

Background: Fatty alcohols produced by fatty acyl reductases (FARs) have important biological roles.
Results: Expression of Arabidopsis FAR5 and FAR8 mutants in yeast revealed amino acids important for their stability or substrate specificity.
Conclusion: Amino acids 355 and 377 are 16:0 versus 18:0 chain length specificity determinants of FAR5 and FAR8.
Significance: Engineered FAR enzymes with desired substrate specificities will enable production of high value products.
Keywords: Enzyme Structure, Lipid Metabolism, Plant Biochemistry, Protein Engineering, Site-directed Mutagenesis, Fatty Acyl Reductase, Fatty Alcohol
Abstract
Fatty alcohols play a variety of biological roles in all kingdoms of life. Fatty acyl reductase (FAR) enzymes catalyze the reduction of fatty acyl-coenzyme A (CoA) or fatty acyl-acyl carrier protein substrates to primary fatty alcohols. FAR enzymes have distinct substrate specificities with regard to chain length and degree of saturation. FAR5 (At3g44550) and FAR8 (At3g44560) from Arabidopsis thaliana are 85% identical at the amino acid level and are of equal length, but they possess distinct specificities for 18:0 or 16:0 acyl chain length, respectively. We used Saccharomyces cerevisiae as a heterologous expression system to assess FAR substrate specificity determinants. We identified individual amino acids that affect protein levels or 16:0-CoA versus 18:0-CoA specificity by expressing in yeast FAR5 and FAR8 domain-swap chimeras and site-specific mutants. We found that a threonine at position 347 and a serine at position 363 were important for high FAR5 and FAR8 protein accumulation in yeast and thus are likely important for protein folding and stability. Amino acids at positions 355 and 377 were important for dictating 16:0-CoA versus 18:0-CoA chain length specificity. Simultaneously converting alanine 355 and valine 377 of FAR5 to the corresponding FAR8 residues, leucine and methionine, respectively, almost fully converted FAR5 specificity from 18:0-CoA to 16:0-CoA. The reciprocal amino acid conversions, L355A and M377V, made in the active FAR8-S363P mutant background converted its specificity from 16:0-CoA to 18:0-CoA. This study is an important advancement in the engineering of highly active FAR proteins with desired specificities for the production of fatty alcohols with industrial value.
Introduction
Primary fatty alcohols are aliphatic compounds that contain a hydroxyl group at the terminal position. In nature, long chain (C16 and C18) and very long chain (>C18) primary fatty alcohols are found either as free alcohols or in a combined state, such as wax esters, alkyl hydroxycinnamates, and ether lipids (1). Primary fatty alcohols and their derivatives have a wide range of biological roles in bacteria, fungi, insects, plants, and animals. Some important functions of free and esterified fatty alcohols in nonplant organisms are as follows: 1) as an energy storage reserve in microorganisms such as Euglena gracilis, Acinetobacter sp., and some marine microalgae; 2) as a constituent of sex pheromones in some insect and reptile species; 3) as a uropygial gland secretion in birds to maintain feather condition; 4) in the heads of sperm whales for echolocation and possibly to regulate buoyancy; and 5) as ether lipids, which play a variety of roles and are highly abundant in animal nervous system tissue (2–10). In plants, fatty alcohols and their derivatives are found primarily as chemical constituents of three extracellular lipid-phenolic barriers as follows: cuticle coating the aerial surfaces of plants, suberin present in the cell walls of various external and internal tissue layers, and sporopollenin found in the outermost layer of the pollen spore coat (1, 11). The functions of these polymeric surface lipid barriers are to protect plants from abiotic and biotic stresses. In a unique example, fatty alcohols are found in the seeds of the jojoba plant (Simmondsia chinensis) in the form of wax esters as a lipid energy reserve (12–14).
Fatty acyl reductases (FARs)2 catalyze the four-electron reduction of fatty acyl-coenzyme A (CoA) or fatty acyl-acyl carrier protein to a primary fatty alcohol in an NADPH-dependent reaction. This is a two-step process involving the production of an unreleased aldehyde intermediate (15–17). FAR proteins share two distinct domains as follows: a Rossmann-fold NAD(P)H binding domain at the N terminus and a fatty acyl-CoA reductase (FAR_C) domain at the C terminus (Fig. 1A) (1). The presence of the Rossmann-fold structure is typical of intermediate short-chain dehydrogenase/reductase proteins (18). Within the Rossmann-fold domain is a conserved GXXGXX(G/A) motif, which is believed to mediate NAD(P)H binding. The Rossmann-fold domain contains the active site motif, YXXXK, where the tyrosine (Y) and lysine (K) residues are predicted to play direct roles in catalysis based on kinetic studies with other reductases (19, 20).
FIGURE 1.

FAR structural domains and protein sequence alignment of Arabidopsis FAR5 and FAR8. A, schematic of the structural domains of FAR proteins. FAR enzymes, minus possible subcellular localization signals (e.g. plastid targeting), are ∼500 amino acids in length and contain an NAD(P)H binding Rossmann-fold domain (light gray) and a FAR_C domain (dark gray) at the N and C termini, respectively. The GXXGXX(G/A) predicted NADPH-binding motif as well as the predicted active site motif YXXXK are indicated (where X represents any amino acid). B, protein sequence alignments of FAR5 and FAR8, which are 85% similar at the amino acid level. Identical amino acids are highlighted in black, and physiochemically similar amino acids are highlighted in gray. Arrows indicate domain swap sites, and asterisks indicate site-specific mutagenesis sites.
FAR enzymes have distinct substrate specificities with regard to acyl chain length and degree of acyl chain saturation (1). The specificity of the FAR is often critical to the physical properties of the final biosynthetic product. An example of this comes from the European corn borer moth, Ostrinia nubilalis, which is represented by two races differentiated by the ratios of cis- and trans-isomers in their sex pheromone chemical mixtures. It is the preference of FAR enzymes, expressed in the pheromone gland of the two moth races, for fatty acyl substrate containing either a cis or a trans double bond that has led to reproductive isolation of the races (21). In recent years, there has been increased interest in engineering FAR proteins that produce fatty alcohols with desired chemical qualities for industrial use; however, little is known about FAR enzymes with respect to the amino acid residues responsible for conferring substrate specificity. A three-dimensional crystal structure of a FAR protein has not yet been elucidated, and no other investigations have yet been carried out to determine the specific residues influencing substrate preference.
The Arabidopsis thaliana genome encodes for eight FAR proteins (FAR1 to FAR8), each with a distinct substrate specificity for saturated fatty acyl precursors with chain lengths ranging from C16 to C30 (17, 22–25). FAR5 (At3g44550) and FAR8 (At3g44560) are located in tandem on chromosome 3 and encode for proteins that are 85% similar at the amino acid level (Fig. 1B). FAR5 generates 18:0 fatty alcohols (18:0-OH) found in the suberin polymer of roots, seed coats, and wounded leaves (23). FAR5 also generates the 18:0 fatty alcohol moiety found in alkyl hydroxycinnamates of Arabidopsis root waxes (26). FAR8 produces low levels of fatty alcohols from endogenous lipid pools when expressed in Escherichia coli (22). However, FAR8 gene expression is nearly undetectable in planta, at least under nonstressed conditions (23). When expressed in yeast, FAR5 and FAR8 produce nearly exclusively 18:0-OH and 16:0-OH, respectively, although the amount of 16:0-OH produced by FAR8 is very low (see below). Because FAR5 and FAR8 have high sequence similarity but strict chain length specificity for different fatty acyl precursors, they are an excellent platform for probing amino acids responsible for dictating substrate specificity. We have engineered chimeric proteins of FAR5 and FAR8 as well as single point mutants of FAR5 and FAR8, and we have evaluated the resulting activities by heterologously expressing them in yeast. We demonstrate that a two-amino acid substitution converts FAR5 specificity from 18:0-CoA to 16:0-CoA, and reciprocal substitutions in FAR8 convert specificity from 16:0-CoA to 18:0-CoA.
