadas TJ, Wong EH, Weisman GR. Q. J. Nucl. Med. Mol. Imaging. 2008;52:185. [PMC free article] [PubMed] [Google Scholar]
- 40.Sprague JE, Peng Y, Sun X, Weisman GR, Wong EH, Achilefu S, Anderson CJ. Clin. Cancer Res. 2004;10:8674. doi: 10.1158/1078-0432.CCR-04-1084. [DOI] [PubMed] [Google Scholar]
- 41.(a) Di Bartolo NM, Sargeson AM, Donlevy TM, Smith SV. J. Chem. Soc., Dalton Trans. 2001:2303. [Google Scholar]; (b) Di Bartolo N, Sargeson AM, Smith SV. Org. Biomol. Chem. 2006;4:3350. doi: 10.1039/b605615f. [DOI] [PubMed] [Google Scholar]
- 42.(a) Smith SV. J. Inorg. Biochem. 2004;98:1874. doi: 10.1016/j.jinorgbio.2004.06.009. [DOI] [PubMed] [Google Scholar]; (b) Barnhatt-Bott A, Green MA. Int. J. Rad Appl Instrum B. 1991;18:865. doi: 10.1016/0883-2897(91)90095-3. [DOI] [PubMed] [Google Scholar]
- 43.Voss SD, Smith SV, Di Bartolo N, McIntosh LJ, Cyr EM, Bonab AA, Dearling JLJ, Carter EA, Fischman AJ, Treves ST, Gillies SD, Sargeson AM, Houston JS, Packard AB. Proc. Natl. Acad. Sci. U. S. A. 2007;104:17489. doi: 10.1073/pnas.0708436104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cai H, Fissekis J, Conti PS. Dalton Trans. 2009:5395. doi: 10.1039/b902210d. [DOI] [PubMed] [Google Scholar]
- 45.Ma MT, Karas JA, White JM, Scanlon D, Donnelly PS. Chem. Commun. 2009;(22):3237. doi: 10.1039/b903426a. [DOI] [PubMed] [Google Scholar]
- 46.Cai H, Li Z, Huang C, Park R, Shahinian A, Conti PS. Nucl. Med. Biol. 2010;37:57. doi: 10.1016/j.nucmedbio.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 47.Cai H, Li Z, Huang C, Shahinian AH, Wang H, Park R, Conti PS. Bioconjugate Chem. 2010;21:1417. doi: 10.1021/bc900537f. [DOI] [PubMed] [Google Scholar]
- 48.Cai W, Chen K, He L, Cao Q, Koong K, Chen X. Eur. J. Nucl. Med. Mol. Imaging. 2007;34:850. doi: 10.1007/s00259-006-0361-6. [DOI] [PubMed] [Google Scholar]
- 49.(a) Takahashi N, Fujibayashi Y, Yonekura Y, Welch MJ, Waki A, Tsuchida T, Sadato N, Sugimoto K, Itoh H. Ann. Nucl. Med. 2000;14:323–328. doi: 10.1007/BF02988690. [DOI] [PubMed] [Google Scholar]; (b) Dehdashti F, Grigsby PW, Mintun MA, Lewis JS, Siegel BA, Welch MJ. Int. J. Radiat. Oncol. Biol., Phys. 2003;55:1233–1238. doi: 10.1016/s0360-3016(02)04477-2. [DOI] [PubMed] [Google Scholar]; (c) Dehdashti F, Mintun MA, Lewis JS, Bradley J, Govindan R, Laforest R, Welch MJ, Siegel BA. Eur. J. Nucl. Med. Mol. Imaging. 2003;30:844–850. doi: 10.1007/s00259-003-1130-4. [DOI] [PubMed] [Google Scholar]; (d) Philpott GW, Schwarz SW, Anderson CJ, Dehdashti F, Connett JM, Zinn KR, Meares CF, Cutler PD, Welch MJ, Siegel BA. J. Nucl. Med. 1995;36:1818. [PubMed] [Google Scholar]
