

Abstract
Importance
The leukodystrophies comprise a clinically and genetically heterogeneous group of progressive hereditary neurological disorders mainly affecting the myelin in the central nervous system. Their onset is variable from childhood to adulthood and presentation can be with a variety of clinical features that include mainly for adult-onset cases cognitive decline, seizures, parkinsonism, muscle weakness, neuropathy, spastic paraplegia, personality/behavioral problems, and dystonia. Recently, Rademakers and colleagues identified mutations in the CSF1R gene as the cause of hereditary diffuse leukoencephalopathy with spheroids (HDLS), offering the possibility for an in-life diagnosis. The detection of mutations in this gene in cases diagnosed with different clinical entities further demonstrated the difficulties in the clinical diagnosis of HDLS.
Objective
To better understand the genetic role of mutations in this gene, we sequenced a large cohort of adult-onset leukodystrophy cases.
Design
Whole-exome sequencing and follow up-screening by Sanger sequencing.
Setting
Collaborative study between the Institute of Neurology, University College London and the Inserm, Paris, France.
Participants
A total of 114 probands, mostly European patients, with a diagnosis of adult-onset leukodystrophy or atypical cases that could fit within a picture of leukodystrophy. These included 3 extended families within the spectrum of leukodystrophy phenotype.
Interventions
Whole-exome sequencing in a family and Sanger sequencing of CSF1R.
Main Outcomes and Measures
Mutations in CSF1R.
Results
We identified 12 probands with mutations in CSF1R. The clinical diagnoses given to these patients included dementia with spastic paraplegia, corticobasal degeneration syndrome, and stroke disorders. Our study shows that CSF1R mutations are responsible for a significant proportion of clinically and pathologically proven HDLS.
Conclusions and Relevance
These results give an indication of the frequency of CSF1R mutations in a European leukodystrophy series and expand the phenotypic spectrum of disorders that should be screened for this gene.
The leukodystrophies are a clinically and pathologically heterogeneous group of white matter diseases of the central nervous system. Recent genotypic and phenotypic advances have allowed a more specific classification of the leukodystrophies but there remain many difficulties in the diagnosis.1 Hereditary diffuse leukoencephalopathy with spheroids (HDLS) is a progressive autosomal dominant leukodystrophy with an onset typically occurring in the fourth or fifth decade of life (although with a wide spread in age at onset between 15-78 years). The clinical presentation is variable and patients have been described with dementia, seizures, parkinsonian features, spastic paraplegia, personality/behavioral changes, and dystonia.2-4 A definitive diagnosis of HDLS has therefore relied on the characteristic pathological features being seen at brain biopsy or post mortem, ie, loss of myelin sheaths and axonal destruction, axonal spheroids, gliosis, and autofluorescent lipid-laden macrophages.4-11 There is definite clinical overlap between HDLS and pigmentary orthochromatic leukodystrophy. The pathological hallmarks of the 2 diseases, axonal spheroids in HDLS and pigmented macrophages in pigmentary orthochromatic leukodystrophy, can be identified in both conditions and this supports HDLS and pigmentary orthochromatic leukodystrophy being referred to collectively, in a review of these 2 conditions, as adult-onset leukoencephalopathy with axonal spheroids and pigmented glia.2
The genetic basis of HDLS has been elusive until very recently when mutations in the colony stimulating factor 1 receptor (CSF1R) gene were identified by Rademakers and colleagues12 as the cause of disease. The mutations identified reside in the CSF1R kinase domain and preliminary functional investigations suggest that mutations disrupt kinase activity most likely by affecting the phosphorylation of downstream targets. After identifying mutations in CSF1R in 1 familial case through exome sequencing, and in an effort to establish genotype-phenotype correlations in this disorder, we sequenced CSF1R in a large cohort of patients with adult-onset leukodystrophy.
METHODS
Human Subjects
Ethical approval and informed consent were obtained from all institutions. In family F375/174, we performed exome sequencing in 3 family members. This French family presented an autosomal dominant pattern of inheritance of a frontotemporal dementia/corticobasal degeneration syndrome phenotype with an age at onset ranging between 40 and 59 years. Secondary screening included 113 cases of adult-onset leukodystrophy. Brain tissue was available for 25 individuals with a clinical or pathological diagnosis of leukodystrophy from the Netherlands; Maryland; Queen Square, London, England; and the Centre de Ressources Biologiques, Angers, France. The main inclusion criterion was white matter abnormalities on magnetic resonance imaging (MRI) consistent with a leukodystrophy, ie, symmetrical, confluent, hypersignal on T2-weighted images and prominent T1 hypointensity of the affected white matter relative to gray matter structure, but excluding those with patchy or asymmetrical features that can be associated with diagnoses such as multiple sclerosis, CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy), metachromatic leukodystrophy, or Alexander disease.