EXPERIMENTAL PROCEDURES
Materials
All chemical reagents were from Sigma unless otherwise stated. [1-14C]Palmitoyl-CoA and [1-14C]stearoyl-CoA were from PerkinElmer Life Sciences and Amersham Biosciences, respectively.
Construction of Yeast Expression Plasmids Containing Arabidopsis FAR5 and FAR8 Variants
The coding regions of FAR5 and FAR8 were cloned between the BamHI and XhoI restriction enzyme sites of a modified version of pYES2 (Invitrogen) called pYES2-His6/T7 (27). This vector allows for galactose-inducible expression in Saccharomyces cerevisiae (yeast) of proteins fused with an N-terminal T7 epitope tag for detection by protein immunoblotting (Western blots). To facilitate these domain swaps, a silent mutation was introduced into the FAR8 coding sequence to create a SalI restriction site, which was present at the equivalent position in the FAR5 coding sequence. The common SalI restriction site was used to generate a pair of reciprocal domain swaps centered at positions corresponding to amino acid residues 283/284 of FAR5 and FAR8. All other FAR5 and FAR8 domain swap constructs and site-directed mutants were made by overlap extension PCR consisting of two sequential DNA amplifications. For each plasmid construct, PCR was first used to amplify two overlapping DNA fragments (<1000 bp) using the primer sequences found in Table 1. The second amplification step used the two overlapping DNA fragments as templates along with flanking primers FAR5_BamHI_Forward or FAR8_BamHI_Forward and FAR5_XhoI_Reverse or FAR8_XhoI_Reverse, such that the entire FAR open reading frames were amplified and flanked by restriction enzyme sites. The amplified products were digested with BamHI and XhoI and ligated into the corresponding restriction sites in pYES2-His6/T7. The insert in each construct was verified by DNA sequencing using flanking sequencing primers.
TABLE 1.
List of oligonucleotide primers used in this study
For means forward, and Rev means reverse.
| Primer name | Sequence (5′ to 3′) |
|---|---|
| FAR5/FAR8 amplification | |
| FAR5_BamHI_For | GAGGGATCCATGGAACTCAATTGTGTTCAAT |
| FAR5_XhoI_Rev | GCGCTCGAGTCACTTCTTAAGCACGTGTG |
| FAR8_BamHI_For | GAGGGATCCATGGAATTCAGTTGTGTTCA |
| FAR8_XhoI_Rev | GCGCTCGAGTTACTTCTTAAGCACGTGAG |
| Domain swap constructs | |
| FAR8-FAR5_388_For | CTGGCTTTGTTCAGCCTCTACATGACCCTA |
| FAR8-FAR5_388_Rev | TAGGGTCATGTAGAGGCTGAACAAAGCCAG |
| FAR5-FAR8_388_For | ATGGCTTTGTTCAGCCTCTACATGACCATA |
| FAR5-FAR8_388_Rev | TATGGTCATGTAGAGGCTGAACAAAGCCAT |
| FAR8-FAR5_353_For | GGAGAGATCCGTGAAATTGCGGTTCGTTAC |
| FAR8-FAR5_353_Rev | GTAACGAACCGCAATTTCACGGATCTCTCC |
| FAR5-FAR8_353_For | GGAGAGATTCATGAGATTTTGTTTTGTTAC |
| FAR5-FAR8_353_Rev | GTAACAAAACAAAATCTCATGAATCTC |
| FAR8-FAR5_344_For | TCATCTCACCAAAACCCAGTCACATTTGGA |
| FAR8-FAR5_344_Rev | TCCAAATGTGACTGGGTTTTGGTGAGATGA |
| Point mutation constructs | |
| FAR8_S363P_For | TCACCAAAAACCCGTTGCGCAGT |
| FAR8_S363P_Rev | ACTGCGCAACGGGTTTTTGGTGA |
| FAR5_P363S_For | TTACGAAAAACTCTTTGCGAAGT |
| FAR5_P363S_Rev | ACTTCGCAAAGAGTTTTTCGTAA |
| FAR8_I347T_For | GGATCTCTCCATATGTTATTGGGTTTTGGT |
| FAR8_I347T_Rev | ACCAAAACCCAATAACATATGGAGAGATCC |
| FAR5_T347I_For | CAGAACCCAGTCATATTTGGAGAGATTCAT |
| FAR5_T347I_Rev | ATGAATCTCTCCAAATATGACTGGGTTCTG |
| FAR5_Y238F_For | GCCTAACACATTTGTTTTCACCA |
| FAR5_Y238F_Rev | TGGTGAAAACAAATGTGTTAGGC |
| FAR5_K242I_For | TGTTTTCACCATATCAATGGGAG |
| FAR5_K242I_Rev | CTCCCATTGATATGGTGAAAACA |
| FAR5_A355L_For | ATTCATGAGATTTTGGTTCGTTACTTTACGAAA |
| FAR5_A355L_Rev | TTTCGTAAAGTAACGAACCAAAATCTCATGAAT |
| FAR5_V377M_For | ACCGTCTCAAAAATGAGGTTCATACCAACCATG |
| FAR5_V377M_Rev | CATGGTTGGTATGAACCTCATTTTTGAGACGGT |
| FAR8_M377V_For | ACTGTCTCGAAAGTGAAGCTGATACCA |
| FAR8_M377V_Rev | TGGTATCAGCTTCACTTTCGAGACAGT |
| FAR8_L355A_For | ATCCGTGAAATTGCGTTTTGTTACTTC |
| FAR8_L355A_Rev | GAAGTAACAAAACGCAATTTCACGGAT |
Expression of FAR5 and FAR8 Variants in Yeast
The pYES2-His6/T7 plasmids containing wild-type and mutant FAR5 and FAR8 coding regions were transformed into S. cerevisiae yeast strain W303-1A (MATa his3Δ1 leu2 trp1-289 ura3-52) using the method described previously (28). Yeast transformants were selected on synthetic dropout (SD) media plates (2% d-glucose, 0.67% yeast nitrogen base, 0.01% adenine, 0.002% histidine, 0.002% tryptophan, 0.01% leucine, and 2% agar) lacking uracil (SD−Ura). Four yeast colonies for each construct, including empty vector, were separately inoculated into 3 ml of SD−Ura liquid media and grown for 24 h at 30 °C and 250 rpm. The A600 for each culture was measured, and a volume of culture required to inoculate 3 ml of induction SG−Ura liquid media (2% galactose, 0.67% yeast nitrogen base, 0.01% adenine, 0.002% histidine, 0.002% tryptophan, 0.01% leucine) at an A600 of 0.4 was removed and centrifuged. The resulting yeast pellets were washed twice with 1.5 ml of sterile water and then resuspended in 3 ml of SG−Ura media. The yeast strains were then grown for 2 days at 30 °C and 250 rpm prior to lipid extraction and analysis of protein levels by Western blotting.
