

Abstract
This Review summarizes recent advances in understanding copper-transporting ATPase 1 (ATP7A), and examines the neurological phenotypes associated with dysfunction of this protein. Involvement of ATP7A in axonal outgrowth, synapse integrity and neuronal activation underscores the fundamental importance of copper metabolism to neurological function. Defects in ATP7A cause Menkes disease, an infantile-onset, lethal condition. Neonatal diagnosis and early treatment with copper injections enhance survival in patients with this disease, and can normalize clinical outcomes if mutant ATP7A molecules retain small amounts of residual activity. Gene replacement rescues a mouse model of Menkes disease, suggesting a potential therapeutic approach for patients with complete loss-of-function ATP7A mutations. Remarkably, a newly discovered ATP7A disorder—isolated distal motor neuropathy—has none of the characteristic clinical or biochemical abnormalities of Menkes disease or its milder allelic variant occipital horn syndrome (OHS), instead resembling Charcot–Marie–Tooth disease type 2. These findings indicate that ATP7A has a crucial but previously unappreciated role in motor neuron maintenance, and that the mechanism underlying ATP7A-related distal motor neuropathy is distinct from Menkes disease and OHS pathophysiology. Collectively, these insights refine our knowledge of the neurology of ATP7A-related copper transport diseases and pave the way for further progress in understanding ATP7A function.
Introduction
Copper is a trace metal with a ready capacity to gain and donate electrons. This property renders copper highly desirable as a cofactor for numerous enzymes—including some enzymes that are critical for proper neurological function—and dangerous as a potential generator of toxic free radicals, which can cause neurodegeneration via oxidative stress. Thus, to regulate copper metabolism, an elaborate system of chaperones and transporters has evolved that enables simultaneous utilization of and protection from this metal (Supplementary Table 1 online).1–3 The P-type ATPase copper-transporting ATPase 1 (ATP7A) is a major component of the intra cellular copper homeostasis apparatus.4–6 In the 17 years since ATP7A was first identified as an X chromosome-linked gene, defects in which are responsible for Menkes disease and occipital horn syndrome (OHS),7–10 new and interesting biological roles for ATP7A have continued to emerge.11–16
Menkes disease is characterized by infantile-onset cerebral and cerebellar neurodegeneration, failure to thrive, coarse hair, and connective tissue abnormalities.17,18 These features are coupled to a biochemical phenotype (low copper levels in the blood, and a copper deficiency in the brain) that denotes abnormal copper metabolism.19–24 The passage of copper across the blood–cerebrospinal fluid (CSF) barrier and the blood–brain barrier (BBB) is mediated by ATP7A, and the response to early treatment in cases of Menkes disease seems to rely on the residual copper transport activity mediated by mutant ATP7A molecules.24,25 The defects in ATP7A that cause Menkes disease encompass a diverse range of genetic mutations,26–30 a considerable proportion of which lead to complete loss of function of the transporter, a circumstance in which even early copper treatment bestows limited benefit.24,31–33 Naturally occurring mouse models of Menkes disease exist34–37 and have been studied to evaluate potential therapies for this disorder.37–40
OHS is a milder allelic variant of Menkes disease, having a later age of onset and being associated with far less severe central neurodegeneration.10,41–43 The milder nature of OHS is often attributable to ‘leaky’ splice junction mutations that allow 20–30% of ATP7A messenger RNA (mRNA) transcripts to be correctly processed. As in cases of Menkes disease, individuals with OHS manifest connective tissue abnormalities resulting from deficient activity of lysyl oxidase, a copper-requiring enzyme that normally deaminates lysine and hydroxylysine in the first step of collagen crosslink formation.44 Such individuals also often endure inconvenient dysautonomic signs and symptoms related to a partial deficiency in dopamine-β-hydroxylase (DBH) activity.45,46 DBH, another copper-dependent enzyme, normally converts dopamine to norepinephrine, a crucial neurotransmitter in norepinephrinergic neurons. A natural mouse model of OHS, the so-called mottled blotchy model, recapitulates the connective tissue abnormalities, DBH deficiency and mild CNS damage seen in humans.35,47,48
ATP7A-related distal motor neuropathy was identified recently by linkage analysis in two unrelated families in which multiple males were affected by a peripheral neuropathy similar to Charcot–Marie–Tooth disease type 2 (CMT2).49–51 Detailed genetic analyses disclosed the cause of the newly recognized disorder to be missense mutations affecting the carboxyl half of ATP7A (leading to one or other of the amino acid substitutions Thr994Ile or Pro1386Ser) that had not been previously reported in patients with Menkes disease or OHS. The dramatic differences in age of presentation and overall clinical–biochemical phenotype between ATP7A-related distal motor neuropathy and Menkes disease or OHS implies a distinct disease mechanism in the former.