Exome Sequencing
Exome sequencing was performed on 3 individuals from family F375/174: 2 affected cousins (patients 001 and 006) and the unaffected mother (case 007) of patient 006 (Figure 1). Mutations in the known frontotemporal genes were previously excluded in this family by Sanger sequencing (granulin[GRN], microtubule-associated protein tau [MAPT], valosin-containing protein [VCP], TAR DNA binding protein [TARDBP], fused in sarcoma [FUS], and prion protein [PRNP]) or repeat-primed polymerase chain reaction (PCR) (chromosome 9 open reading frame 72 [C9ORF72]). Large structural variants were also excluded by analysis of whole-genome genotyping arrays.
Figure 1. Pedigrees of Families F375/174, F662, and F232.
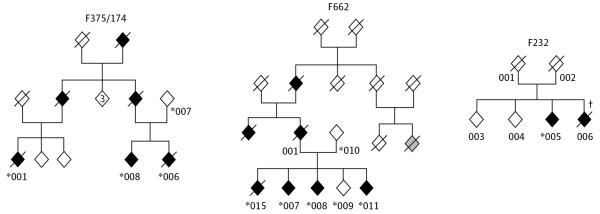
Sex is masked for confidentiality purposes. Affected individuals are represented with black, filled diamonds; unaffected family members, white diamonds. Exome sequencing was performed in individuals 001, 006, and 007 of family F375/174. *Individuals tested for the respective mutation found in CSF1R. †Individual in whom brain biopsy was performed.
The approach used assumed a dominant mode of inheritance where shared variants could be determined in the affected cases and subsequently filtered out in the unaffected mother. Genomic DNA was prepared according to Illumina TruSeq Sample Preparation version 3 (Illumina) and whole-exome capture was performed with Illumina TruSeq Exome Enrichment according to the manufacturer’s instructions. Sequencing was performed in Illumina HiSeq2000 using 100–base pair paired-end reads.
Following quality-control procedures, samples yielded between 7.2 and 9.3 Gb of high-quality, aligned data. This amount of data represented mean target coverage between 48.6 and 66.4 times, percentages of targets covered at 10 times of 87.4% to 88.3% or greater and less than 0.5% of targets not being covered at least once. Sequence alignment and variant calling were performed against the reference human genome (UCSC hg19) using bwa13 and the Genome Analysis Toolkit.14 The PCR duplicates were removed prior to variant calling using Picard software (http://picard.sourceforge.net/index.shtml). Based on the hypothesis that the mutation underlying this rare familial disease was not present in the general population, we excluded all single-nucleotide polymorphisms identified in the 1000 Genomes Project (http://www.1000genomes.org/) or dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/).
Sanger Sequencing
ExonPrimer was used to generate primers for amplification of CSF1R exons 1 to 22 plus exon-intron boundaries (eTable 1 in the Supplement). These exons were PCR amplified using Roche FastStart PCR Master Mix polymerase (Roche Diagnostics) and Sanger sequenced using the standard Applied Biosystems protocol. Mutations were named based on sequences with accession numbers NM_005211.3 and NP_005202.2. In silico analyses using PolyPhen-215 and SIFT16 were performed to predict mutation pathogenicity.
Neuropathological Study
Postmortem neuropathological examination was carried out in all 25 cases in the respective brain banks, and additional pathological analysis was performed in all mutation-positive cases by 2 neuropathologists at the Queen Square Brain Bank (T. R. and R. T.). Frozen cortical tissue blocks from one of the frontal lobes was available for analysis in 5 cases (cases 1, 2, 5, 6, and 8) and paraffin sections from representative brain regions, including major cortical areas with subcortical and deep white matter, hippocampal formation with the parahippocampus, basal ganglia, thalamus, midbrain, pons, medulla, and cerebellum, were available for analysis from case 4. Detailed methods used in the neuropathological study of these cases are given in the eAppendix in the Supplement.