Analysis of Protein Levels in Yeast by Protein Immunoblotting (Western Blots)
A volume of each galactose-induced yeast culture equal to an A600 of 2.5 was harvested by centrifugation. The supernatants were removed and the pellets stored at −80 °C. Protein samples were prepared according to Ref. 29 using phosphate-containing loading buffer (62.5 mm sodium phosphate buffer, pH 7.0, 2% SDS, 10% glycerol, 5% β-mercaptoethanol, 0.0001% bromphenol blue) prior to separation on a 12% SDS-polyacrylamide gel at 150 V in 1× running buffer (25 mm Tris, pH 8.3, 186 mm glycine, 0.1% SDS). The proteins were transferred to nitrocellulose membranes by wet-transfer electrophoresis. The nitrocellulose membranes were blocked overnight at 4 °C in blocking solution consisting of 5% fat-free skim milk in TBST (137 mm NaCl, 25 mm Tris base, pH 7.6, 0.1% Tween 20). A 1:50,000 dilution of T7 Tag monoclonal mouse antibody (catalogue number 69522-3, EMD4Biosciences) was added to detect the T7 epitope-tagged proteins; the membranes were washed four times for 5 min each with TBST. The membranes were then incubated with a 1:50,000 dilution of horseradish peroxidase-conjugated anti-mouse secondary antibody (EMD4Biosciences) for 1 h and then rinsed four times with TBST for 5 min each. The membranes were then incubated with a 1:1 mixture of Lumigen TMA-6 Solution A (contains Tris buffer in 3.2% v/v ethanol) and Lumigen TMA-6 Solution B (proprietary substrate in Tris buffer) (GE Healthcare) for 5 min in the dark and imaged with a FluorChemQ (Alpha Innotech). Western blots with yeast microsomes were done with 15 μg of protein and revealed with the ECL Western blotting detection kit (Amersham Biosciences).
Analysis of Fatty Alcohol Content of Transgenic Yeast by Gas Chromatography (GC)
Yeast cells from 2 ml of each galactose-induced culture were harvested by centrifugation, and the supernatant was poured into a separate glass tube. 10 μg of 1-pentadecanol (15:0-OH) was added as an internal standard to both the pellet and supernatant. The yeast supernatant was extracted twice with 1 ml of 2:1 chloroform/methanol and once with 1 ml of chloroform, and the organic phases were combined and washed with 2.5 ml of 0.9% NaCl (w/v) before being evaporated under nitrogen gas at 37 °C. Both the yeast pellet and supernatant samples were resuspended in 3 ml of 1 m methanolic-HCl, and lipids were transmethylated at 80 °C for 90 min. 0.9% NaCl was added to each sample, and the lipids were extracted twice into 500 μl of hexane. The pooled hexane extracts were dried under nitrogen gas at 37 °C. The lipids were then resuspended in 100 μl of N,O-bis(trimethylsilyl) trifluoroacetamide plus 100 μl of pyridine and incubated at 110 °C for 15 min for silylation of free hydroxyl groups. The samples were then re-dried down as before and resuspended in a 1:1 (v/v) mixture of hexane/toluene. Lipids were quantified with a Varian GC450 equipped with a split-splitless injector, HP-1 column (30 m length, 0.25 mm inner diameter, 0.10 μm film thickness), and a flame ionization detector. One μl of each sample was injected using a 10:1 split ratio. The carrier gas was helium with a constant flow rate of 2 ml/min. The column oven was held initially at 150 °C for 5 min, then ramped at 10 °C/min to 300 °C, and held for 8 min, for a total run time of 28 min.
Yeast Microsome Preparations and FAR in Vitro Enzyme Assays
Yeast microsomes were prepared according to Ref. 30, and FAR activity was assayed with 5 μg of protein for 5 min at 30 °C in 50 μl of sodium phosphate buffer (50 mm, pH 6.5) containing 5 mm NADPH, 20 μm 14C-labeled fatty acyl-CoA (palmitoyl- or stearoyl-CoA), 1 mm MgCl2, and 2 mg/ml fatty acid-deficient bovine serum albumin. Reactions were stopped by adding 600 μl of 1% HClO4, and lipids were extracted by the successive addition of 2 ml of chloroform/methanol (1:2; v/v), 1 ml of chloroform, and 0.8 ml of 1% HClO4. The lower organic phases were dried under nitrogen and analyzed by thin layer chromatography using HPTLC Silica Gel 60 plates (Merck) and hexane/ether/acetic acid (70:30:2, v/v/v) as the solvent. Radiolabeled products were identified by co-migration with unlabeled standards and quantified by autoradiography using a Storm 860 molecular imager (GE Healthcare).
RESULTS
Amino Acids of FAR5 and FAR8 Important for Protein Stability and Enzymatic Activity
We examined the activities and substrate specificities of the FAR5 and FAR8 enzymes using S. cerevisiae as a heterologous system. The open reading frames for both genes were cloned into the yeast expression vector pYES2-His6/T7 downstream of the GAL1 promoter to allow high level protein induction by galactose. The expressed FAR proteins contained a T7 epitope tag at the N terminus to enable detection using Western blots. The empty pYES2-His6/T7 vector was used as a negative control. The endogenous acyl-CoA pool of S. cerevisiae has high and nearly equal amounts of 16:0-CoA and 18:0-CoA (31), thus providing relevant substrates for heterologously expressed FAR5 and FAR8 variants.
Analysis of the internal lipid content of 2-day-old galactose-induced yeast cultures by GC demonstrated that FAR5 produced high levels of 18:0 primary fatty alcohol (2.07 μg per unit of absorbance) and that the protein was highly expressed in yeast as determined by Western blot analysis (Figs. 2 and 3). In contrast, FAR8 did not produce any fatty alcohols under the same conditions, and the protein was barely detected in the Western blot (Figs. 2 and 3). However, when transgenic yeasts expressing FAR8 were grown for 4 days prior to lipid analysis, low levels of 16:0 fatty alcohol were detected (data not shown). Similarly, very small amounts of 16:0-OH were sometimes detected in yeast expressing FAR5 for 2 days (e.g. Fig. 5A), and these amounts increased after 4 days of FAR5 expression albeit to levels less than 4% of the accumulated 18:0-OH (data not shown).
FIGURE 2.

Gas chromatograms of internal lipids of yeast expressing Arabidopsis FAR5, FAR8, or FAR8-S363P. The empty vector pYES2-His6/T7 tag acted as a negative control. Transformants were cultured in galactose media to induce protein expression. Fatty acids were transmethylated to their corresponding methyl esters, and fatty alcohol hydroxyl groups were derivatized to trimethylsilyl ethers before separation by GC and detection by flame ionization. The peaks corresponding to pentadecanol (15:0-OH) internal standard (IS), 16:0-OH and 18:0-OH are indicated, as well as saturated and monounsaturated C16 and C18 fatty acids (FA).
FIGURE 3.

Amino acids important for FAR5 and FAR8 enzyme stability and activity. A, left, schematics of FAR5 and FAR8 variants. The FAR5 variants are in black, and FAR8 variants are in gray, with positions of site-specific mutations indicated above each protein schematic. Right, the amounts of nonsecreted fatty alcohols produced by yeast-expressing FARs, where values are expressed in μg/A600 unit ± S.D. (n = 4). B, protein levels of FAR5 and FAR8 variants expressed in yeast. Western blots (top) were performed using anti-T7 mouse antibody to detect the N-terminal T7 epitope in the protein fusions. The Coomassie-stained gel is shown at the bottom to indicate equal loading. The positions of the protein size markers (in kDa) are indicated to the left of the Western blot and stained gel.
FIGURE 5.

