

One of the abiding mysteries of all multi-cellular organisms is the requirement for controlled death —apoptosis — of unwanted cells. It has been estimated that without apoptosis an 80 year old person would have two tons of bone marrow and lymph nodes and an intestine 16 kilometers long.1 Progress in defining pathways of apoptosis has revealed complex interconnections between various cell death programs that may affect the treatment of a wide range of diseases.2–10 This article reviews advances in our understanding of mechanisms of cell death and highlights current and potential therapies based upon these concepts.
Perhaps the most widely used classification of mammalian cell death consists of two types: apoptosis and necrosis.3,4,11 Autophagy, which has recently been proposed as a third distinct mode of cell death, is a process by which cells generate energy and metabolites by digesting organelles or macromolecules.12–15. Normally, autophagy allows a starving cell, or a cell deprived of growth factors to survive.12–15 Ultimately, however, cells deprived of nutrients for extended periods will digest all available substrates and die an ‘autophagy-associated cell death’. Distinctions between apoptosis, necrosis, and autophagy entail differences in mode-specific or selective morphologic, biochemical, and molecular attributes (Fig. 1).3,4,11
Figure 1. Schematic diagram showing 3 possible pathways of cell death.
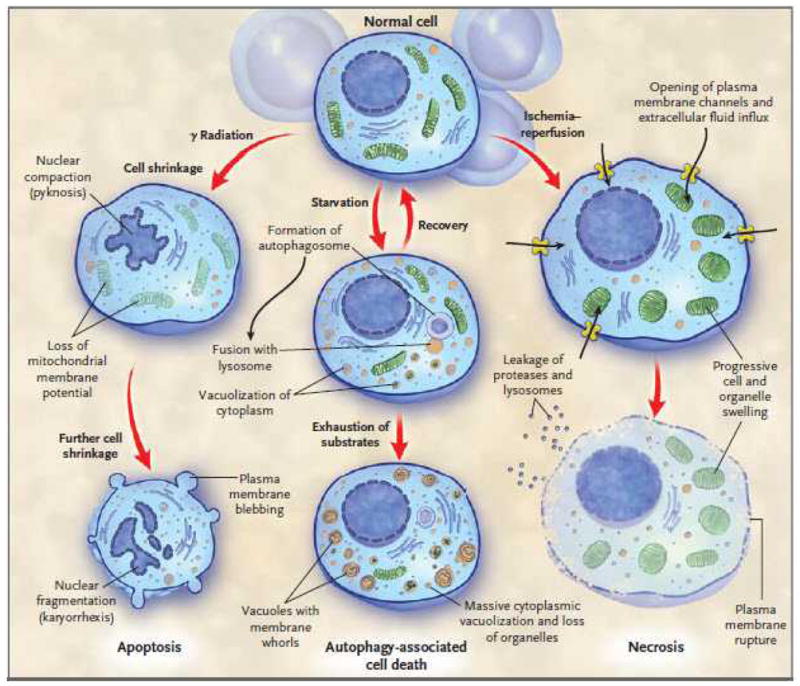
Note the characteristic differences in the 3 types of death. Depending upon the injury and the type of cell, a particular mode of cell death may predominate. Crosstalk between the different types of cell death pathways exists at multiple levels and is not shown.
An important concept embodied in part by these attributes is “programmed” cell death. Cell death is “programmed” if it is genetically controlled. The two fundamental types of programmed cell death are apoptosis and autophagy-associated cell death.3,12 The recognition that cell death can occur by genetically controlled processes has enabled advances in unraveling the mechanisms of many diseases. As a result, we now have improved knowledge of the initiation of cell death programs and the relevant signaling pathways. This information has facilitated development of pharmacologic agents that initiate or inhibit programmed cell death.6–8,16 Moreover, there is now evidence that necrosis, traditionally considered an accidental form of cell death, can, in certain instances, be initiated or modulated under programmed control mechanisms.17–21
Apoptosis
Apoptosis, (derived from άπóπτωσις, a falling off) is a word that suggests ‘leaves falling from a tree’.22–24 In contrast to the cell and organelle swelling that defines necrosis, the morphological features of apoptosis are cell and nuclear shrinkage (Figs.2,3 & Supplement-1–4). This distinction between necrosis and apoptosis is due in part to differences in how the plasma membrane participates in the two processes. In necrosis, there is early loss of integrity of the plasma membrane, which allows an influx of extracellular ions and fluid, with resultant cell and organellar swelling.17–20,25,26 By contrast, plasma membrane integrity is preserved until late in apoptosis. The key feature of apoptosis is cleavage of cytoskeletal proteins by pro-apoptotic aspartate-specific proteases (caspases), thereby causing collapse of subcellular components.2,5,8,23 Other characteristic features are chromatin condensation, nuclear fragmentation, and the formation of plasma membrane blebs.
Figure 2. Apoptotic cells in thymus, liver, and intestine.
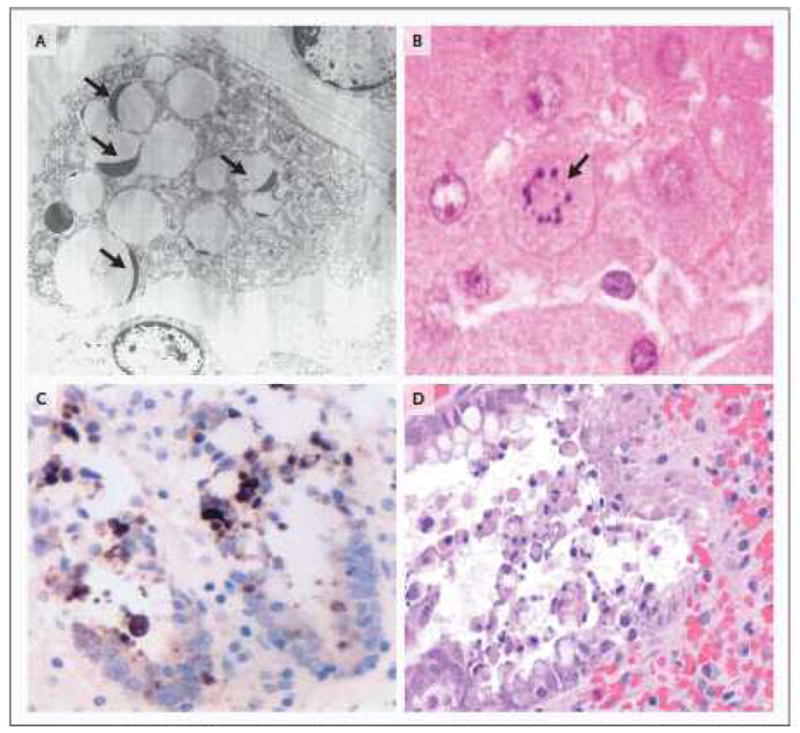
2A. Electron microscopic image of a phagocytic cell that has engulfed multiple apoptotic thymocytes. The compacted thymocyte nuclei have a classic ‘crescent shaped’ appearance, due to ‘layering’ of chromatin along the nuclear membrane. Normal appearing nuclei are present at six and one o’clock in the field of view (uranyl acetate/lead citrate; x2500). Thymic tissue section obtained from a 26 y.o. female who died following a motor vehicle accident complicated by ARDS and sepsis. 2B. A single apoptotic hepatocyte (identified by arrow) contains multiple compacted nuclear fragments indicative of apoptosis (hematoxylin and eosin [H&E]; x1000). Sample from 81 y.o. male in motor vehicle accident complicated by ventilator associated pneumonia. 2C Two adjacent crypts in colon mucosa immunohistochemically stained for cytokeratin 18 cleavage fragments (a positive reaction is brown in this color image). Cytokeratin 18 is cleaved by active caspases in both intrinsic and extrinsic apoptotic pathways. Detached cells in crypt lumens and epithelial cells still integrated into the crypt lining are positive; these cells also have classic apoptotic nuclear morphology. (see also 2D) (cytokeratin 18 immunostain [clone M30]/DAB with hematoxylin counterstain; x600). Sample from a 24 y.o. male who had aortic dissection and bowel ischemia following motor vehicle accident. 2D. A H&E stain of a colon section from a 23 y.o. patient with ischemic injury to bowel following intestinal surgery. The colonic intestinal epithelial cells show characteristic apoptotic features of nuclear compaction and fragmentation and have been sloughed into the bowel lumen (x400).
Figure 3. Apoptotic and necrotic cell death.
