

Abstract
Ten to fifteen percent of couples are confronted with infertility and a male factor is involved in approximately half the cases. A genetic etiology is likely in most cases yet only few genes have been formally correlated with male infertility. Homozygosity mapping was carried out on a cohort of 20 North African individuals, including 18 index cases, presenting with primary infertility resulting from impaired sperm motility caused by a mosaic of multiple morphological abnormalities of the flagella (MMAF) including absent, short, coiled, bent, and irregular flagella. Five unrelated subjects out of 18 (28%) carried a homozygous variant in DNAH1, which encodes an inner dynein heavy chain and is expressed in testis. RT-PCR, immunostaining, and electronic microscopy were carried out on samples from one of the subjects with a mutation located on a donor splice site. Neither the transcript nor the protein was observed in this individual, confirming the pathogenicity of this variant. A general axonemal disorganization including mislocalization of the microtubule doublets and loss of the inner dynein arms was observed. Although DNAH1 is also expressed in other ciliated cells, infertility was the only symptom of primary ciliary dyskinesia observed in affected subjects, suggesting that DNAH1 function in cilium is not as critical as in sperm flagellum.
Main Text
Male infertility affects more than 20 million men worldwide and represents a real health concern.1 It is a typical multifactorial disorder with a strong genetic basis and additional etiological factors such as urogenital infections, immunological or endocrine diseases, attack from reactive oxygen species (ROS), or perturbations from endocrine disruptors. To date, despite substantial efforts made to identify genes specifically involved in male infertility by many teams including ours,2,3 only a handful of genes have been formally correlated with human sperm defects. Male infertility caused by impaired sperm motility (asthenozoospermia) is also often observed in men with primary ciliary dyskinesia (PCD), a group of mainly autosomal-recessive disorders caused by dysfunctions of motile cilia leading primarily to respiratory infections and often to situs invertus. Recent research on PCD has been extremely prolific and allowed the identification and characterization of numerous proteins necessary for adequate axonemal molecular structure and assembly (Table S1 available online). The axoneme is a highly evolutionarily conserved structure found in motile cilia and in sperm flagella, mainly composed of an intricate network of microtubules and dyneins. Sperm parameters have not been systematically explored and are often only scarcely described in manuscripts investigating PCD-affected individuals. Although sperm flagella and motile cilia have a similar axonemal structure based on the presence of nine peripheral microtubule doublets plus two central ones, they present several differences that might explain why PCDs are not always associated with asthenozoospermia.4 We note that no mutations in axonemal genes have been described as being involved exclusively in infertility without also inducing PCD.
In the present study, we analyzed 20 subjects presenting with asthenozoospermia resulting from a combination of five morphological defects of the sperm flagella (absent, short, bent, and coiled flagella and flagella of irregular width) without any of the other PCD-associated symptoms. Similar phenotypes have been previously described and named “dysplasia of the fibrous sheath,” “short tails,” or “stump tails.”5–15 We propose to call this syndrome “multiple morphological anomalies of the flagella (MMAF),” a name that provides a more accurate description of this phenotype. We carried out a SNP whole-genome scan on 20 individuals presenting with severe MMAF. The study was approved by our local ethics committee; all individuals gave their signed informed consent and national laws and regulations were respected. All individuals originated from North Africa (11 Tunisians, 7 Algerians, and 2 Libyans) and were treated in Tunis (Clinique des Jasmins, Tunis, Tunisia) for primary infertility. Twelve of the subjects were born from related parents, usually first cousins. None of the subjects were related to one another apart from three individuals (P1–P3) who were brothers. All subjects had normal somatic karyotypes. All sperm analyses were performed at least twice, in accordance with the World Health Organization recommendations.16 Subjects were recruited on the basis of the identification of >5% of at least four of the aforementioned flagellar morphological abnormalities (absent, short, coiled, bent, and irregular flagella) (Table 1). All subjects presented with severe asthenozoospermia: 11 out of 20 subjects had no (0%) motility, 8 had sperm motility <10%, and one (P6) had 35% motility. Saliva was obtained from all participants via Oragene DNA Self-Collection Kit (DNAgenotech) but only one subject (P3) agreed to donate sperm and blood samples for research use. During their medical consultation for infertility, all subjects answered a health questionnaire focused on PCD manifestations, and none indicated suffering from any of the other symptoms encountered in PCD.
Table 1.
Semen Parameters of the 20 Subjects and the 7 Subjects Carrying DNAH1 Homozygous Variants
| Semen Parameters | Average of 20 Subjectsa | P1 | P2 | P3 | P6 | P8 | P9 | P17 |
|---|---|---|---|---|---|---|---|---|
| DNAH1 mutations | c.11788−1G>A (p.Gly3930Alafs∗120) | c.11788−1G>A (p.Gly3930Alafs∗120) | c.11788−1G>A (p.Gly3930Alafs∗120) | c.3877G>A (p.Asp1293Asn) | c.12796 T>C (p.4266Glnext∗21) | c.5094+1G>A (p.Leu1700Serfs72) | c.11788−1G>A (p.Gly3930Alafs∗120) | |
| Consanguinity | yes | yes | yes | no | yes | yes | no | |
| Origin of the subject | Tunisia | Tunisia | Tunisia | Algeria | Algeria | Algeria | Tunisia | |
| Sperm volume (ml) | 3.2 (1–5.5) | 5 | 2.5 | 2 | 5 | 4.5 | 3.5 | 2.5 |
| Sperm concentration 106/ml | 22 (0–59) | 45 | 0 | 2.8 | 57 | 11 | 53 | 31 |
| Motility (A+B) 1 hr | 2.5 (0–35) | 0 | NA | 2 | 35 | 0 | 0.5 | 0 |
| Vitality | 44 (6–73) | 22 | NA | NA | 73 | 61 | 48 | NA |
| Normal spermatozoa | 0.35 (0–6) | 0 | NA | 0 | 6 | 0 | 0 | 0 |
| Absent flagella | 30 (8–46) | 34 | + | 34 | + | + | 16 | 30 |
| Short flagella | 44 (16–70) | 38 | + | 44 | + | + | 70 | 20 |
| Coiled flagella | 13 (2–32) | 14 | NA | 14 | + | + | 12 | 32 |
| Angulation | 12 (2–19) | 4 | NA | 2 | + | + | 8 | 6 |
| Flagella of irregular caliber | 55 (16–92) | 48 | NA | 50 | + | + | 54 | 16 |
| Multiple anomalies index | 2.9 (1.9–3.9) | 3.1 | NA | 2.4 | NA | NA | 3 | 2.6 |
Values are expressed in percents, unless specified otherwise. Abbreviations are as follows: NA, not available; plus sign, anomalies reported (>5%) but not accurately quantified.