Domain swaps between FAR5 and FAR8. A, left, schematics of FAR5 and FAR8 domain swap chimeras, with the portions from FAR5 in black and the portions from FAR8 in gray. The active FAR8-S363P mutant, denoted as FAR8R, was used in all domain swaps. Right, amounts of nonsecreted fatty alcohols produced by yeast expressing FAR5, FAR8R, or a FAR5/FAR8 chimera, where values are expressed in μg/A600 unit ± S.D. (n = 4). B, protein levels of FAR5, FAR8R, and FAR5/FAR8 chimeras expressed in yeast. Western blots (top) and Coomassie-stained gel (bottom) are as described in Fig. 3 legend.
With the exception of Arabidopsis FAR8, all plant FAR proteins reported have a proline at relative position 363 of FAR5 and FAR8. FAR8 instead has a serine at this position (Fig. 1B). To investigate the significance of this conserved proline residue, serine 363 from FAR8 was converted to proline (FAR8-S363P). Unlike native FAR8 protein, the FAR8-S363P variant generated substantial amounts of 16:0-OH (0.85 μg per unit of absorbance) (Figs. 2 and 3). A small amount of 18:0-OH was sometimes detected, but this quantity was negligible compared with that of 18:0-OH produced by FAR5 (<1%) or that of 16:0-OH produced by FAR8-S363P. Analysis of protein levels in yeast revealed that FAR8-S363P accumulated to higher levels than its native counterpart (Fig. 3B). For much of our subsequent mutational analyses, this active FAR8-S363P mutant was used as the model of 16:0 fatty alcohol production. We subsequently refer to this active FAR8 variant as FAR8R, where R stands for “resurrected.” The reciprocal amino acid conversion in FAR5, proline to serine conversion at position 363, resulted in a 30% decrease in 18:0-OH production (Fig. 3A), although the FAR5-P363S and native FAR5 protein levels in yeast were comparable (Fig. 3B). It was possible that FAR8 or FAR8R had activity for a shorter acyl chain length than 16:0, with the most likely possibility being 14:0. Therefore, we also tested whether FAR5, FAR8, and/or FAR8R could generate 14:0-OH by feeding yeast 14:0 fatty acid using a previously described protocol (32). No 14:0-OH was produced by FAR5 or FAR8 and only a very small amount by FAR8R in our feeding experiments (data not shown). We cannot rule out other potential substrates for FAR8, but the most likely scenario is that FAR8 is indeed a low activity FAR due to the serine rather than proline at position 363 and that the specificity of FAR8R is nearly strict for 16:0-CoA.
The 18:0 and 16:0 fatty alcohols produced by the FAR5 and FAR8R variants, respectively, were found not only within the yeast cells but also secreted into the media (Fig. 4). Of the total fatty alcohols produced by FAR5 and FAR8R in yeast, 47 and 38%, respectively, were found secreted into the media (Fig. 4). For ease of screening purposes (below), we only quantified the fatty alcohols found in the yeast pellet. We verified that no bias was introduced by excluding the secreted fatty alcohols in our screens by measuring the internal and secreted fatty alcohols of FAR5 and FAR8R, as well as some key FAR5 and FAR8 variants that alter substrate specificity as described below (Fig. 4). The chain length distributions of fatty alcohols produced by these FARs expressed in yeast were nearly identical whether examining just internal fatty alcohols or examining total fatty alcohols (internal plus secreted).
FIGURE 4.
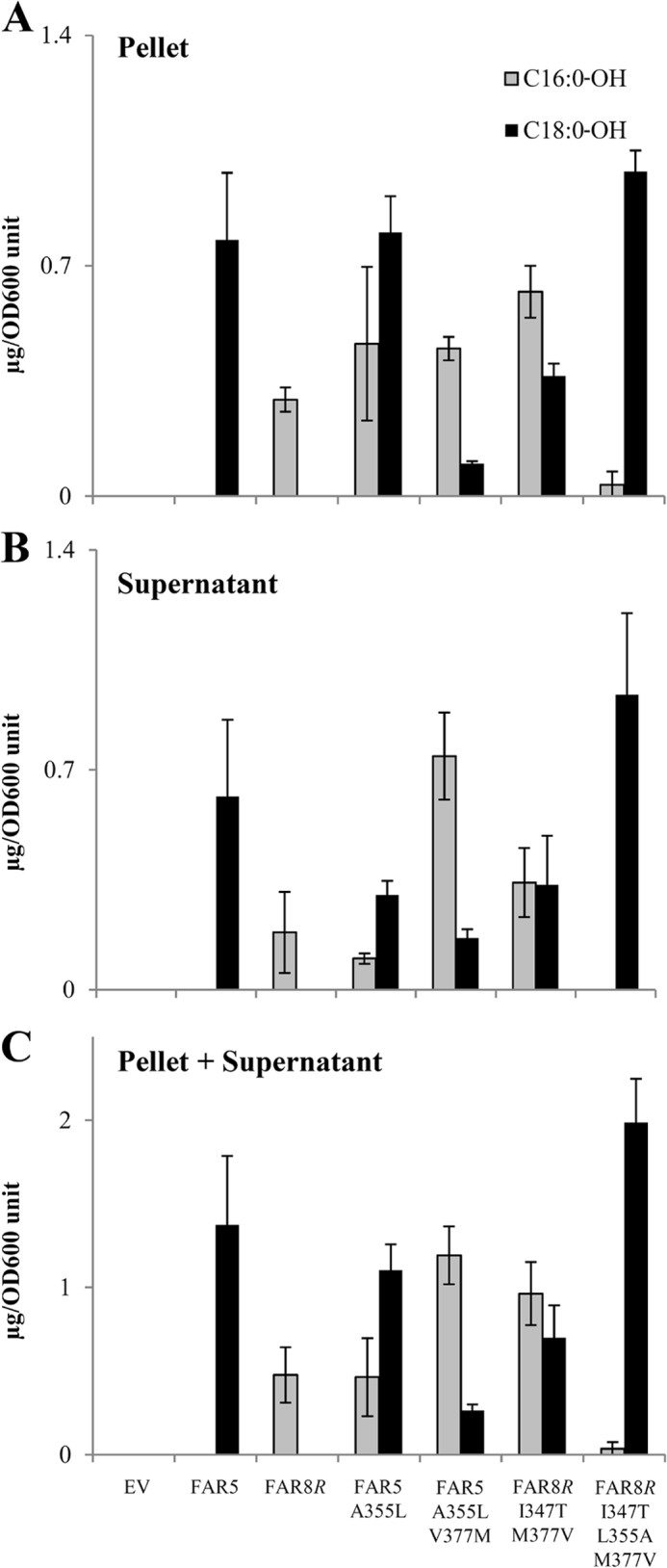
Amounts of total fatty alcohols produced by FAR5 and FAR8 variants expressed in yeast. A, internal fatty alcohol content of yeast cells (nonsecreted fatty alcohols). B, fatty alcohols found in supernatant of yeast cultures (secreted fatty alcohols). C, total fatty alcohols produced by yeast expressing a FAR variant (combined nonsecreted and secreted fatty alcohol content). Values are expressed in μg/A600 unit ± S.D. (n = 4). Yeast cultures were grown for 48 h in galactose media for protein induction before lipid extraction and analysis.
We next investigated the proposed roles of tyrosine 238 and lysine 242 in the predicted YXXXK active site motif of the Rossmann-fold in FAR proteins (Fig. 1) (1, 22). Two separate site-specific substitutions were made in the FAR5 coding sequence to convert tyrosine 238 to a phenylalanine (FAR5-Y238F) and separately lysine 242 to an isoleucine (FAR5-K242I) in the encoded protein. These same amino acid substitutions were used to investigate the active site of alcohol dehydrogenase from Drosophila melanogaster (19), revealing the importance of the hydroxy group of tyrosine and the amino group of lysine in catalysis. Phenylalanine differs from tyrosine only by the absence of a hydroxyl group on the aromatic ring. Lysine and isoleucine are similarly sized amino acids, but isoleucine lacks the positive charge conferred by the amino group on lysine. Lipid analysis of both the FAR5-Y238F and FAR5-K242I variants expressed in yeast revealed no fatty alcohol production (Fig. 3A), although Western blot analysis of the mutant proteins indicated that they were expressed to the same levels as the wild-type FAR5 (Fig. 3B).
