

Abstract
Reactive stroma initiates during early prostate cancer development and co-evolves with prostate cancer progression. Previous studies have defined the key markers of reactive stroma and have established that reactive stroma biology influences prostate tumorigenesis and progression. The stem/progenitor cells of origin and the mechanisms that regulate their recruitment and activation to myofibroblasts or carcinoma-associated fibroblasts are essentially unknown. Key regulatory factors have been identified, including transforming growth factor beta, interleukin-8, fibroblast growth factors, connective tissue growth factor, wingless homologs-Wnts, and stromal cell-derived factor-1, among others. The biology of reactive stroma in cancer is similar to the more predictable biology of the stroma compartment during wound repair at sites where the epithelial barrier function is breached and a stromal response is generated. The co-evolution of reactive stroma and the biology of how reactive stroma - carcinoma interactions regulate cancer progression and metastasis are targets for new therapeutic approaches. Such approaches are strategically designed to inhibit cancer progression by uncoupling the reactive stroma niche.
Keywords: Myofibroblasts, Carcinoma-Associated Fibroblasts, Prostate Cancer, Microenvironment
I. Introduction
The epithelium in the human prostate gland is composed of cuboidal to columnar secretory epithelial cells with apical junctional complexes, a continuous layer of basal cells (El-Alfy, et al. 2000), and sparse neuroendocrine cells, each attached to a basal lamina. The epithelium is organized as glandular acini that secrete into the luminal space that converges upon a duct and into the urethra. A fibromuscular stroma is situated on the opposite side of the basal lamina. This stroma is composed of fibroblasts, smooth muscle cells, and an extracellular matrix rich in collagen fibers that intervenes between the secretory acini (Figure 1). Other key cells in the stroma compartment include endothelial cells, autonomic nerve fibers and associated ganglia, and various immune cells. After peak reproductive age, the histological architecture of the prostate begins to undergo age-related changes that continue throughout life. Although key mechanisms are not yet understood, these changes might be attributed to altered androgen action and inflammatory processes that lead to either an unabated trophic effect on the gland and/or a chronic inflammation. It is possible that repeated epithelial insult sustained throughout the aging process, when coupled with the critical need for androgen action to regulate prostate gland biology, results in a change of biology from one of differentiated reproductive function to one of chronic wound repair (Rowley 1998; Schauer and Rowley 2011; Tuxhorn, et al. 2001). We suggest that the repair state biology is key in promoting different prostate diseases, all of which have reactive stroma and inflammation in common. Indeed, there is a well-established association between prostate cancer and inflammation, with concomitant changes in the stromal compartment typified by expression of key markers of reactive stroma (Figure 2) (De Marzo, et al. 2003; De Marzo, et al. 2007; Nelson, et al. 2004).
Figure 1. Cellular components of the human prostate gland.

Secretory epithelial cells are situated on a basal lamina / basement membrane and secretes products into the acinar lumen. Basal cells and sparse neuroendocrine cells are also present in the epithelial compartment. The stromal compartment immediately surrounding the epithelial acini is complex, and consists of smooth muscle, trace fibroblasts, blood vessels, autonomic nerve fibers, inflammatory cells, and extracellular matrix components.
Figure 2. Stromal cell phenotypes and associated markers.
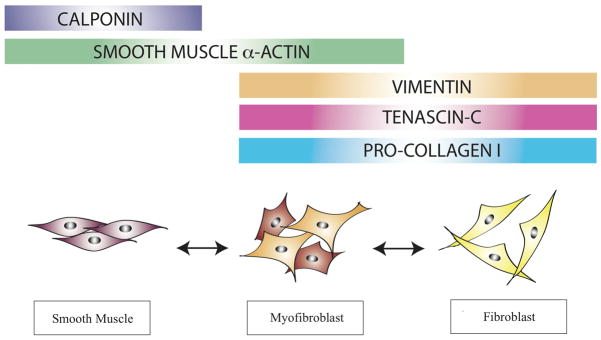
Stromal cell populations found in the prostate gland and other epithelial-lined organs are distinguished based on their morphology and expression of marker proteins. Shown here are three major stromal cell types and associated markers used to identify them in the prostate gland. Importantly, these markers are not lineage-specific.
Prostate cancer is the most commonly diagnosed non-cutaneous malignancy in men in the United States. The incidence increases rapidly with age, particularly after the age of 50, although prostate cancer can occur in men less than 50 years of age and has also been observed in autopsies of men this age. It is generally accepted that organ-confined prostate adenocarcinoma initiates from preneoplastic lesions known as prostatic intraepithelial neoplasia (PIN), whereby a combination of cellular events initiates a cascade of genomic instability (Bettendorf, et al. 2008; De Marzo et al. 2003). The histological changes associated with PIN are reminiscent of cancer, evidenced by a loss of cellular polarity, nuclear atypia, and focal dysplasia that results in cellular tufts lining the acinar space of normal ducts and glands (Epstein 2009; Zynger and Yang 2009). Other histologic changes include loss of neuroendocrine and secretory differentiation, nuclear and nucleolar abnormalities, neovascularity, increased proliferative potential and genetic instability with variation of DNA content (Ayala and Ro 2007). With increasing degrees of PIN, more nuclear aberration and basal cell disruption are observed. For the most part, the basal lamina remains intact during PIN, although changes in epithelial cell polarity, alterations in junctional complexes and some changes in basal lamina may indicate a breach in epithelial layer integrity, even as early as PIN. We suggest that this breach in the epithelial barrier is a key event for the initiation and genesis of reactive stroma observed in prostatic diseases.
II. Reactive stroma in prostate carcinogenesis
The normal stromal compartment has evolved with an inherent plasticity to respond rapidly to emerging situations, such as in wound repair and disrupted homeostasis, which may result from the genesis of cancer. As such, stromal components within the microenvironment immediately adjacent to epithelium act coordinately when the epithelium sustains damage or is breached. Microorganisms are potentially present in the lumen and ducts of epithelial acini and on the apical surface of all epithelial layers. Hence, the rapid and coordinate stromal responses subsequent to epithelial damage likely occur to prevent these microorganisms from invading the underlying stroma and gaining access to the microvasculature, which could result in systemic spread and death.
The phenotypic and genotypic alterations that occur during this damage response have collectively been referred to as reactive stroma. This altered stroma is very similar to that found in a generic wound repair scenario and includes matrix remodeling and altered expression of repair-associated growth factors and cytokines (Desmouliere, et al. 2005; Gabbiani 2003). In the prostate gland, reactive stroma initiates during pre-malignant PIN and co-evolves with cancer through high-grade organ confined disease (Ayala, et al. 2003; Rowley 1998; Tuxhorn et al. 2001; Tuxhorn, et al. 2002a). Reactive stroma in prostate cancer is composed of carcinoma-associated fibroblasts and myofibroblasts. The cell type or types of origin is not understood. Reactive stroma might evolve from activation of existing fibroblasts, from vimentin-positive periacinar cells, from circulating marrow-derived progenitors, from vessel-associated pericytes, or from other tissue-resident mesenchymal stem/progenitor cells (Rowley 1998; Tuxhorn et al. 2002a) (Figure 3). The precise origin and biology of these cells is still an unresolved issue, although several groups have now identified various sources of these unique reactive stroma cells in different model systems and disease states (Kalluri and Neilson 2003; Placencio, et al. 2010; Potenta, et al. 2008; Zeisberg and Kalluri 2010).
Figure 3. Prostate gland homeostasis and reactive stroma formation in cancer.

The histopathologic changes in inflammatory conditions of the prostate (i.e. proliferative inflammatory atrophy or PIA) are similar to changes observed in wound repair. Similarly, prostate cancer can be viewed as an equivalent of chronic wounding with a prototypical reactive stroma response. It is hypothesized that the inflammatory cell populations recruited to lesions work coordinately with factors secreted by the carcinoma cells to generate a reactive stroma response. As cancer progresses, this reactive stroma co-evolves with foci of adjacent carcinoma. The net effect is likely to be tumor-promoting within the context of early and organ-confined disease.
The stromal microenvironment has emerged as a key player in the growth and development of cancer (Hanahan and Weinberg 2011; Olumi, et al. 1999; Ronnov-Jessen, et al. 1996; Ronnov-Jessen, et al. 1995; Tuxhorn et al. 2002a). It is well understood that the progression of organ-confined tumors is influenced by angiogenesis and inflammatory cells, as has already been described elsewhere (Coussens and Werb 2002; de Visser, et al. 2005; Folkman 1971; Kalluri 2003; Kalluri and Neilson 2003; Kim, et al. 1993; Tuxhorn, et al. 2002b; Yang, et al. 2005). Coordinately, the biology of reactive stromal cells in the tumor microenvironment modulates the progression and severity of cancer, as well as aspects of angiogenesis and inflammation (Ao, et al. 2007; Hanahan and Weinberg 2011; Kiskowski, et al. 2011; Li, et al. 2008; Olumi et al. 1999; Tuxhorn et al. 2002b; Yang et al. 2005). Fibroblasts can exhibit several different phenotypic and genotypic properties. The previous categorization of either resting or activated fibroblasts is likely an oversimplification. Similar to the phenotypic diversity of macrophages in the tumor microenvironment, the plasticity of activated or reactive fibroblasts is only beginning to be elucidated. Several groups have carefully studied an activated form of fibroblasts, termed myofibroblasts, or carcinoma-associated fibroblasts (CAFs) in prostate cancer (Franco, et al. 2011; Kiskowski et al. 2011; Li et al. 2008; Olumi et al. 1999; Singh, et al. 2004; Tuxhorn, et al. 2002c; Yang et al. 2005). There remains some debate about whether CAFs and myofibroblasts represent different cell types or are the same cell type with differential gene expression profiles. Even less is understood about the origin of these cells and the mechanisms that lead to their activation in cancer. Irrespective, it is widely accepted that these activated fibroblasts are important modulators of tumorigenesis (Mueller and Fusenig 2004).