RESULTS
Genetic Sequencing
Exome sequencing in a French family (F375/174) revealed the CSF1R mutation p.R777Q to be shared between the affected cousins and absent in the mother. This finding and the publication of mutations in CSF1R as the cause of HDLS12 led us to screen the gene in additional families and cases with similar phenotypes. A total of 12 heterozygous mutations (eTable 2 in the Supplement and Figure 2) were found in the 114 probands studied (11%); 5 of 25 cases (20%) with pathological HDLS diagnosis; and 6 of 88 (7%) of the clinical series of patients with white matter abnormalities on brain MRI. The main clinical and neuropathological characteristics of the 12 probands carrying CSF1R mutations and 7 relatives are described in the Table. Mean (SD) age at onset was 42.4 (11.4) years (range, 20-60 years), with mean (SD) disease duration of 10.4 (8.9) years (range, 3-29 years) and a mean (SD) age at death of 56 (8.6) years (range, 36-65 years).
Figure 2. Mutations Identified in CSF1R Associated With Hereditary Diffuse Leukoencephalopathy With Spheroids.

A, The CSF1R gene structure. B, Exons 12 to 22, which code the protein kinase domain of the protein, with all the mutations identified. The bottom of part B shows mutations described by Rademakers et al,12 and the top of part B shows mutations described in the present study. Red arrows represent novel mutations described herein. Black arrows represent previously reported mutations.
Table. Main Clinical and Neuropathological Characteristics of the 12 Probands Carrying CSF1R Mutations and 7 Relatives.
| Family | Individual ID | Origin | Sex | Age at Onset, y | Age at Death, y | Family History | Mutation | Clinical Diagnosis | Main Clinical Features | Main Neuropathology Findings |
|---|---|---|---|---|---|---|---|---|---|---|
| F375/174 | 001 | France | F | 60 | 65 | + | P.R777Q | FTD/CBDS | Progressive dementia; severe apraxia; dysarthric and hypophonic speech; myoclonic jerks; leukodystrophy | NA |
| F375/174 | 006 | France | M | 44 | 48 | + | P.R777Q | FTD/CBDS | Frontal dementia; apraxia; myoclonic jerks; postural tremor; dysarthric speech; seizures; leukodystrophy | NA |
| F375/174 | 008 | France | M | 58 | Alive (60 y) | + | P.R777Q | FTD | Behavioral and cognitive impairment; discrete WM lesions | NA |
| F662 | 007 | France | F | 47 | Alive (51 y) | + | P.Y856H | FTD/HDLS | Severe frontal dementia; gait apraxia; dysarthria and hypophonia; stuttering; palilalia/echolalia; leukodystrophy | NA |
| F662 | 015 | France | F | 39 | 42 | + | P.Y856H | Pallido-ponto-nigral degeneration | Severe frontal dementia; parkinsonism; dysarthria and hypophonia; stuttering; palilalia and echolalia; myoclonic jerks; dystonia; seizures; leukodystrophy | NA |
| F662 | 008 | France | F | 42 | Alive (48 y) | + | P.Y856H | CBDS | Frontal dementia; apraxia; dystonia; parkinsonian rigidity; myoclonic jerks; seizures | NA |
| F662 | 011 | France | M | 42 | Alive (43 y) | + | P.Y856H | FTD | Behavioral and cognitive impairment | NA |
| F662 | 001 | France | M | 55 | 64 | + | p.Y856Ha | Parkinsonism | Parkinsonism; dysarthria; cognitive impairment | NA |
| F232 | 5 | France | F | 42 | Alive (44 y) | + | P.I827T | Leukodystrophy | Frontal syndrome; tremor; cerebellar ataxia; leukodystrophy | NA |
| F232 | 6b | France | F | 39 | 41 | + | NA | Leukodystrophy | Frontal dementia; ataxia; leukodystrophy | HDLS |
| Case 1 | US | F | 24 | 53 | ? | p.T567fsX44 | Progressive terminal MS | Weakness; spasticity; severe peripheral neuropathy | Occasional axonal spheroids, severe myelin and axonal pallor, axonal loss, gliosis, and pigmented macrophages | |
| Case 2 | The Netherlands | M | 45 | 52 | − | P.L630R | FTD | Mixed pyramidal and extrapyramidal signs | Frequent axonal spheroids, moderate myelin and axonal pallor, pigmented macrophages | |
| Case 3 | Greece | F | 46 | 54 | − | P.E633K | Spasticity | Rapid progression; cognitive decline, spasticity, seizures, and dystonia; behavioral symptoms; leukodystrophy | NA | |
| Case 4 | UK | F | 46 | 52 | + | P.R777W | FTD | Progressive behavioral and cognitive impairment; generalized tonic-clonic seizures | Severe myelin and axonal loss in cerebral WM with frequent axonalspheroids, gliosis, and pigmented macrophages | |