Values are expressed as the mean with the lower and higher values in parentheses.
Homozygosity mapping was carried out with 250K Sty1 SNP mapping arrays (Affymetrix) on DNA extracted from the 20 studied subjects’ saliva samples. Common regions of homozygosity were identified with the homoSNP software. After exclusion of the centromeric regions, we identified two regions located on chromosomes 3 and 20 with a region of homozygosity > 1 Mb common to 10/20 analyzed individuals (Figure S1). In addition, 4 and 9 subjects presented with a stretch of homozygosity > 15 Mb overlapping chromosomes 20 and 3 regions, respectively. All three brothers (P1–P3) were homozygous at the chromosome 3 region, although only two of them were homozygous at the chromosome 20 region. We excluded all other regions of homozygosity because they did not fulfil the following criteria: (1) more than eight individuals including at least two of the brothers sharing a region of homozygosity > 1 Mb and (2) presence of a potential candidate gene in the region according to its expression profile and/or presumed function. Finer analysis of the chromosome 3 region showed that 15 individuals were homozygous for two smaller subregions located at chr3: 46,745,396–47,606,570 and chr3: 52,111,974–53,028,375 (UCSC Genome Browser human reference genome build hg17, Figure S1). Sixteen genes are annotated in the first subregion (Table S2), among which only one gene (KIF9 [MIM 607910]) appeared as a good candidate; indeed, studies in the protist Trypanosoma brucei showed that kif9A (the mouse ortholog of human KIF9) is located in the axoneme and that its depletion alters motility.17 The second subregion in chromosome 3 includes 28 genes (Table S2). The dynein heavy chain 1 gene (DNAH1 [MIM 603332]) appeared as the best candidate gene because it codes for an axonemal dynein heavy chain and is expressed in various tissues including testis.18 Furthermore, asthenozoospermia was described in mice lacking Dnahc1, the DNAH1 mouse ortholog (previously named Mdhc7).19 Finally, among the ten genes located in the selected region of chromosome 20 (chr20: 33,572,687–34,070,415), only SPAG4 (MIM 603038) appeared as a good candidate: it was described in rat to be associated with the axoneme in elongating spermatids and epididymal sperm.20 We therefore decided to sequence KIF9 (RefSeq accession number NM_001134878.1), DNAH1 (RefSeq NM_015512.4), and SPAG4 (RefSeq NM_003116.1).
We sequenced the 12 exons and the intron boundaries of SPAG4 in the 13 individuals homozygous at this locus, and the 19 exons and intron boundaries of KIF9 in the 15 relevant individuals. We did not identify any likely pathogenic variants in these two genes. We then sequenced the 78 exons and intron boundaries of DNAH1 in P3 (primer sequences available in Table S3). We identified one homozygous splicing mutation (c.11788−1G>A) in intron 73. The same homozygous mutation was identified in the two other brothers (Figure S2). We then sequenced DNAH1 for the 17 remaining subjects. The same homozygous mutation (c.11788−1G>A) was identified in one additional individual (P17). We identified three other homozygous variants: another splicing mutation (c.5094+1G>A) in individual P9, a homozygous no-stop mutation disrupting the stop codon in exon 78 (c.12796T>C [p.4266Glnext∗21]) in individual P8, and a homozygous missense variant in exon 23 in individual P6 (c.3877G>A [p.Asp1293Asn]). The localization of the DNAH1 mutations is presented in Figure 1. If we consider only index cases, we identified 5 homozygous variants in 18 unrelated individuals (28%). None of these variants were detected in our control cohort of 100 individuals of North African origin. We note that the parents of the subjects could not be analyzed to confirm the transmission of the variants. We therefore cannot formally exclude the possibility that some of the identified variants may be hemizygous with a deletion on the other allele. However, depending on its size, its position, and its effect on the reading frame, a deleted allele would be at least as deleterious as the identified variants.
Figure 1.

Location of DNAH1 Mutations in the Intron-Exon Structure and in the Protein Representation of DNAH1
(A) DNAH1 genomic structure.
(B) DNAH1 domain map showing the location of the four identified mutations. The red boxes indicate the six known AAA-ATPase domains (AAA 1 to 6) as detected by homology (Uniprot server). The microtubule-binding domain (MBT) lies between AAA4 and AAA5. The N-terminal part of the protein binds to the intermediate, light-intermediate dynein chains. The position of the stalk and the microtubule-binding domain (MTB) are indicated.