Our data has shown that both CAFs and myofibroblasts compose the reactive stroma in well-differentiated foci of prostate cancer (Tuxhorn et al. 2002a). Myofibroblasts function during classic wound repair responses in most tissues. The presence of cells defined as myofibroblasts was first identified over 30 years ago as vimentin-positive, desmin-negative cells within the fibrillar meshwork of granulation tissue (Gabbiani, et al. 1971; Hirschel, et al. 1971). These cells were later identified within desmoplastic reactions of human breast cancer, within the reactive stroma compartment of several other adenocarcinomas, and in fibrotic diseases (Ahmed 1990; Barsky, et al. 1984; Brown, et al. 2005; Desmouliere, et al. 2003; Hinz 2007). The biology of myofibroblasts favors survival by providing a pro-wound repair environment. Myofibroblasts are proliferative, synthetic, and exhibit contractile properties, all features they share with either fibroblasts or smooth muscle (Powell, et al. 1999). However, these unique cells have the added capability of forming granulation tissue fibers and matrix while also contracting to close the wound. This suturing effect provides an obvious adaptive advantage during wound repair. This inherent versatility places the myofibroblast at a pivotal position in the tightly orchestrated process of maintaining tissue homeostasis.
Reactive stroma is associated with an increased number of myofibroblasts/CAFs, greater capillary density, and deposition of mature type-I collagen and other ECM-associated substrates (Ronnov-Jessen et al. 1996; Tuxhorn et al. 2001; Tuxhorn et al. 2002a; Yanagisawa, et al. 2007). In human prostate carcinoma tissue, the reactive stroma in Gleason 3 prostate cancer foci was composed of approximately 50% fibroblasts and 50% myofibroblasts, whereas the stroma in Gleason 4 foci was composed primarily of myofibroblasts (Tuxhorn et al. 2001; Tuxhorn et al. 2002a). Mesenchymal markers such as pro-collagen type I, tenascin, fibroblast activation protein (FAP), vimentin, and smooth muscle alpha-actin, are all expressed in myofibroblasts relatively early in the genesis of reactive stroma in prostate cancer (Tuxhorn et al. 2002a). This is coordinate with the biology exhibited in response to tissue damage, as each of the aforementioned biomarkers is typical of reactive stroma at sites of wound repair (Gabbiani 2003; Rowley 1998). While normal fibroblasts are viewed as being instrumental in maintaining tissue homeostasis in the absence of insult, their activated counterpart (myofibroblasts/CAFs) promote tumor progression most likely via their repair-centric and pro-survival biology that would include new growth and angiogenesis.
The role of androgen receptor in regulating homeostasis in prostate gland stromal cells is not well understood and a role for androgen receptor in regulating reactive stroma in cancer progression cannot be ruled out. Recent studies suggest that androgen receptor is expressed in a subset of fibroblasts in the prostate gland and regulate expression of several growth factors (Tanner, et al. 2011). Moreover, secreted factors from androgen stimulation of prostate stromal cells in this study functioned to stimulate prostate cancer cell proliferation. Knockout of androgen receptor in prostate stromal cells resulted in a lower proliferation of epithelial cells, altered deposition of collagen and decreased expression of several growth factors (Yu, et al. 2012). Androgen action also functioned to stimulate myodifferentiation of prostate fibroblasts and functioned to enhance the effects of TGF-β induced myodifferentiation (Gerdes, et al. 2004). Together, these studies suggest that androgen action in prostate stroma is important for maintenance of homeostasis and may play a role in reactive stroma biology in prostate cancer.
Many studies have focused on the biology of CAFs and myofibroblasts in cancer. Experimentally, CAFs from human prostate cancer were shown to affect the proliferation and phenotype of “initiated” prostatic epithelial cells, whereas normal fibroblasts did not (Olumi et al. 1999). Likewise, colon cancer CAFs were shown to facilitate the invasiveness of cancer cells when co-injected into mice (Dimanche-Boitrel, et al. 1994). Similarly, xenografts containing breast cancer cells grew faster when supplemented with CAFs as compared with xenografts containing normal fibroblasts (Orimo, et al. 2005). Additional studies have shown that myofibroblasts at the metastatic site promote local proliferation of cancer cells, mimicking their tumor promoting behavior in the primary tumor (Olaso, et al. 1997). The role of stromal cell populations in prostate cancer progression has been explored in xenograft and tissue recombination models. Remarkably, Hayward and colleagues have shown that nontumorigenic prostate epithelial cells from BPH tissue were transformed and formed cancer when recombined with prostate cancer-derived CAFs (Hayward, et al. 2001). Moreover, xenografts constructed with recombined human prostate cancer cells and stromal cells had differential degrees of tumor incidence and growth rate using several different prostate stromal cell lines derived from different cadaver donors (Tuxhorn et al. 2002b). Additional studies have shown that the loss of transforming growth factor beta (TGF-β) signaling in stroma and a resulting stimulation of Wnt3a signaling is involved, further defining the complexity of the biological functions of TGF-β in the tumor microenvironment (Li et al. 2008). Together, these studies suggest that the inductive nature seems to correspond to altered expression of key genes that are regulated by TGF-β, including FGF-2, CTGF, SDF-1(CXCL12), and Wnt3a (Ao et al. 2007; Ao, et al. 2006; Li et al. 2008; Tuxhorn et al. 2002b; Tuxhorn et al. 2002c; Yang, et al. 2008a; Yang et al. 2005). Other studies that were focused on mammary cancer have shown that TGF-β induced SDF-1 was responsible for the generation of myofibroblasts in the tumor microenvironment (Kojima, et al. 2010). Subsequent gene expression profiling studies using laser captured or microdissected reactive stroma from prostate cancer or from urogenital sinus mesenchyme have identified other candidate factors that may mediate the pro-tumorigenic or inductive nature of myofibroblasts/CAFs, thereby providing insight into potentially targetable pathways for new therapeutics (Dakhova, et al. 2009; Richardson, et al. 2007; Vanpoucke, et al. 2007). More recent computational modeling of the potential interactions between prostate cancer cells, TGF-β, and myofibroblasts/CAFs has been used as a new tool to help delineate potential pathways and mechanisms that may affect both the cancer and the microenvironment (Basanta, et al. 2012; Basanta, et al. 2009).
It is becoming clear that the activation of myofibroblasts/CAFs is a predictable biological response to a disrupted or damaged epithelial layer, irrespective of whether this damage results from wounding, benign prostatic hyperplasia, chronic inflammation, or early development of cancer. Furthermore, activation of a reactive stroma is pro-repair and, hence, may affect the biology of adjacent epithelium. The activation of reactive stroma biology in these foci facilitates granulation tissue formation and tissue remodeling through several mechanisms, including ECM deposition and growth factor production (Barron, et al. 2010; Desmouliere et al. 2005; Gabbiani 2003; Schauer, et al. 2009). This generalized reactive stroma response is adaptive and functions to preserve tissue integrity and homeostasis by promoting tissue repair. Our early studies showed that reactive stroma initiates at foci of early premalignant PIN in the human prostate gland and co-evolves with the development of cancer (Tuxhorn et al. 2001; Tuxhorn et al. 2002a). Subsequently, we reported that reactive stroma initiates at focal sites of benign prostatic hyperplasia that overexpress interleukin-8 (IL-8) and have enhanced deposition of tenascin-C (Schauer, et al. 2008). These observations were confirmed using different in vivo rodent modeling studies. These studies showed that elevated IL-8 or keratinocyte chemokine (KC, the murine paralog of IL-8) expression in prostate epithelial cells in either an orthotopic xenograft (IL-8) or in a transgenic mouse (KC) induced a tenascin-C positive reactive stroma with markers nearly identical to those observed in prostate cancer (Schauer et al. 2009; Schauer and Rowley 2011). Together, these studies suggest that several factors that affect tissue homeostasis, inflammatory responses, and angiogenesis are involved in the activation and biology of reactive stroma. Moreover, these studies suggest that the damage response biology of reactive stroma is likely to be tumor-promoting. As such, the pro-tumorigenic mechanisms of the factors that mediate this biology could be the focus of future therapeutic approaches. Of these factors, perhaps more has been published about TGF-β, although the biology regulated by TGF-β signaling is complex and not fully understood. Appropriately, TGF-β has been termed the “Jekyll and Hyde of cancer” (Bierie and Moses 2006).
III. TGF-β signaling in Reactive Stroma
The TGF-β superfamily family regulates a vast array of biological processes with respect to prostate homeostasis (Gerdes, et al. 1998; Jones, et al. 2009; Salm, et al. 2005; Stover, et al. 2007; Zhu and Kyprianou 2005). The various TGF-β isoforms have similar but not identical biologic actions in cells. All three can stimulate chemotaxis of inflammatory cells and production of extracellular matrix proteins through increased synthesis of collagens and proteoglycans. In addition, the TGF-β isoforms generally downregulate the synthesis of matrix metalloproteinases (MMPs) and upregulate synthesis of the natural inhibitors of MMPs, the tissue inhibitors of metalloproteinases (TIMPs) in stromal cells. These properties make the TGF-β isoforms important regulators of the deposition and removal of extracellular matrix. However, excess or prolonged action of TGF-β has been implicated in several fibroproliferative diseases, such as scleroderma, hepatic sclerosis, and interstitial pulmonary fibrosis (Kalluri and Han 2008; Menke and Adler 2002; Prud'homme 2007; Sanderson, et al. 1995). Studies on keloid and hypertrophic scars have also showed increased expression of TGF-β1 mRNA in these lesions (Jagadeesan and Bayat 2007). Importantly, the expression of TGF-β1 is elevated in most carcinomas and many proliferative diseases including benign prostatic hyperplasia, prostate cancer, and prostatitis (Alonso-Magdalena, et al. 2009; Ao et al. 2007; Gann, et al. 1999; Shoskes, et al. 2002). Moreover, each of these disorders is associated with inflammation along with altered proliferation and tissue remodeling.
The activity of TGF-β induces multiple effects on various signaling pathways that result in both tumor inhibiting and promoting actions (Bierie and Moses 2006; Dvorak 1986; Stover et al. 2007). In normal tissues for example, TGF-β signaling exerts an anti-proliferative and apoptotic effect on epithelial cells, which would be expected to limit the emergence and growth of malignant carcinomas (Hanahan and Weinberg 2000; Siegel and Massague 2003). TGF-β also facilitates the interactions between fibroblasts and epithelial cells to further suppress cancer initiation events (Bhowmick, et al. 2004). Paradoxically, in advanced cancers the anti- proliferative properties of TGF-β are not apparent and TGF-β becomes a significant factor in inducing EMT, which is usually associated with cancer progression (Sugimoto, et al. 2006). Pathways responsible for these actions involve the canonical Smads as signaling mediators and to a lesser extent, the noncanonical PI3K and p38 MAPK mediators (Derynck, et al. 2001; Kalluri and Neilson 2003). Indeed, Hayward and colleagues showed that TGF-β induced EMT in prostate carcinoma cells was mediated through constitutively active Akt, which functioned to inhibit Smad3 and p21 translocation to the nucleus (Ao et al. 2006). Accordingly, whereas normal prostate epithelial cells were growth inhibited by TGF-β, prostate carcinoma cells avoided cell cycle arrest and were alternatively induced to undergo EMT by TGF-β.