| Case 5 | The Netherlands | M | 46 | 49 | − | P.I794T | Presenile dementia | Severe dementia; leukodystrophy | Frequent axonal spheroids, moderate myelin and axonal pallor, pigmented macrophages | |
| Case 6 | US | M | 21 | 36 | − | P.L817P | Possible stroke disorder | Leukodystrophy with cognitive decline and headaches | Frequent axonal spheroids, moderate myelin and axonal pallor, pigmented macrophages | |
| Case 7 | UK | F | 44 | Alive (49 y) | + | P.E847D | Leukodystrophy of unclear cause | Progressive cognitive impairment and spastic paraparesis | NA | |
| Case 8 | US | NA | 20 | 45 | − | P.P901S | Possible MS | Asymmetrical increased tone; cognitive decline; leukodystrophy | Severe myelin pallor, axonal loss, gliosis, and pigmented macrophages | |
| Case 9 | UK | F | 50 | 52 | + | c.2655-2A>G | FTD | Progressive aphasia; later frontal dementia; motor signs; upper limb myoclonus | NA |
Abbreviations: CBDS, corticobasal degeneration syndrome; FTD, frontotemporal dementia; HDLS, hereditary diffuse leukoencephalopathy with spheroids; MS, multiple sclerosis; NA, not available; UK, United Kingdom; US, United States; WM, white matter; +, positive; −, negative; ?, possible.
Obligate carrier.
Represents individual in whom a brain biopsy was performed but no genetic screening was completed.
Neuropathological Findings
In all cases, the microscopical findings included moderate to severe myelin and axonal loss; astrogliosis; variable numbers of macrophages, which were frequently pigment laden; and axonal spheroids in cerebral white matter (Figure 3A-C). The axonal spheroids varied between 10 and 50 μm in diameter, were readily recognizable on the hematoxylin-eosin–stained histological sections; and were argyrophilic on the Bielschowsky silver impregnation. The spheroids were uniformly immunoreactive for phosphorylated neurofilaments and amyloid precursor protein, with variable positivity for ubiquitin and p62 (Figure 3A). In case 4, where several tissue blocks from representative anatomical areas were available for neuropathological assessment, extensive involvement of the centrum semiovale and periventricular white matter could be ascertained, with the frontal white matter showing the most severe changes followed by the temporal and parietal white matter. Involvement of long tracts, such as the optic radiation, the corticospinal tract in the internal capsule, or the frontopontine and temporopontine tracts in the midbrain cerebral peduncle, was noted. The myelinated axons of the cortical arcuate fibers, which also contained significant numbers of axonal spheroids, were frequently better preserved than the axons of the underlying subcortical and deep white matter, which were often severely depleted of axons. The presence of occasional spheroids in the deeper cortical laminae was also noted. Scattered cortical neurons had swollen cytoplasm, which was positively stained with antibodies to phosphorylated neurofilaments (ballooned neurons). The basal ganglia and cerebellum showed minimal involvement. Additional immunohistochemical studies demonstrated sparse Aβ-positive parenchymal plaques in case 2 and sparse neurofibrillary tangles in the transentorhinal cortex in case 4, in whom tissue blocks from major representative anatomical areas were available. α-Synuclein and TDP-43 immunohistochemical studies were negative in all cases.
Figure 3. Axonal Spheroids Stained With an Antibody to Phosphorylated Neurofilaments in Case 4 With Hereditary Diffuse Leukoencephalopathy With Spheroids.

Where the CSF1R p.R777W mutation was identified (A). Scattered macrophages (B), often with pigment-laden cytoplasm (C), were also characteristic. For comparison, axonal spheroids from the spinal cord of a case with PLA2G6 mutation (D) and the external globus pallidus from a case with PANK2 mutation (E) are also given. The bar in part A represents 45 μm for parts A, B, and D; 22.5 μm in part E; and 16 μm in part C. A, D, and E, RT97; B, C68; and C, Luxol fast blue, cresyl violet.
Detailed Case Description From 2 Families
These descriptions illustrate the range of symptoms and signs that patients with HDLS and CSF1R mutations are given. The overall diagnosis in these patients varies, suggesting that a range of clinical phenotypes, when associated with a leukodystrophy on imaging, should be screened for CSF1R mutations.