This Review examines recent advances in our understanding of the mechanisms whereby ATP7A facilitates normal neurological function in the CNS and PNS, and provides updates on the neurogenetic diseases that result from dysfunction of this copper transporter. The article also highlights progress in therapeutic approaches for these disorders, and outlines the questions that remain to be answered.
Copper metabolism and ATP7A
Systemic copper metabolism
Copper is an essential trace element that is necessary for numerous critical biological processes, including the following: iron transport; connective tissue and blood vessel formation; pigmentation of hair, retina, and skin; detoxification of reactive oxygen species; mitochondrial electron transport chain function; amidation of neuroendocrine peptides; and catecholamine biosynthesis. Sources of intestinal copper for absorption include dietary intake, as well as pancreatic and biliary secretions.52 Approximately 50% of intestinal copper is absorbed across the apical membranes of enterocytes—a process mediated by the copper uptake proteins copper transporter 1 (CTR1) and divalent metal transporter 1 (DMT1)—and enters the circulation by the pumping action of ATP7A at the basolateral membrane (Supplementary Figure 1a online). In the blood, copper binds predominantly to the serum proteins albumin and α2-macroglobulin53 and is conveyed to tissues for cellular uptake and utilization. In the liver, a major organ for maintaining copper homeostasis, hepatocytes direct copper to one of several destinations: copper may be stored within cells (typically bound to small proteins called metallothioneins), secreted back into the circulation (mostly incorporated within ceruloplasmin), or excreted into the bile (Supplementary Figure 1b online). The latter process is mediated by copper-transporting ATPase 2 (ATP7B), a copper transporter closely related to ATP7A. Mutations in ATP7B are responsible for Wilson disease.1–6 Supplementary Table 1 online provides expanded descriptions of proteins involved in human copper metabolism.
In polarized cells, ATP7A and ATP7B reside in the trans-Golgi network and transport cytoplasmic copper to this compartment for incorporation into copper enzymes, but they relocate towards the plasma membrane to mediate an exodus of copper from the cell in response to an increase in the intra cellular concentration of this metal.54 ATP7A typically traffics to the basolateral membrane, while ATP7B moves towards the apical surface (Supplementary Figure 1 online).4–6 The molecular domains responsible for the intracellular relocalization and trafficking of ATP7A, along with their effects, are summarized in Box 1.
Box 1. ATP7A molecular traffic signals and effects.
38 amino acid segment in transmembrane domain 3 is associated with movement from endoplasmic reticulum to trans-Golgi100,156
Amino-terminal copper-binding motifs mediate movement from trans-Golgi network to plasma membrane157–159
Carboxy-terminal di-leucine motif (positions 1487–1488) regulates endosomal retrieval of ATP7A from the plasma membrane160-162
Carboxy-terminal PDZ motif (DTAL; positions 1497–1500) mediates basolateral (instead of apical) localization of ATP7A in polarized cells162
CPC motif in transmembrane domain 6 (positions 1000–1002) enables copper-induced relocalization from post-Golgi vesicles to plasma membrane151
Phosphorylation motif (DKTG; positions 1044–1047) regulates copper-induced relocalization from post-Golgi vesicles to plasma membrane151
Phosphatase motif (LITGEA; positions 873–878) regulates endosomal retrieval of ATP7A from the plasma membrane151
Abbreviation: ATP7A, copper-transporting ATPase 1.