The actions of TGF-β appear to be critical in maintaining stromal compartment biology and overall tissue homeostasis. Interestingly, Bhowmick and colleagues reported that knockout of TGF-β signaling in mouse fibroblasts resulted in a PIN phenotype in adjacent prostate epithelial cells, indicating that normal fibroblasts with intact TGF-β signaling have the potential to be cancer-inhibitory at baseline and that loss of TGF-β responsiveness is permissive or promotive for PIN development (Bhowmick et al. 2004). However, further evaluation showed that receptor was knocked out in only 40–60% of fibroblasts in the prostate gland (Kiskowski et al. 2011). Additional tissue recombination studies showed that elevated expression of Wnt3a in TGF-β receptor 2 null fibroblasts was responsible for stimulated tumor growth (Li et al. 2008). More recent tissue recombination studies showed that a mixture of TGF-β receptor null fibroblasts and receptor intact fibroblasts resulted in a more potent induction of adenocarcinoma (Franco et al. 2011; Kiskowski et al. 2011). Moreover, a heterogeneous mixture of TGF-β responsive and nonresponsive fibroblasts resulted in higher expression of chemokines and growth factors, including TGF-β, which are each associated with inflammation and tumor promotion (Franco et al. 2011). Our earlier studies had shown that expression of TGF-β stimulated genes, FGF-2 and CTGF, in TGF-β responsive human prostate stromal cells/fibroblasts promoted angiogenesis and was tumor promoting when these cells were recombined with human prostate cancer cells in xenografts (Tuxhorn et al. 2002b; Tuxhorn et al. 2002c; Yang et al. 2008a; Yang et al. 2005). Moreover, recombinations with stromal cells null for TGF-β signaling resulted a reduced tumor mass and a lower rate of angiogenesis (Yang et al. 2008a). This is consistent with many reports showing that TGF-β can stimulate angiogenesis, alter immune surveillance, and induce tumor stromal cells to secrete growth factors and matrix associated proteins (Li, et al. 2007). In vitro, TGF-β1 can induce resting fibroblasts to develop stress fibers and express smooth muscle alpha-actin, reinforcing the idea that this growth factor has a direct effect on the induction of a myofibroblast-like reactive stroma (Gerdes et al. 2004; Peehl and Sellers 1997, 2000; Ronnov-Jessen and Petersen 1993). In addition, it is well documented that TGF-β exhibits pluripotent activities that are usually modified or dependent on the activity of other growth factors in the local environment (Roberts and Sporn 1996; Sporn and Roberts 1988; Wakefield and Roberts 2002). Accordingly, differential TGF-β activity and function in tissues and tumors is complicated and may sometimes seem paradoxical. The key observations that heterogeneous mixes of TGF-β unresponsive and responsive fibroblasts results in stimulated tumorigenesis and expression of factors is very interesting and may ultimately explain some of these seemingly paradoxical results (Franco et al. 2011; Kiskowski et al. 2011). Hence, the specific role of TGF-β in the initiation and promotion of early cancer is complex and not entirely understood.
As discussed earlier, TGF-β also regulates key immune functions that affect tumor promotion. It should be noted that TGF-β1 is a key factor released by platelets at sites of wound repair where it regulates both inflammatory responses and angiogenesis (Roberts and Sporn 1996; Roberts, et al. 1986). TGF-β1 can affect several pathways known to mediate rapid host immune cell modulation through cytostatic, chemotactic, and fibrotic induction of different cell populations. Again, TGF-β exhibits multiple functions that may seem paradoxical. Historically, TGF-β has been known to be chemoattractive to immune cells, specifically to monocytes and neutrophils (Brandes, et al. 1991; Wahl, et al. 1987). Whereas these cell populations are instrumental in maintaining a relatively aseptic environment during primary wound healing in normal tissue, evidence from tumor microenvironment studies suggests that the myeloid compartment generates tumor-associated macrophages (TAMs) that have a role in promoting cancer growth and metastasis via elevated expression of CSF-1 among other factors (Condeelis and Pollard 2006; Pollard 2004). Importantly, deletion of TGF-β receptor 2 in a mammary carcinoma mouse model stimulated recruitment of myeloid derived suppressor cells that expressed higher levels of TGF-β and MMPs resulting in elevated tumor invasion and metastasis (Bierie and Moses 2010; Yang, et al. 2008b; Yang and Moses 2008). It appears likely that the modulation from tumor inhibiting to tumor-promoting activities may lie in the different immune cell populations that are recruited and regulated by TGF-β and the interactions with other growth factors and downstream mediators of action.
In the development of prostate cancer, overexpression of TGF-β1 was first noted in PIN-associated epithelial cells in a heterogeneous focal pattern during the evolution of PIN, where epithelial cells had lost polarity, suggesting defects in acini wall integrity (Tuxhorn et al. 2002a). Interestingly, we showed that the overexpression of TGF-β1 targeted to the mouse prostate gland resulted in an age-dependent phenotypic alteration characterized by attenuation of epithelium thickness in acini walls, induction of fibroplasia, and inflammation in vessels and nerve (Barron et al. 2010). These mice also exhibited an age-dependent increase in the frequency of unique fibrotic collagenous micronodules, with histopathology nearly identical to collagenous micronodules that are associated with human prostate cancer (Epstein 2004). The nodules in the TGF-β1 transgenic mice exhibited elevated expression of tenascin-C and collagen, both prototypical markers of reactive stroma. Accordingly, these nodules likely form as a result of age-associated disruption of epithelial and basal lamina integrity, together with access of epithelial expressed TGF-β1 to the stromal compartment, resulting in an adaptive homeostasis to protect from further damage and to promote repair. We proposed that the evolution of the TGF-β induced micronodules was part of a reactive stroma response, designed to impinge on a damaged acini in order to close off the lumen from prostatic ducts and thereby limit access to microbial agents potentially present in the lumen and ducts (Barron et al. 2010). Again, we interpret these data as part of a local host adaptive response to a damaged or breached epithelial barrier. In this context, it is not surprising that this phenotype was age dependent.
Less understood, was the observed TGF-β induced inflammation in local nerve ganglia in these studies and the role of neural regulation of reactive stroma. Whereas the interaction between prostate cancer cells and nerves leading the perineural invasion was stimulated by the presence of prostate stroma (Cornell, et al. 2003), the regulation of normal or reactive stroma in prostate cancer by autonomic nerves is poorly understood. However, it is quite likely that neural regulation of reactive stroma during wound repair and in the tumor microenvironment is important in the evolving biology of reactive stroma. In this regard, Ayala has shown that nerve density is elevated in both pre-neoplastic lesions and in prostate cancer, confirming the process of axonogenesis in human prostate cancer (Ayala, et al. 2008). These studies also showed an elevated number of neurons, suggesting neurogenesis is also associated with development and progression of prostate cancer as this was correlated with more aggressive cancers and with recurrent disease. Semaphorin 4F was implicated as a putative mediator of neurogenesis in these studies, although specific mechanisms are not fully understood.
The generation of focal fibroplasia, inflammation and collagenous micronodules in the stromal microenvironment of TGF-β1 overexpressing mice is consistent with the role of reactive stroma in wound repair biology (Desmouliere et al. 2005; Gabbiani 2003). These important adaptive responses whereby fibrosis and inflammation act in coordinate manners may provide additional insight into the tumor promoting nature of the reactive stroma microenvironment observed in most carcinomas. Overexpression of TGF-β1 in other murine epithelial organ systems also produced reactive and fibrotic responses, albeit somewhat different from what we reported (Kopp, et al. 1996; Sanderson et al. 1995; Sanvito, et al. 1995). Understanding these responses and mechanisms is important for developing novel therapeutic targets for disorders where the stromal microenvironment plays a pivotal role in progression and clinical outcome. Further insight into downstream pathways of TGF-β1 and mechanisms in prostate tissue homeostasis is important for understanding the role of this factor in prostate disease progression.
IV. Modeling the Putative Origins of Reactive Stroma
The origin of reactive stroma fibroblasts and myofibroblasts has been an active area of research for the past several years. Depending on the tissue type remodeled, precursors to the myofibroblast are recruited from different cellular compartments, the most common of which appears to be locally residing fibroblasts (Hinz 2007; Sugimoto et al. 2006). Other mesenchymal cells that may serve as myofibroblast progenitors are pericytes and smooth muscle cells from the vasculature. These seem to play an important role in blood vessel repair and in the pathogenesis of diseases such as scleroderma (Hao, et al. 2006; Rajkumar, et al. 2005). The work of Gary Owens and others has shed light on the importance of local stromal cells in responding to injury in cardiovascular models (Hoofnagle, et al. 2004). Their studies demonstrated that the origin of neointimal smooth muscle in hyperlipidemia induced atherosclerotic lesions was from a local population of cells, rather than from a marrow-derived source (Bentzon, et al. 2006; Hoofnagle, et al. 2006). Epithelial cells have also been proposed as a source of reactive stroma via epithelial-to-mesenchymal transition (EMT) events (Kalluri 2009; Kalluri and Neilson 2003; Zavadil, et al. 2008). This has been stratified into different classifications depending on the biological process in question (Kalluri and Weinberg 2009).
Bone marrow derived circulating cells known as fibrocytes may represent an alternative source for myofibroblasts during skin wound healing, and in liver, lung, and kidney fibrosis (Abe, et al. 2001; Direkze, et al. 2003; Forbes, et al. 2004; Ishii, et al. 2003; Schmidt, et al. 2003). In particular, studies of asthma showed in both mouse models and human tissues the recruitment of circulating CD34+ progenitors to bronchial tissue, where they subsequently expressed collagen type I and smooth muscle alpha-actin, two markers that are consistent with the myofibroblast phenotype (Schmidt et al. 2003). Other studies focused on mammary cancer, colorectal cancer, and pancreatic cancer have suggested the recruitment of circulating cells of hematopoietic origin to sites of reactive stroma formation, although whether these cells are contributing more towards an inflammatory state or directly to reactive stroma remains unknown.