Family F662 (France)
The proband (patient 007, Figure 1) was a 48-year-old woman who presented with a 1-year history of behavioral symptoms and cognitive impairment. She had become apathetic and disinhibited, with inappropriate laughing and emotional lability. She had also been laid off from her work because of repeated errors. On examination, frontal release signs were present, micrographia was noted, and in the limbs, there was bradykinesia and rigidity. Reflexes were brisk throughout with an extensor plantar on the right. She had abnormal speech consisting of dysarthria, stuttering, and hypophonia. Her Mini-Mental State Examination score was 18 of 30 and detailed neuropsychological evaluation showed severe executive dysfunction, episodic memory impairment, and limb apraxia. The initial diagnosis was of an atypical frontotemporal dementia. At 4 years from symptom onset, speech output had become severely reduced, limited to verbal stereotypies and echolalic productions. She had severe limb and gait apraxia and there was myoclonus. Brain MRI revealed frontal and callosal atrophy predominantly, with diffuse white matter hyperintensity bilaterally with relative sparing of U fibers (Figure 4). Brain single-photon emission computed tomography revealed marked hypoperfusion of prefrontal dorsolateral and orbitoventral cortices bilaterally.
Figure 4. Brain Magnetic Resonance Imaging of Patients With CSF1R Mutations From Families F375/174 and F662.

A and B, Axial T2-weighted imaging for patient F375/174-008 (disease duration of 2 years) showing moderate frontal atrophy and discrete periventricular hyperintensity (arrows). C and D, Axial fluid-attenuated inversion recovery (C) and coronal T2-weighted (D) imaging for patient F662-008 (disease duration of 3 years) showing severe white matter hyperintensity and callosal atrophy. E, Sagittal T1-weighted imaging for patient F662-008.
Her sister (patient 015, Figure 1) presented with a parkinsonian phenotype at age 39 years, was diagnosed with pallido-ponto-nigral degeneration, and died at the age of 42 years. Two other siblings were also affected. A sister (patient 008, Figure 1) developed left upper limb rigidity and dystonia with rest tremor and myoclonic jerks at the age of 42 years and a younger brother (patient 011, Figure 1) developed impairment of behavior and cognition at the age of 42 years. The mother (case 010, Figure 1) was healthy at age 80 years. However, the father (patient 001, Figure 1) had developed a gait disorder with rigidity and bradykinesia in the upper limbs at age 55 years and died at the age of 64 years. A paternal aunt had parkinsonism and dementia at the age of 55 years and died at age 58 years. The paternal grandfather had developed a gait disorder and repeated strokelike episodes at the age of 40 years, dying at the age of 48 years.
Case 4 (England)
The proband was a 48-year-old, right-handed woman initially admitted to an inpatient ward following 2 generalized tonic-clonic seizures. She was found to have a 2-year history of progressive change in personality. She had become disinhibited, often laughing inappropriately, and had developed a craving for boiled sweets with an associated increase in her weight. She had also become less empathetic in her job within the caring profession. At home she had been increasingly apathetic and had lost her libido. Her memory was relatively intact and there were no concerns of problems in other cognitive domains. She had no past medical problems but her father had died of dementia at the age of 57 years. On examination, she had a Mini-Mental State Examination score of 20 of 30 with little insight into her problems. She had little spontaneous speech but the content of her speech was normal. Her main cognitive impairment was executive dysfunction with impaired verbal fluency, concrete thinking, and poor cognitive estimates. Her MRI brain scan showed predominantly frontal lobe atrophy, with temporal and parietal atrophy to a lesser extent as well as extensive bilateral white matter changes. Her clinical syndrome and the pattern of brain atrophy were felt to be consistent with a frontotemporal dementia, although the white matter changes were considered unusual for this diagnosis. Over the following years, she had progressive behavioral and cognitive impairment and died at the age of 52 years. Postmortem brain examination showed severe atrophy affecting the frontal, temporal, and parietal lobes with relative preservation of the occipital lobes. The cortical ribbon appeared to be of normal thickness; however, the underlying white matter showed severe changes.
DISCUSSION
Hereditary diffuse leukoencephalopathy with spheroids and orthochromatic leukodystrophy are overlapping disorders that display clinical and genetic heterogeneity. The recent finding of mutations in CSF1R as the genetic cause in several cases with pathologically proven HDLS12 has advanced our understanding of this form of leukodystrophy and provided an important diagnostic test.