- 50.Yoo J, Tang L, Perkins TA, Rowland DJ, Laforest R, Lewis JS, Welch MJ. Nucl. Med. Biol. 2005;32:891. doi: 10.1016/j.nucmedbio.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 51.Germastani K, Milenic DE, Plascjak PS, Brechbiel MW. Nucl. Med. Biol. 2002;29:599. doi: 10.1016/s0969-8051(02)00322-0. [DOI] [PubMed] [Google Scholar]
- 52.Brechbiel MW. Q. J. Nucl. Med. Mol. Imaging. 2008;52:166. [PMC free article] [PubMed] [Google Scholar]
- 53.Sharkey RM, Motta-Hennessy C, Gansow OA, Brechbiel MW, Fand I, Griffiths GL, Jones AL, Goldenberg DM. Int. J. Cancer. 1990;46:79. doi: 10.1002/ijc.2910460116. [DOI] [PubMed] [Google Scholar]
- 54.Brechbiel MW, Gansow OA. Bioconjugate Chem. 1991;2:187. doi: 10.1021/bc00009a008. [DOI] [PubMed] [Google Scholar]
- 55.Camera L, Kinuya S, Germastani K, Wu C, Brechbiel MW, Pai LH, McMurry TJ, Gansow OA, Pastan I, Paik CH, et al. J. Nuc. Med. 1994;35:882. [PubMed] [Google Scholar]
- 56.(a) Deshpande SV, DeNardo SJ, Kukis DL, Moi MK, McCall MJ, DeNardo GL, Meares CF. J. Nucl. Med. 1990;31:473. [PubMed] [Google Scholar]; (b) Harrison A, Walker CA, Parker D, Jankowski KJ, Cox JP, Craig AS, Sansom JM, Beeley NR, Boyce RA, Chaplin L. Int. J. Rad. Appl. Instrum. B. 1991;18:469. doi: 10.1016/0883-2897(91)90107-v. [DOI] [PubMed] [Google Scholar]
- 57.(a) Wong JY, Chu DZ, Williams LE, Liu A, Zhan J, Yamauchi DM, Wilczynski S, Wu AM, Yazaki PJ, Shively JE, Leong L, Raubitschek AA. Cancer Biother. Radiopharm. 2006;21:88. doi: 10.1089/cbr.2006.21.88. [DOI] [PubMed] [Google Scholar]; (b) Palm S, Enmon RM, Jr, Matei C, Kolbert KS, Xu S, Zanzonico PB, Finn RL, Koutcher JA, Larson SM, Sgouros G. J. Nucl. Med. 2003;44:1148. [PubMed] [Google Scholar]; (c) Lovqvist A, Humm JL, Sheikh A, Finn RD, Koziorowski J, Ruan S, Pentlow KS, Jungbluth A, Welt S, Lee FT, Brechbiel MW, Larson SM. J. Nucl. Med. 2001;42:1281. [PubMed] [Google Scholar]
- 58.Palm S, Enmon RM, Jr, Matei C, Kolbert KS, Xu S, Zanzonico PB, Finn RL, Koutcher JA, Larson SM, Sgouros G. J. Nucl. Med. 2003;44:1148. [PubMed] [Google Scholar]
- 59.Jamar F, Barone R, Mathieu I, Walrand S, Labar D, Carlier P, de Camps J, Schran H, Chen T, Smith MC, Bouterfa H, Valkema R, Krenning EP, Kvols LK, Pauwels S. Eur. J. Nucl. Med. Mol. Imaging. 2003;30:510. doi: 10.1007/s00259-003-1117-1. [DOI] [PubMed] [Google Scholar]
- 60.(a) Holland JP, Sheh Y, Lewis JS. Nucl. Med. Biol. 2009;36:729. doi: 10.1016/j.nucmedbio.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mustafa MG, West HIJ, O’Brien H, Lanier RG, Benhamou M, Tamura T. Phys. Rev. C: Nucl. Phys. 1988;38:1624. doi: 10.1103/physrevc.38.1624. [DOI] [PubMed] [Google Scholar]
- 61.Szajek LP, Plascjak P, Meyer W, Herscovitch P. J. Labelled Comp. 2007;50:S93. [Google Scholar]