Altogether, these data showed that FAR5 and FAR8 nearly exclusively produce 18:0-OH and 16:0-OH, respectively, that proline 363 is important for protein stability, and that tyrosine 238 and lysine 242 are necessary for catalytic activity.
Domain Swaps between FAR5 and FAR8
A series of domain swap chimeras involving FAR5 and FAR8 were then made to investigate the protein region responsible for conferring substrate specificity. An initial reciprocal pair of domain swaps were made between FAR5 and FAR8R at positions corresponding to amino acid residues 283/284 (Figs. 1B and 5A). The FAR5(1–283)FAR8R(284–496) {1} chimera produced predominantly 16:0-OH. Conversely, the FAR8(1–283)FAR5(284–496) chimera {2} produced substantial amounts of 18:0-OH and very little 16:0-OH (Fig. 5A). This indicated that C16/C18 chain length specificity is dictated by amino acids between position 284 and the C terminus of the FAR5/FAR8 proteins. To further investigate this, domain swaps between FAR5 and FAR8 were made at amino acid residues 388/389 (Figs. 1B and 5A). We found that the FAR5(1–388)FAR8(389–496) chimera {3} produced nearly exclusively 18:0-OH, whereas the FAR8R(1–388)FAR5(389–496) chimera {4} had low activity, but it produced more 16:0-OH than 18:0-OH (Fig. 5A). Collectively, the data from these four domain swaps indicate that C16/C18 chain length specificity is dictated by amino acids between positions 284 and 388 of the FAR5 and FAR8 proteins.
To examine this further, we made two FAR5/FAR8 internal domain swap mutants spanning residues 284–388, inclusively (Fig. 5A). The FAR5(1–283)FAR8R(284–388)FAR5(389–496) {5} and FAR8(1–283)FAR5(284–388)FAR8(389–496) {6} internal domain swap chimeras produced nearly exclusively 16:0-OH and 18:0-OH, respectively (Fig. 5A). This verified that the amino acid residues responsible for 16:0 fatty acyl-CoA versus 18:0 fatty acyl-CoA substrate specificity are between residues 284 and 388. To further narrow down the protein region responsible for conferring chain length specificity, two more reciprocal internal domain swap constructs were made spanning residues 354–388 (Fig. 5A). The FAR5(1–353)FAR8R(354–388)FAR5(389–496) chimera {7} produced nearly exclusively 16:0-OH. The FAR8(1–353)FAR5(354–388)FAR8(389–496) chimera {8} appeared to have very low activity for either fatty acyl-CoA substrate, but it produced more 18:0-OH than 16:0-OH. This suggests that the amino acid residues responsible for dictating 16:0 versus 18:0 acyl chain length specificity in FAR5 and FAR8 are found within the 35 amino acids interval spanning residues 354–388.
The chimeras that contained FAR5 sequence at the N-terminal end {1 + 3} accumulated to higher levels in yeast than the chimeras that contained FAR8 at the N-terminal end {2 + 4} (Fig. 5B). The internal domain swap chimeras were generally expressed to similar levels as the FAR8R protein, with the exception of FAR8(1–353)FAR5(354–388)FAR8(389–496) {8}, which gave very little fatty alcohol production and had near undetectable protein accumulation, and FAR5(1–353)FAR8R(354–388)FAR5(389–496) {7}, which generated high amounts of fatty alcohols and was expressed at high levels similar to that of FAR5 (Fig. 5B). Additionally, this latter chimera {7} accumulated to higher levels than the FAR5(1–283)FAR8R(284–388)FAR5(389–496) chimera {5}, which had FAR8 sequence between residues 284 and 353. We therefore speculated that some residues immediately N-terminal to position 354 are critical for protein stability. To test this, another FAR8-FAR5-FAR8 domain swap chimera was expressed that contained residues 345–388 of FAR5. This FAR8(1–344)FAR5(345–388)FAR8(389–496) chimera {9} accumulated to relatively high levels (Fig. 5B), indicating that one or more amino acids between positions 345 and 353 are important for protein stability. Of the four amino acids that are different than FAR5 and FAR8 in this region, only position 347 is a nonconservative amino acid difference (Fig. 1B). Residue 347 is a threonine in FAR5, and it is an isoleucine in FAR8. We found that converting the isoleucine present at position 347 in FAR8 to the reciprocal threonine of FAR5 caused the resultant FAR8R-I347T protein to accumulate to higher levels in yeast than the parent FAR8R (Fig. 3B). The amount of 16:0-OH produced by FAR8R-I347T was 1.9 times that of FAR8R (Fig. 3A). The presence of a threonine at position 347 in FAR5 thus likely promotes protein stability. To further investigate the role of threonine at position 347, we made the reverse mutation in FAR5 converting threonine 347 to isoleucine. The resultant FAR5-T347I mutant had a 40% decrease in 18:0-OH production (Fig. 3A). This was paralleled by decreased FAR5-T347I protein levels compared with wild-type FAR5 (Fig. 3B), again indicating that threonine 347 plays an important role in protein stability.
Reciprocal Amino Acid Substitutions in FAR5 and FAR8
To determine individual amino acid residues of FAR5 and FAR8 that are responsible for dictating chain length substrate specificity, site-specific substitutions were made within the 44-amino acid region (residues 345–388) of FAR5 and FAR8 identified by internal domain swaps to “flip” substrate specificity and be important for protein stability (Fig. 6A). Within this region, there are 13 amino acids that differ between FAR5 and FAR8, and we targeted eight of these residues for site-specific mutagenesis. Of these eight amino acids, positions 347 and 363 had already been examined (see above) and found to be important for protein stability/activity but not influencing chain length specificity. In all cases, we converted a given amino acid found at a certain position in one FAR to the reciprocal amino acid found at that same position in the other FAR.
FIGURE 6.

Reciprocal amino acid substitutions in FAR5 and FAR8. A, protein alignment of amino acid residues 345–388 of FAR5 and FAR8. Identical amino acids are highlighted in black, and physiochemically similar amino acids are highlighted in gray. Asterisks indicate the four amino acids (347, 355, 363, and 377) targeted for analysis in this region by site-specific mutagenesis. B, analysis of amino acids 355 and 377 affecting substrate specificity of FAR5. C, analysis of amino acids 355 and 377 affecting substrate specificity of FAR8. The S363P mutation in FAR8, denoted as FAR8R, was present in all FAR8 variants. The top parts of B and C are graphs reporting the amounts of nonsecreted fatty alcohols produced by FAR5 and FAR8 variants, where values are expressed in μg/A600 unit ± S.D. (n = 4). The bottom parts of B and C report the protein levels of FAR5 and FAR8 variants. Western blots (top) and Coomassie-stained gel (bottom) are as described in Fig. 3 legend.