Copper transport to the nervous system
The precise mechanisms of copper uptake and utilization in the mammalian CNS are poorly understood. The BBB comprises polarized brain capillary endothelial cells in which ATP7A, ATP7B, CTR1 and DMT1 are all expressed.55 These cells presumably localize ATP7A to their basolateral surfaces to accomplish copper delivery from the blood to the brain (Figure 1a). The epithelial cells of the choroid plexus (Figure 1b), which comprise the blood–CSF barrier, are also polarized. In the rodent brain, these specialized cells mediate rapid entrance of the trace metal manganese following intravenous infusion.56 A similar process has been suggested to occur for copper.25 Mammalian choroid plexus epithelial cells express levels of ATP7A57,58 that are fivefold higher than those of ATP7B.55 The orientation of the choroid plexus epithelial cells is such that their apical but not basolateral membranes protrude into the CSF (Figure 1b). On the basis of this topology, ATP7A in these specialized epithelia might localize towards the apical rather than the basolateral membranes for its copper transport function. A precedent for such a phenomenon exists from studies of the Na+–K+-ATPase, which is located on the basolateral surface in most polarized epithelia, but resides at the apical membrane in choroids plexus epithelium.59,60
Figure 1.

Proposed mechanisms of copper transport to the CNS. a ∣ Copper delivery across the blood-brain barrier. ATP7A, ATP7B and CTR1 are expressed in brain capillaries.55 Copper delivery might occur via CTR1 and ATP7A, with return to the blood via ATP7B. Brain copper overload in Wilson disease (caused by mutations in ATP7B) is consistent with this hypothesis. b ∣ Proposed orientations of ATP7A and ATP7B for copper delivery across the blood–CSF barrier. c ∣ Activation of NMDARs at glutamatergic synapses triggers rapid, reversible ATP7A trafficking to axonal and dendritic processes, and copper efflux.11,65 Synaptic release of copper might competitively inhibit NMDAR-mediated calcium uptake, and modulate NMDAR activity in a neuroprotective fashion. Impaired ATP7A function might lead to prolonged, potentially deleterious NMDAR activation. d ∣ In norepinephrinergic neurons, ATP7A provides copper to DBH for conversion of dopamine to norepinephrine.22 A series of norepinephrine receptors and NETs handle the binding or reuptake of this neurotransmitter following synaptic release. Abbreviations: AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; ATP7A, copper-transporting ATPase 1; ATP7B, copper-transporting ATPase 2; CSF, cerebrospinal fluid; CTR1, copper transporter 1; DBH, dopamine-β-hydroxylase; MAO, monoamine oxidase; NET, norepinephrine transporter; NMDAR, N-methyl-d-aspartate receptor; PAM, peptidyl α-amidating monooxygenase; VMAT2, vesicular monoamine transporter 2.
Once delivered to the CSF, copper can bind to albumin (which is abundant in CSF) or form small-molecule complexes with amino acids. The copper importers CTR1 and DMT1 can then receive and deliver copper into the cytoplasm of neurons, where the metal is transferred to metallochaperones61 and delivered to ATP7A and ATP7B (Supplementary Figure 1c online). This process enables metallation of ceruloplasmin, hephaestin, copper–zinc superoxide dismutase (SOD1), cytochrome c oxidase (CCO), DBH, peptidyl-α-amidating monooxygenase (PAM) and tyrosinase. These enzymes are all expressed throughout the brain and serve important functions in the vertebrate CNS.62,63
ATP7A is a multitasking protein
In addition to providing copper across the BBB and blood–CSF barrier for metallation of cuproenzymes, evidence of ATP7A trafficking in neuronal cells of the CNS and PNS implies additional functions for this protein. Using the olfactory receptor neuron system as a neurodevelopmental model, El Meskini et al. demonstrated that murine Atp7a shifted location from the cell bodies of developing neurons to their extending axons, with axonal expression of Atp7a peaking during synaptogenesis.12,64 These researchers further showed that axonal outgrowth and synapse integrity were disrupted in the mottled brindled mouse model of Menkes disease, proving that regulated expression of Atp7a was required for normal neuronal development.58,64
Schlief et al. showed that Atp7a traffics to neuronal processes of hippocampal glutamatergic neurons in response to activation of synaptic N-methyl-d-aspartate receptors.11,65 Activation of these receptors, which induces calcium entry through the receptor after glutamate binding (Figure 1c), was also associated with rapid copper efflux from these cells. Atp7a trafficking and copper release were impaired in mottled brindled mice.