In our series of leukodystrophy cases, we identified 12 probands or cases with mutations in the CSF1R gene. CSF1R mutations were, therefore, responsible for 11% (12 of 114) of the overall series; 7% (6 of 88) of the clinical series of patients with leukodystrophy on brain MRI, and 20% (5 of 25) of the patients with a pathological diagnosis of HDLS. Age at onset ranged from 20 to 60 years; disease duration, from 3 to 29 years; and age at death, from 36 to 65 years. Marked intrafamilial variability was observed, with ages at onset differing by 16 years among family members in both F375/174 and F662 and clinical features like parkinsonism, dystonia, and seizures being present in only some cases within a family (F662). Cognitive decline was the presenting and most prominent feature in the majority of cases, whereas spasticity and other movement disorders were reported in the remaining cases. The clinical diagnosis given to the patients included multiple sclerosis, presenile dementia, and multi-infarct dementia, reflecting the difficulty in the clinical diagnosis of HDLS, which has been previously exemplified.4,12 With the exception of family F662, none of these cases has been clinically diagnosed with HDLS, showing the importance of including HDLS in the differential diagnosis of familial presenile dementia and molecular screenings of early-onset dementia cases.
So far, all the mutations in CSF1R associated with HDLS12,17 are located in the tyrosine kinase domain of the CSF1R protein, encoded by exons 12 to 22 of the gene, suggesting a crucial role of the CSF1R-dependent phosphorylation of tyrosine residues in the development of HDLS. We identified several polymorphisms throughout the whole gene (eTable 3 in the Supplement), but all mutations thought to be associated with disease were also found to be located in these exons. With the exception of families F375/174 and F662, where segregation of the respective mutations with the disease was demonstrated, in all other cases, it is not possible to establish a definite pathogenicity. Nonetheless, both PolyPhen-2 and SIFT in silico analyses of pathogenicity predict these mutations to be pathogenic. Additionally, none of these changes has been reported in the large-scale genome and exome sequencing databases (dbSNP132; 1000 Genomes Project, March 2012 release; and the National Heart, Lung, and Blood Institute Exome Sequencing Project [http://evs.gs.washington.edu/EVS/]).
There seems to be no correlation between mutations’ location in the gene or the type of amino acid change with the age at onset or disease duration. However, in our series of cases with CSF1R mutations, an earlier age at onset of the disease (in the early 20s) seems to be associated with longer disease duration (average of 23 years). Also, until now, no disease-associated mutations have been found in exons 15 or 16 of the gene. These exons code the kinase insert domain of the protein, which splits the kinase domain and has been shown not to be necessary for intrinsic activity, although its role in receptor internalization and degradation and in protein-protein interaction during signal transduction is not completely elucidated.18,19 The fact that no mutations have been found in the kinase insert domain further suggests that this domain is not essential for the pathological pathways in HDLS. Further studies identifying mutations in this gene are necessary to prove this hypothesis.
All our cases with a CSF1R mutation that were reviewed pathologically showed microscopic features of a leukoencephalopathy with variable loss of myelin and axons in cerebral white matter. In addition, axonal spheroids, immunoreactive for phosphorylated neurofilaments, variably for ubiquitin and p62, were a prominent feature and lipid- or pigment-laden macrophages and astrogliosis were also seen. These microscopic findings are in keeping with the diagnosis of HDLS, which was originally described in a large Swedish pedigree.3 Data from morphological studies suggest that HDLS and familial pigmentary orthochromatic leukodystrophy, in which the presence of prominent sudanophilic lipopigment (ceroid) in macrophages and glia is an additional feature, are variants of the same disorder.2,4,20 However, it remains to be proven whether the 2 conditions share a common genetic background.12
Apart from HDLS, axonal spheroids are a major morphological feature of a number of other hereditary neurodegenerative conditions, including neurodegeneration with brain iron accumulation types 1 and 2, associated with mutations in the PANK2 and PLA2G6 genes, respectively.21-23 Ultrastructurally, axonal spheroids contain neurofilaments, dense and vesicular bodies, and degenerating mitochondria.17,20 Because CSF1R plays a major role in proliferation and differentiation of microglia in the brain, microglial dysfunction could be a major factor in HDLS pathogenesis.12 Recent data from CSF1R-deficient (CSF1R−/−) mice also suggest novel roles for CSF1R such as regulation of corticogenesis by influencing proliferation and apoptosis of neocortical progenitors and differentiation of specific excitatory neuronal subtypes, which may also be relevant for HDLS pathogenesis.24
In conclusion, based on the limited information available to date on HDLS, a phenotype-genotype correlation is difficult to determine. Detailed guidelines for genetic testing are also difficult to establish; we suggest that CSF1R sequencing should be considered in all patients with adult-onset disease with white matter abnormalities on T2-weighted MRI combined with unspecific symptoms of cognitive decline and movement disorders (mainly lower limb spasticity) and a history of depression and headaches, irrespective of family history.