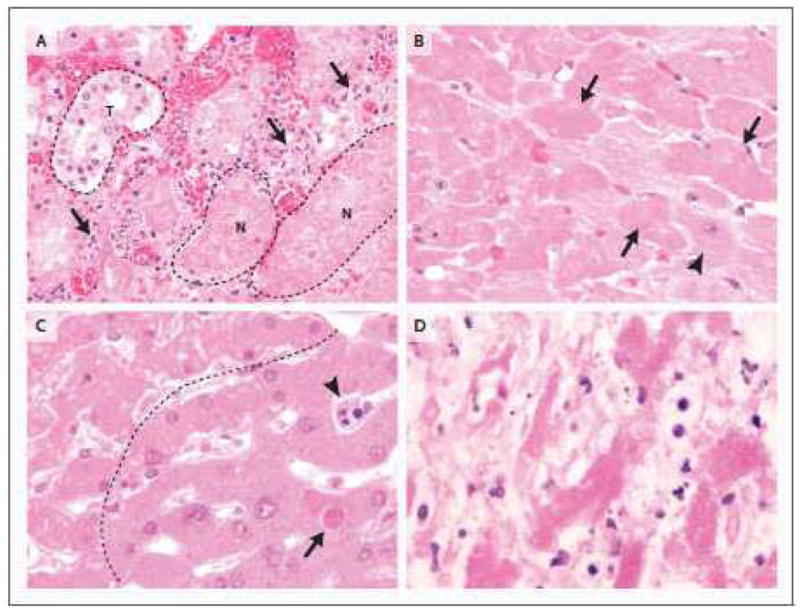
3A. The transition from viable cells to ischemic parenchyma is recapitulated in renal cortical necrosis, in this case from a 42 y.o. male who had undergone renal arterial embolization for renal cell carcinoma (not shown). Hypereosinophilia, loss of distinct nuclear detail and cytoplasmic vacuolization again characterize necrotic cells. Note the relatively intact tubule (t, dashed outline), compared with representative necrotic tubules (n; dashed outline). Note also that in the interface between viable and necrotic tubules (arrows), apoptotic cells (presumably neutrophils and mononuclear inflammatory cells) are abundant (H&E; x600). 3B. At higher magnification, the characteristic features of necrosis are apparent. Compared to viable cardiomyocytes with pale cytoplasm and distinct nuclear features (lower right of image with representative normal cell identified by arrowhead), necrotic cells are hypereosinophilic with uneven cytoplasmic vacuolization and either loss of nuclear detail or nuclear absence (identified by arrows). This morphology typifies an early manifestation of so-called ‘coagulative’ necrosis (H&E; x600). 3C. In this image, hepatocytes above and to the left of the dashed line exhibit changes of early necrosis, including vacuolization of hypereosinophilic cytoplasm and loss of nuclear detail. Heavy and narrow black arrows identify a sinusoidal inflammatory cell and a hepatocyte, respectively, with compacted and fragmented nuclei indicative of apoptosis. (H&E; x600). 53 y.o. male with pneumonia and bacteremia due to Streptococcus pneumoniae. (600X). 3D. Similar to 3C, but from an 81 y.o. male with ventilator associated pneumonia; this liver shows features typical of more advanced necrosis, with remnants of hepatocytes (eosinophilic cords) that lack obvious cell borders or recognizable nuclei. Cells and cell fragments admixed with hepatocytes are products of apoptosis; they most likely represent apoptotic lymphocytes or neutrophils in sinusoids (H&E; x600).
Caspase activation commits cells to one of two distinct but convergent apoptotic pathways, the death receptor and mitochondrial pathways. (Fig. 4). The death receptor pathway is triggered by binding of specific members of the tumor necrosis factor (TNF) superfamily to cell surface ‘death receptors’, members of the TNF receptor family (TNF-R).27–31 Ligation of these receptors initiates the formation of the multi-protein death-inducing signaling complex (DISC).5,32 c-FLIP is a regulatory molecule that resembles caspase-8 in sequence and structure; depending upon its concentration, it can either enhance or inhibit capase-8 activation.33 Aggregation of the DISC causes conformational changes that unleash the catalytic activity of caspase-8, which mediates cell destruction.
Figure 4. Pathways of cellular apoptosis.
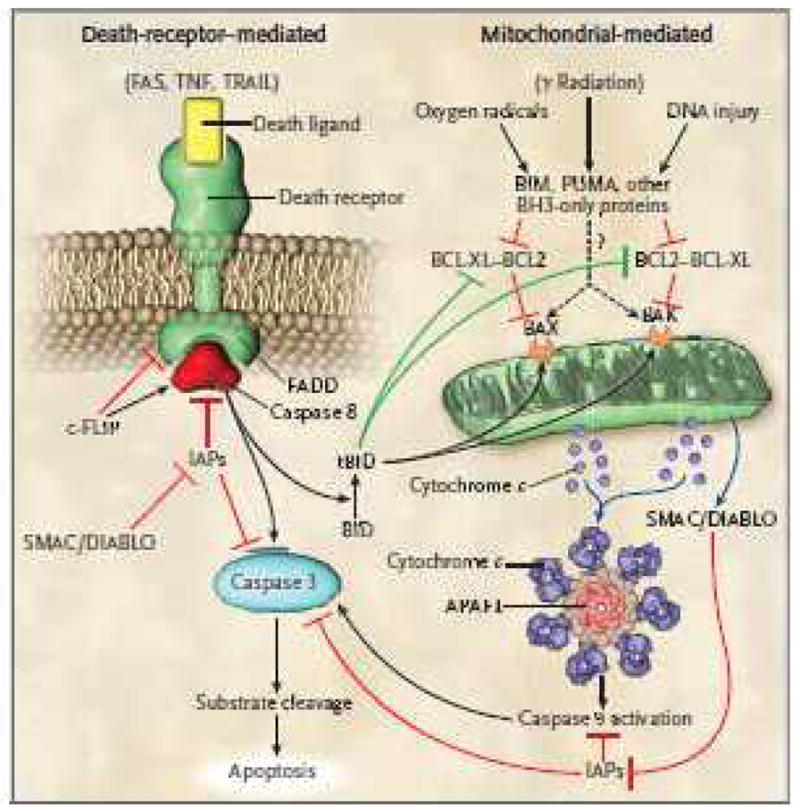
There are two major pathways of apoptosis, i.e, the death receptor pathway which is mediated by activation of death receptors and the Bcl-2 regulated mitochondrial pathway which is mediated by noxious stimuli that ultimately lead to mitochondrial injury. Ligation of death receptors recruits the adaptor protein FADD (Fas associated death domain) via its death domain. FADD in turn recruits caspase-8 which ultimately activates caspase-3, the key ‘executioner’ caspase. c-FLIP can either inhibit or potentiate binding of FADD and caspase-8 depending upon its concentration. In the intrinsic pathway, pro-apoptotic BH3 proteins are activated by noxious stimuli which interact with and inhibit anti-apoptotic Bcl-2 or Bcl-xL. Thus, Bax and Bax are free to induce mitochondrial permeabilization with release of cytochrome-C which ultimately results in activation of caspase-9 (via the apoptosome). Caspase-9 then activates caspase-3. SMAC/DIABLO is also released following mitochondrial permeabilization and acts to block the action of IAPs (inhibitors of apoptosis protein) which inhibit caspase activation. Note that there is potential cross-talk between the 2 pathways which is mediated by tBid that is produced by caspase-8 mediated Bid cleavage. tBid acts to inhibit Bcl-2/Bcl-xL and to activate Bax and Bak. There is debate about whether pro-apoptotic BH3 molecules Bim, PUMA, etc. can act directly on Bax and Bak (indicated by the question mark) to induce mitochondrial permeability or whether they only act on Bcl-2/Bcl-xL.