In norepinephrinergic neurons, the importance of ATP7A relates to provision of copper to DBH (Figure 1d). DBH is present only in the vesicles of norepinephrinergic neurons and converts dopamine to the neurotransmitter norepinephrine.22 The recent discovery that certain ATP7A missense mutations cause adult-onset distal motor neuron disease revealed for the first time that ATP7A is also required in cholinergic neurons for PNS maintenance and function.51 Other known functions of ATP7A include roles in angiotensin II-induced hypertension,13 cisplatin resistance,14 macrophage bactericidal activity,15 and hepatic mobilization of copper in response to cardiac copper deficiency (Supplementary Table 2 online).16
ATP7A-related diseases
Three distinct and well-characterized disorders related to mutations in ATP7A have been described: Menkes disease, OHS and ATP7A-related distal motor neuropathy (Figure 2).10,18,51 The neurological, biochemical and molecular features of the three disorders are explored below and summarized in Table 1.
Figure 2.
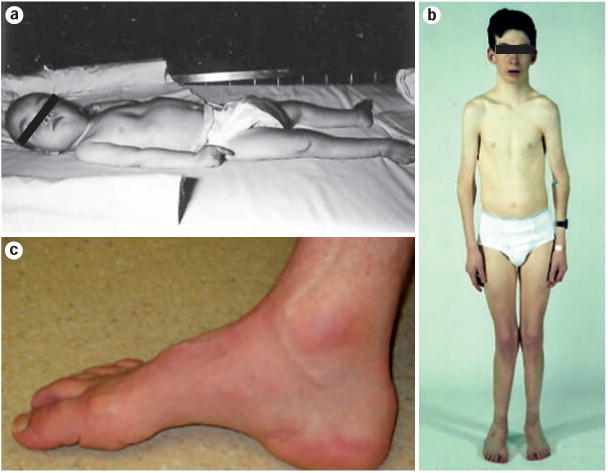
Clinical phenotypes associated with mutations at the ATP7A locus.
a ∣ Classic Menkes disease in a 20-month-old infant. Pectus excavatum deformity of the thorax and decorticate posturing can be observed. b ∣ Occipital horn syndrome in a 14-year-old boy. Narrow thorax, dislocated elbows, genu valgum and pes planus are all features of this disease. c ∣ Pes cavus foot deformity in a 43-year-old individual with the Pro1386Ser ATP7A missense mutation associated with Charcot–Marie–Tooth type 2-like peripheral neuropathy. Permission for part b obtained from Nature Publishing group © Kaler, S. G. et al. Nat. Genet. 8, 195–202 (1994). Part c reprinted from Am. J. Hum. Genet. 86, Kennerson, M. L. et al., Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy, 343–352 © 2010, with permission from The American Society of Human Genetics.
Table 1. Features of ATP7A-related copper transport diseases.
| Condition | Age of onset (years) | Neurological and other clinical signs | Biochemical findings | Molecular defects | Treatment options and prognosis | Future directions |
|---|---|---|---|---|---|---|
| Menkes disease | 0–1 | Hypotonia; seizures; developmental delay; coarse hair; jowly facies; lax skin and joints; decreased bone density; bladder diverticula; gastric polyps; vascular tortuousity and distension | Low serum copper and ceruloplasmin; abnormal plasma and CSF neurochemical levels; increased urine β2-microglobulin | Diverse mutations in ATP7A; 0–15% residual ATP7A function | Early copper replacement; poor prognosis, unless early diagnosis and treatment (within 2 weeks of birth) | Newborn screening to enable early detection; gene transfer plus copper therapy in selected patients |
| Occipital horn syndrome | 3–10 | Dysautonomia;* slight reduction in muscle strength; coarse hair; occipital exostoses; hammer-shaped clavicular heads; lax skin and joints; bladder diverticula; vascular tortuousity and distension | Low to normal serum copper and ceruloplasmin; abnormal plasma and CSF neurochemical levels | Leaky splice junction or hypofunctional ATP7A missense mutations; 20–30% residual ATP7A function | Copper replacement; l-dihydroxy-phenylserine for dysautonomia; fair prognosis; long-term natural history not known; may be at risk of vascular problems | Newborn screening to enable early detection and treatment |
| ATP7A-related distal motor neuropathy | 5–50 | Atrophy and weakness of distal muscles; foot drop; decreased or absent deep tendon reflexes; abnormal nerve conduction studies;‡ pes cavus foot deformity; no other specific clinical abnormalities | No specific abnormalities | Missense mutations causing substitutions in or near transmembrane segments in carboxyl half of ATP7A; 60–70% residual ATP7A function | No treatment currently recommended (pending improved understanding of disease mechanism); prognosis uncertain; natural history unknown; slow disease progression likely | Copper replacement or chelation; motor neuron-directed gene therapy |
Syncope, dizziness, orthostatic hypotension, abnormal sinoatrial conduction, nocturnal bradycardia, and bowel or bladder dysfunction.