To evaluate the association of the variants with the pathology, we compared their frequency in our cohort with that in the Exome Variant Server (EVS) database. At the four genomic positions of interest, the EVS data are of sufficient coverage to provide genotype calls for at least 6,200 individuals, corresponding to 12,400 alleles. There were no variant nucleotides identified at positions c.5094+1, c.11788−1, or c.12796, and only one A allele was identified out of 12,460 alleles at position c.3877. We performed Fisher exact tests (with fisher.test in R) to evaluate whether each observed SNV was statistically overrepresented in our cohort of 18 unrelated individuals, compared to EVS. All four individual SNVs are significantly enriched (p values: 5.9 × 10−11 for c.11788−1G>A, 8.1 × 10−6 for c.5094+1G>A and c.12796T>C, and 2.4 × 10−5 for c.3877G>A). We then investigated whether DNAH1 as a whole was significantly enriched in damaging SNVs in our cohort. Overall, EVS contains five nonsense and two splice-site SNVs, all observed heterozygously in a total of ten individuals. By using the coverage data available on the EVS website, we find that 20,737 positions covering the DNAH1 exons and intron boundaries had sufficient sequence coverage to be genotyped in 6,189 individuals on average. By contrast, counting only the two splice-site mutations as damaging, we observe 6 damaging alleles among 36 in our cohort: this represents a highly significant enrichment (Fisher exact test, p value: 3 × 10−12). Furthermore, we note that there were no homozygous damaging variants observed in the 6,189 EVS individuals compared to 3 in our cohort of 18 (Fisher exact test, p value: 3 × 10−8). Altogether we believe that these genetic results convincingly demonstrate that mutations in DNAH1 are associated with MMAF.
The c.5094+1G>A variant found in individual P9 affects DNAH1 intron 31 consensus donor splice site. The abnormal splicing is predicted to cause the prolongation of exon 31 until the introduction of a nonsense codon (p.Leu1700Serfs72). The position of the next donor site was predicted by “Splice Site Prediction by Neural Network.” Unfortunately we could not obtain any leukocytes from this subject to validate this prediction and observe whether this variant also led to nonsense-mediated mRNA decay (NMD). Because of the location of the variant on a consensus splice site and the unambiguous predictions of splice prediction software, we did not synthesize a minigene to verify the effect of this variant in vitro. A missense change, p.Asp1293Asn, was identified in P6. Interestingly, the Asp1293 amino acid is well conserved across species (Figure S3). This missense change is also predicted to be possibly damaging by SIFT and PolyPhen-2, two prediction softwares for nonsynonymous SNPs. It affects the N-ter of the protein (Figure 1B), known to be important for the structure of dynein arms.21 Variant p.4266Glnext∗21 found in individual P8 abolishes the stop codon in exon 78, leading to the addition of 21 codons at the 3′ end of the coding sequence. The role of the C-terminal domain is uncertain, but based on the D. discoideum structures, it may participate in long-range allosteric communication between microtubule-binding and ATPase regions.22,23 The addition of 21 extra amino acids to this region is likely to disrupt these interactions.
The c.11788−1G>A variant identified in four subjects (P1–P3 and P17) affects the final G nucleotide of DNAH1 intron 73, one of the consensus splice acceptor nucleotides. The resulting abnormal splicing is predicted to recognize a new CG acceptor site located just one nucleotide further, thus shifting the reading frame and inducing a premature stop codon (p.Gly3930Alafs∗120). As could be expected, P1–P3 share a common haplotype (Table S4). P17 also shares a common haplotype of 30 SNPs with P1–P3, suggesting a founder effect for this mutation. To assess the functional impact of the DNAH1 splice acceptor site mutation c.11788−1G>A, we studied mRNA products isolated from control and P3 lymphocytes (primer sequences available in Table S5). RT-PCR of P3’s samples yielded no product despite repeated attempts, whereas the three amplification attempts from control lymphocytes yielded the expected product (Figure 2A). RT-PCR targeting GAPDH (MIM 138400) and RPLP0 (MIM 180510) confirmed the integrity of P3’s RNA (Figure 2B). This suggests a specific degradation of the mutant DNAH1 transcripts by NMD. To further validate the pathogenicity of this variant, we analyzed DNAH1 localization in sperm from P3 by immunofluorescence and the ultrastucture of the flagella by electron microscopy. In control individuals, DNAH1 antisera decorated the full length of the sperm flagellum (Figure 2C), suggesting a putative role in the tethering of the inner dynein arms along the entire axoneme. In contrast, in sperm from individual P3 carrying the c.11788−1G>A mutation, DNAH1 immunostaining was absent, confirming that the splicing defect results in the degradation of the transcripts by NMD (Figure 2D). We next tested the integrity of the outer and inner dynein arms by using antibodies directed against DNALI1 and DNAI2, two well-established diagnostic markers of the inner and outer dynein arms, respectively. Staining with DNALI1 was strongly reduced in the sperm of individual P3, suggesting that inner arms were mostly absent in this individual (Figure 2F). On the other hand, the antibodies directed against DNAI2 stained the sperm flagella in both control and individual P3, suggesting that the outer dynein arms were not affected by the absence of DNAH1 (Figures 2E and 2F). In order to confirm that the inner arms were disorganized, we studied the ultrastructure of individual P3’s sperm by transmission electron microscopy (TEM) (Figure 3). We could observe 40 doublets of microtubules in cross sections presenting a sufficient quality to observe the dynein arms. Fifteen outer dynein arms (ODA) and only 4 inner dynein arms (IDA) were observed, confirming the complete disorganization of the IDA. Moreover, approximately one third of the microtubule doublets were malformed or absent in the observed sections. Furthermore, the central singlet of microtubules was missing (9+0) in 47% of these sections. The fibrous sheath was also strongly disorganized in 90% of the sections (Figure 3).
Figure 2.