Interplay between pro- and anti-apoptotic members of the Bcl-2 family controls the mitochondrial apoptotic pathway (supplemental Table-1). Caspase-9 regulates this pathway, which comes into play after intracellular sensors indicate overwhelming cell damage.5,23,34 Increased intracellular reactive oxygen species, DNA damage, the unfolded protein response, and deprivation of growth factors trigger the mitochondrial pathway. These initiating events increase mitochondrial permeability, promoting release of pro-apoptotic proteins (-including cytochrome C and SMAC/DIABLO), from the inter-mitochondrial membrane space into the cytosol (Fig. 4).35–38 SMAC/DIABLO antagonizes cytosolic inhibitors of apoptosis protein (IAPs), thereby allowing activation of caspases and apoptosis to proceed. Activated caspase-8 (death receptor pathway) and caspase-9 (mitochondrial pathway) in turn mobilize caspases-3, -6 and -7, proteases that cleave numerous proteins and activate DNAses heralding cellular demolition. 23,38
Several factors determine which death pathway(s) is (are) activated including the cell cycle stage, the type and magnitude of the apoptotic stimulus and, for immune cells, the stage of cellular activation.23,31,34 In certain diseases such as sepsis, blocking the death receptor or the mitochondrial pathway decreases cell death in a percentage of the cells in the tissue, whereas blocking both pathways protects a larger number of cells. Multiple pathologic stimuli triggering different apoptotic pathways may thus occur concomitantly.30
The Bcl-2 Protein Family
The interplay between pro- and anti-apoptotic Bcl-2 protein family members controls the mitochondrial apoptotic pathway (supplemental Table-1).2,23,24 Bcl-2, originally identified as the gene deregulated by the t(14;18) chromosomal translocation in follicular center B-cell lymphomas, is an inhibitor of apoptosis.5 Bcl-2 is normally present in those cells that routinely undergo apoptotic turnover, i.e., hematopoietic lineages, intestinal epithelial cells, as well as glandular epithelium in which hormones regulate hyperplasia or involution.39 Inclusion in the Bcl-2 family requires at least one conserved Bcl-2 homology (BH) domain in the candidate protein. The pro-survival members – Bcl-2, Bcl-xL, Bcl-w, Mcl-1, A1, and Boo/Diva – share up to 4 BH regions; these proteins are essential for cell survival, functioning in a cell-type and stimulus-specific manner – (see supplemental Table-1).40–43 There are two groups of pro-apoptotic Bcl-2 family proteins, which differ in function and the number of BH domains they possess. Bax and Bak, which have three BH domains, are critical for the mitochondrial permeabilization and release of cytochrome-C that lead to caspase-9 activation. Members of the other pro-apoptotic group have only the “BH3” domain.44,45 The “BH3-only” proteins bind to and inhibit the anti-apoptotic Bcl-2 family members, liberating the pro-apoptotic Bax and Bak to induce loss of mitochondrial membrane permeability and subsequent cell death.44–46
The balance between the pro-apoptotic BH3-only proteins anti-apoptotic Bcl-2 family members is the critical determinant of the life or death decision of a cell.34,47 BH3-only proteins differ in their ability to trigger apoptosis.47,48 Three of these –(Bim, PUMA, and Bid)- bind with high affinity to all pro-survival Bcl-2 family members. Moreover, different apoptotic stimuli preferentially activate distinct BH3-only proteins; Bim is essential for apoptosis induced by deprivation of growth factors, while Puma is critical for apoptosis induced by DNA damage.49–51 The concordant loss of Bim and PUMA is more protective against apoptotic stimuli than loss of either alone, revealing functional overlap between these apoptosis initiators.52
Diseases Associated with Abnormally Decreased Apoptosis
Over 50% of neoplasms have defects in apoptotic machinery. Among the best characterized of these are increased expression of pro-survival Bcl-2 family proteins and mutations in the tumor suppressor gene TP53.53–55 TP53, often described as “guardian of the genome” normally functions to initiate apoptosis in response to DNA damage (induced by radiation, chemical agents, oxidative stress, etc.) by transcriptional induction of many pro-apoptotic proteins including PUMA, Noxa, and Bax. Inherited defects in TP53 (Li-Fraumeni syndrome) result in numerous neoplasia including gliomas and sarcomas.55 Most chemotherapeutic agents induce apoptosis in tumor cells (Table-1). The tyrosine kinase inhibitor imatinib (Gleevec) kills chronic myelogenous leukemia cells by up-regulating the pro-apoptotic Bcl-2 family members Bim and Bad.56 ABT-737, a small molecule mimic of BH3-only proteins that binds to anti-apoptotic Bcl-2 and Bcl-xL is able to kill certain tumor cells on its own and greatly enhances the efficacy of other anti-cancer drugs.57,58 An orally available analog of ABT-737 (ABT-263) has entered clinical trials as a single agent in hematologic malignancies and as an adjuvant therapy in lung and solid organ tumors.58 Other pro-apoptotic chemotherapeutic agents that are in multiple clinical cancer trials target survivin and XIAP, endogenous inhibitors of apoptosis (IAPs) that block key executioner caspases.59
Table 1.
Pharmacologic Modulators of Cell Death in Clinical Trials.*
| Disease | Compound | Mechanism of Action |
|---|---|---|
| Cancer | ||
| Leukemia, multiple myeloma, non-Hodgkin’s lymphoma, lung, solid-organ | ABT-263, gossypol, GX15-070 (obatoclax) | Induces apoptosis by inhibiting antiapoptotic BCL2 family members |
| Colorectal, non–small-cell lung, non-Hodgkin’s lymphoma | Recombinant human Apo2L/TRAIL (dulanermin) and anti-TRAIL R1-mAb | Induces apoptosis by activation of TRAIL death receptors |
| Breast, pancreatic, ovarian, solid-organ, glioma | PARP inhibitors | Prevents repair of DNA strand breaks leading to apoptosis |
| Multiple myeloma, breast, prostate | Hydroxychloroquine | Inhibits autophagy |
| Ovarian, small-cell lung, cervical | Topotecan (Hycantin) | Induces apoptosis by inhibiting topoisomerase I, an enzyme essential for DNA replication |
| Breast, renal, rectal, large-B-cell lymphoma | Temsirolimus and sirolimus | Inhibits mTOR, resulting in autophagy; other actions |
| Non–small-cell lung, pancreas, breast | XIAP antisense (AEG35156) | Induces apoptosis by knockdown of endogenous caspase inhibitors |
| Acute myeloid leukemia | Survivin antagonist | Induces apoptosis by inhibiting survivin, an endogenous caspase inhibitor |
| Ischemia–reperfusion injury | ||
| Stroke | Minocycline | Reduces apoptosis |
| Myocardial infarction | Mitochondrial ATP-sensitive potassium-channel agonist (nicorandil) | Reduces necrosis by preventing cell ionic disequilibrium; may also reduce apoptosis by acting on MPTP |
| PARP inhibitors | Reduces necrosis by prevention of cell energy failure | |
| Cyclosporine | Reduces apoptosis by blocking opening of MPTP | |
| Neurodegenerative disease | ||
| Amyotrophic lateral sclerosis | Arimoclomol | Reduces apoptosis, improves elimination of misfolded proteins by heat-shock protein chaperone–mediated disposal |
| Huntington’s disease and Alzheimer’s disease | Ursodiol | Reduces apoptosis and oxidation; other effects |
| Parkinson’s disease and Alzheimer’s disease | Rasagiline | Reduces apoptosis and other effects |
| Other | ||
| Hepatitis C | Caspase inhibitors IDN-6556, GS-9450 | Reduces apoptosis by blocking caspases |
Details regarding the status of all clinical trials are available in Table 1 in the Supplementary Appendix and at ClinicalTrials.gov. The majority of drugs that are used in cancer trials are administered in combination with chemotherapeutic drugs, which induce apoptosis. MPTP denotes mitochondrial permeability transition pore, mTOR mammalian target of rapamycin (now known as sirolimus), PARP poly–ADP–ribose polymerase, and XIAP X-linked inhibitor of apoptosis.
Abnormalities in apoptosis can cause autoimmune disease.60 During development, B- and T- lymphocyte clones that express auto-reactive antigen receptors are deleted from the immune repertoire. This “negative selection” of self-reactive lymphocytes relies on the pro-apoptotic BH3-only protein Bim.61 Moreover, killing of mature, antigen-activated T and B lymphocytes during shutdown of immune responses is mediated by both Bim and the death receptor Fas.30 Patients with defective Fas ligand or Fas receptor develop autoimmune lymphoproliferative syndrome (ALPS) typified by massive lymphadenopathy, hypersplenism, and autoimmune cytopenias.60 Apoptosis of intestinal epithelial cells and basal keratinocytes in graft-versus-host-disease is a functionally related phenomenon. In type I diabetes, loss of beta cells is thought to be Fas death receptor-mediated; Fas ligand expressing CD8 T cells interact with Fas receptors on the insulin secreting cells to induce death.62
Disease States Related to Abnormally Increased Apoptosis
There is growing evidence that neuronal apoptosis plays a key pathogenic role in neonatal brain disorders.63 Developing neurons are particularly susceptible to apoptosis in response to many noxious stimuli during the period of synaptogenesis.64 In neonatal hypoxic brain injury, the cell death phenotype changes over time from early necrosis to apoptosis. This evolution has been termed the “necrosis-apoptosis” continuum. Evidence suggests that apoptosis is a more important mechanism of neonatal brain injury than necrosis.63 The fetal-alcohol-syndrome is due to apoptogenic neurodegeneration that results from ethanol-induced NMDA receptor blockade and GABA receptor activation.65 General anesthetics also modulate NMDA and GABA receptors and animal studies showing that general anesthetics induce extensive neuronal apoptosis in neonates have raised considerable concern that clinical use of general anesthetics in neonates might cause long-term neurocognitive defects (A&A 106: 1659 2008, A&A 108:90 2009).66
Hepatocytes are particularly prone to apoptosis in response to various types of stress including infections.29 A trial of a potent caspase inhibitor (IDN-6556) in patients with chronic hepatitis-C showed a highly significant lowering or serum ALT and AST.67 IDN-6556 is also being clinically evaluated to reduce ischemia/reperfusion injury in patients following liver transplantation (Table-1).