Decreased peroneal and median muscle amplitudes with normal conduction velocities.
Abbreviations: ATP7A, copper-transporting ATPase 1; CSF, cerebrospinal fluid.
Menkes disease
Discovery
The critical role of copper in mammalian neurodevelopment was established as early as 1937, when an association was noted between copper deficiency and demyelinating disease in ataxic lambs.23 The mothers of these lambs grazed in copper-deficient pastures throughout pregnancy, leading to cerebral demyelination, porencephaly and brain cavitation in their offspring. In 1972, Danks et al. identified Menkes kinky hair syndrome as a human example of abnormal neurodevelopment resulting from a copper deficiency.19 This discovery was based on the recognition that the unusual hair of infants with Menkes syndrome resembled the texture of the brittle wool of sheep raised on copper-deficient soil in Australia, where Danks resided. The documentation of low serum copper and ceruloplasmin concentrations in patients with Menkes disease sparked renewed interest in the clinical phenotype, which had been carefully delineated 10 years earlier.17 Menkes and colleagues had described five male infants in an Irish American family with a distinctive syndrome of neurological degeneration, peculiar hair and failure to thrive. The infants had seemed normal at birth and throughout their first few months of life, but had subsequently experienced seizures and developmental regression and, ultimately, died between the ages of 7 months and 3.5 years. The family pedigree strongly suggested that the condition was an X-linked genetic disease.17 In 1993, identification of the Menkes disease gene by positional cloning was reported.7–9 This landmark discovery disclosed that the gene product—ATP7A—was a member of a highly conserved family of cation-transporting ATPases.66,67
Clinical features and natural history
Menkes disease typically presents in males at 2–3 months of age, and features loss of previously obtained developmental milestones and the onset of hypotonia, seizures and failure to thrive (Figure 2a). This disorder often escapes attention at birth as a result of the highly subtle nature of the disease manifestations in neonates,18 and the fact that healthy, unaffected newborns have low serum copper levels that overlap with those levels found in affected infants. Premature labor and delivery, large cephalohematomas, hypothermia, hypoglycemia and jaundice are common but nonspecific features of new-borns with Menkes disease. The skin of these neonates often appears loose and redundant, particularly at the nape of the neck, in the axillae, and on the trunk. Occasionally, hair hypopigmentation might suggest a diagnosis of Menkes disease in affected newborns,18 but the twisted hair shafts (pili torti) found on light microscopy at later ages (2–4 months of age) are generally not evident. Neurologically, neonates with this disease seem normal. If Menkes disease remains untreated, however, brain abnormalities, including diffuse atrophy, ventriculomegaly, and tortuosity of cerebral blood vessels, become evident on brain MRI several months after birth.68 In addition, subdural hematomas are common.18 EEG in these patients is usually moderately to severely abnormal, and reflects high rates of status epilepticus and infantile spasms.69,70 Indeed, three large surveys together revealed the presence of clinical seizures and EEG abnormalities in 27 of 29 (93%) symptomatic patients with Menkes disease who were diagnosed at ≥2 months of age.69–71
Later in infancy, pelvic ultrasonography reveals diverticula of the urinary bladder in nearly all patients with Menkes disease.18 Radiographs often disclose abnormalities of bone formation in the skull (wormian bones), long bones (metaphyseal spurring) and ribs (anterior flaring and/or multiple fractures).72 More recently identified problems include gastrointestinal polyps, pulmonary emphysema, arterial aneurysms, and lateral neck masses resulting from dilation of internal jugular veins.73–76 Deficiency of the secreted cuproenzyme lysyl oxidase explains the myriad of connective tissue abnormalities.18,44
The natural history of untreated Menkes disease typically leads to death within 2–3 years after birth, although some individuals live beyond this age. In the latter cases, however, profound neurodevelopmental delays complicate patients' quality of life.