Within the 345–388 region, there are two triplet blocks of amino acids, residues 355–357 (triplet I) and 377–379 (triplet II), that are not conserved between FAR5 and FAR8 (Fig. 6A). We individually mutated each of the six amino acids in triplets I and II of FAR5 to the corresponding FAR8 amino acid, and we found that amino acid conversions at positions 355 and 377 of triplet I and II, respectively, resulted in the most 16:0-OH production (Fig. 6B), with the other four conversions causing little or no 16:0-OH production (data not shown). The FAR5-A355L single mutant produced 16:0-OH to levels similar to that of FAR8R and produced 18:0-OH to about 66% of that observed with wild-type FAR5 (Fig. 6B). The FAR5-V377M single mutant also produced both 16:0-OH and 18:0-OH, but the amount of 16:0-OH produced was about 25% of FAR8R, whereas the amount of 18:0-OH produced was similar to wild-type FAR5 (Fig. 6B). By combining the mutations, we were able to generate a FAR5 variant, FAR5-A355L/V377M, which produced mostly 16:0-OH (Fig. 6B). The FAR5-A355L/V377M mutant had 37% greater 16:0-OH yield than FAR8R, although it produced a small amount of 18:0-OH, ∼6% of the amount produced by wild-type FAR5. These single and double FAR5 variants were all expressed to similar levels as wild-type FAR5 in yeast (Fig. 6B).
We then examined the effects of reciprocal amino substitutions at positions 355 and 377 of FAR8 (Fig. 6C). The FAR8R-L355A and FAR8R-M377V mutants had greatly reduced levels of 16:0 fatty alcohol production, ∼8 and 18% of FAR8R levels, respectively. These mutants also produced 18:0-OH, albeit at very low levels compared with FAR5. Because the I347T mutation had a major effect on FAR8 protein stability/activity (see above), we combined this change with the M377V change. The resultant FAR8R-I347T/M377V double mutant produced about twice the amount of 16:0-OH than the parent FAR8R, although slightly less than FAR8R-I347T (Figs. 3A and 6C). FAR8R-I347T/M377V additionally produced high levels of 18:0-OH (0.63 μg per unit of absorbance). As predicted, FAR8R-I347T/M377V protein accumulated at much higher levels in yeast than FAR8R-M377V, likely accounting for the increased fatty alcohol production. In an attempt to further convert FAR8 substrate preference from 16:0-CoA to 18:0-CoA, we added a third L355A mutation to create FAR8R-I347T/L355A/M377V. This triple mutation nearly completely converted FAR8R specificity to 18:0-CoA (Fig. 6C). The FAR8R-I347T/L355A/M377V mutant produced 18:0-OH to levels about 60% of wild-type FAR5. The FAR8R-I347T/L355A/M377V mutant produced a small amount of 16:0-OH, but it was only about 30% of that found with FAR8R. In summary, we altered FAR8 chain length specificity from 16:0-CoA nearly completely to 18:0-CoA by changing amino acids at two positions, 355 and 377, to that of the corresponding FAR5 amino acids, but amino acids changes were also required at positions 347 and 363 to allow for high protein accumulation.
In Vitro FAR Assays Using Yeast Microsomes
To provide additional support of our findings, we prepared microsomes from different transgenic yeast strains and performed in vitro assays using either [1-14C]palmitoyl-CoA (16:0-CoA) or [1-14C]stearoyl-CoA (18:0-CoA) as substrate. After 5 min of incubation in the presence of 5 μg of microsomal proteins, lipids were extracted and analyzed by thin layer chromatography. Microsomes containing FAR5 produced labeled fatty alcohols in the presence of [1-14C]18:0-CoA, whereas no radioactivity associated with fatty alcohols was observed using microsomes from yeast transformed with empty vector, regardless of the labeled fatty acyl-CoA used (Fig. 7A). In the presence of [1-14C]16:0-CoA, FAR5 produced very little fatty alcohol. Conversely, FAR5-A355L/V377M produced significant amounts of fatty alcohols with [1-14C]16:0-CoA as substrate, in agreement with the results obtained in vivo. In contrast to the microsomes containing FAR5 variants, no fatty alcohols were detected when using microsomes from yeast transformed with FAR8R, FAR8R-I347T/M377V, or FAR8R-I347T/L355A/M377V, whatever the labeled fatty acyl-CoA used (Fig. 7A). The reason that these microsomes were inactive in vitro was most probably due to protein stability, as none of the FAR8 variants could be detected in the microsomes by Coomassie Blue staining or by Western blots in contrast to the FAR5 variants (Fig. 7B). The FAR8 variants may also be deficient in their ability to associate with membranes. These in vitro assays nevertheless allowed for quantification of the specific activities of FAR5, FAR5-A355L and FAR5-A355L/V377M for 16:0-CoA and 18:0-CoA (Table 2). FAR5 was 20 times more active in vitro with 18:0-CoA than with 16:0-CoA. The FAR5-A355L single mutant was able to convert both 16:0-CoA and 18:0-CoA to the corresponding fatty alcohols, with its specific activity for 16:0-CoA being about half of that for 18:0-CoA. The FAR5-A355L/V377M double mutant had a clear preference for 16:0-CoA, with its specific activity for 16:0-CoA being 4.7 times that for 18:0-CoA (Table 2). Altogether, the in vitro assays using FAR5, FAR5-A355L, and FAR5-A355L/V377M were in good agreement with the results obtained in vivo.
FIGURE 7.

FAR in vitro assays with yeast microsomes containing FAR5 and FAR8 variants. A, representative experiment showing separation of lipids recovered from in vitro FAR assays by TLC. Assays were conducted in the presence of 14C-radiolabeled 18:0-CoA (top) or 14C-radiolabeled 16:0-CoA (bottom), and microsomes were extracted from yeast expressing the FAR indicated below the plate images. The radiolabeled fatty alcohols were identified by co-migration with unlabeled standards. B, protein levels of FAR5 and FAR8 variants in yeast microsomes. Western blots (top) and Coomassie-stained gel (bottom) are as described in Fig. 3 legend.
TABLE 2.
Specific activities of FAR5, FAR5-A355L, and FAR5-A355L/V377M
In vitro FAR assays were done using yeast microsomes in the presence of NADPH and either [14C]16:0-CoA or [14C]18:0-CoA. Data are presented as nmol/mg/min ± S.D. (n = 5).
| FAR5 variant | [14C]16:0-CoA | [14C]18:0-CoA |
|---|---|---|
| FAR5 (wild type) | 0.49 ± 0.39 | 10.14 ± 1.61 |
| FAR5-A355L | 3.67 ± 0.97 | 7.23 ± 1.99 |
| FAR5-A355L/V377M | 12.6 ± 1.63 | 2.7 ± 0.94 |
DISCUSSION
FAR5 and FAR8 are located in tandem on the Arabidopsis genome, and the encoded proteins are 85% identical in sequence. It is likely that FAR5 and FAR8 are the result of a recent genome duplication event. We therefore speculate that the low native activity of FAR8 is a result of recently acquired mutations through its evolutionary history and that it may be on the way to being a pseudogene. The serine at position 363 of FAR8 rather than the conserved proline at this position may be the result of such a mutation. Although FAR5 and FAR8 are highly similar, they have distinct and nonoverlapping substrate specificities. Expression of Arabidopsis FAR5 in yeast produced relatively large quantities of 18:0-OH and very small amounts of 16:0-OH, although expression of the native Arabidopsis FAR8 produced exclusively 16:0-OH in very small quantities. To date, 16:0 fatty alcohols have not yet been detected, either in free or combined form, in Arabidopsis (25), although they are expected to be part of pollen exine based on the high activity of MS2/FAR2 for 16:0-acyl carrier protein (24). MS2/FAR2 is essential for pollen exine development (24, 33). FAR8 may be contributing to 16:0-OH in pollen exine as low levels of FAR8 transcript were detected in developing and mature pollen using DNA microarray analysis (34). Regardless of the endogenous role of FAR8, it provided an ideal partnership with FAR5 to investigate amino acids important for dictating their distinct chain length substrate specificities.