Although necrosis predominates in ischemic injury, apoptotic cells are often found in the hypoxic penumbra, (so-called “watershed zone” as in myocardial infarction or stroke) or in globally hypoxic zones after reperfusion. Importantly, in those situations in which apoptosis is precipitated by hypoxia-induced premature activation of the apoptotic program, cell loss might be prevented if apoptosis is blocked (e.g. by caspase inhibitors) and if adequate cellular oxygenation is maintained/restored. Cyclosporine, which inhibits apoptosis by blocking mitochondrial permeability-transition pores, decreased infarct size in patients with acute myocardial infarction.68 Acute stroke patients treated with minocycline, an anti-apoptotic compound with multiple actions, had superior neurologic outcomes.69 Sepsis is perhaps the most remarkable clinical setting for apoptosis. Septic patients develop massive apoptosis of immune effector cells and gastrointestinal epithelial cells (Figs.2,3 & Supplement 1–4).70–72 The profound loss of immune effector cells in patients with sepsis inhibits their ability to eradicate the primary infection and renders them more susceptible to nosocomial infections. The key role of apoptosis in sepsis is illustrated by numerous animal studies showing that prevention of sepsis-induced apoptosis improves survival.73,74
Autophagy
“Autophagy” [derived from the Greek - to eat (“phagy”) oneself (“auto”)] was first applied to observations of novel single- or double-membrane lysosomal-derived vesicles that contained parts of cytoplasm including organelles in various stages of disintegration by electron microscopy (Fig. 5).75 Autophagy is the process by which cells recycle their own non-essential, redundant, or damaged organelles and macro-molecular components.12–14 It is an adaptive response to sub-lethal stress, such as nutrient deprivation, to supply the cell with metabolites for fuel. However, autophagy is also involved in tumor suppression (see discussion on autophagy in cancer - below), deletion of toxic misfolded proteins, elimination of intracellular microorganisms, and antigen presentation.12–14 Three forms of autophagy (based on how material is delivered to the lysosomes for degradation) have been defined.12 In macro-autophagy, cargo is sequestered within a double-membrane structure, (“autophagosome”), which then fuses with lysosomes. In micro-autophagy, cargo is engulfed by invagination of the lysosomal membrane. In chaperone-mediated autophagy, substrates are delivered to lysosomes by heat shock cognate proteins.
Figure 5. Electron microscopic detection of autophagic and necrotic cell death.

5A. Two large autophagosomes are present and identified by arrows. The autophagosome on the lower right (double arrow) contains mitochondria and other organelles in varying stages of degradation. The autophagosome in the left of the image encompasses organelle fragments with more extensive degradation (uranyl acetate/lead citrate; x40,000). Sample obtained from an 85 y.o. female with peritonitis. 5B. This cell also demonstrates extensive autophagic vacuolization with few remaining intact organelles. The arrow identifies an autophagosome containing mitochondrial fragments. Note that the cell nucleus (identified by lower arrow) has features of nuclear condensation and that it is unlikely that this cell would have remained viable - an example of ‘autophagy-associated cell death’ (uranyl acetate/lead citrate; x10,000). Sample obtained from a 73 y.o. female with urosepsis. 5C. A cell in which the autophagosomes have assumed a more complex appearance of with redundant whorls of membrane derived material. This complex lysosomal structure is juxtaposed to and focally invaginates into an adjacent mitochondrion (uranyl acetate/lead citrate;x30,000). Specimen obtained from a 73 y.o. female with urosepsis. 5D. Image of a necrotic proximal renal tubular from a 44 y.o. male who presented with acute renal failure. The patient had a history of vancomycin toxicity and cirrhosis. The cells show marked organellar and cytoplasmic swelling, loss of brush border and loss of cytoplasmic detail. (uranyl acetate/lead citrate; x5000). Figures 5A, 5B, and 5C are reproduced with permission from Laboratory Investigation (Feb 2. 2009 Epub ahead of print – Watanabe E, et al. Sepsis induces extensive vacuolization in hepatocytes: a clinical and laboratory based study.)
Autophagosomes are the hallmark of autophagy and they are best visualized by electron microscopy (Fig. 5). Formation of autophagosomes is regulated by a complex set of autophagy-related (atg) proteins (supplement Fig. 6).12,14 Once formed, autophagosomes fuse with lysosomes where acid hydrolases catabolize the ingested material into metabolic substrates. The membrane remnants in autophagic vacuoles often assume a “whorl-like” appearance due to residual membrane components (Fig. 5).75,76 Autophagosome formation is initiated by Class III phosphatidylinositol-3-kinase (PI-3K) and atg-6 (also known as Beclin-1). Additional control of autophagy is exerted by the mammalian target of rapamycin (mToR), a serine/threonine protein kinase that integrates input from multiple factors, e.g., cellular nutrients, growth factors, and cellular redox state to inhibit autophagosome formation (supplement Fig. 6).
As previously noted, there is controversy regarding the role of autophagy in cell death.77 Although it is accepted that autophagy is generally an adaptive response, only some investigators believe that unbridled autophagy can result in cell death by depleting the cell of organelles and critical proteins resulting in a caspase-independent cell death. In this view, cells that exhibit massive numbers of cytoplasmic autophagic vacuoles but lack signs of apoptosis are undergoing autophagic cell death (see Fig. 5C). However, while studies do demonstrate significant increases in the number of autophagosomes in some dying cells, it is unclear whether these autophagosomes facilitate cell death or represent a cell that can no longer compensate by sacrificing vital components. This latter process of cell death has been referred to as ‘autophagy-associated cell death’ rather than ‘autophagy-induced’ cell death. Importantly, genetic deletion of key autophagic genes accelerates rather than inhibits cell death, emphasizing the predominant survival role of autophagy.3,11
Autophagy in Health and Disease
Autophagy is a survival mechanism that is rapidly induced during cellular stress and provides alternative sources of substrates when nutrients are limited.12,15,77 Theoretically, autophagy also protects against cell death by eliminating damaged mitochondria (which may trigger apoptosis by generating excess reactive oxygen species) or toxic misfolded proteins, including those believed to induce neurodegeneration. Drugs that activate autophagy can clear toxic protein aggregates including mutant Huntingtin protein and mutant Tau in models of neurologic disease.78–80 Rapamycin analogs (which induce autophagy by inhibiting mToR) decrease polyglutamine proteins and have been effective in animal models of Huntington’s disease and are also being examined in acute brain injury.78–80
Autophagy plays a complex role in cancer.81–83 Although currently data are only associative, autophagy presumably functions as a suppressor of neoplasia. Many oncogenes (including PI3k/Akt, Bcl-2, and mToR) suppress autophagy while tumor suppressors e.g., PTEN, TSC2, and HIF1α, generally promote autophagy.83 In addition, loss of individual autophagy-related genes (especially beclin-1, UVRAG and Bif-1) results in lymphomas and gastrointestinal tumors in mouse models and these same genes are frequently mutated in human cancers including bowel and hepatocellular carcinomas.84–86 It is thus seemingly paradoxical that hydroxychloroquine, an anti-malarial drug that blocks autophagy by raising intralysosomal pH, is under evaluation in multiple cancer trials (Table-1).81 However, in the setting of chemotherapy, autophagy can promote resistance to cell death, especially to DNA-damaging agents, and hydroxychloroquine blocks this cellular adaptive response thereby resulting in increased tumor killing..
Necrosis
Necrosis (Greek “nekros” for corpse) is best defined by light or electron microscopic detection of cell and organelle swelling or surface membrane rupture with spillage of intracellular contents (Fig. 3,5, Supplement Fig. 6).4,29,87 ‘Oncosis’ (Greek for swelling) is preferred by some investigators and ‘oncotic necrosis’ has also been used.4 Necrosis usually results from metabolic failure, coincident with rapid depletion of ATP; it classically occurs with tissue ischemia.26,88 In contrast to apoptosis, necrosis is characterized by early loss of plasma membrane and organelle integrity, probably related to osmotic forces resulting from opening of non-specific, glycine-inhibitable anion channels.25 Because organellar membranes are compromised, degradative enzymes escape from lysosomes and enter the cytosol causing cell demolition.17,20,87–92
Necrosis is usually considered an ‘accidental’ (i.e., non-programmed) cell death that occurs in response to acute hypoxic/ischemic injury such as myocardial infarction or stroke. Necrosis occurs spontaneously in neoplasms when cell proliferation outpaces neoangiogenesis. Exposure of cells to supra-physiologic conditions, e.g., mechanical force, heat, cold, or membrane-permeabilizing toxins, also precipitates necrosis. Necrosis exhibits varied morphologic patterns in affected tissues (so-called ‘liquefactive,’ ‘coagulative,’ and ‘caseating’) dependant upon the underlying etiology and temporal sequence of cell death.