Biochemical findings
The biochemical phenotype of Menkes disease involves low levels of copper in serum, the liver and the brain, owing to impaired intestinal absorption, reduced activities of copper-dependent enzymes and, paradoxically, accumulation of copper in certain tissues (duodenum, kidney, spleen, pancreas, skeletal muscle and/or placenta). Copper retention is also evident in patients' cultured fibroblasts, in which a decrease in radiolabeled copper egress is demonstrable in pulse–chase experiments.10
A deficiency in the activity of tyrosinase, a copper enzyme needed for melanin biosynthesis, causes reductions in hair and skin pigmentation in patients with Menkes disease,18,77 while a partial deficiency in DBH activity accounts for the distinctively abnormal plasma and CSF neurochemical patterns observed in such individuals.18,22,24,78 PAM—a copper-requiring enzyme similar to DBH that is also metallated in the trans-Golgi network—removes the carboxy-terminal glycine residues of neuroendocrine peptide precursors (including gastrin, cholecystokinin, vasoactive intestinal peptide, corticotropin releasing hormone, thyrotropin releasing hormone, calcitonin, vasopressin, neuropeptide Y and pituitary adenylyl cyclase-activating polypeptide).79 Impairment of PAM activity in Menkes disease leads to diminished bioactivity of these hormones and, as a result, potentially detrimental effects on neurogenesis and neuronal survival.80 Deficient CCO and SOD1 activities might also contribute to the neuropathology of this disorder.18
Genetics
A diverse range of mutations in ATP7A causes Menkes disease.24,26–32,68,74,81–88 Molecular defects producing this phenotype include small deletions or insertions (22% of cases), nonsense mutations (18% of cases), splice junction mutations (18% of cases), large gene deletions (17% of cases), and missense mutations (17% of cases).89 In six large case series, totaling 128 unrelated patients with Menkes disease who had alterations other than large ATP7A deletions (which occur across all regions of this gene) or duplications of contiguous exons,30 43 of the mutations (relating to 34% of patients) occurred within a 700 nucleotide region corresponding to exons 7–10 (15% of the gene).26,28,30,85,86,88 Mutations in this region, therefore, seem most likely to cause disease. Missense alterations cluster in the 3′ half of the gene (Figure 3 and Supplementary Table 3 online). ATP7A mutations associated with Menkes disease cause a profound reduction in the quantity and/or functional capacity of ATP7A molecules, which, in turn, results in an estimated reduction of copper transport to 0–17% of that exhibited under healthy conditions.24,84,90,91 Misfolding or altered trafficking of mutant ATP7A may also contribute to disease effects.83,84,88
Figure 3.

Topology of ATP7A missense mutations. ATP7A has eight transmembrane segments and six copper-binding domains. The protein also has phosphatase, phosphorylation, transduction and ATP-binding domains. The majority of ATP7A missense mutations affect the carboxy-terminal half of the protein. Early diagnosis and treatment is currently rare in Menkes disease; thus, little is known about the treatment responsivity of the missense mutations26,28,85,86,88 associated with this phenotype. Of these mutations, only two (Gly666Arg [G666R] and Gly727Arg [G727R]) have been evaluated in terms of early intervention, and each proved responsive to treatment.24,84 Thus, newborn screening for Menkes disease that would detect affected infants in the first week of life is urgently needed. The locations of missense mutations that cause occipital horn syndrome and isolated distal motor neuropathy are also noted. Further details of the 47 ATP7A missense mutations are provided in Supplementary Table 3 online. Abbreviation: ATP7A, copper-transporting ATPase 1.