While this highlights the importance of understanding the mechanisms of recruitment of circulating fibrocytes to sites of wound repair, very little is known about the existence and recruitment potential of these cells in prostate cancer, much less any potential mechanisms that might mediate such biology. In fact, much work has focused on establishing the existence of tissue resident cell populations capable of giving rise to reactive stroma in the progression from PIN to organ-confined disease (Kaminski, et al. 2006; Sheffer, et al. 2007; Tuxhorn et al. 2002b; Verona, et al. 2007). Certainly, these reports underscore the importance of tissue resident stroma in the genesis of reactive stroma. It is possible that reactive fibroblasts that evolve at sites of tissue injury may originate from different sources, depending on the nature of the wound and the type of injury sustained. In minor wounding, fibroblasts might predominantly migrate from surrounding undamaged tissue, whereas in deep tissue wounds, fibrocytes might be recruited that can differentiate into fibroblasts. Investigations by Bucala in the early 1990s that led to the discovery of these fibrocytes, were based on the hypothesis that specialized cells reminiscent of fibroblasts in morphology and function were found in the circulation and had the capacity to home to experimentally implanted wound chambers much the same as cells of the innate immune system (Bucala, et al. 1994). This discovery led to a thorough characterization of a distinct population of CD34+/Col 1+ fibroblast-like cells that rapidly entered sites of tissue injury where they differentiated to myofibroblasts that expressed smooth muscle alpha-actin (Abe et al. 2001; Quan, et al. 2004; Schmidt et al. 2003).
The precise origin of peripheral blood fibrocytes has puzzled investigators since their discovery. Early studies using sex-mismatched, bone marrow chimeric mice together with DNA amplification of the male-specific SRY gene showed that fibrocytes originated from a bone marrow progenitor cell population (Bucala et al. 1994). Additional studies showed that fibrocytes isolated from peripheral blood differentiate ex vivo from an adherent CD14+ cell population (Abe et al. 2001). It has been postulated that circulating fibrocyte progenitors undergo phenotypic and gene expression changes and differentiate to fibrocytes that are subsequently recruited to wound sites where they become mature fibroblasts and play a role in wound contracture and healing (Metz 2003). Very little is known about the potential role of these unique cells in cancer-associated reactive stroma. Our preliminary data using bone marrow transplant approaches suggest that few reactive stroma myofibroblast/CAF cells originate from bone marrow and that the majority are derived local stem or progenitor cells. Understanding the origin of reactive stroma stem/progenitor cells and mechanisms that regulate differentiation to myofibroblasts/CAFs is important, since these cells may be targets for novel therapeutic approaches.
Although reactive stroma in wound repair may be similar to reactive stroma in cancer, the self-limiting nature of wound repair appears to be absent in cancer. In cancer, the stromal and epithelial compartments seem to co-evolve and is self-perpetuating during the progression of cancer (Rowley 1998). The cancer-associated factors work in coordination with the complement of proteins and factors secreted by vessels, immune-associated cells, and even nerves, to regulate the stromal compartment. The end result is a response by both marrow-derived cells and local fibroblasts to re-establish homeostasis. The main difference in cancer would seem to be the potential for these stromal cell populations to co-evolve with the carcinoma cells in an orchestrated indolent process that occurs on the order of months or years, far longer than a typical wound repair scenario. It is possible that as the growing tumor co-opts more stromal cellular elements to develop into reactive stroma, the demands on the microenvironment are increased, leading to a sustained inflammatory response observed in the prostate cancer model systems (Condeelis and Pollard 2006; Pollard 2009). Therefore, it is possible that marrow-derived progenitor cells recruited to these sites may evolve either into myofibroblast/CAF-like stromal cells or follow alternate differentiation pathways to become more macrophage-like cells (Figure 4) (de Visser and Coussens 2006; DeNardo, et al. 2009; Qian and Pollard 2010; Ruffell, et al. 2010).
Figure 4. Model of potential origin and regulators of reactive stroma.

It is hypothesized that reactive stroma in prostate cancer may be derived from both bone marrow derived cells as well as non-marrow sources. The latter cell population is most likely tissue-resident mesenchymal/stromal stem cells or existing fibroblasts. Carcinoma-produced factors, including TGF-beta have the potential to reach and regulate the stromal compartment when the epithelial layer integrity and/or basement membrane integrity is disrupted during cancer progression.
V. New Perspectives on the Origin and Evolution of Reactive Stroma
Understanding the cellular origin and the key regulators of reactive stroma that evolve during progression are all active areas of investigation. Our preliminary studies suggest that the majority of reactive stroma is derived from local, tissue resident progenitor cells and a minority from circulating marrow-derived cells. It is possible that role of bone marrow derived cells in the tumor niche is different from their normal role in re-establishing homeostasis in damaged tissue such as wounds and infection. Whereas inflammatory cells are typically viewed as being beneficial to primary wounds in preventing a potential nidus of infection from spreading throughout the organism, tumor-associated inflammation seems to foster the growth and spread of the cancer. This inflammation can take on a variety of different forms, including those subtypes rich in lymphoid and myeloid cells. If we expand the concept of inflammation to include cells that contribute directly to reactive stroma, then this tumor-associated inflammatory cell recruitment could have even broader implications for the progression of cancer. Indeed, there have been several reports of marrow-derived myofibroblasts seen in human tissue as well as a variety of other model systems (Brittan, et al. 2002; Campbell, et al. 2000; Direkze et al. 2003; Epperly, et al. 2003; Haudek, et al. 2006). In line with the tumor-promoting aspects of reactive stroma, these myofibroblasts could contribute to the already developing local microenvironment to further promote a process of stromal and epithelial co-evolution that has clear growth advantages for the primary tumor. In addition, there are now suggestions that infectious agents and sexually transmitted diseases elevate the risk of prostate cancer. One study has reported that prostate cancer risk was lower in men circumcised before the first sexual contact, suggesting that the elevated risk of sexually transmitted disease in uncircumcised men is associated with elevated risk of prostate cancer (Wright, et al. 2012).
The involvement of cells from the myeloid lineage in reactive stroma compartment is interesting due to their plasticity in vivo to differentiate to a wide range of cell populations in normal tissues, but also because of their striking tumor-promoting ability in the context of macrophages (Condeelis and Pollard 2006; Gordon and Taylor 2005; Pollard 2004; Treves 1984). Recently, a primitive cell population termed monocyte-derived multipotential cells (MOMCs) was discovered (Seta and Kuwana 2010). These cells possess a fibroblast-like morphology in culture and a unique phenotype positive for CD14, CD45, CD34, and type I collagen, a similar profile to what has been defined in the fibrocyte literature. MOMCs are likely derived from circulating CD14+ monocytes, however precursors for MOMCs still remain undetermined although they are likely from myeloblastic cells within the bone marrow. It was suggested that MOMCs contain progenitors with capacity to differentiate into a variety of nonphagocytes, including bone, cartilage, fat, skeletal and cardiac muscle, neuron, and endothelium, suggesting that circulating monocytes are more multipotent than previously thought. This plasticity is certainly observed in a variety of cell populations of the hematopoietic system (Colvin, et al. 2004; Heike and Nakahata 2004; Ogawa 2010; Zardo, et al. 2008; Zubair, et al. 2002). The potential role of MOMCs in the generation of reactive stroma in cancer and their biology in wound repair is unknown. The identification of these cells adds to the growing list of potential cell types that may contribute to the composition of reactive stroma in cancer. The putative plasticity of cells recruited to the tumor microenvironment may explain the heterogeneity observed in the content and composition of reactive stroma in human prostate cancer.
The recruitment model of reactive stroma connotes a scenario of inflammatory cell “education” at the tissue destination. If the tissue is infected with a sexually transmitted virus, for example, then a unique subset of cells is generated from the myeloid compartment that is recruited to the site of infection. Most of these may be mature monocytes and macrophages designed to control the level of infection during an innate immune response to establish a scenario similar to that observed in proliferative inflammatory atrophy (PIA) observed in the prostate gland. As the tissue is remodeled and altered over time, a fundamentally different growth factor and extracellular matrix profile may develop, thereby educating the recruited myeloid progenitors or local mesenchymal stem/progenitor cells to differentiate towards reactive stroma forming cells. This putative model is indeed consistent with the remodeling-PIN-prostate adenocarcinoma spectrum (De Marzo et al. 2007) (Figure 3). If this model is true, it presents a very attractive therapeutic target that focuses on the nature of the inflammatory and microenvironment stromal cell recruitment in additional to the carcinoma cells.
The potential biological significance of tissue resident prostate cells contributing to reactive stroma has been demonstrated in other studies of prostate cancer that correlated the levels of expression of CD90 with the degree of tumor-promoting capacity within the stroma (True, et al. 2010; Zhao and Peehl 2009). Importantly, these studies indicated the need to include CD90 as a biomarker of prostate cancer based on its association with more aggressive forms of reactive stroma. The origin of these cells is not understood but several local cell types are candidate progenitors. The potential for local endothelial cells to contribute to stromal lineages was recently shown in a mechanism termed “endothelial-to-mesenchymal transition” (Nataraj, et al. 2010; Zeisberg, et al. 2007). Similarly, local pericytes associated with the vasculature may be contributing to the evolution of reactive stroma in tumors. Notably, the phenotypic and expression profiles of pericytes are very similar to myofibroblasts, which places them on the continuum of cells that express prototypical markers of reactive stroma (Eyden, et al. 2009; Sugimoto et al. 2006). Some studies have even linked the expression of CD34 to adipose stromal cells that have characteristics of pericytes by virtue of their perivascular localization and ability to stabilize endothelial networks (Suga, et al. 2009; Traktuev, et al. 2008). Whereas key advancements have been made in understanding the stem cell types for prostate epithelium (Wang and Shen 2011), the origins of reactive stroma CAFs and myofibroblasts remains poorly understood.