Mediators of Necrosis
Reactive oxygen species (ROS), Ca2+, poly-ADP-ribose-polymerase (PARP), calcium-activated non-lysosomal proteases (calpains), and cathepsins mediate necrosis.89,92 PARP is a DNA repair enzyme that may deplete cellular ATP stores in repairing the multiple DNA strand breaks that occur with cell injury. In apoptosis, PARP is rapidly cleaved and inactivated (detection of cleaved PARP is a diagnostic test for apoptosis) and therefore ATP stores are preserved. ATP is necessary for numerous effector processes in apoptosis while exhaustion of ATP shifts the cell from apoptosis toward a necrotic cell death. PARP inhibition mitigates necrosis in ischemia-reperfusion injury and other insults93,94 Increased intracellular Ca2+, a central feature of necrosis, activates proteases that degrade critical proteins. Intriguingly, the source and amount of increased Ca2+ may induce different types of cell death: Ca2+ influx across the plasma membrane triggers necrosis whereas Ca2+ released from endoplasmic reticulum more readily induces apoptosis.17,95
Necrosis – programmed and/or regulated?
Accumulating evidence indicates that necrosis is more ordered than originally thought. When cells die by necrosis, damage-associated-molecular-pattern molecules (DAMPs), such as HMGB-1, enter the circulation and activate innate immune cells.96 Thus, the first cells dying in trauma or infection may function as sentinels, alerting the host to the need for defensive/reparative responses. Additionally, necrosis can be initiated by activation of selected cell surface receptors. For example, high concentrations of TNF induce hepatocyte necrosis.21,26 The identification of an intracellular serpin that prevents necrosis caused by multiple noxious stimuli indicates that necrosis can be regulated, programmed, and driven by a peptidase stress-response pathway.20
Additional Forms of Cell Death
Other less well-characterized forms of cell death including mitotic catastrophe,97 pyroptosis,98 paraptosis,99 entosis,100 and anoikis101 may represent distinct modes of cell death or nuanced variations of apoptosis, necrosis, or autophagy. Because of space limitations, they are discussed in supplemental materials.
Crosstalk Between Cell Death Mechanisms
The type and intensity of noxious signals, ATP concentration, cell type, and other factors determine how cell death occurs.11 For example, acute myocardial ischemia (which precipitates rapid and profound decreases in ATP) induces necrosis (Fig. 3) while chronic congestive heart failure (with more modest yet chronic decreases in ATP) induces apoptosis.102 Blocking a particular pathway of cell death may not prevent cell death but may instead recruit an alternative path.103,104 Consequently, anti-apoptotic caspase inhibitors cause hyperacute TNF–α induced necrosis of hepatocytes and kidney tubular cells.105 Over-expression of anti-apoptotic proteins may allow injured cells to survive and autophagy may assist by providing critical metabolites.15 However, if death stimuli persist, anti-apoptotic pathways and autophagy are unlikely to support indefinite cell survival and necrosis may ensue. Furthermore, cells may be more susceptible to apoptosis if autophagy is inhibited.106,107 NF-kB, Atg 5, ATP, and PARP putatively function as molecular switches that determine whether a cell undergoes apoptosis, necrosis, or autophagy.26,88,93,108–110 p53, a key regulator of DNA damage-induced apoptosis, also modulates autophagy and other responses to cell stress. Recent work indicates that basal p53 activity suppresses autophagy whereas activation of p53 by certain stimuli induces autophagy and activation of p53 by different stimuli results in the Puma/Noxa-mediated engagement of apoptosis.111–113
It is not only the internal state of the individual cell but also signals from a cell’s community that affects cell fate. Some of the most important survival signals come from contact with neighboring cells or the extracellular matrix or via paracrine signals.101 Even systemic administration of apoptotic or necrotic cells has been found to alter the extent of cell death due to certain noxious stimuli.114
Immunomodulatory Effects of Dying Cells
The impact of dying cells on immunity is a particularly exciting area of investigation.96,115,116 Apoptotic cells induce anergy or an immunosuppressive phenotype whereas necrotic cells augment inflammation in part by binding the receptor CLEC9A on dendritic cells.117 Severe infections can induce extensive apoptotic and necrotic cell death. Accordingly, administration of apoptotic cells to mice prior to parasite challenge greatly increased eventual blood parasite numbers compared to controls, while mice receiving necrotic cells had greatly decreased parasitemia.118 Similarly, mice that received apoptotic cells prior to peritonitis had a significantly worse mortality compared to control mice, whereas mice that received necrotic cells prior to peritonitis had improved survival.119 These important data demonstrate the effect of the type of cell death on host defenses and may lead to methods to therapeutically modulate host immunity.
Future Directions and Therapeutic Implications
Initial attempts at therapeutic modulation of cell death have yielded some surprising and paradoxical findings.2,6–8,16 Ironically, some ‘pro-death’ cellular proteins are also essential for cell survival. Although caspase-8 and its activator FADD are essential for death receptor-mediated apoptosis, they are also critical for antigen receptor activation-induced T-cell proliferation and macrophage differentiation.27,32,120 Cytolocalization of caspase-8 may determine which substrates are processed.120 Thus, prudence is justified to avoid unexpected adverse outcomes of planned therapies. Additionally, blockade of multiple death pathways may keep susceptible cells alive but survivors may be clonogenically and functionally dead and therefore useless (“zombie cells”).121,122 Finally, therapy to prevent cell death may need to be of limited duration to prevent abnormal survival of pre-neoplastic cells leading to cancer.
Induction of cell death
Many examples of cell death-modulating therapies have already been presented and a more complete listing is provided in Table-1. A highly promising area of cancer therapy today involves death receptor activation. Unlike normal cells, many cancer cells are sensitive to TRAIL and clinical trials employing the physiological ligand TRAIL or antibodies to TRAIL receptor are underway in colorectal cancer, non-small-cell lung cancer, and non-Hodgkin’s lymphoma.123 As discussed earlier, pro-apoptotic BH3-mimetics are being tested in leukemia, multiple myeloma, and other malignancies.57,58 Drugs that block endogenous inhibitors of apoptosis (IAPs), e.g., XIAP and survivin, have been employed in clinical trials of leukemia as well as pancreatic, pulmonary and other parenchymal malignancies.124,125 PARP inhibitors are also in multiple clinical trials; these peptides dramatically sensitize cancer cells to chemotherapy by preventing DNA repair.126
Prevention of cell death
Prevention of cell death is more technically challenging than cell death induction. Multiple forms of cell death may occur simultaneously due to coordinate release of multiple death-inducing stimuli. In ischemia/reperfusion injury, ROS, Ca2+ overload, and destructive protease activation may induce cell death independently. Thus, it may be necessary to target multiple death pathways or identify and block common ‘funnel’ points of cell death signaling to enable survival. Despite these challenges, meaningful clinical advances are emerging (Table-1). Inhibition of calpains and cathepsins, potent proteases responsible for necrosis, mitigates disease progression in animal models.127,128 Cyclosporin, which preserves mitochondrial membrane potential, reduces myocardial infarct size.68 Nicorandil, a cardioprotective drug that acts on mitochondrial ATP-sensitive potassium channels, lowers serum troponin-T in cardiac bypass surgery patients.129 Huntington’s, Parkinson’s, Alzheimer’s, and amyotrophic lateral sclerosis are current investigational targets of anti-apoptotic and autophagy-enhancing drugs (Table-1).130,131 Therapeutic induction of metabolic arrest may also prove valuable. Although the means by which cells enter a ‘hibernating’ state remain unknown, important mediators of this low-energy state are now better understood; 5′-AMP is one such mediator that allows non-hibernating animals to safely enter a hypothermic condition.121
In conclusion, our evolving appreciation of cell death pathways has led to development of promising novel therapies. Despite these advances, answers to a number of intriguing questions remain. Why are some cells, such as neurons, much more vulnerable to ischemic cell death than most others? How does a cell select a particular type of death? How does a cell switch from a stress recovery program to cell death? What criteria drive the selection of cell death pathway? When is a cell irrevocably committed to death? Answers to these and other questions will ultimately lead to a more profound understanding of cell death, an expanding foundation on which increasingly effective therapeutic interventions may be modeled and introduced into clinical practice.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Dr. Eizo Watanabe who performed electron microscopy of patient liver samples and Dr. Helen Liapis who provided electron microscopic images of patient kidneys. The authors also acknowledge Dr. Kevin Tinsley who performed immunohistochemical staining of many tissue sections, and Drs. Craig Coopersmith, Timothy Buchman, and J. Perren Cobb who have provided many stimulating discussions and assisted in patient tissue sampling. The authors apologize to the many outstanding cell death investigators whose work was not cited due to space limitations. This work was supported by NIH grants GM44118, GM55194, the Leukemia and Lymphoma Society of America, National Health and Medical Research Council (Australia), and by the Alan A. and Edith L. Wolff Foundation.