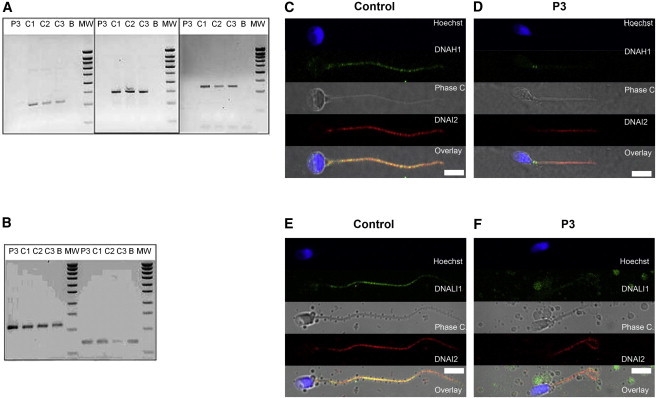
Analysis of P3 Carrying the c.11788−1G>A Variant Evidencing DNAH1 mRNA Decay by RT-PCR and the Absence of DNAH1 in Sperm by Immunolocalization
(A) RT-PCR analyses of subject P3 (c.11788−1G>A homozygote) and control individuals from the general population (C1–C3). Electrophoresis showing the RT-PCR amplification of DNAH1 exons 30–32, 64–66, 73–75. C1, C2, and C3 yield a normal fragment of 228, 293, and 241 bp, whereas subject P3 shows no amplification. There is no amplification from the RT-negative blank control (column B).
(B) Electrophoresis showing the amplification of the same cDNAs with GAPDH and RPL0 primers. Bands of equivalent intensity are obtained from all samples including P3. Reverse transcription was carried out with 500 ng of extracted RNA and oligo dT priming. Two microliters of the obtained cDNA mix was used for the subsequent PCR. PCR amplification was carried out with three couples of primers located in exons 30–32, 64–66, and 73–75 of DNAH1 at an elongation temperature of 57°C (40 cycles), in parallel to amplification of the same samples with the control housekeeping GAPDH and RPL0, respectively, at an elongation temperature of 60°C (35 cycles). RT-PCR primers are listed in Table S4.
(C and D) Immunofluorescence staining of human spermatozoa with DNAH1 antibodies (green) and DNAI2 (red). DNAH1 is observed throughout the flagellum in control sperm, whereas it is absent from P3’s sperm. In both control and P3 sperm, ODA is present as witnessed by the immunostaining of DNAI2.
(E and F) Immunofluorescence staining of human spermatozoa with DNALI1 antibodies (green) and DNAI2 (red). DNALI1, a marker of IDA, is localized throughout flagella in control sperm, whereas it is strongly reduced in sperm from P3. No difference is noticed in the coimmunostaining of DNAI2. Sperm were counterstained with Hoechst 33342 (blue) as nuclei marker. White scale bars represent 5 μm. Sperm cells were washed in phosphate-buffered saline (PBS), fixed in 4% PFA for 2 min at room temperature (RT), and washed twice in PBS. Fixed spermatozoa were allowed to air-dry on poly-L-lysine coated slides followed by permeabilization with 0.5% Triton X-100. Samples were then blocked with (PBS)/1% bovine serum albumin (BSA)/2% normal goat serum (NGS) for 30 min at RT. Slides were incubated with the primary antibodies 2 hr followed by an incubation with the secondary antibodies for 45 min at RT and mounting in Dako mounting medium (Dako). Appropriate controls were performed, omitting the primary antibodies. Polyclonal mouse DNALI1 and monoclonal mouse DNAI2 were purchased from Abcam (UK) and Abnova Corporation (Taiwan), respectively. Polyclonal DNAH1 antibodies were purchased from Prestige Antibodies (Sigma-Aldrich). Monoclonal mouse anti-acetylated-α-tubulin were purchased from Sigma-Aldrich. Highly cross-adsorbed secondary antibodies (Alexa Fluor 488 and Alexa Fluor 546) were obtained from Molecular Probes (Invitrogen).
Figure 3.

Electron Microscopy Analysis of Spermatozoa from P3, Carrying the c.11788−1G>A Variant, Reveals Numerous Ultrastructural Defects
(A) EM cross-section of a flagellum from a control individual sperm sample.
(B) Cross-section from the individual P3 sample showing numerous defects: lack of IDA (red arrow) and axonemal disorganization with mislocalized peripheral doublets (green arrows) associated with a displacement of the central pair (blue asterisk).
(C) Cross-section from individual P3 sperm flagellum showing a complete absence of the central pair.
(D) Cross-section from individual P3 sperm flagellum showing supernumerary dense fibers with absence of mitochondrion on the right side of the mid-piece.
(E and F) Drawings describing the normal sperm axoneme ultrastructure with their different components (E) and different defects observed in DNAH1 mutated subjects (F).
Sperm cells were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) during 2 hr at room temperature. Details of transmission electron microscopy technique were detailed previously.3
After complete DNA sequencing of DNAH1, we identified two variants altering a consensus splice site, highly likely to have a damaging effect, in 3 out of 18 unrelated individuals. The predicted effect of the other two identified variants is not as clear but the addition of 21 residues caused by the stop-loss variant is probably pathogenic. Interestingly, P1, P2, P3, P8, P9, and P17—who have inherited the variants predicted to have most severe effects—present 0% morphologically normal spermatozoa with a motility <2%, in contrast to P6, carrying the p.Asp1293Asn variant, who presents a milder phenotype with 35% motility and 6% morphologically normal spermatozoa (Table 1). These data therefore suggest that the c.3877G>A variant might be a hypomorphic allele, which is consistent with a single amino acid substitution in a large protein. Unfortunately we could not obtain any additional biological material from the other mutated subjects and in particular from P6 and could not assess the effects of the other variants on protein expression/localization and on the ultrastructure of the flagella. In addition, no mutations were identified in DNAH1 in 13 subjects, suggesting that MMAF is genetically heterogeneous. We are currently sequencing the exomes of these 13 subjects in order to identify other genes involved in MMAF. We note that with the exception of P6, who carried a missense mutation and presented a milder form of the pathology, we included here only individuals with the most severe phenotypes. We can therefore expect that individuals with intermediate asthenozoospermia and low levels of morphological anomalies could also harbor homozygous or compound heterozygous DNAH1 mutations of moderate severity.