Although TGF-β seems to be a major regulator of reactive stroma, other interactive factors and processes are also likely to be important regulators. Regulatory events may be mediated by a variety of processes, ranging from direct stromal-epithelial interactions, to microvesicle secretion from bone marrow derived cells, to even direct fusion of progenitors with local stromal cells. The nature of these interactions may involve epigenetic reprogramming of the local stroma genome to one more consistent with a CAF (Hu, et al. 2010; Maherali, et al. 2007; Rodriguez-Canales, et al. 2007). Direct communication between marrow-derived cells and local stroma may also mediate reciprocal conversion to cells with more ‘reactive’ phenotypes, similar to what has been observed in tissue recombination studies done with prostate epithelium and stroma (Ao et al. 2006; Basanta et al. 2009; Cunha, et al. 2002; Cunha, et al. 2003; Franco, et al. 2010; Grompe 2003; Placencio, et al. 2008). Interestingly, microvesicles and fusion mechanisms may be involved, although these are not yet understood with respect to prostate reactive stroma biology. Although cell fusion has mainly been demonstrated in other tissues such as liver and bone marrow, it might be a potential mechanism of interactions of bone marrow derived cells with more mesenchymal cells in vivo via fusion with local fibroblasts, pericytes, or smooth muscle (Almeida-Porada, et al. 2010; Grompe 2003; Quesenberry, et al. 2010). Interestingly, the existence of microvesicles in prostate tissue was demonstrated by Michael Freeman (Di Vizio, et al. 2009). These studies show that prostate cancer cells have the capacity to secrete oncosomes, membrane-bound microvesicle particles containing several signal transduction proteins, which significantly altered the proliferation and migration of recipient tumor cells. The role of such microvesicle shedding from prostate cancer cells in the genesis of reactive stroma has not been investigated, but will likely yield some interesting findings.
VI. Future Directions
The reactive stroma tumor microenvironment in carcinomas is very complex and provides a regulatory function during cancer progression. Specific mechanisms are largely unknown, however, recent studies are providing new information on key pathways. The core biology that has evolved to maintain tissue homeostasis is likely to be important in the biology of reactive stroma in cancer. It is becoming clear that many different cell types interact within the tumor microenvironment and these interactions result in focal and heterogeneous nature of reactive stroma reported in most carcinomas. It is now becoming clear that the heterogeneity of the reactive stroma compartment and the apparent interactions of different types of reactive stroma cells result in a much more potent tumor-inducing microenvironment. Growth factors and chemokines/cytokines including TGF-βs, interleukins, fibroblast growth factors, Wnts, and essentially any factor or pathway that regulates inflammatory processes and tissue repair biology are likely involved in these complex interactions. Understanding reactive stroma stem or progenitor cells and mechanisms that regulate their recruitment, activation, and biology is an important issue. Although not yet understood, it is likely that the paracrine interactions between epithelium and reactive stroma is involved in the evolution of the castration resistant phenotype during prostate cancer progression. Moreover, there is very little understood about reactive stroma at sites of tumor metastasis. It is possible that new prognostics can be developed that evaluate markers of reactive stroma composition and biology. There is also a clear need to develop novel therapeutic approaches designed to target the reactive stroma microenvironment in prostate cancer. Since reactive stroma initiates early during pre-neoplastic disease, it may be possible to target the genesis of early reactive stroma formation, as an attempt to uncouple the tumor-promoting effects early in the evolution of cancer. In addition, targeting reactive stroma biology in well-developed tumors may inhibit the rate of tumor progression to invasive and metastatic cancer. Finally, there is a clear need to understand the possible role of reactive stroma at sites of metastasis. It is possible that reactive stroma provides a key biology in maintenance of the niche that supports active or dormant prostate cancer stem cells. The targeting of this niche would represent a key therapeutic advancement. It is anticipated that future efforts will result in improved early diagnostics and novel therapeutic approaches designed to target the tumor microenvironment as an additional tool to improve direct cancer therapeutic outcomes. The complex nature of reactive stroma and the involvement of multiple signaling pathways and cell types provide much opportunity to develop such novel therapeutic approaches.
Acknowledgments
Funding: Supported by NIH R01 CA58093 (DRR) and a Predoctoral Prostate Cancer Training Grant W81XWH-08-10059 (DAB) from the U.S. Department of Defense.
Footnotes
Declaration of Interest: The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported
Author Contributions: Each author contributed equally to this manuscript.
Disclaimer. This is not the definitive version of the record of this article
References
- Abe R, Donnelly SC, Peng T, Bucala R, Metz CN. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol. 2001;166:7556–7562. doi: 10.4049/jimmunol.166.12.7556. [DOI] [PubMed] [Google Scholar]
- Ahmed A. The myofibroblast in breast disease. Pathol Annu. 1990;25(Pt 2):237–286. [PubMed] [Google Scholar]
- Almeida-Porada G, Zanjani ED, Porada CD. Bone marrow stem cells and liver regeneration. Exp Hematol. 2010;38:574–580. doi: 10.1016/j.exphem.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Magdalena P, Brossner C, Reiner A, Cheng G, Sugiyama N, Warner M, Gustafsson JA. A role for epithelial-mesenchymal transition in the etiology of benign prostatic hyperplasia. Proc Natl Acad Sci U S A. 2009;106:2859–2863. doi: 10.1073/pnas.0812666106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ao M, Franco OE, Park D, Raman D, Williams K, Hayward SW. Cross-talk between paracrine-acting cytokine and chemokine pathways promotes malignancy in benign human prostatic epithelium. Cancer Res. 2007;67:4244–4253. doi: 10.1158/0008-5472.CAN-06-3946. [DOI] [PubMed] [Google Scholar]
- Ao M, Williams K, Bhowmick NA, Hayward SW. Transforming growth factor-beta promotes invasion in tumorigenic but not in nontumorigenic human prostatic epithelial cells. Cancer Res. 2006;66:8007–8016. doi: 10.1158/0008-5472.CAN-05-4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala AG, Ro JY. Prostatic intraepithelial neoplasia: recent advances. Arch Pathol Lab Med. 2007;131:1257–1266. doi: 10.5858/2007-131-1257-PINRA. [DOI] [PubMed] [Google Scholar]
- Ayala G, Tuxhorn JA, Wheeler TM, Frolov A, Scardino PT, Ohori M, Wheeler M, Spitler J, Rowley DR. Reactive stroma as a predictor of biochemical-free recurrence in prostate cancer. Clin Cancer Res. 2003;9:4792–4801. [PubMed] [Google Scholar]
- Ayala GE, Dai H, Powell M, Li R, Ding Y, Wheeler TM, Shine D, Kadmon D, Thompson T, Miles BJ, et al. Cancer-related axonogenesis and neurogenesis in prostate cancer. Clin Cancer Res. 2008;14:7593–7603. doi: 10.1158/1078-0432.CCR-08-1164. [DOI] [PubMed] [Google Scholar]
- Barron DA, Strand DW, Ressler SJ, Dang TD, Hayward SW, Yang F, Ayala GE, Ittmann M, Rowley DR. TGF-beta1 induces an age-dependent inflammation of nerve ganglia and fibroplasia in the prostate gland stroma of a novel transgenic mouse. PLoS One. 2010;5:e13751. doi: 10.1371/journal.pone.0013751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsky SH, Green WR, Grotendorst GR, Liotta LA. Desmoplastic breast carcinoma as a source of human myofibroblasts. Am J Pathol. 1984;115:329–333. [PMC free article] [PubMed] [Google Scholar]
- Basanta D, Scott JG, Fishman MN, Ayala G, Hayward SW, Anderson AR. Investigating prostate cancer tumour-stroma interactions: clinical and biological insights from an evolutionary game. Br J Cancer. 2012;106:174–181. doi: 10.1038/bjc.2011.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basanta D, Strand DW, Lukner RB, Franco OE, Cliffel DE, Ayala GE, Hayward SW, Anderson AR. The role of transforming growth factor-beta-mediated tumor-stroma interactions in prostate cancer progression: an integrative approach. Cancer Res. 2009;69:7111–7120. doi: 10.1158/0008-5472.CAN-08-3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentzon JF, Weile C, Sondergaard CS, Hindkjaer J, Kassem M, Falk E. Smooth muscle cells in atherosclerosis originate from the local vessel wall and not circulating progenitor cells in ApoE knockout mice. Arterioscler Thromb Vasc Biol. 2006;26:2696–2702. doi: 10.1161/01.ATV.0000247243.48542.9d. [DOI] [PubMed] [Google Scholar]
- Bettendorf O, Schmidt H, Staebler A, Grobholz R, Heinecke A, Boecker W, Hertle L, Semjonow A. Chromosomal imbalances, loss of heterozygosity, and immunohistochemical expression of TP53, RB1, and PTEN in intraductal cancer, intraepithelial neoplasia, and invasive adenocarcinoma of the prostate. Genes Chromosomes Cancer. 2008;47:565–572. doi: 10.1002/gcc.20560. [DOI] [PubMed] [Google Scholar]
- Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, Washington MK, Neilson EG, Moses HL. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–851. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6:506–520. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- Bierie B, Moses HL. Transforming growth factor beta (TGF-beta) and inflammation in cancer. Cytokine Growth Factor Rev. 2010;21:49–59. doi: 10.1016/j.cytogfr.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes ME, Mai UE, Ohura K, Wahl SM. Type I transforming growth factor-beta receptors on neutrophils mediate chemotaxis to transforming growth factor-beta. J Immunol. 1991;147:1600–1606. [PubMed] [Google Scholar]