Diagnosis and treatment
Accurate prenatal diagnosis is available for prospective mothers with a positive family history of Menkes disease;92 however, presymptomatic detection of affected newborns has, historically, been difficult. Infants with this disorder are usually indistinguishable from unaffected infants, with no specific physical features or neurological signs that highlight the condition during the first 2 weeks of life—the critical time frame for treatment.18 Fortunately, neonatal diagnosis of Menkes disease through measurement of neurochemical levels in plasma is now established as a rapid and reliable diagnostic approach.22,24,78 This advance is important since early treatment of Menkes disease with copper injections has been shown to enhance survival,24 reduce seizures85 and, in some cases, normalize neurodevelopmental outcomes.24,68,84,91,93
Various small-molecule copper complexes—including copper chloride, copper gluconate, copper histidine and copper sulfate25—have been used for treatment of Menkes disease, with variable clinical outcomes.18,24 As highlighted above, the success of treatment with small copper complexes depends heavily on early diagnosis and early treatment administration,24,31–33,68,78,84 with recent findings suggesting that this approach generally improves brain electrical activity and decreases seizure occurrence in classic Menkes disease irrespective of the precise molecular defect.85 Of note, early intervention seems to confer particularly good protection against epilepsy in individuals with certain ATP7A mutations.24,85
The best clinical responses to treatment seem to occur in newborns with Menkes disease who harbor mutations that do not completely abrogate ATP7A activity, which can be predicted by a yeast complementation assay (Figure 4). Differences in treatment outcomes probably relate to the fact that in addition to copper entry into the cell, intracellular barriers must be crossed for copper to reach its final destinations, a function that ATP7A normally mediates (Supplementary Figure 1 and Supplementary Table 1 online).3,52 In conjunction with early copper treatment, as little as 5–10% total ATP7A activity seems to be adequate for successful clinical outcomes, which are defined as achievement of normal or near-normal neurodevelopment by 3 years of age.84,91 Four patients with missense mutations affecting one or other of the first two trans membrane segments of ATP7A (Figure 3) responded well to early treatment,24,84 as did two others with leaky splice junction mutations,24,68,82 one with a nonsense mutation associated with inefficient translational read-through,91 and one with a frameshift mutation affecting the amino-terminal region93 that may have involved translation reinitiation.32,94 Serial brain imaging data for three patients with Menkes disease who received early copper treatment are presented in Figure 5.24 In patients with severe loss-of-function ATP7A mutations, for whom outcomes are predicted to be sub optimal even in the context of very early diagnosis and treatment,24,31,33 copper treatment remains a relevant consideration.24,85,95
Figure 4.

Yeast complementation assay predicts treatment response in Menkes disease. Plating pattern (clockwise from 12 o'clock) comprises yeast copper transport mutant ccc2Δ; ccc2Δ transformed with wild-type ATP7A; mock-transformed ccc2Δ; ccc2Δ transformed with mutant ATP7A harboring deletion of exons 20–23; ccc2Δ transformed with the Gly666Arg mutant ATP7A allele; and ccc2Δ transformed with the Asn1304Ser mutant ATP7A allele (associated with occipital horn syndrome).43 Only yeast expressing wild-type, Gly666Arg or Asn1304Ser ATP7A alleles showed growth on copper–iron-limited media, indicating that the proteins encoded by these variants retain some copper transport activity. The wild-type ATP7A allele-transformed yeast showed the most robust growth. The exon 20–23 deletion allele failed to complement ccc2Δ. Permission obtained from the Massachusetts Medical Society © Kaler, S. G. et al. N. Engl. J. Med. 358, 605–614 (2008).
Figure 5.

Serial brain MRI scans from three patients with Menkes disease diagnosed and treated during the newborn period. a ∣ T2-weighted brain MRI scans from a patient with a Gly666Arg ATP7A mutation. b ∣ Axial FLAIR images from a patient with a splice junction mutation (IVS9 DS+6T>G) that had caused severe Menkes disease in an older sibling.24 c ∣ FLAIR images from a second patient with a Gly666Arg ATP7A mutation (this individual was unrelated to the other patient harboring this mutation). In all three individuals, mild signal changes on initial MRI examinations at 7–12 months disappeared in subsequent studies, indicating progressive white matter myelination. No evidence of cortical atrophy was seen in these patients. Abbreviations: ATP7A, copper-transporting ATPase 1; FLAIR, fluid-attenuated inversion recovery.