Supplementary Material
Acknowledgments
Funding/Support: This work was supported in part by Alzheimer’s Research UK, the Medical Research Council, the Wellcome Trust, Parkinson’s Disease Foundation USA, the Brain Research Trust, the National Organization for Rare Disorders, the Motor Neurone Disease Association, the Wellcome Trust/MRC Joint Call in Neurodegeneration award (WT089698) to the UK Parkinson’s Disease Consortium (whose members are from the Institute of Neurology, University College London; the University of Sheffield; and the MRC Protein Phosphorylation Unit, University of Dundee), the program “Investissements d’avenir” ANR-10-IAIHU-06, and an anonymous donor. Some of this work was funded/supported by the National Institute for Health Research Biomedical Research Centre at University College London Hospitals NHS Foundation Trust, the National Institute for Health Research Biomedical Research Unit in Dementia based at University College London Hospitals, University College London, and an Alzheimer’s Research UK Fellowship (Dr Guerreiro).
Additional Contributions: We are grateful to France Alzheimer; Instituts Hospitalo-Universitaires; Frédérique Etcharry-Bouix, MD, PhD, and Annick Barthelaix-Pouplard, MD, PhD, CHU Angers, Angers, France; Serge Belliard, MD, PhD, CHU Rennes, Rennes, France; Claudie Schleisch, MD, Rennes; Roger Gil, MD, CHU Poitiers, Poitiers, France; Bertrand Fontaine, MD, PhD, Pitié-Salpêtrière Hospital, Paris, France; Jean-Jacques Hauw, MD, PhD, Laboratoire de neuropathologie Escourolle, Hopital de la Salpêtrière, Paris; the Centre de Ressource Biologiques, CHU Angers; and the DNA and Cell Bank, ICM, Pitié-Salpêtrière Hospital and to all referring clinicians and patients for participation in the study. We thank the MRC London Neurodegenerative Diseases Brain Bank, the Eunice Kennedy Shriver National Institute of Child Health and Human Development Brain and Tissue Bank for Developmental Disorders (Maryland Brain Bank), and the Netherlands Brain Bank for providing tissue samples. We also thank the National Heart, Lung, and Blood Institute Grand Opportunity (GO) Exome Sequencing Project and its ongoing studies, which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926), and the Heart GO Sequencing Project (HL-103010).
Footnotes
Conflict of Interest Disclosures: None reported.
Disclaimer: The views expressed are those of the authors and not necessarily those of the National Health Service, National Institute for Health Research, or Department of Health.
REFERENCES
- 1.Costello DJ, Eichler AF, Eichler FS. Leukodystrophies: classification, diagnosis, and treatment. Neurologist. 2009;15(6):319–328. doi: 10.1097/NRL.0b013e3181b287c8. [DOI] [PubMed] [Google Scholar]
- 2.Wider C, Van Gerpen JA, DeArmond S, Shuster EA, Dickson DW, Wszolek ZK. Leukoencephalopathy with spheroids (HDLS) and pigmentary leukodystrophy (POLD): a single entity? Neurology. 2009;72(22):1953–1959. doi: 10.1212/WNL.0b013e3181a826c0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Axelsson R, Röyttä M, Sourander P, Akesson HO, Andersen O. Hereditary diffuse leucoencephalopathy with spheroids. Acta Psychiatr Scand Suppl. 1984;314:1–65. [PubMed] [Google Scholar]
- 4.Sundal C, Lash J, Aasly J, et al. Hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS): a misdiagnosed disease entity. J Neurol Sci. 2012;314(1-2):130–137. doi: 10.1016/j.jns.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boissé L, Islam O, Woulfe J, Ludwin SK, Brunet DG. Neurological picture: hereditary diffuse leukoencephalopathy with neuroaxonal spheroids. novel imaging findings. J Neurol Neurosurg Psychiatry. 2010;81(3):313–314. doi: 10.1136/jnnp.2009.180224. [DOI] [PubMed] [Google Scholar]