The data we present here are consistent with the phenotype described for Dnahc1 knockout (KO) mice (the ortholog of DNAH1), which display asthenozoospermia and male infertility.19 In this model, however, no structural defects of the axoneme were observed either by optical or by transmission electronic microscopy.19 This contrasts with the strong axonemal disorganization we observed in sperm from P3 carrying the homozygous c.11788−1G>A mutation, where the inner dynein arms and the central pair of microtubules were mostly absent. In the Dnahc1 KO, however, the authors describe that the targeted deletion did not lead to a complete disruption of the gene and resulted in a truncated protein with a preserved N terminus.19 Because the N-terminal part of the DyHCs plays a crucial role in the assembly and stabilization of the inner dynein arms, as shown in Chlamydomonas mutants,24 it is likely that the formation of the base of the inner dynein arm is preserved and that the described KO animals could ensure correct axonemal biogenesis and organization. Alternatively, it is possible that the DNAH1 role in axonemal structure is not as central in mouse as it is in human.
Apart from infertility, none of the 20 individuals declared suffering from any of the principal PCD symptoms such as an impairment of the respiratory functions. This suggests that DNAH1 function in cilia is probably compensated by other HC dyneins. Previous phylogenetic studies indicate that DNAH3 (MIM 603334), DNAH7 (MIM 610061), and DNAH12 (MIM 603340) are close paralogs to DNAH1, DNAH12 being the closest.18 We therefore measured the relative expression of these four IDA heavy chains to assess whether the expression of these proteins could compensate the absence of DNAH1 in other ciliated tissues. By using qPCR (primer sequences available in Table S6), we showed that DNAH1 is expressed at a much higher level (8-fold) in the testis compared to the trachea (Figure 4). Conversely, the other IDA heavy chains are expressed at higher levels (DNAH3, DNAH7) or at a similar level (DNAH12) in control tracheal cells as in testis (Figure 4). Data available from public expression databases show a similar expression pattern to what was observed here. Moreover, these data show that whereas DNAH3 and DNAH7 expression is restricted to ciliated cells, DNAH1 and DNAH12 expression is rather atypical, because it is almost ubiquitous (EST profile viewer and GeneHub-GEPIS). We therefore note that not only are DNAH1 and DNAH12 closest to one another from a phylogenetic point of view, but they also share a broad expression pattern. We therefore believe that DNAH12 is the most likely candidate for a potential functional compensation of DNAH1 in ciliated cells. In addition and/or alternatively to this compensation, we cannot exclude the possibility that some of the affected individuals might retain expression of DNAH1 in the ciliated cells of the trachea, although this is not expected for the most severe variants identified. Alternatively, we cannot exclude a reduction of ciliary beats, which could lead to a small decrease of cilia function in respiratory epithelium or in other ciliated tissues without pathological consequences, or at least none that have been noticed by the affected men themselves. We could not obtain nasal brushings or curette biopsies from affected individuals and therefore cannot formally exclude this possibility. Future work on DNAH1 mutated subjects should include a thorough analysis of PCD symptoms including nasal nitric oxide measurements, video microscopy, and transcription electron microscopy. This would provide valuable information regarding the role of DNAH1 in ciliated cells as well as indicate whether mutated men might be at risk of developing PCD symptoms, perhaps as late onset.
Figure 4.
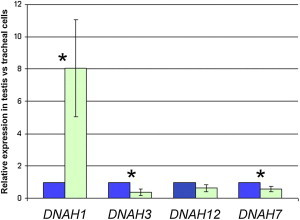
Relative mRNA Expression of DNAH1, DNAH3, DNAH7, and DNAH12 in Testis and in Tracheal Cells
Expression of DNAH1 mRNA (in green) in testis is significantly higher (8-fold) than in tracheal cells (in blue). Tracheal cell expression is set to 1. Expression of DNAH3 and DNAH7 mRNA are significantly lower in testis than in tracheal cells, whereas DNAH12 expression is not significantly different. Data are presented as mean ± standard deviation of three independent quantitative real-time PCR experiments. Statistical tests (paired t test) with a two-tailed p value ≤ 0.05 were considered as significant (∗). Human testis and trachea cDNA were obtained from Amsbio (Abingdon). mRNA expression were assessed by qPCR with a Biorad CFX9 (Biorad). PCR primers used to amplify DNAH1 and three other inner dynein arm heavy chain genes (DNAH3, DNAH7, and DNAH12) and the reference gene ACTB are listed in Table S5. The PCR cycle was as follows: 10 min 95°C, 1 cycle; 10 s 95°C, 30 s 58°C + fluorescence acquisition, 55 cycles. Analysis was performed with Biorad software CFX Manager v.3.0, with advanced relative quantification mode. Values for each gene were normalized to expression level of beta-actin gene (ACTB) via the 2-ΔΔCT method.25 The 2-ΔΔCT value was set at 0 in tracheal cells, resulting in an arbitrary expression of 1.
Inner dynein arms are organized in seven molecular complexes, viewed in electronic microscopy as globular heads arranged in 3-2-2 groups and corresponding to three different types of inner arms (IDA1 to IDA3, see Figure 5). In Dnahc1 KO mice, electron microscopy studies indicated that one head of the IDA3 was missing, leading to a 3-2-1 globular head arrangement, suggesting that DNAH1 is a component of IDA3. Radial spokes are present on microtubule doublets and interact with the inner arms. They allow a connection between external doublets of the microtubules and the two central microtubules. They are multiprotein complexes of more than 20 proteins. In mammals, there are three different radial spokes (RS1, RS2, and RS3) binding tightly to the inner arm bases of different IDAs. Among the different proteins involved in axonemal formation and organization, only mutations in CCDC39 (MIM 613798) and CCDC40 (MIM 613799) lead to a disorganization of the axonemal structure, a phenotype similar to what we observe in subjects with DNAH1 mutations. CDCC39 and CDCC40 control the assembly of the dynein regulatory complex (DRC), a major regulatory node interacting with numerous axonemal structures.27–29 In the absence of DRC, RS2 anchoring is weakened, leading to the displacement or the absence of the central pair and the mislocalization of the peripheral doublets. Interestingly, in T. thermophila, the RS3 stalk is directly connected to the dynein d/a tail through an arc-like structure (Figure 5).26 It can therefore be speculated that the absence of DNAH1 removes the anchoring site of the radial spoke 3. As a consequence, the attachment of the two central singlet microtubules should be weakened. This scheme is in agreement with our data that show 47% of 9+0 axonemes (Figure 3).