- Brittan M, Hunt T, Jeffery R, Poulsom R, Forbes SJ, Hodivala-Dilke K, Goldman J, Alison MR, Wright NA. Bone marrow derivation of pericryptal myofibroblasts in the mouse and human small intestine and colon. Gut. 2002;50:752–757. doi: 10.1136/gut.50.6.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol. 2005;45:657–687. doi: 10.1146/annurev.pharmtox.45.120403.095802. [DOI] [PubMed] [Google Scholar]
- Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med. 1994;1:71–81. [PMC free article] [PubMed] [Google Scholar]
- Campbell JH, Efendy JL, Han C, Girjes AA, Campbell GR. Haemopoietic origin of myofibroblasts formed in the peritoneal cavity in response to a foreign body. J Vasc Res. 2000;37:364–371. doi: 10.1159/000025752. [DOI] [PubMed] [Google Scholar]
- Colvin GA, Lambert JF, Moore BE, Carlson JE, Dooner MS, Abedi M, Cerny J, Quesenberry PJ. Intrinsic hematopoietic stem cell/progenitor plasticity: Inversions. J Cell Physiol. 2004;199:20–31. doi: 10.1002/jcp.10436. [DOI] [PubMed] [Google Scholar]
- Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Cornell RJ, Rowley D, Wheeler T, Ali N, Ayala G. Neuroepithelial interactions in prostate cancer are enhanced in the presence ofprostatic stroma. Urology. 2003;61:870–875. doi: 10.1016/s0090-4295(02)02426-3. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha GR, Hayward SW, Wang YZ. Role of stroma in carcinogenesis of the prostate. Differentiation. 2002;70:473–485. doi: 10.1046/j.1432-0436.2002.700902.x. [DOI] [PubMed] [Google Scholar]
- Cunha GR, Hayward SW, Wang YZ, Ricke WA. Role of the stromal microenvironment in carcinogenesis of the prostate. Int J Cancer. 2003;107:1–10. doi: 10.1002/ijc.11335. [DOI] [PubMed] [Google Scholar]
- Dakhova O, Ozen M, Creighton CJ, Li R, Ayala G, Rowley D, Ittmann M. Global gene expression analysis of reactive stroma in prostate cancer. Clin Cancer Res. 2009;15:3979–3989. doi: 10.1158/1078-0432.CCR-08-1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Marzo AM, Meeker AK, Zha S, Luo J, Nakayama M, Platz EA, Isaacs WB, Nelson WG. Human prostate cancer precursors and pathobiology. Urology. 2003;62:55–62. doi: 10.1016/j.urology.2003.09.053. [DOI] [PubMed] [Google Scholar]
- De Marzo AM, Platz EA, Sutcliffe S, Xu J, Gronberg H, Drake CG, Nakai Y, Isaacs WB, Nelson WG. Inflammation in prostate carcinogenesis. Nat Rev Cancer. 2007;7:256–269. doi: 10.1038/nrc2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Visser KE, Coussens LM. The inflammatory tumor microenvironment and its impact on cancer development. Contrib Microbiol. 2006;13:118–137. doi: 10.1159/000092969. [DOI] [PubMed] [Google Scholar]
- de Visser KE, Korets LV, Coussens LM. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell. 2005;7:411–423. doi: 10.1016/j.ccr.2005.04.014. [DOI] [PubMed] [Google Scholar]
- DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, Coussens LM. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009;16:91–102. doi: 10.1016/j.ccr.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- Desmouliere A, Chaponnier C, Gabbiani G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen. 2005;13:7–12. doi: 10.1111/j.1067-1927.2005.130102.x. [DOI] [PubMed] [Google Scholar]
- Desmouliere A, Darby IA, Gabbiani G. Normal and pathologic soft tissue remodeling: role of the myofibroblast, with special emphasis on liver and kidney fibrosis. Lab Invest. 2003;83:1689–1707. doi: 10.1097/01.lab.0000101911.53973.90. [DOI] [PubMed] [Google Scholar]
- Di Vizio D, Kim J, Hager MH, Morello M, Yang W, Lafargue CJ, True LD, Rubin MA, Adam RM, Beroukhim R, et al. Oncosome formation in prostate cancer: association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res. 2009;69:5601–5609. doi: 10.1158/0008-5472.CAN-08-3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimanche-Boitrel MT, Vakaet L, Jr, Pujuguet P, Chauffert B, Martin MS, Hammann A, Van Roy F, Mareel M, Martin F. In vivo and in vitro invasiveness of a rat colon-cancer cell line maintaining E-cadherin expression: an enhancing role of tumor-associated myofibroblasts. Int J Cancer. 1994;56:512–521. doi: 10.1002/ijc.2910560410. [DOI] [PubMed] [Google Scholar]
- Direkze NC, Forbes SJ, Brittan M, Hunt T, Jeffery R, Preston SL, Poulsom R, Hodivala-Dilke K, Alison MR, Wright NA. Multiple organ engraftment by bone-marrow-derived myofibroblasts and fibroblasts in bone-marrow-transplanted mice. Stem Cells. 2003;21:514–520. doi: 10.1634/stemcells.21-5-514. [DOI] [PubMed] [Google Scholar]
- Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- El-Alfy M, Pelletier G, Hermo LS, Labrie F. Unique features of the basal cells of human prostate epithelium. Microsc Res Tech. 2000;51:436–446. doi: 10.1002/1097-0029(20001201)51:5<436::AID-JEMT6>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Epperly MW, Guo H, Gretton JE, Greenberger JS. Bone marrow origin of myofibroblasts in irradiation pulmonary fibrosis. Am J Respir Cell Mol Biol. 2003;29:213–224. doi: 10.1165/rcmb.2002-0069OC. [DOI] [PubMed] [Google Scholar]
- Epstein JI. Diagnosis and reporting of limited adenocarcinoma of the prostate on needle biopsy. Mod Pathol. 2004;17:307–315. doi: 10.1038/modpathol.3800050. [DOI] [PubMed] [Google Scholar]
- Epstein JI. Precursor lesions to prostatic adenocarcinoma. Virchows Arch. 2009;454:1–16. doi: 10.1007/s00428-008-0707-5. [DOI] [PubMed] [Google Scholar]
- Eyden B, Banerjee SS, Shenjere P, Fisher C. The myofibroblast and its tumours. J Clin Pathol. 2009;62:236–249. doi: 10.1136/jcp.2008.061630. [DOI] [PubMed] [Google Scholar]
- Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- Forbes SJ, Russo FP, Rey V, Burra P, Rugge M, Wright NA, Alison MR. A significant proportion of myofibroblasts are of bone marrow origin in human liver fibrosis. Gastroenterology. 2004;126:955–963. doi: 10.1053/j.gastro.2004.02.025. [DOI] [PubMed] [Google Scholar]
- Franco OE, Jiang M, Strand DW, Peacock J, Fernandez S, Jackson RS, 2nd, Revelo MP, Bhowmick NA, Hayward SW. Altered TGF-beta signaling in a subpopulation of human stromal cells promotes prostatic carcinogenesis. Cancer Res. 2011;71:1272–1281. doi: 10.1158/0008-5472.CAN-10-3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco OE, Shaw AK, Strand DW, Hayward SW. Cancer associated fibroblasts in cancer pathogenesis. Semin Cell Dev Biol. 2010;21:33–39. doi: 10.1016/j.semcdb.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003;200:500–503. doi: 10.1002/path.1427. [DOI] [PubMed] [Google Scholar]
- Gabbiani G, Ryan GB, Majne G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia. 1971;27:549–550. doi: 10.1007/BF02147594. [DOI] [PubMed] [Google Scholar]
- Gann PH, Klein KG, Chatterton RT, Ellman AE, Grayhack JT, Nadler RB, Lee C. Growth factors in expressed prostatic fluid from men with prostate cancer, BPH, and clinically normal prostates. Prostate. 1999;40:248–255. doi: 10.1002/(sici)1097-0045(19990901)40:4<248::aid-pros6>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Gerdes MJ, Dang TD, Larsen M, Rowley DR. Transforming growth factor-beta1 induces nuclear to cytoplasmic distribution of androgen receptor and inhibits androgen response in prostate smooth muscle cells. Endocrinology. 1998;139:3569–3577. doi: 10.1210/endo.139.8.6138. [DOI] [PubMed] [Google Scholar]
- Gerdes MJ, Larsen M, Dang TD, Ressler SJ, Tuxhorn JA, Rowley DR. Regulation of rat prostate stromal cell myodifferentiation by androgen and TGF-beta1. Prostate. 2004;58:299–307. doi: 10.1002/pros.10327. [DOI] [PubMed] [Google Scholar]
- Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- Grompe M. The role of bone marrow stem cells in liver regeneration. Semin Liver Dis. 2003;23:363–372. doi: 10.1055/s-2004-815560. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hao H, Gabbiani G, Camenzind E, Bacchetta M, Virmani R, Bochaton-Piallat ML. Phenotypic modulation of intima and media smooth muscle cells in fatal cases of coronary artery lesion. Arterioscler Thromb Vasc Biol. 2006;26:326–332. doi: 10.1161/01.ATV.0000199393.74656.4c. [DOI] [PubMed] [Google Scholar]
- Haudek SB, Xia Y, Huebener P, Lee JM, Carlson S, Crawford JR, Pilling D, Gomer RH, Trial J, Frangogiannis NG, et al. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc Natl Acad Sci U S A. 2006;103:18284–18289. doi: 10.1073/pnas.0608799103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward SW, Wang Y, Cao M, Hom YK, Zhang B, Grossfeld GD, Sudilovsky D, Cunha GR. Malignant transformation in a nontumorigenic human prostatic epithelial cell line. Cancer Res. 2001;61:8135–8142. [PubMed] [Google Scholar]
- Heike T, Nakahata T. Stem cell plasticity in the hematopoietic system. Int J Hematol. 2004;79:7–14. doi: 10.1007/BF02983527. [DOI] [PubMed] [Google Scholar]
- Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127:526–537. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- Hirschel BJ, Gabbiani G, Ryan GB, Majno G. Fibroblasts of granulation tissue: immunofluorescent staining with antismooth muscle serum. Proc Soc Exp Biol Med. 1971;138:466–469. doi: 10.3181/00379727-138-35920. [DOI] [PubMed] [Google Scholar]
- Hoofnagle MH, Thomas JA, Wamhoff BR, Owens GK. Origin of neointimal smooth muscle: we've come full circle. Arterioscler Thromb Vasc Biol. 2006;26:2579–2581. doi: 10.1161/01.ATV.0000249623.79871.bc. [DOI] [PubMed] [Google Scholar]
- Hoofnagle MH, Wamhoff BR, Owens GK. Lost in transdifferentiation. J Clin Invest. 2004;113:1249–1251. doi: 10.1172/JCI21761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Gharaee-Kermani M, Wu Z, Phan SH. Epigenetic regulation of myofibroblast differentiation by DNA methylation. Am J Pathol. 2010;177:21–28. doi: 10.2353/ajpath.2010.090999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii G, Sangai T, Oda T, Aoyagi Y, Hasebe T, Kanomata N, Endoh Y, Okumura C, Okuhara Y, Magae J, et al. Bone-marrow-derived myofibroblasts contribute to the cancer-induced stromal reaction. Biochem Biophys Res Commun. 2003;309:232–240. doi: 10.1016/s0006-291x(03)01544-4. [DOI] [PubMed] [Google Scholar]