- 6.Freeman SH, Hyman BT, Sims KB, et al. Adult onset leukodystrophy with neuroaxonal spheroids: clinical, neuroimaging and neuropathologic observations. Brain Pathol. 2009;19(1):39–47. doi: 10.1111/j.1750-3639.2008.00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baba Y, Ghetti B, Baker MC, et al. Hereditary diffuse leukoencephalopathy with spheroids: clinical, pathologic and genetic studies of a new kindred. Acta Neuropathol. 2006;111(4):300–311. doi: 10.1007/s00401-006-0046-z. [DOI] [PubMed] [Google Scholar]
- 8.Itoh K, Shiga K, Shimizu K, Muranishi M, Nakagawa M, Fushiki S. Autosomal dominant leukodystrophy with axonal spheroids and pigmented glia: clinical and neuropathological characteristics. Acta Neuropathol. 2006;111(1):39–45. doi: 10.1007/s00401-005-1113-6. [DOI] [PubMed] [Google Scholar]
- 9.Terada S, Ishizu H, Yokota O, et al. An autopsy case of hereditary diffuse leukoencephalopathy with spheroids, clinically suspected of Alzheimer’s disease. Acta Neuropathol. 2004;108(6):538–545. doi: 10.1007/s00401-004-0920-5. [DOI] [PubMed] [Google Scholar]
- 10.Marotti JD, Tobias S, Fratkin JD, Powers JM, Rhodes CH. Adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia: report of a family, historical perspective, and review of the literature. Acta Neuropathol. 2004;107(6):481–488. doi: 10.1007/s00401-004-0847-x. [DOI] [PubMed] [Google Scholar]
- 11.van der Knaap MS, Naidu S, Kleinschmidt-Demasters BK, Kamphorst W, Weinstein HC. Autosomal dominant diffuse leukoencephalopathy with neuroaxonal spheroids. Neurology. 2000;54(2):463–468. doi: 10.1212/wnl.54.2.463. [DOI] [PubMed] [Google Scholar]
- 12.Rademakers R, Baker M, Nicholson AM, et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet. 2011;44(2):200–205. doi: 10.1038/ng.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11(5):863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kinoshita M, Yoshida K, Oyanagi K, Hashimoto T, Ikeda S. Hereditary diffuse leukoencephalopathy with axonal spheroids caused by R782H mutation in CSF1R: case report. J Neurol Sci. 2012;318(1-2):115–118. doi: 10.1016/j.jns.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 18.Reedijk M, Liu X, van der Geer P, et al. Tyr721 regulates specific binding of the CSF-1 receptor kinase insert to PI 3′-kinase SH2 domains: a model for SH2-mediated receptor-target interactions. EMBO J. 1992;11(4):1365–1372. doi: 10.1002/j.1460-2075.1992.tb05181.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carlberg K, Tapley P, Haystead C, Rohrschneider L. The role of kinase activity and the kinase insert region in ligand-induced internalization and degradation of the c-fms protein. EMBO J. 1991;10(4):877–883. doi: 10.1002/j.1460-2075.1991.tb08020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ali ZS, Van Der Voorn JP, Powers JM. A comparative morphologic analysis of adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia: a role for oxidative damage. J Neuropathol Exp Neurol. 2007;66(7):660–672. doi: 10.1097/nen.0b013e3180986247. [DOI] [PubMed] [Google Scholar]
- 21.Kruer MC, Hiken M, Gregory A, et al. Novel histopathologic findings in molecularly-confirmed pantothenate kinase-associated neurodegeneration. Brain. 2011;134(pt 4):947–958. doi: 10.1093/brain/awr042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paisán-Ruiz C, Li A, Schneider SA, et al. Widespread Lewy body and tau accumulation in childhood and adult onset dystonia-parkinsonism cases with PLA2G6 mutations. Neurobiol Aging. 2012;33(4):814–823. doi: 10.1016/j.neurobiolaging.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li A, Paudel R, Johnson R, et al. Pantothenate kinase-associated neurodegeneration is not a synucleinopathy [published online March 15, 2012] Neuropathol Appl Neurobiol. doi: 10.1111/j.1365-2990.2012.01269.x. doi:10.1111/j.1365-2990.2012.01269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nandi S, Gokhan S, Dai XM, et al. The CSF-1 receptor ligands IL-34 and CSF-1 exhibit distinct developmental brain expression patterns and regulate neural progenitor cell maintenance and maturation. Dev Biol. 2012;367(2):100–113. doi: 10.1016/j.ydbio.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.