References
- 1.Melino G. The Sirens’ song. Nature. 2001;412:23. doi: 10.1038/35083653. [DOI] [PubMed] [Google Scholar]
- 2.Adams JM, Cory S. Bcl-2-regulated apoptosis: mechanism and therapeutic potential. Curr Opin Immunol. 2007;19:488–96. doi: 10.1016/j.coi.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kroemer G, Galluzzi L, Vandenabeele P, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- 5.Marsden VS, Strasser A. Control of apoptosis in the immune system: Bcl-2, BH3-only proteins and more. Annu Rev Immunol. 2003;21:71–105. doi: 10.1146/annurev.immunol.21.120601.141029. [DOI] [PubMed] [Google Scholar]
- 6.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–62. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 7.Reed JC. Drug insight: cancer therapy strategies based on restoration of endogenous cell death mechanisms. Nat Clin Pract Oncol. 2006;3:388–98. doi: 10.1038/ncponc0538. [DOI] [PubMed] [Google Scholar]
- 8.Green DR, Kroemer G. Pharmacological manipulation of cell death: clinical applications in sight? J Clin Invest. 2005;115:2610–7. doi: 10.1172/JCI26321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller JB, Girgenrath M. The role of apoptosis in neuromuscular diseases and prospects for anti-apoptosis therapy. Trends Mol Med. 2006;12:279–86. doi: 10.1016/j.molmed.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 10.Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov. 2008;7:989–1000. doi: 10.1038/nrd2658. [DOI] [PubMed] [Google Scholar]
- 11.Galluzzi L, Maiuri MC, Vitale I, et al. Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 2007;14:1237–43. doi: 10.1038/sj.cdd.4402148. [DOI] [PubMed] [Google Scholar]
- 12.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–7. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 13.Kroemer G, Jaattela M. Lysosomes and autophagy in cell death control. Nat Rev Cancer. 2005;5:886–97. doi: 10.1038/nrc1738. [DOI] [PubMed] [Google Scholar]
- 14.Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767–77. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levine B, Abrams J. p53: The Janus of autophagy? Nat Cell Biol. 2008;10:637–9. doi: 10.1038/ncb0608-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bouchier-Hayes L, Lartigue L, Newmeyer DD. Mitochondria: pharmacological manipulation of cell death. J Clin Invest. 2005;115:2640–7. doi: 10.1172/JCI26274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zong WX, Thompson CB. Necrotic death as a cell fate. Genes Dev. 2006;20:1–15. doi: 10.1101/gad.1376506. [DOI] [PubMed] [Google Scholar]
- 18.Golstein P, Kroemer G. Cell death by necrosis: towards a molecular definition. Trends Biochem Sci. 2007;32:37–43. doi: 10.1016/j.tibs.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 19.Festjens N, Vanden Berghe T, Vandenabeele P. Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim Biophys Acta. 2006;1757:1371–87. doi: 10.1016/j.bbabio.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 20.Luke CJ, Pak SC, Askew YS, et al. An intracellular serpin regulates necrosis by inhibiting the induction and sequelae of lysosomal injury. Cell. 2007;130:1108–19. doi: 10.1016/j.cell.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laster SM, Wood JG, Gooding LR. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol. 1988;141:2629–34. [PubMed] [Google Scholar]
- 22.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. British Journal of Cancer. 1972;26:239–57. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strasser A. The role of BH3-only proteins in the immune system. Nat Rev Immunol. 2005;5:189–200. doi: 10.1038/nri1568. [DOI] [PubMed] [Google Scholar]
- 24.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 25.Nishimura Y, Lemasters JJ. Glycine blocks opening of a death channel in cultured hepatic sinusoidal endothelial cells during chemical hypoxia. Cell Death Differ. 2001;8:850–8. doi: 10.1038/sj.cdd.4400877. [DOI] [PubMed] [Google Scholar]
- 26.Malhi H, Gores GJ, Lemasters JJ. Apoptosis and necrosis in the liver: a tale of two deaths? Hepatology. 2006;43:S31–44. doi: 10.1002/hep.21062. [DOI] [PubMed] [Google Scholar]
- 27.Salmena L, Lemmers B, Hakem A, et al. Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 2003;17:883–95. doi: 10.1101/gad.1063703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sheridan C, Martin SJ. Commitment in apoptosis: slightly dead but mostly alive. Trends Cell Biol. 2008;18:353–7. doi: 10.1016/j.tcb.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 29.Lemasters JJ. Dying a thousand deaths: redundant pathways from different organelles to apoptosis and necrosis. Gastroenterology. 2005;129:351–60. doi: 10.1053/j.gastro.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 30.Hughes PD, Belz GT, Fortner KA, Budd RC, Strasser A, Bouillet P. Apoptosis regulators Fas and Bim cooperate in shutdown of chronic immune responses and prevention of autoimmunity. Immunity. 2008;28:197–205. doi: 10.1016/j.immuni.2007.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Green DR. Apoptotic pathways: ten minutes to dead. Cell. 2005;121:671–4. doi: 10.1016/j.cell.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 32.Newton K, Harris AW, Bath ML, Smith KG, Strasser A. A dominant interfering mutant of FADD/MORT1 enhances deletion of autoreactive thymocytes and inhibits proliferation of mature T lymphocytes. Embo J. 1998;17:706–18. doi: 10.1093/emboj/17.3.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu JW, Shi Y. FLIP and the death effector domain family. Oncogene. 2008;27:6216–27. doi: 10.1038/onc.2008.299. [DOI] [PubMed] [Google Scholar]
- 34.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–19. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 35.Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–89. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 36.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 37.Wu H, Tschopp J, Lin SC. Smac mimetics and TNFalpha: a dangerous liaison? Cell. 2007;131:655–8. doi: 10.1016/j.cell.2007.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goldstein JC, Munoz-Pinedo C, Ricci JE, et al. Cytochrome c is released in a single step during apoptosis. Cell Death Differ. 2005;12:453–62. doi: 10.1038/sj.cdd.4401596. [DOI] [PubMed] [Google Scholar]
- 39.Hockenbery DM, Zutter M, Hickey W, Nahm M, Korsmeyer SJ. BCL2 protein is topographically restricted in tissues characterized by apoptotic cell death. Proc Natl Acad Sci U S A. 1991;88:6961–5. doi: 10.1073/pnas.88.16.6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yin XM, Wang K, Gross A, et al. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature. 1999;400:886–91. doi: 10.1038/23730. [DOI] [PubMed] [Google Scholar]
- 41.McKenzie MD, Carrington EM, Kaufmann T, et al. Proapoptotic BH3-only protein Bid is essential for death receptor-induced apoptosis of pancreatic beta-cells. Diabetes. 2008;57:1284–92. doi: 10.2337/db07-1692. [DOI] [PubMed] [Google Scholar]
- 42.Kaufmann T, Tai L, Ekert PG, et al. The BH3-only protein bid is dispensable for DNA damage- and replicative stress-induced apoptosis or cell-cycle arrest. Cell. 2007;129:423–33. doi: 10.1016/j.cell.2007.03.017. [DOI] [PubMed] [Google Scholar]
- 43.Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 2001;15:1481–6. doi: 10.1101/gad.897601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim H, Rafiuddin-Shah M, Tu HC, et al. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8:1348–58. doi: 10.1038/ncb1499. [DOI] [PubMed] [Google Scholar]
- 45.Kuwana T, Bouchier-Hayes L, Chipuk JE, et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17:525–35. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 46.Willis SN, Fletcher JI, Kaufmann T, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–9. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- 47.Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18:157–64. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen L, Willis SN, Wei A, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 49.Bouillet P, Metcalf D, Huang DC, et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–8. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- 50.Villunger A, Michalak EM, Coultas L, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–8. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 51.Jeffers JR, Parganas E, Lee Y, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–8. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- 52.Erlacher M, Michalak EM, Kelly PN, et al. BH3-only proteins Puma and Bim are rate-limiting for {gamma}-radiation- and glucocorticoid-induced apoptosis of lymphoid cells in vivo. Blood. 2005;106:4131–8. doi: 10.1182/blood-2005-04-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–2. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 54.Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–3. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- 55.Vazquez A, Bond EE, Levine AJ, Bond GL. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov. 2008;7:979–87. doi: 10.1038/nrd2656. [DOI] [PubMed] [Google Scholar]