- 62.(a) Meijs WE, Haisma HJ, Klok RP, van Gog FB, Kievit E, Pinedo HM, Herscheid JD. J. Nucl. Med. 1997;38:112. [PubMed] [Google Scholar]; (b) Meijs WE, Haisma HJ, Van der Schors R, Wijbrandts R, Van den Oever K, Klok RP, Pinedo HM, Herscheid JD. Nucl. Med. Biol. 1996;23:439. doi: 10.1016/0969-8051(96)00020-0. [DOI] [PubMed] [Google Scholar]
- 63.(a) Verel I, Visser GWM, Boellaard R, Stigter-van Walsum M, Snow GB, van Dongen GAMS. J. Nucl. Med. 2003;44:1271. [PubMed] [Google Scholar]; (b) Holland JP, Caldas-Lopes E, Divilov V, Longo VA, Taldone T, Zatorska D, Chiosis G, Lewis JS. PLoS One. 2010;5:e8859. doi: 10.1371/journal.pone.0008859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.(a) Perk LR, Visser GWM, Budde M, Vosjan M, Jurek P, Keifer G, Dongen GV. Nat. Protoc. 2008:22. doi: 10.1038/nprot.2010.13. [DOI] [PubMed] [Google Scholar]; (b) Perk LR, Vosjan M, Visser GWM, Budde M, Jurek P, Keifer G, Dongen GV. Eur. J. Nucl. Med. Mol. Imaging. 2009;37:250. doi: 10.1007/s00259-009-1263-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tinianow JN, Gill HS, Ogasawara A, Flores JE, Vanderbilt AN, Luis E, Vandlen R, Darwish M, Junutula JR, Williams S, Marik J. Nucl. Med. Biol. 2010;37:289. doi: 10.1016/j.nucmedbio.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 66.Perk LR, Visser OJ, Stigter-Van MW, Maria JWDV, Gerard WMV, Josée MZ, Huijgens PC, van Dongen GAMS. Eur. J. Nucl. Med. Mol. Imaging. 2006;33:1337. doi: 10.1007/s00259-006-0160-0. [DOI] [PubMed] [Google Scholar]
- 67.Börjesson PKE, Jauw YWS, Boellaard R, de Bree R, Comans EFI, Roos JC, Castelijns JA, Vosjan MJWD, Kummer JA, Leemans CR, Lammertsma AA, van Dongen GAMS. Clin. Cancer Res. 2006;12:2133. doi: 10.1158/1078-0432.CCR-05-2137. [DOI] [PubMed] [Google Scholar]
- 68.(a) Lewis JS, Laforest R, Buettner TL, Song S-K, Fujibayashi Y, Connett JM, Welch MJ. Proc. Natl. Acad. Sci. U. S. A. 2001;98:1206. doi: 10.1073/pnas.98.3.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lewis JS, Lewis MR, Cutler PD, Srinivasan A, Schmidt MA, Schwarz SW, Morris MM, Miller JP, Anderson CJ. Clin. Cancer Res. 1999;5:3608. [PubMed] [Google Scholar]; (c) Obata A, Kasamatsu S, Lewis JS, Furukawa T, Takamatsu S, Toyohara J, Asai T, Welch MJ, Adams SG, Saji H, Yonekura Y, Fujibayashi Y. Nucl. Med. Biol. 2005;32:21. doi: 10.1016/j.nucmedbio.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 69.Castillo AX, Perez-Malo M, Isaac-Olive K, Mukhallalati H, Gonzalez EC, Berdequez MT, Diaz NC. Nucl. Med. Biol. 2010;37:935. doi: 10.1016/j.nucmedbio.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 70.(a) Marcus R. Semin. Oncol. 2005;32:36. doi: 10.1053/j.seminoncol.2005.01.012. [DOI] [PubMed] [Google Scholar]; (b) Mohsin H, Jia F, Sivaguru G, Hudson MJ, Shelton TD, Hoffman TJ, et al. Bioconjugate Chem. 2006;17:485. doi: 10.1021/bc0502356. [DOI] [PubMed] [Google Scholar]; (c) Knogler K, Grunberg J, Novak-Hofer I, Zimmermann K, Schubiger PA. Nucl. Med. Biol. 2006;33:883. doi: 10.1016/j.nucmedbio.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 71.Knapp FF, Jr, Beets AL, Guhlke S, Zamora PO, Bender H, Palmedo H, Biersack HJ. Anticancer Res. 17:1783. [PubMed] [Google Scholar]