Figure 5.

Proposed Schematic Model for the Location and the Function of the Inner Arm Heavy Chain DNAH1 in the Axoneme of Human Sperm Flagellum
(A) Simplified representation showing a cross-sectional view of one microtubule doublet of an axoneme surrounding the central pair complex; the viewing is from the flagellar base. Light gray, central pair complex; light blue, outer doublets; dark blue, outer arm dyneins (OAD); dark pink, inner arm dynein heavy chain DNAH1; dark gray, radial spoke 3.
(B) Longitudinal view illustrating the approximate localization on the outer doublet A-tubule of the various dyneins and regulatory structures within a single 96 nm axonemal repeat. RS3 stalk is directly connected to the DNAH1 tail through an arc-like structure. DNAH1 may therefore stabilize the RS3.26 Light pink, inner arm dyneins (IAD); yellow, IC/LC, intermediate chain/light chain; orange, nexin-dynein regulatory complex (N-DRC); green, modifier of inner arms (MIA) complex; blue, calmodulin- and spoke-associated complex (CSC); purple, distal protrusion (DP).
As illustrated in Table S1, not all PCDs are associated with an infertility phenotype. For instance, subjects with mutations in CCDC114 (MIM 615038)30,31 and DNAH11 (MIM 603339)4 are fully fertile and could procreate spontaneously whereas subjects with DNAAF2 (MIM 612517), DNAH5 (MIM 603335), DNAI1 (MIM 604366), or HYDIN (MIM 610812)4,32–34 present complete sperm immobility. Also, mutations in genes involved in the preassembly of the dynein arms DNAAF1 (MIM 611390), DNAAF2 (MIM 612517), DNAAF3 (MIM 614566),32,35–37 and LRRC6 (MIM 614930),38 described to induce IDA loss, did not present axonemal microtubule disorganization. Consistently, IDAs are not or are only partially affected in deficient Chlamydomonas mutants for ODA7 (DNAAF1), ktu/pf13 (DNAAF2), and pf22 (DNAAF3): in ODA7 mutants, inner arms are not affected, in ktu/pf13 mutants only IDA dynein c is missing, and in pf22 mutants IDA dynein b and c are absent.27,29,36,37,39 Most importantly, IDA dynein f and p28 remain located in the flagella. It can thus be speculated that there are several pathways for the preassembly and/or the targeting of the different mammalian IDAs, thus explaining the absence of axonemal microtubule disorganization in subjects presenting with mutations in DNAAF1, DNAAF2, DNAAF3, and LRRC6. Here we observed that DNALI1 immunostaining was strongly reduced along the whole flagellum (Figure 2E), suggesting that DNALI1 may be located mainly in IDA3.40 In agreement with this result, p28, the Chlamydomonas ortholog of DNAIL1, is associated with the inner dynein arm located in IDA3. Interestingly, DNALI1 has an expression pattern similar to DNAH1: it presents a predominant testis expression and also a remarkable expression in nonciliated cells.41 Altogether, these facts suggest a close molecular partnership between DNAH1 and DNALI1.
Mutations affecting axonemal components and/or axoneme assembly often result in PCD, which frequently includes a male infertility phenotype. In this study we describe that mutations in DNAH1, which codes for an axonemal component, leads to male infertility only with no other apparent PCD-associated syndromes. Our data indicate that DNAH1 is required in spermatozoa for the formation of the inner dynein arms and that its absence is deleterious for the organization and biogenesis of the axoneme. Overall our data confirm that despite close structural similarities, sperm flagella and cilia present important divergences in axonemal organization and biogenesis.
Acknowledgments
This work was supported by the research grant ICG2I funded by the program GENOPAT 2009 from the French Research Agency (ANR). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We are grateful to Frédéric Plewniak, IGBMC, Strasbourg, who developed the homoSNP software used in this study (the software is available on request to plewniak@igbmc.u-strasbg.fr).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
Berkeley Drosophila Genome Project NNSplice 0.9, http://www.fruitfly.org/seq_tools/splice.html
dbSNP v.137, http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi
GeneHub-GEPIS, http://research-public.gene.com/Research/genentech/genehub-gepis/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
UniGene, http://www.ncbi.nlm.nih.gov/unigene
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Boivin J., Bunting L., Collins J.A., Nygren K.G. International estimates of infertility prevalence and treatment-seeking: potential need and demand for infertility medical care. Hum. Reprod. 2007;22:1506–1512. doi: 10.1093/humrep/dem046. [DOI] [PubMed] [Google Scholar]
- 2.Dieterich K., Soto Rifo R., Faure A.K., Hennebicq S., Ben Amar B., Zahi M., Perrin J., Martinez D., Sèle B., Jouk P.S. Homozygous mutation of AURKC yields large-headed polyploid spermatozoa and causes male infertility. Nat. Genet. 2007;39:661–665. doi: 10.1038/ng2027. [DOI] [PubMed] [Google Scholar]
- 3.Harbuz R., Zouari R., Pierre V., Ben Khelifa M., Kharouf M., Coutton C., Merdassi G., Abada F., Escoffier J., Nikas Y. A recurrent deletion of DPY19L2 causes infertility in man by blocking sperm head elongation and acrosome formation. Am. J. Hum. Genet. 2011;88:351–361. doi: 10.1016/j.ajhg.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwabe G.C., Hoffmann K., Loges N.T., Birker D., Rossier C., de Santi M.M., Olbrich H., Fliegauf M., Failly M., Liebers U. Primary ciliary dyskinesia associated with normal axoneme ultrastructure is caused by DNAH11 mutations. Hum. Mutat. 2008;29:289–298. doi: 10.1002/humu.20656. [DOI] [PubMed] [Google Scholar]