- 56.Kuroda J, Puthalakath H, Cragg MS, et al. Bim and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc Natl Acad Sci U S A. 2006;103:14907–12. doi: 10.1073/pnas.0606176103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov. 2008;7:989–1000. doi: 10.1038/nrd2658. [DOI] [PubMed] [Google Scholar]
- 58.Cragg MS, Harris C, Strasser A, Scott CL. Unleashing the power of inhibitors of oncogenic kinases through BH3 mimetics. Nat Rev Cancer. 2009;9:321–6. doi: 10.1038/nrc2615. [DOI] [PubMed] [Google Scholar]
- 59.Tolcher AW, Mita A, Lewis LD, et al. Phase I and pharmacokinetic study of YM155, a small-molecule inhibitor of survivin. J Clin Oncol. 2008;26:5198–203. doi: 10.1200/JCO.2008.17.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Oliveira JB, Gupta S. Disorders of apoptosis: mechanisms for autoimmunity in primary immunodeficiency diseases. J Clin Immunol. 2008;28 (Suppl 1):S20–8. doi: 10.1007/s10875-007-9161-4. [DOI] [PubMed] [Google Scholar]
- 61.Bouillet P, Purton JF, Godfrey DI, et al. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature. 2002;415:922–6. doi: 10.1038/415922a. [DOI] [PubMed] [Google Scholar]
- 62.Foulis AK. Pancreatic pathology in type 1 diabetes in human. Novartis Found Symp. 2008;292:2–13. discussion 13–8, 122–9, 202–3. [PubMed] [Google Scholar]
- 63.Ferriero DM. Neonatal brain injury. N Engl J Med. 2004;351:1985–95. doi: 10.1056/NEJMra041996. [DOI] [PubMed] [Google Scholar]
- 64.Barinaga M. Neurobiology. A new clue to how alcohol damages brains. Science. 2000;287:947–8. doi: 10.1126/science.287.5455.947. [DOI] [PubMed] [Google Scholar]
- 65.Ikonomidou C, Bittigau P, Ishimaru MJ, et al. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–60. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- 66.Loepke AW, Istaphanous GK, McAuliffe JJ, 3rd, et al. The effects of neonatal isoflurane exposure in mice on brain cell viability, adult behavior, learning, and memory. Anesth Analg. 2009;108:90–104. doi: 10.1213/ane.0b013e31818cdb29. [DOI] [PubMed] [Google Scholar]
- 67.Pockros PJ, Schiff ER, Shiffman ML, et al. Oral IDN-6556, an antiapoptotic caspase inhibitor, may lower aminotransferase activity in patients with chronic hepatitis C. Hepatology. 2007;46:324–9. doi: 10.1002/hep.21664. [DOI] [PubMed] [Google Scholar]
- 68.Piot C, Croisille P, Staat P, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359:473–81. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- 69.Lampl Y, Boaz M, Gilad R, et al. Minocycline treatment in acute stroke: an open-label, evaluator-blinded study. Neurology. 2007;69:1404–10. doi: 10.1212/01.wnl.0000277487.04281.db. [DOI] [PubMed] [Google Scholar]
- 70.Hotchkiss RS, Swanson PE, Freeman BD, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Critical Care Medicine. 1999;27:1230–51. doi: 10.1097/00003246-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 71.Hotchkiss RS, Tinsley KW, Swanson PE, et al. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. Journal of Immunology. 2001;166:6952–63. doi: 10.4049/jimmunol.166.11.6952. [DOI] [PubMed] [Google Scholar]
- 72.Hotchkiss RS, Schmieg RE, Jr, Swanson PE, et al. Rapid onset of intestinal epithelial and lymphocyte apoptotic cell death in patients with trauma and shock. [see comments] Crit Care Med. 2000;28:3207–17. doi: 10.1097/00003246-200009000-00016. [DOI] [PubMed] [Google Scholar]
- 73.Hotchkiss RS, Nicholson DW. Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol. 2006;6:813–22. doi: 10.1038/nri1943. [DOI] [PubMed] [Google Scholar]
- 74.Ayala A, Perl M, Venet F, Lomas-Neira J, Swan R, Chung CS. Apoptosis in sepsis: mechanisms, clinical impact and potential therapeutic targets. Curr Pharm Des. 2008;14:1853–9. doi: 10.2174/138161208784980617. [DOI] [PubMed] [Google Scholar]
- 75.Ashford TP, Porter KR. Cytoplasmic components in hepatic cell lysosomes. J Cell Biol. 1962;12:198–202. doi: 10.1083/jcb.12.1.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Espert L, Denizot M, Grimaldi M, et al. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J Clin Invest. 2006;116:2161–72. doi: 10.1172/JCI26185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–88. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 79.Sarkar S, Rubinsztein DC. Small molecule enhancers of autophagy for neurodegenerative diseases. Mol Biosyst. 2008;4:895–901. doi: 10.1039/b804606a. [DOI] [PubMed] [Google Scholar]
- 80.Bossy B, Perkins G, Bossy-Wetzel E. Clearing the Brain’s Cobwebs: The Role of Autophagy in Neuroprotection. Curr Neuropharmacol. 2008;6:97–101. doi: 10.2174/157015908784533897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Amaravadi RK, Thompson CB. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin Cancer Res. 2007;13:7271–9. doi: 10.1158/1078-0432.CCR-07-1595. [DOI] [PubMed] [Google Scholar]
- 82.Marx J. Autophagy: is it cancer’s friend or foe? Science. 2006;312:1160–1. doi: 10.1126/science.312.5777.1160. [DOI] [PubMed] [Google Scholar]
- 83.Maiuri MC, Tasdemir E, Criollo A, et al. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ. 2009;16:87–93. doi: 10.1038/cdd.2008.131. [DOI] [PubMed] [Google Scholar]
- 84.Takahashi Y, Coppola D, Matsushita N, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9:1142–51. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ahn CH, Jeong EG, Lee JW, et al. Expression of beclin-1, an autophagy-related protein, in gastric and colorectal cancers. Apmis. 2007;115:1344–9. doi: 10.1111/j.1600-0463.2007.00858.x. [DOI] [PubMed] [Google Scholar]
- 86.Liang C, Feng P, Ku B, et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–99. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 87.Robbins, Cotran . Pathologic Basis of Disease: Saunders. 2005. Cellular Adaptation, Cell Injury, and Cell Death; pp. 4–46. [Google Scholar]
- 88.Leist M, Single B, Castoldi AF, Kuhnle S, Nicotera P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med. 1997;185:1481–6. doi: 10.1084/jem.185.8.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Conus S, Simon HU. Cathepsins: key modulators of cell death and inflammatory responses. Biochem Pharmacol. 2008;76:1374–82. doi: 10.1016/j.bcp.2008.07.041. [DOI] [PubMed] [Google Scholar]
- 90.Liu X, Van Vleet T, Schnellmann RG. The role of calpain in oncotic cell death. Annu Rev Pharmacol Toxicol. 2004;44:349–70. doi: 10.1146/annurev.pharmtox.44.101802.121804. [DOI] [PubMed] [Google Scholar]
- 91.Turk B, Stoka V. Protease signalling in cell death: caspases versus cysteine cathepsins. FEBS Lett. 2007;581:2761–7. doi: 10.1016/j.febslet.2007.05.038. [DOI] [PubMed] [Google Scholar]
- 92.Conus S, Perozzo R, Reinheckel T, et al. Caspase-8 is activated by cathepsin D initiating neutrophil apoptosis during the resolution of inflammation. J Exp Med. 2008;205:685–98. doi: 10.1084/jem.20072152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Los M, Mozoluk M, Ferrari D, et al. Activation and caspase-mediated inhibition of PARP: a molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol Biol Cell. 2002;13:978–88. doi: 10.1091/mbc.01-05-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jagtap P, Szabo C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005;4:421–40. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- 95.Vanlangenakker N, Berghe TV, Krysko DV, Festjens N, Vandenabeele P. Molecular mechanisms and pathophysiology of necrotic cell death. Curr Mol Med. 2008;8:207–20. doi: 10.2174/156652408784221306. [DOI] [PubMed] [Google Scholar]
- 96.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–42. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 97.Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death Differ. 2008;15:1153–62. doi: 10.1038/cdd.2008.47. [DOI] [PubMed] [Google Scholar]