- Jagadeesan J, Bayat A. Transforming growth factor beta (TGFbeta) and keloid disease. Int J Surg. 2007;5:278–285. doi: 10.1016/j.ijsu.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Jones E, Pu H, Kyprianou N. Targeting TGF-beta in prostate cancer: therapeutic possibilities during tumor progression. Expert Opin Ther Targets. 2009;13:227–234. doi: 10.1517/14728220802705696. [DOI] [PubMed] [Google Scholar]
- Kalluri R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer. 2003;3:422–433. doi: 10.1038/nrc1094. [DOI] [PubMed] [Google Scholar]
- Kalluri R. EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Invest. 2009;119:1417–1419. doi: 10.1172/JCI39675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Han Y. Targeting TGF-beta and the extracellular matrix in Marfan's syndrome. Dev Cell. 2008;15:1–2. doi: 10.1016/j.devcel.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski A, Hahne JC, Haddoutiel M, Florin A, Wellmann A, Wernert N. Tumour-stroma interactions between metastatic prostate cancer cells and fibroblasts. Int J Mol Med. 2006;18:941–950. [PubMed] [Google Scholar]
- Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, Ferrara N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 1993;362:841–844. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- Kiskowski MA, Jackson RS, 2nd, Banerjee J, Li X, Kang M, Iturregui JM, Franco OE, Hayward SW, Bhowmick NA. Role for stromal heterogeneity in prostate tumorigenesis. Cancer Res. 2011;71:3459–3470. doi: 10.1158/0008-5472.CAN-10-2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, Onder TT, Wang ZC, Richardson AL, Weinberg RA, et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A. 2010;107:20009–20014. doi: 10.1073/pnas.1013805107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp JB, Factor VM, Mozes M, Nagy P, Sanderson N, Bottinger EP, Klotman PE, Thorgeirsson SS. Transgenic mice with increased plasma levels of TGF-beta 1 develop progressive renal disease. Lab Invest. 1996;74:991–1003. [PubMed] [Google Scholar]
- Li H, Fan X, Houghton J. Tumor microenvironment: the role of the tumor stroma in cancer. J Cell Biochem. 2007;101:805–815. doi: 10.1002/jcb.21159. [DOI] [PubMed] [Google Scholar]
- Li X, Placencio V, Iturregui JM, Uwamariya C, Sharif-Afshar AR, Koyama T, Hayward SW, Bhowmick NA. Prostate tumor progression is mediated by a paracrine TGF-beta/Wnt3a signaling axis. Oncogene. 2008;27:7118–7130. doi: 10.1038/onc.2008.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maherali N, Sridharan R, Xie W, Utikal J, Eminli S, Arnold K, Stadtfeld M, Yachechko R, Tchieu J, Jaenisch R, et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell. 2007;1:55–70. doi: 10.1016/j.stem.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Menke A, Adler G. TGFbeta-induced fibrogenesis of the pancreas. Int J Gastrointest Cancer. 2002;31:41–46. doi: 10.1385/IJGC:31:1-3:41. [DOI] [PubMed] [Google Scholar]
- Metz CN. Fibrocytes: a unique cell population implicated in wound healing. Cell Mol Life Sci. 2003;60:1342–1350. doi: 10.1007/s00018-003-2328-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller MM, Fusenig NE. Friends or foes - bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4:839–849. doi: 10.1038/nrc1477. [DOI] [PubMed] [Google Scholar]
- Nataraj D, Ernst A, Kalluri R. Idiopathic pulmonary fibrosis is associated with endothelial to mesenchymal transition. Am J Respir Cell Mol Biol. 2010;43:129–130. doi: 10.1165/rcmb.2010-0044ED. [DOI] [PubMed] [Google Scholar]
- Nelson WG, De Marzo AM, DeWeese TL, Isaacs WB. The role of inflammation in the pathogenesis of prostate cancer. J Urol. 2004;172:S6–11. doi: 10.1097/01.ju.0000142058.99614.ff. discussion S11–12. [DOI] [PubMed] [Google Scholar]
- Ogawa M. Hematopoietic stem cell plasticity. Introduction. Exp Hematol. 2010;38:527–528. doi: 10.1016/j.exphem.2010.04.009. [DOI] [PubMed] [Google Scholar]
- Olaso E, Santisteban A, Bidaurrazaga J, Gressner AM, Rosenbaum J, Vidal-Vanaclocha F. Tumor-dependent activation of rodent hepatic stellate cells during experimental melanoma metastasis. Hepatology. 1997;26:634–642. doi: 10.1002/hep.510260315. [DOI] [PubMed] [Google Scholar]
- Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59:5002–5011. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- Peehl DM, Sellers RG. Induction of smooth muscle cell phenotype in cultured human prostatic stromal cells. Exp Cell Res. 1997;232:208–215. doi: 10.1006/excr.1997.3525. [DOI] [PubMed] [Google Scholar]
- Peehl DM, Sellers RG. Cultured stromal cells: an in vitro model of prostatic mesenchymal biology. Prostate. 2000;45:115–123. doi: 10.1002/1097-0045(20001001)45:2<115::aid-pros5>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Placencio VR, Li X, Sherrill TP, Fritz G, Bhowmick NA. Bone marrow derived mesenchymal stem cells incorporate into the prostate during regrowth. PLoS One. 2010;5:e12920. doi: 10.1371/journal.pone.0012920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Placencio VR, Sharif-Afshar AR, Li X, Huang H, Uwamariya C, Neilson EG, Shen MM, Matusik RJ, Hayward SW, Bhowmick NA. Stromal transforming growth factor-beta signaling mediates prostatic response to androgen ablation by paracrine Wnt activity. Cancer Res. 2008;68:4709–4718. doi: 10.1158/0008-5472.CAN-07-6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol. 2009;9:259–270. doi: 10.1038/nri2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potenta S, Zeisberg E, Kalluri R. The role of endothelial-to-mesenchymal transition in cancer progression. Br J Cancer. 2008;99:1375–1379. doi: 10.1038/sj.bjc.6604662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB. Myofibroblasts. I. Paracrine cells important in health and disease. Am J Physiol. 1999;277:C1–9. doi: 10.1152/ajpcell.1999.277.1.C1. [DOI] [PubMed] [Google Scholar]
- Prud'homme GJ. Pathobiology of transforming growth factor beta in cancer, fibrosis and immunologic disease, and therapeutic considerations. Lab Invest. 2007;87:1077–1091. doi: 10.1038/labinvest.3700669. [DOI] [PubMed] [Google Scholar]
- Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan TE, Cowper S, Wu SP, Bockenstedt LK, Bucala R. Circulating fibrocytes: collagen-secreting cells of the peripheral blood. Int J Biochem Cell Biol. 2004;36:598–606. doi: 10.1016/j.biocel.2003.10.005. [DOI] [PubMed] [Google Scholar]
- Quesenberry PJ, Dooner MS, Aliotta JM. Stem cell plasticity revisited: the continuum marrow model and phenotypic changes mediated by microvesicles. Exp Hematol. 2010;38:581–592. doi: 10.1016/j.exphem.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkumar VS, Howell K, Csiszar K, Denton CP, Black CM, Abraham DJ. Shared expression of phenotypic markers in systemic sclerosis indicates a convergence of pericytes and fibroblasts to a myofibroblast lineage in fibrosis. Arthritis Res Ther. 2005;7:R1113–1123. doi: 10.1186/ar1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson AM, Woodson K, Wang Y, Rodriguez-Canales J, Erickson HS, Tangrea MA, Novakovic K, Gonzalez S, Velasco A, Kawasaki ES, et al. Global expression analysis of prostate cancer-associated stroma and epithelia. Diagn Mol Pathol. 2007;16:189–197. doi: 10.1097/PDM.0b013e3180de20ac. [DOI] [PubMed] [Google Scholar]
- Roberts AB, Sporn MB. Transforming growth factor-β. In: Clark RAF, editor. The Molecular and Cellular Biology of Wound Repair. New York, NY: Plenum Press; 1996. pp. 275–308. [Google Scholar]
- Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, Heine UI, Liotta LA, Falanga V, Kehrl JH, et al. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci U S A. 1986;83:4167–4171. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Canales J, Hanson JC, Tangrea MA, Erickson HS, Albert PS, Wallis BS, Richardson AM, Pinto PA, Linehan WM, Gillespie JW, et al. Identification of a unique epigenetic sub-microenvironment in prostate cancer. J Pathol. 2007;211:410–419. doi: 10.1002/path.2133. [DOI] [PubMed] [Google Scholar]
- Ronnov-Jessen L, Petersen OW. Induction of alpha-smooth muscle actin by transforming growth factor-beta 1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab Invest. 1993;68:696–707. [PubMed] [Google Scholar]
- Ronnov-Jessen L, Petersen OW, Bissell MJ. Cellular changes involved in conversion of normal to malignant breast: importance of the stromal reaction. Physiol Rev. 1996;76:69–125. doi: 10.1152/physrev.1996.76.1.69. [DOI] [PubMed] [Google Scholar]
- Ronnov-Jessen L, Petersen OW, Koteliansky VE, Bissell MJ. The origin of the myofibroblasts in breast cancer. Recapitulation of tumor environment in culture unravels diversity and implicates converted fibroblasts and recruited smooth muscle cells. J Clin Invest. 1995;95:859–873. doi: 10.1172/JCI117736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley DR. What might a stromal response mean to prostate cancer progression? Cancer Metastasis Rev. 1998;17:411–419. doi: 10.1023/a:1006129420005. [DOI] [PubMed] [Google Scholar]
- Ruffell B, DeNardo DG, Affara NI, Coussens LM. Lymphocytes in cancer development: polarization towards pro-tumor immunity. Cytokine Growth Factor Rev. 2010;21:3–10. doi: 10.1016/j.cytogfr.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salm SN, Burger PE, Coetzee S, Goto K, Moscatelli D, Wilson EL. TGF-{beta} maintains dormancy of prostatic stem cells in the proximal region of ducts. J Cell Biol. 2005;170:81–90. doi: 10.1083/jcb.200412015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson N, Factor V, Nagy P, Kopp J, Kondaiah P, Wakefield L, Roberts AB, Sporn MB, Thorgeirsson SS. Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc Natl Acad Sci U S A. 1995;92:2572–2576. doi: 10.1073/pnas.92.7.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanvito F, Nichols A, Herrera PL, Huarte J, Wohlwend A, Vassalli JD, Orci L. TGF-beta 1 overexpression in murine pancreas induces chronic pancreatitis and, together with TNF-alpha, triggers insulin-dependent diabetes. Biochem Biophys Res Commun. 1995;217:1279–1286. doi: 10.1006/bbrc.1995.2906. [DOI] [PubMed] [Google Scholar]