- 5.Rawe V.Y., Galaverna G.D., Acosta A.A., Olmedo S.B., Chemes H.E. Incidence of tail structure distortions associated with dysplasia of the fibrous sheath in human spermatozoa. Hum. Reprod. 2001;16:879–886. doi: 10.1093/humrep/16.5.879. [DOI] [PubMed] [Google Scholar]
- 6.Olmedo S.B., Rawe V.Y., Nodar F.N., Galaverna G.D., Acosta A.A., Chemes H.E. Pregnancies established through intracytoplasmic sperm injection (ICSI) using spermatozoa with dysplasia of fibrous sheath. Asian J. Androl. 2000;2:125–130. [PubMed] [Google Scholar]
- 7.Chemes H.E., Brugo S., Zanchetti F., Carrere C., Lavieri J.C. Dysplasia of the fibrous sheath: an ultrastructural defect of human spermatozoa associated with sperm immotility and primary sterility. Fertil. Steril. 1987;48:664–669. doi: 10.1016/s0015-0282(16)59482-5. [DOI] [PubMed] [Google Scholar]
- 8.Neugebauer D.C., Neuwinger J., Jockenhövel F., Nieschlag E. ‘9 + 0’ axoneme in spermatozoa and some nasal cilia of a patient with totally immotile spermatozoa associated with thickened sheath and short midpiece. Hum. Reprod. 1990;5:981–986. doi: 10.1093/oxfordjournals.humrep.a137232. [DOI] [PubMed] [Google Scholar]
- 9.Escalier D., Albert M. New fibrous sheath anomaly in spermatozoa of men with consanguinity. Fertil. Steril. 2006;86:e1–e9. doi: 10.1016/j.fertnstert.2005.12.042. [DOI] [PubMed] [Google Scholar]
- 10.Escalier D. Arrest of flagellum morphogenesis with fibrous sheath immaturity of human spermatozoa. Andrologia. 2006;38:54–60. doi: 10.1111/j.1439-0272.2006.00711.x. [DOI] [PubMed] [Google Scholar]
- 11.Escalier D., Gallo J.M., Schrével J. Immunochemical characterization of a human sperm fibrous sheath protein, its developmental expression pattern, and morphogenetic relationships with actin. J. Histochem. Cytochem. 1997;45:909–922. doi: 10.1177/002215549704500701. [DOI] [PubMed] [Google Scholar]
- 12.David G., Feneux D., Serres C., Escalier D., Jouannet P. [A new entity of sperm pathology: peri-axonemal flagellar dyskinesia] Bull. Acad Natl. Med. 1993;177:263–271. discussion 272–275. [PubMed] [Google Scholar]
- 13.Stalf T., Sánchez R., Köhn F.M., Schalles U., Kleinstein J., Hinz V., Tielsch J., Khanaga O., Turley H., Gips H. Pregnancy and birth after intracytoplasmic sperm injection with spermatozoa from a patient with tail stump syndrome. Hum. Reprod. 1995;10:2112–2114. doi: 10.1093/oxfordjournals.humrep.a136244. [DOI] [PubMed] [Google Scholar]
- 14.Moretti E., Geminiani M., Terzuoli G., Renieri T., Pascarelli N., Collodel G. Two cases of sperm immotility: a mosaic of flagellar alterations related to dysplasia of the fibrous sheath and abnormalities of head-neck attachment. Fertil. Steril. 2011;95:e19–e23. doi: 10.1016/j.fertnstert.2010.11.027. [DOI] [PubMed] [Google Scholar]
- 15.Marmor D., Grob-Menendez F. Male infertility due to asthenozoospermia and flagellar anomaly: detection in routine semen analysis. Int. J. Androl. 1991;14:108–116. doi: 10.1111/j.1365-2605.1991.tb01072.x. [DOI] [PubMed] [Google Scholar]
- 16.World Health Organization (2010). WHO Laboratory Manual for the Examination and Processing of Human Semen, fifth ed.
- 17.Demonchy R., Blisnick T., Deprez C., Toutirais G., Loussert C., Marande W., Grellier P., Bastin P., Kohl L. Kinesin 9 family members perform separate functions in the trypanosome flagellum. J. Cell Biol. 2009;187:615–622. doi: 10.1083/jcb.200903139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maiti A.K., Mattéi M.G., Jorissen M., Volz A., Zeigler A., Bouvagnet P. Identification, tissue specific expression, and chromosomal localisation of several human dynein heavy chain genes. Eur. J. Hum. Genet. 2000;8:923–932. doi: 10.1038/sj.ejhg.5200555. [DOI] [PubMed] [Google Scholar]
- 19.Neesen J., Kirschner R., Ochs M., Schmiedl A., Habermann B., Mueller C., Holstein A.F., Nuesslein T., Adham I., Engel W. Disruption of an inner arm dynein heavy chain gene results in asthenozoospermia and reduced ciliary beat frequency. Hum. Mol. Genet. 2001;10:1117–1128. doi: 10.1093/hmg/10.11.1117. [DOI] [PubMed] [Google Scholar]
- 20.Shao X., Tarnasky H.A., Lee J.P., Oko R., van der Hoorn F.A. Spag4, a novel sperm protein, binds outer dense-fiber protein Odf1 and localizes to microtubules of manchette and axoneme. Dev. Biol. 1999;211:109–123. doi: 10.1006/dbio.1999.9297. [DOI] [PubMed] [Google Scholar]
- 21.Habura A., Tikhonenko I., Chisholm R.L., Koonce M.P. Interaction mapping of a dynein heavy chain. Identification of dimerization and intermediate-chain binding domains. J. Biol. Chem. 1999;274:15447–15453. doi: 10.1074/jbc.274.22.15447. [DOI] [PubMed] [Google Scholar]
- 22.Moore D.J., Onoufriadis A., Shoemark A., Simpson M.A., Zur Lage P.I., de Castro S.C., Bartoloni L., Gallone G., Petridi S., Woollard W.J. Mutations in ZMYND10, a gene essential for proper axonemal assembly of inner and outer dynein arms in humans and flies, cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2013;93:346–356. doi: 10.1016/j.ajhg.2013.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Höök P., Vallee R. Dynein dynamics. Nat. Struct. Mol. Biol. 2012;19:467–469. doi: 10.1038/nsmb.2290. [DOI] [PubMed] [Google Scholar]