As with other NAD(P)H-dependent oxidoreductases, the FAR enzymes possess a predicted YXXXK active site motif (35). The tyrosine and lysine residues present at this position, along with a serine residue that comes from another part of the protein, have been shown to play an important role in catalysis (19, 20). Deletion of the YXXXK motif in MS2/FAR2 results in a protein that cannot complement the pollen exine defects of ms2/far2 mutants (24). However, the individual roles of the tyrosine and lysine predicted to be directly involved in catalysis had not been previously investigated. We made two site-specific FAR5 mutants in which we converted tyrosine 238 to phenylalanine and lysine 242 to isoleucine (FAR5-Y238F and FAR5-K242I). The same substitutions were used to investigate the catalytic residues of Drosophila alcohol dehydrogenase (19). We found that both FAR5-Y238F and FAR5-K242I produced no fatty alcohol but were expressed to the same level as wild-type FAR5 in yeast. This confirms the essential functions of these two residues for FAR enzyme activity; however, a detailed catalytic mechanism for the FAR enzymes is not yet known.
We found that substitution of the serine at position 363 to proline in FAR8 (FAR8-S363P) greatly increased its production of 16:0 fatty alcohol in yeast. We hypothesize that a proline at this position plays an important structural role, influencing protein stability and activity. Proline is commonly found in tight turns within proteins, and such a turn of the backbone may be important for the structural integrity of FAR8. The “resurrected” FAR8 was expressed to higher levels than the native FAR8, likely due to enhanced protein stability. The reciprocal conversion of proline 363 to serine in FAR5 (FAR5-P363S) reduced but did not abolish 18:0-OH production in yeast. This suggests that although proline 363 is conserved and strongly influences FAR protein activity and stability, it is not necessarily essential in all FAR enzymes. The presence of threonine at position 347 also had a positive effect on protein accumulation, although this is not a strictly conserved amino acid in the FAR enzyme family. Native FAR8 has an isoleucine at this position, and conversion to threonine significantly enhanced FAR8 accumulation in yeast and correspondingly the amount of 16:0-OH produced. Conversely, replacement of threonine in FAR5 with isoleucine dramatically decreased its protein levels. However, the amount of FAR8-S363P/I347T that was expressed in yeast was still far less than native FAR5. There are likely other amino acids in FAR8 that would need to be altered to increase protein accumulation further.
Through domain swaps between FAR5 and FAR8, we found that an internal span of 35 residues (354–388) dictated the 16:0-CoA versus 18:0-CoA specificity of these two enzymes. Using site-specific substitutions, we identified residues 355 and 377 as the two key residues dictating 16:0-CoA versus 18:0-CoA chain length substrate specificity in FAR5 and FAR8. Individual reciprocal substitution at either of these positions broadened enzyme specificity of either FAR5 or FAR8 such that both 16:0 and 18:0 fatty alcohols were produced in yeast. Although single amino acid conversions at position 355 and 377 were not sufficient to fully convert substrate specificity of FAR5 or FAR8, we found that combining the two amino acid conversions nearly completely shifted substrate preference. For FAR5, this phenomenon was confirmed using in vitro enzyme assays with radiolabeled 16:0-CoA and 18:0-CoA substrates. Therefore, to fully convert FAR5/FAR8 specificity from one chain length to another, the enzyme's preference for a particular chain length must be “switched off,” otherwise the enzyme exhibits activity for both substrates.
In summary, the results of this study indicate that specific amino acids at the junction of the Rossmann-fold domain and the FAR_C domain govern FAR chain length specificity and that it is possible to alter this substrate specificity by just two amino acid changes. However, without the benefit of an x-ray or NMR structure of the FAR substrate-binding site, it is difficult to determine precisely how amino acids 355 and 377 in FAR5 and FAR8 control binding of 16:0-CoA versus 18:0-CoA substrates. We attempted to use comparative homology modeling of FAR5 and FAR8 to gain further insights into the roles of these residues, but this approach was not feasible due to the lack of a similar enough modeling template. Because fatty alcohols are used in a variety of human applications, such as in cleaning detergents, cosmetics, and pharmaceutical formulations, food products, textiles and coatings, or high performance industrial lubricants (1), our study is a step forward in the engineering of FAR enzymes to produce fatty alcohols with desired specificities and to allow the production of renewable fatty alcohol-containing lipid products of high commercial value.
Acknowledgment
We thank Dr. Shelley Hepworth (Carleton University) for critical reading of the manuscript.
This work was supported by a Discovery grant from the Natural Sciences and Engineering Research Council of Canada (to O. R.), by a France-Canada Research Fund grant from the Embassy of France in Canada (to F. D. and O. R.), and by European Commission Framework 7 Project entitled “Industrial Crops Producing Added Value Oils for Novel Chemicals” (ICON).
- FAR
- fatty acyl reductase
- CoA
- coenzyme A
- GC
- gas chromatography
- 16:0-OH
- 16:0 primary fatty alcohol
- 18:0-OH
- 18:0 primary fatty alcohol.
REFERENCES
- 1. Rowland O., Domergue F. (2012) Plant fatty acyl reductases: enzymes generating fatty alcohols for protective layers with potential for industrial applications. Plant Sci. 193, 28–38 [DOI] [PubMed] [Google Scholar]
- 2. Antia N. J., Lee R. F., Nevenzel J. C., Cheng J. Y. (1974) Wax ester production by the marine cryptomonad Chroomonas salina grown photoheterotrophically on glycerol. J. Protozool. 21, 768–771 [DOI] [PubMed] [Google Scholar]
- 3. Teerawanichpan P., Robertson A. J., Qiu X. (2010) A fatty acyl-CoA reductase highly expressed in the head of honey bee (Apis mellifera) involves biosynthesis of a wide range of aliphatic fatty alcohols. Insect Biochem. Mol. Biol. 40, 641–649 [DOI] [PubMed] [Google Scholar]
- 4. Reiser S., Somerville C. (1997) Isolation of mutants of Acinetobacter calcoaceticus deficient in wax ester synthesis and complementation of one mutation with a gene encoding a fatty acyl-coenzyme A reductase. J. Bacteriol. 179, 2969–2975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Moto K., Yoshiga T., Yamamoto M., Takahashi S., Okano K., Ando T., Nakata T., Matsumoto S. (2003) Pheromone gland-specific fatty-acyl reductase of the silkmoth, Bombyx mori. Proc. Natl. Acad. Sci. U.S.A. 100, 9156–9161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liénard M. A., Hagström A. K., Lassance J. M., Löfstedt C. (2010) Evolution of multicomponent pheromone signals in small ermine moths involves a single fatty acyl reductase gene. Proc. Natl. Acad. Sci. U.S.A. 107, 10955–10960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Khannoon E. R., El-Gendy A., Hardege J. D. (2011) Scent marking pheromones in lizards: cholesterol and long chain alcohols elicit avoidance and aggression in male Acanthodactylus boskianus (Squamata: Lacertidae). Chemoecology 21, 143–149 [Google Scholar]