- Schauer IG, Ressler SJ, Rowley DR. Keratinocyte-derived chemokine induces prostate epithelial hyperplasia and reactive stroma in a novel transgenic mouse model. Prostate. 2009;69:373–384. doi: 10.1002/pros.20886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer IG, Ressler SJ, Tuxhorn JA, Dang TD, Rowley DR. Elevated epithelial expression of interleukin-8 correlates with myofibroblast reactive stroma in benign prostatic hyperplasia. Urology. 2008;72:205–213. doi: 10.1016/j.urology.2007.11.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer IG, Rowley DR. The functional role of reactive stroma in benign prostatic hyperplasia. Differentiation. 2011;82:200–210. doi: 10.1016/j.diff.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Sun G, Stacey MA, Mori L, Mattoli S. Identification of circulating fibrocytes as precursors of bronchial myofibroblasts in asthma. J Immunol. 2003;171:380–389. doi: 10.4049/jimmunol.171.1.380. [DOI] [PubMed] [Google Scholar]
- Seta N, Kuwana M. Derivation of multipotent progenitors from human circulating CD14+ monocytes. Exp Hematol. 2010;38:557–563. doi: 10.1016/j.exphem.2010.03.015. [DOI] [PubMed] [Google Scholar]
- Sheffer Y, Leon O, Pinthus JH, Nagler A, Mor Y, Genin O, Iluz M, Kawada N, Yoshizato K, Pines M. Inhibition of fibroblast to myofibroblast transition by halofuginone contributes to the chemotherapy-mediated antitumoral effect. Mol Cancer Ther. 2007;6:570–577. doi: 10.1158/1535-7163.MCT-06-0468. [DOI] [PubMed] [Google Scholar]
- Shoskes DA, Albakri Q, Thomas K, Cook D. Cytokine polymorphisms in men with chronic prostatitis/chronic pelvic pain syndrome: association with diagnosis and treatment response. J Urol. 2002;168:331–335. [PubMed] [Google Scholar]
- Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3:807–821. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- Singh H, Dang TD, Ayala GE, Rowley DR. Transforming growth factor-beta1 induced myofibroblasts regulate LNCaP cell death. J Urol. 2004;172:2421–2425. doi: 10.1097/01.ju.0000138082.68045.48. [DOI] [PubMed] [Google Scholar]
- Sporn MB, Roberts AB. Peptide growth factors are multifunctional. Nature. 1988;332:217–219. doi: 10.1038/332217a0. [DOI] [PubMed] [Google Scholar]
- Stover DG, Bierie B, Moses HL. A delicate balance: TGF-beta and the tumor microenvironment. J Cell Biochem. 2007;101:851–861. doi: 10.1002/jcb.21149. [DOI] [PubMed] [Google Scholar]
- Suga H, Matsumoto D, Eto H, Inoue K, Aoi N, Kato H, Araki J, Yoshimura K. Functional implications of CD34 expression in human adipose-derived stem/progenitor cells. Stem Cells Dev. 2009;18:1201–1210. doi: 10.1089/scd.2009.0003. [DOI] [PubMed] [Google Scholar]
- Sugimoto H, Mundel TM, Kieran MW, Kalluri R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol Ther. 2006;5:1640–1646. doi: 10.4161/cbt.5.12.3354. [DOI] [PubMed] [Google Scholar]
- Tanner MJ, Welliver RC, Jr, Chen M, Shtutman M, Godoy A, Smith G, Mian BM, Buttyan R. Effects of androgen receptor and androgen on gene expression in prostate stromal fibroblasts and paracrine signaling to prostate cancer cells. PLoS One. 2011;6:e16027. doi: 10.1371/journal.pone.0016027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traktuev DO, Merfeld-Clauss S, Li J, Kolonin M, Arap W, Pasqualini R, Johnstone BH, March KL. A population of multipotent CD34-positive adipose stromal cells share pericyte and mesenchymal surface markers, reside in a periendothelial location, and stabilize endothelial networks. Circ Res. 2008;102:77–85. doi: 10.1161/CIRCRESAHA.107.159475. [DOI] [PubMed] [Google Scholar]
- Treves AJ. The origin of monocyte-macrophage heterogeneity: possible alternatives. Med Hypotheses. 1984;14:335–346. doi: 10.1016/0306-9877(84)90138-5. [DOI] [PubMed] [Google Scholar]
- True LD, Zhang H, Ye M, Huang CY, Nelson PS, von Haller PD, Tjoelker LW, Kim JS, Qian WJ, Smith RD, et al. CD90/THY1 is overexpressed in prostate cancer-associated fibroblasts and could serve as a cancer biomarker. Mod Pathol. 2010;23:1346–1356. doi: 10.1038/modpathol.2010.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuxhorn JA, Ayala GE, Rowley DR. Reactive stroma in prostate cancer progression. J Urol. 2001;166:2472–2483. [PubMed] [Google Scholar]
- Tuxhorn JA, Ayala GE, Smith MJ, Smith VC, Dang TD, Rowley DR. Reactive stroma in human prostate cancer: induction of myofibroblast phenotype and extracellular matrix remodeling. Clin Cancer Res. 2002a;8:2912–2923. [PubMed] [Google Scholar]
- Tuxhorn JA, McAlhany SJ, Dang TD, Ayala GE, Rowley DR. Stromal cells promote angiogenesis and growth of human prostate tumors in a differential reactive stroma (DRS) xenograft model. Cancer Res. 2002b;62:3298–3307. [PubMed] [Google Scholar]
- Tuxhorn JA, McAlhany SJ, Yang F, Dang TD, Rowley DR. Inhibition of transforming growth factor-beta activity decreases angiogenesis in a human prostate cancer-reactive stroma xenograft model. Cancer Res. 2002c;62:6021–6025. [PubMed] [Google Scholar]
- Vanpoucke G, Orr B, Grace OC, Chan R, Ashley GR, Williams K, Franco OE, Hayward SW, Thomson AA. Transcriptional profiling of inductive mesenchyme to identify molecules involved in prostate development and disease. Genome Biol. 2007;8:R213. doi: 10.1186/gb-2007-8-10-r213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verona EV, Elkahloun AG, Yang J, Bandyopadhyay A, Yeh IT, Sun LZ. Transforming growth factor-beta signaling in prostate stromal cells supports prostate carcinoma growth by up-regulating stromal genes related to tissue remodeling. Cancer Res. 2007;67:5737–5746. doi: 10.1158/0008-5472.CAN-07-0444. [DOI] [PubMed] [Google Scholar]
- Wahl SM, Hunt DA, Wakefield LM, McCartney-Francis N, Wahl LM, Roberts AB, Sporn MB. Transforming growth factor type beta induces monocyte chemotaxis and growth factor production. Proc Natl Acad Sci U S A. 1987;84:5788–5792. doi: 10.1073/pnas.84.16.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield LM, Roberts AB. TGF-beta signaling: positive and negative effects on tumorigenesis. Curr Opin Genet Dev. 2002;12:22–29. doi: 10.1016/s0959-437x(01)00259-3. [DOI] [PubMed] [Google Scholar]
- Wang ZA, Shen MM. Revisiting the concept of cancer stem cells in prostate cancer. Oncogene. 2011;30:1261–1271. doi: 10.1038/onc.2010.530. [DOI] [PubMed] [Google Scholar]
- Wright JL, Lin DW, Stanford JL. Circumcision and the risk of prostate cancer. Cancer. 2012 doi: 10.1002/cncr.26653. Epub Date: 2012/03/14 In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa N, Li R, Rowley D, Liu H, Kadmon D, Miles BJ, Wheeler TM, Ayala GE. Stromogenic prostatic carcinoma pattern (carcinomas with reactive stromal grade 3) in needle biopsies predicts biochemical recurrence-free survival in patients after radical prostatectomy. Hum Pathol. 2007;38:1611–1620. doi: 10.1016/j.humpath.2007.04.008. [DOI] [PubMed] [Google Scholar]
- Yang F, Strand DW, Rowley DR. Fibroblast growth factor-2 mediates transforming growth factor-beta action in prostate cancer reactive stroma. Oncogene. 2008a;27:450–459. doi: 10.1038/sj.onc.1210663. [DOI] [PubMed] [Google Scholar]
- Yang F, Tuxhorn JA, Ressler SJ, McAlhany SJ, Dang TD, Rowley DR. Stromal expression of connective tissue growth factor promotes angiogenesis and prostate cancer tumorigenesis. Cancer Res. 2005;65:8887–8895. doi: 10.1158/0008-5472.CAN-05-1702. [DOI] [PubMed] [Google Scholar]
- Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, Carbone DP, Matrisian LM, Richmond A, Lin PC, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008b;13:23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Moses HL. Transforming growth factor beta: tumor suppressor or promoter? Are host immune cells the answer? Cancer Res. 2008;68:9107–9111. doi: 10.1158/0008-5472.CAN-08-2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Yeh CR, Niu Y, Chang HC, Tsai YC, Moses HL, Shyr CR, Chang C, Yeh S. Altered prostate epithelial development in mice lacking the androgen receptor in stromal fibroblasts. Prostate. 2012;72:437–449. doi: 10.1002/pros.21445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zardo G, Cimino G, Nervi C. Epigenetic plasticity of chromatin in embryonic and hematopoietic stem/progenitor cells: therapeutic potential of cell reprogramming. Leukemia. 2008;22:1503–1518. doi: 10.1038/leu.2008.141. [DOI] [PubMed] [Google Scholar]
- Zavadil J, Haley J, Kalluri R, Muthuswamy SK, Thompson E. Epithelial-mesenchymal transition. Cancer Res. 2008;68:9574–9577. doi: 10.1158/0008-5472.CAN-08-2316. [DOI] [PubMed] [Google Scholar]
- Zeisberg EM, Kalluri R. Origins of cardiac fibroblasts. Circ Res. 2010;107:1304–1312. doi: 10.1161/CIRCRESAHA.110.231910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007;67:10123–10128. doi: 10.1158/0008-5472.CAN-07-3127. [DOI] [PubMed] [Google Scholar]
- Zhao H, Peehl DM. Tumor-promoting phenotype of CD90hi prostate cancer-associated fibroblasts. Prostate. 2009;69:991–1000. doi: 10.1002/pros.20946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B, Kyprianou N. Transforming growth factor beta and prostate cancer. Cancer Treat Res. 2005;126:157–173. doi: 10.1007/0-387-24361-5_7. [DOI] [PubMed] [Google Scholar]
- Zubair AC, Silberstein L, Ritz J. Adult hematopoietic stem cell plasticity. Transfusion. 2002;42:1096–1101. doi: 10.1046/j.1537-2995.2002.00156.x. [DOI] [PubMed] [Google Scholar]
- Zynger DL, Yang X. High-grade prostatic intraepithelial neoplasia of the prostate: the precursor lesion of prostate cancer. Int J Clin Exp Pathol. 2009;2:327–338. [PMC free article] [PubMed] [Google Scholar]