- 98.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sperandio S, Poksay K, de Belle I, et al. Paraptosis: mediation by MAP kinases and inhibition by AIP-1/Alix. Cell Death Differ. 2004;11:1066–75. doi: 10.1038/sj.cdd.4401465. [DOI] [PubMed] [Google Scholar]
- 100.Overholtzer M, Mailleux AA, Mouneimne G, et al. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell. 2007;131:966–79. doi: 10.1016/j.cell.2007.10.040. [DOI] [PubMed] [Google Scholar]
- 101.Valentijn AJ, Zouq N, Gilmore AP. Anoikis. Biochem Soc Trans. 2004;32:421–5. doi: 10.1042/BST0320421. [DOI] [PubMed] [Google Scholar]
- 102.Schneider MD. Cyclophilin D: knocking on death’s door. Sci STKE. 2005;2005:pe26. doi: 10.1126/stke.2872005pe26. [DOI] [PubMed] [Google Scholar]
- 103.Vandenabeele P, Vanden Berghe T, Festjens N. Caspase inhibitors promote alternative cell death pathways. Sci STKE. 2006;2006:pe44. doi: 10.1126/stke.3582006pe44. [DOI] [PubMed] [Google Scholar]
- 104.Sato T, Machida T, Takahashi S, et al. Apoptosis supercedes necrosis in mitochondrial DNA-depleted Jurkat cells by cleavage of receptor-interacting protein and inhibition of lysosomal cathepsin. J Immunol. 2008;181:197–207. doi: 10.4049/jimmunol.181.1.197. [DOI] [PubMed] [Google Scholar]
- 105.Cauwels A, Janssen B, Waeytens A, Cuvelier C, Brouckaert P. Caspase inhibition causes hyperacute tumor necrosis factor-induced shock via oxidative stress and phospholipase A2. Nat Immunol. 2003;4:387–93. doi: 10.1038/ni914. [DOI] [PubMed] [Google Scholar]
- 106.Ravikumar B, Berger Z, Vacher C, O’Kane CJ, Rubinsztein DC. Rapamycin pre-treatment protects against apoptosis. Hum Mol Genet. 2006;15:1209–16. doi: 10.1093/hmg/ddl036. [DOI] [PubMed] [Google Scholar]
- 107.Boya P, Gonzalez-Polo RA, Casares N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–40. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: balancing life and death--a new approach to cancer therapy. J Clin Invest. 2005;115:2625–32. doi: 10.1172/JCI26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yousefi S, Perozzo R, Schmid I, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–32. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 110.Moubarak RS, Yuste VJ, Artus C, et al. Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol Cell Biol. 2007;27:4844–62. doi: 10.1128/MCB.02141-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tasdemir E, Maiuri MC, Galluzzi L, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676–87. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Maclean KH, Dorsey FC, Cleveland JL, Kastan MB. Targeting lysosomal degradation induces p53-dependent cell death and prevents cancer in mouse models of lymphomagenesis. J Clin Invest. 2008;118:79–88. doi: 10.1172/JCI33700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Murray-Zmijewski F, Slee EA, Lu X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat Rev Mol Cell Biol. 2008;9:702–12. doi: 10.1038/nrm2451. [DOI] [PubMed] [Google Scholar]
- 114.Schwulst SJ, Davis CG, Coopersmith CM, Hotchkiss RS. Adoptive transfer of dying cells causes bystander-induced apoptosis. Biochem Biophys Res Commun. 2007;353:780–5. doi: 10.1016/j.bbrc.2006.12.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Martinon F, Tschopp J. NLRs join TLRs as innate sensors of pathogens. Trends Immunol. 2005;26:447–54. doi: 10.1016/j.it.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 116.Johnson GB, Brunn GJ, Platt JL. Activation of mammalian Toll-like receptors by endogenous agonists. Crit Rev Immunol. 2003;23:15–44. doi: 10.1615/critrevimmunol.v23.i12.20. [DOI] [PubMed] [Google Scholar]
- 117.Sancho D, Joffre OP, Keller AM, et al. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature. 2009;458:899–903. doi: 10.1038/nature07750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Freire-de-Lima CG, Nascimento DO, Soares MB, et al. Uptake of apoptotic cells drives the growth of a pathogenic trypanosome in macrophages. Nature. 2000;403:199–203. doi: 10.1038/35003208. [DOI] [PubMed] [Google Scholar]
- 119.Hotchkiss RS, Chang KC, Grayson MH, et al. Adoptive transfer of apoptotic splenocytes worsens survival, whereas adoptive transfer of necrotic splenocytes improves survival in sepsis. Proc Natl Acad Sci U S A. 2003;100:6724–9. doi: 10.1073/pnas.1031788100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Koenig A, Russell JQ, Rodgers WA, Budd RC. Spatial differences in active caspase-8 defines its role in T-cell activation versus cell death. Cell Death Differ. 2008;15:1701–11. doi: 10.1038/cdd.2008.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lee CC. Is human hibernation possible? Annu Rev Med. 2008;59:177–86. doi: 10.1146/annurev.med.59.061506.110403. [DOI] [PubMed] [Google Scholar]
- 122.Narula J, Arbustini E, Chandrashekhar Y, Schwaiger M. Apoptosis and the systolic dysfunction in congestive heart failure. Story of apoptosis interruptus and zombie myocytes. Cardiol Clin. 2001;19:113–26. doi: 10.1016/s0733-8651(05)70198-3. [DOI] [PubMed] [Google Scholar]
- 123.Plummer R, Attard G, Pacey S, et al. Phase 1 and pharmacokinetic study of lexatumumab in patients with advanced cancers. Clin Cancer Res. 2007;13:6187–94. doi: 10.1158/1078-0432.CCR-07-0950. [DOI] [PubMed] [Google Scholar]
- 124.Hunter AM, LaCasse EC, Korneluk RG. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis. 2007;12:1543–68. doi: 10.1007/s10495-007-0087-3. [DOI] [PubMed] [Google Scholar]
- 125.LaCasse EC, Mahoney DJ, Cheung HH, Plenchette S, Baird S, Korneluk RG. IAP-targeted therapies for cancer. Oncogene. 2008;27:6252–75. doi: 10.1038/onc.2008.302. [DOI] [PubMed] [Google Scholar]
- 126.Lord CJ, Ashworth A. Targeted therapy for cancer using PARP inhibitors. Curr Opin Pharmacol. 2008;8:363–9. doi: 10.1016/j.coph.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 127.Jiang SX, Lertvorachon J, Hou ST, et al. Chlortetracycline and demeclocycline inhibit calpains and protect mouse neurons against glutamate toxicity and cerebral ischemia. J Biol Chem. 2005;280:33811–8. doi: 10.1074/jbc.M503113200. [DOI] [PubMed] [Google Scholar]
- 128.Koumura A, Nonaka Y, Hyakkoku K, et al. A novel calpain inhibitor, ((1S)-1((((1S)-1-benzyl-3-cyclopropylamino-2,3-di-oxopropyl)amino)carbonyl )-3-methylbutyl) carbamic acid 5-methoxy-3-oxapentyl ester, protects neuronal cells from cerebral ischemia-induced damage in mice. Neuroscience. 2008;157:309–18. doi: 10.1016/j.neuroscience.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 129.Yamamoto S, Yamada T, Kotake Y, Takeda J. Cardioprotective effects of nicorandil in patients undergoing on-pump coronary artery bypass surgery. J Cardiothorac Vasc Anesth. 2008;22:548–53. doi: 10.1053/j.jvca.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 130.Cudkowicz ME, Shefner JM, Simpson E, et al. Arimoclomol at dosages up to 300 mg/day is well tolerated and safe in amyotrophic lateral sclerosis. Muscle Nerve. 2008;38:837–44. doi: 10.1002/mus.21059. [DOI] [PubMed] [Google Scholar]
- 131.Weinreb O, Mandel S, Bar-Am O, et al. Multifunctional neuroprotective derivatives of rasagiline as anti-Alzheimer’s disease drugs. Neurotherapeutics. 2009;6:163–74. doi: 10.1016/j.nurt.2008.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chalmers AJ. The potential role and application of PARP inhibitors in cancer treatment. Br Med Bull. 2009;89:23–40. doi: 10.1093/bmb/ldp005. [DOI] [PubMed] [Google Scholar]
- 133.Teicher BA. Next generation topoisomerase I inhibitors: Rationale and biomarker strategies. Biochem Pharmacol. 2008;75:1262–71. doi: 10.1016/j.bcp.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 134.Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271–81. doi: 10.1056/NEJMoa066838. [DOI] [PubMed] [Google Scholar]
- 135.Tolcher AW, Mita A, Lewis LD, et al. Phase I and pharmacokinetic study of YM155, a small-molecule inhibitor of survivin. J Clin Oncol. 2008;26:5198–203. doi: 10.1200/JCO.2008.17.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Min W, Wang ZQ. Poly (ADP-ribose) glycohydrolase (PARG) and its therapeutic potential. Front Biosci. 2009;14:1619–26. doi: 10.2741/3329. [DOI] [PubMed] [Google Scholar]
- 137.Rodrigues CM, Stieers CL, Keene CD, et al. Tauroursodeoxycholic acid partially prevents apoptosis induced by 3-nitropropionic acid: evidence for a mitochondrial pathway independent of the permeability transition. J Neurochem. 2000;75:2368–79. doi: 10.1046/j.1471-4159.2000.0752368.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.