- 8. Hellenbrand J., Biester E. M., Gruber J., Hamberg M., Frentzen M. (2011) Fatty acyl-CoA reductases of birds. BMC Biochem. 12, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Spencer G. F. (1979) Alkoxy-acyl combinations in the wax esters from winterized sperm whale oil by gas chromatography-mass spectrometry. J. Am. Oil Chem. Soc. 56, 642–646 [Google Scholar]
- 10. Hajra A. K. (1983) in Ether Lipids: Biochemical and Biomedical Aspects (Mangold H. K., Paltauf F., eds) pp. 85–106, Academic Press, New York [Google Scholar]
- 11. Pollard M., Beisson F., Li Y., Ohlrogge J. B. (2008) Building lipid barriers: biosynthesis of cutin and suberin. Trends Plant Sci. 13, 236–246 [DOI] [PubMed] [Google Scholar]
- 12. Pollard M. R., McKeon T., Gupta L. M., Stumpf P. K. (1979) Studies on biosynthesis of waxes by developing jojoba seed II. The demonstration of wax biosynthesis by cell-free homogenates. Lipids 14, 651–662 [Google Scholar]
- 13. Lardizabal K. D., Metz J. G., Sakamoto T., Hutton W. C., Pollard M. R., Lassner M. W. (2000) Purification of a jojoba embryo wax synthase, cloning of its cDNA, and production of high levels of wax in seeds of transgenic Arabidopsis. Plant Physiol. 122, 645–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Metz J. G., Pollard M. R., Anderson L., Hayes T. R., Lassner M. W. (2000) Purification of a jojoba embryo fatty acyl-coenzyme A reductase and expression of its cDNA in high erucic acid rapeseed. Plant Physiol. 122, 635–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kolattukudy P. E. (1970) Reduction of fatty alcohols to acids by cell-free preparations of. Euglena gracilis. Biochemistry 9, 1095–1102 [DOI] [PubMed] [Google Scholar]
- 16. Cheng J. B., Russell D. W. (2004) Mammalian wax biosynthesis. I. Identification of two fatty acyl-coenzyme A reductases with different substrate specificities and tissue distributions. J. Biol. Chem. 279, 37789–37797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rowland O., Zheng H., Hepworth S. R., Lam P., Jetter R., Kunst L. (2006) CER4 encodes an alcohol-forming fatty acyl-coenzyme A reductase involved in cuticular wax production in Arabidopsis. Plant Physiol. 142, 866–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kavanagh K. L., Jörnvall H., Persson B., Oppermann U. (2008) Medium- and short-chain dehydrogenase/reductase gene and protein families: the SDR superfamily: functional and structural diversity within a family of metabolic and regulatory enzymes. Cell. Mol. Life Sci. 65, 3895–3906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen Z., Jiang J. C., Lin Z. G., Lee W. R., Baker M. E., Chang S. H. (1993) Site-specific mutagenesis of Drosophila alcohol dehydrogenase: evidence for involvement of tyrosine 152 and lysine 156 in catalysis. Biochemistry 32, 3342–3346 [DOI] [PubMed] [Google Scholar]
- 20. Fujimoto K., Hara M., Yamada H., Sakurai M., Inaba A., Tomomura A., Katoh S. (2001) Role of the conserved Ser-Tyr-Lys triad of the SDR family in sepiapterin reductase. Chem. Biol. Interact. 130, 825–832 [DOI] [PubMed] [Google Scholar]
- 21. Lassance J. M., Groot A. T., Liénard M. A., Antony B., Borgwardt C., Andersson F., Hedenström E., Heckel D. G., Löfstedt C. (2010) Allelic variation in a fatty-acyl reductase gene causes divergence in moth sex pheromones. Nature 466, 486–489 [DOI] [PubMed] [Google Scholar]
- 22. Doan T. T., Carlsson A. S., Hamberg M., Bülow L., Stymne S., Olsson P. (2009) Functional expression of five Arabidopsis fatty acyl-CoA reductase genes in Escherichia coli. J. Plant Physiol. 166, 787–796 [DOI] [PubMed] [Google Scholar]
- 23. Domergue F., Vishwanath S. J., Joubès J., Ono J., Lee J. A., Bourdon M., Alhattab R., Lowe C., Pascal S., Lessire R., Rowland O. (2010) Three Arabidopsis fatty acyl-coenzyme A reductases, FAR1, FAR4, and FAR5, generate primary fatty alcohols associated with suberin deposition. Plant Physiol. 153, 1539–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen W., Yu X.-H., Zhang K., Shi J., De Oliveira S., Schreiber L., Shanklin J., Zhang D. (2011) Male Sterile2 encodes a plastid-localized fatty acyl carrier protein reductase required for pollen exine development in Arabidopsis. Plant Physiol. 157, 842–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Doan T. T., Domergue F., Fournier A. E., Vishwanath S. J., Rowland O., Moreau P., Wood C. C., Carlsson A. S., Hamberg M., Hofvander P. (2012) Biochemical characterization of a chloroplast localized fatty acid reductase from Arabidopsis thaliana. Biochim. Biophys. Acta 1821, 1244–1255 [DOI] [PubMed] [Google Scholar]
- 26. Kosma D. K., Molina I., Ohlrogge J. B., Pollard M. (2012) Identification of an Arabidopsis fatty alcohol:caffeoyl-coenzyme A acyltransferase required for the synthesis of alkyl hydroxycinnamates in root waxes. Plant Physiol. 160, 237–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pulsifer I. P., Kluge S., Rowland O. (2012) Arabidopsis long-chain acyl-CoA synthetase 1 (LACS1), LACS2, and LACS3 facilitate fatty acid uptake in yeast. Plant Physiol. Biochem. 51, 31–39 [DOI] [PubMed] [Google Scholar]
- 28. Gietz R. D., Woods R. A. (2002) Transformation of yeast by the LiAc/ss carrier DNA/PEG method. Methods Enzymol. 350, 87–96 [DOI] [PubMed] [Google Scholar]
- 29. Kushnirov V. V. (2000) Rapid and reliable protein extraction from yeast. Yeast 16, 857–860 [DOI] [PubMed] [Google Scholar]
- 30. Yang W., Pollard M., Li-Beisson Y., Beisson F., Feig M., Ohlrogge J. (2010) A distinct type of glycerol-3-phosphate acyltransferase with sn-2 preference and phosphatase activity producing 2-monoacylglycerol. Proc. Natl. Acad. Sci. U.S.A. 107, 12040–12045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Domergue F., Abbadi A., Ott C., Zank T. K., Zähringer U., Heinz E. (2003) Acyl carriers used as substrates by the desaturases and elongases involved in very long-chain polyunsaturated fatty acids biosynthesis reconstituted in yeast. J. Biol. Chem. 278, 35115–35126 [DOI] [PubMed] [Google Scholar]
- 32. Teerawanichpan P., Qiu X. (2010) Fatty acyl-CoA reductase and wax synthase from Euglena gracilis in the biosynthesis of medium-chain wax esters. Lipids 45, 263–273 [DOI] [PubMed] [Google Scholar]
- 33. Aarts M. G., Hodge R., Kalantidis K., Florack D., Wilson Z. A., Mulligan B. J., Stiekema W. J., Scott R., Pereira A. (1997) The Arabidopsis Male Sterility 2 protein shares similarity with reductases in elongation/condensation complexes. Plant J. 12, 615–623 [DOI] [PubMed] [Google Scholar]
- 34. Schmid M., Davison T. S., Henz S. R., Pape U. J., Demar M., Vingron M., Schölkopf B., Weigel D., Lohmann J. U. (2005) A gene expression map of Arabidopsis thaliana development. Nat. Genet. 37, 501–506 [DOI] [PubMed] [Google Scholar]
- 35. Labesse G., Vidal-Cros A., Chomilier J., Gaudry M., Mornon J.-P. (1994) Structural comparisons lead to the definition of a new superfamily of NAD(P)(H)-accepting oxidoreductases: the single-domain reductases/epimerases/dehydrogenases (the “RED” family). Biochem. J. 304, 95–99 [DOI] [PMC free article] [PubMed] [Google Scholar]