- 72.El-Mabhouh A, Mercer JR. Appl. Radiat. Isot. 2005;62:541. doi: 10.1016/j.apradiso.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 73.(a) Hashimoto K, Yoshihara K. Top. Curr. Chem. 2003;176:275. [Google Scholar]; (b) Boschi A, Bolzati C, Uccelli L, Duatti A. Nucl. Med. Biol. 2003;30:381. doi: 10.1016/s0969-8051(03)00002-7. [DOI] [PubMed] [Google Scholar]
- 74.Boswell CA, Brechbiel MW. Nucl. Med. Biol. 2007;34:757. doi: 10.1016/j.nucmedbio.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. http://www.clinicaltrials.gov/
- 76.Gabriel M, Oberauer A, Dobrozemsky G, Decristoforo C, Putzer D, Kendler D, Uprimny C, Kovacs P, Bale R, Virgolini IJ. J. Nucl. Med. 1427;50 doi: 10.2967/jnumed.108.053421. [DOI] [PubMed] [Google Scholar]
- 77.Forrer F, Lohri A, Schmid P, Hermann R, Maecke HR, Muller-Brand J. Proceedings of the 52nd Annual Meeting of the Society of Nuclear Medicine. 2005:445. [Google Scholar]
- 78.Li S, Liu J, Zhang H, Tian M, Wang J, Zheng X. Clin. Nucl. Med. 2001;26:919. doi: 10.1097/00003072-200111000-00006. [DOI] [PubMed] [Google Scholar]
- 79.Bruland ø. S, Jonasdottir TJ, Fisher DR, Larsen RH. Curr. Radiopharm. 2008;1:203. [Google Scholar]
- 80.(a) Zalutsky MR, Vaidyanathan G. Curr. Pharm. Des. 2000;6:1433. doi: 10.2174/1381612003399275. [DOI] [PubMed] [Google Scholar]; (b) Zalutsky MR, Reardon DA, Akabani G, Coleman RE, Friedman AH, Friedman HS, McLendon RE, Wong TZ, Bigner DD. J. Nucl. Med. 2008;49:30. doi: 10.2967/jnumed.107.046938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Madsen MT, Anderson JA, Halama JR. Med. Phys. 2006;33:4. doi: 10.1118/1.2135911. [DOI] [PubMed] [Google Scholar]
- 82.Lever SZ. J. Cell. Biochem. 2002;87(S39):60. [Google Scholar]
- 83.Hasimoto K, Matsuoka H, Uchida S. J. Radioanal. Nucl. Chem. 2003;255:575. [Google Scholar]
- 84.Ramamoorthy M, Saraswati P, Das MK, Mehra KS, Ananthakrishnan M. Nucl. Med. Commun. 2002;23:83. doi: 10.1097/00006231-200201000-00013. [DOI] [PubMed] [Google Scholar]
- 85.Guseva LI, Dogadkin NN. Radiochemistry. 2009;51:169. [Google Scholar]
- 86.Matsumoto K, Fujibayashi Y, Konishi J, Yokoyama A. Radioiso-topes. 1990;39:482. doi: 10.3769/radioisotopes.39.11_482. [DOI] [PubMed] [Google Scholar]
- 87.Henricksen G, Hoff P, Alstad J, Larsen RH. Radiochim. Acta. 2001;89:661. [Google Scholar]
- 88.An entire issue of Inorg. Chim. Acta. 2010;Vol. 363(issue 12) has been dedicated to Prof. Animesh Chakravorty