- 24.Myster S.H., Knott J.A., Wysocki K.M., O’Toole E., Porter M.E. Domains in the 1alpha dynein heavy chain required for inner arm assembly and flagellar motility in Chlamydomonas. J. Cell Biol. 1999;146:801–818. doi: 10.1083/jcb.146.4.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26.King S.M. A solid-state control system for dynein-based ciliary/flagellar motility. J. Cell Biol. 2013;201:173–175. doi: 10.1083/jcb.201302077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Antony D., Becker-Heck A., Zariwala M.A., Schmidts M., Onoufriadis A., Forouhan M., Wilson R., Taylor-Cox T., Dewar A., Jackson C., Uk10k Mutations in CCDC39 and CCDC40 are the major cause of primary ciliary dyskinesia with axonemal disorganization and absent inner dynein arms. Hum. Mutat. 2013;34:462–472. doi: 10.1002/humu.22261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Becker-Heck A., Zohn I.E., Okabe N., Pollock A., Lenhart K.B., Sullivan-Brown J., McSheene J., Loges N.T., Olbrich H., Haeffner K. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat. Genet. 2011;43:79–84. doi: 10.1038/ng.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Merveille A.C., Davis E.E., Becker-Heck A., Legendre M., Amirav I., Bataille G., Belmont J., Beydon N., Billen F., Clément A. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 2011;43:72–78. doi: 10.1038/ng.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Onoufriadis A., Paff T., Antony D., Shoemark A., Micha D., Kuyt B., Schmidts M., Petridi S., Dankert-Roelse J.E., Haarman E.G., UK10K Splice-site mutations in the axonemal outer dynein arm docking complex gene CCDC114 cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2013;92:88–98. doi: 10.1016/j.ajhg.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knowles M.R., Leigh M.W., Ostrowski L.E., Huang L., Carson J.L., Hazucha M.J., Yin W., Berg J.S., Davis S.D., Dell S.D., Genetic Disorders of Mucociliary Clearance Consortium Exome sequencing identifies mutations in CCDC114 as a cause of primary ciliary dyskinesia. Am. J. Hum. Genet. 2013;92:99–106. doi: 10.1016/j.ajhg.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Omran H., Kobayashi D., Olbrich H., Tsukahara T., Loges N.T., Hagiwara H., Zhang Q., Leblond G., O’Toole E., Hara C. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature. 2008;456:611–616. doi: 10.1038/nature07471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fliegauf M., Olbrich H., Horvath J., Wildhaber J.H., Zariwala M.A., Kennedy M., Knowles M.R., Omran H. Mislocalization of DNAH5 and DNAH9 in respiratory cells from patients with primary ciliary dyskinesia. Am. J. Respir. Crit. Care Med. 2005;171:1343–1349. doi: 10.1164/rccm.200411-1583OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olbrich H., Schmidts M., Werner C., Onoufriadis A., Loges N.T., Raidt J., Banki N.F., Shoemark A., Burgoyne T., Al Turki S., UK10K Consortium Recessive HYDIN mutations cause primary ciliary dyskinesia without randomization of left-right body asymmetry. Am. J. Hum. Genet. 2012;91:672–684. doi: 10.1016/j.ajhg.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loges N.T., Olbrich H., Becker-Heck A., Häffner K., Heer A., Reinhard C., Schmidts M., Kispert A., Zariwala M.A., Leigh M.W. Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am. J. Hum. Genet. 2009;85:883–889. doi: 10.1016/j.ajhg.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duquesnoy P., Escudier E., Vincensini L., Freshour J., Bridoux A.M., Coste A., Deschildre A., de Blic J., Legendre M., Montantin G. Loss-of-function mutations in the human ortholog of Chlamydomonas reinhardtii ODA7 disrupt dynein arm assembly and cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2009;85:890–896. doi: 10.1016/j.ajhg.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mitchison H.M., Schmidts M., Loges N.T., Freshour J., Dritsoula A., Hirst R.A., O’Callaghan C., Blau H., Al Dabbagh M., Olbrich H. Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nat. Genet. 2012;44:381–389. doi: 10.1038/ng.1106. S1–S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kott E., Duquesnoy P., Copin B., Legendre M., Dastot-Le Moal F., Montantin G., Jeanson L., Tamalet A., Papon J.F., Siffroi J.P. Loss-of-function mutations in LRRC6, a gene essential for proper axonemal assembly of inner and outer dynein arms, cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2012;91:958–964. doi: 10.1016/j.ajhg.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Freshour J., Yokoyama R., Mitchell D.R. Chlamydomonas flagellar outer row dynein assembly protein ODA7 interacts with both outer row and I1 inner row dyneins. J. Biol. Chem. 2007;282:5404–5412. doi: 10.1074/jbc.M607509200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mazor M., Alkrinawi S., Chalifa-Caspi V., Manor E., Sheffield V.C., Aviram M., Parvari R. Primary ciliary dyskinesia caused by homozygous mutation in DNAL1, encoding dynein light chain 1. Am. J. Hum. Genet. 2011;88:599–607. doi: 10.1016/j.ajhg.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rashid S., Breckle R., Hupe M., Geisler S., Doerwald N., Neesen J. The murine Dnali1 gene encodes a flagellar protein that interacts with the cytoplasmic dynein heavy chain 1. Mol. Reprod. Dev. 2006;73:784–794. doi: 10.1002/mrd.20475. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.