In the future, gene therapy that restores at least low levels of functional ATP7A perhaps represents the best hope for patients with ATP7A mutations who are unresponsive to conventional treatment. Gene therapy can be defined as the treatment of diseases by transfer of DNA or RNA to affected cells. Gene addition—the delivery of the correct version of a defective gene without removing the endogenous mutant gene—is a useful and practical approach for monogenic neurometabolic disorders, such as Menkes disease, that are caused by loss-of-function mutations. In the case of Menkes disease, the main target organ of such therapy would be the brain. Whether strict control of ATP7A expression would be needed for beneficial clinical and biochemical effects remains unknown. Gene therapy raises unique safety and risk-benefit issues, although adeno-associated viral (AAV) vectors have emerged as attractive vehicles for gene delivery into the CNS, owing to a reasonable safety profile coupled to reliable transduction efficiency and long duration of transgene expression.96 Preliminary results of brain-directed ATP7A gene therapy using AAV serotype 5 (AAV5) in a mouse model of Menkes disease appear highly promising.97
Occipital horn syndrome
Clinical features
OHS is a neurologically milder allelic variant of Menkes disease. This syndrome takes its name from the wedge-shaped calcifications that form bilaterally within the trapezius and sternocleidomastoid muscle tendons at their point of attachment to the occiput in affected individuals, generally by the second decade of life (Figure 2b).41–43 Such protuberances can be palpated in some patients and are demonstrable radiographically on lateral and Towne view skull X-rays,10 or appropriate sagittal images from CT or MRI (Supplementary Figure 2 online). OHS shares the hair and connective tissue abnormalities observed in classic Menkes disease, which are attributable to a lysyl oxidase deficiency98 However, since the neurological phenotype in OHS is mild (slight generalized muscle weakness, and dysautonomia that includes syncope, orthostatic hypotension and chronic diarrhea), affected individuals often escape detection until mid-childhood or later. Patients with this disease have low to normal levels of serum copper and ceruloplasmin, and abnormal plasma and CSF catecholamine levels, which together reflect a deficiency in DBH activity, as in Menkes disease.
The natural history of patients with OHS is poorly under stood, owing to the scarcity of patients for whom long-term follow-ups have been reported. Potential vascular complications might be anticipated for these patients, although no reports of catastrophic vascular rupture, stroke or cardiac events exist in patients with this phenotype.
Genetics and mechanism of disease
The molecular basis of OHS often involves exon skipping and a reduction in the level of correctly spliced ATP7A mRNA.10,47,99-101 Eight of the 15 identified OHS ATP7A mutations (Supplementary Table 3 online), as well as the molecular defect in the mottled blotchy mouse model of OHS,47,102 are associated with such aberrant splicing. Eight cases of OHS involving missense mutations with or without abnormal splicing have been reported (Figure 3 and Supplementary Table 3 online).10,30,43,88,90,99,101 Two other mutations associated with OHS are a deletion in the upstream gene promoter,103 and a 1 bp deletion that causes a frameshift near the 3′ end of ATP7A.104 This frameshift, which resides in exon 23, removes the dileucine motif that is necessary for endocytic recycling of ATP7A (Box 1). The milder neurological phenotype of OHS implies that the ability of mutant ATP7A to traffic to the plasma membrane and pump copper is not completely sabotaged, and that either the levels of wild-type ATP7A are reduced by 70–80%,10 or that mutant ATP7A levels are normal and the copper transport function of the mutant molecules is reduced to 20–30% of normal activity.43
Treatment
Limited information is available concerning copper replacement treatment for OHS.10 Patients with this disease typically have serum copper levels that are only slightly below normal levels of this metal. Nevertheless, these individuals could potentially benefit from copper treatment if such therapy enhanced metallation of lysyl oxidase and DBH. The autonomic symptoms that inconvenience many of these individuals (and which also occur in patients with Menkes disease who are success fully treated) should be amenable to treatment with l-threo-3,4- dihydroxyphenylserine (l-DOPS),45,46 a compound that is converted to norepinephrine via decarboxyla tion by l-aromatic-amino-acid decarboxylase in a copper-independent manner.105 Oral treatment with l-DOPS could provide a direct approach to management of dysautonomic symptoms in patients with OHS. A pilot clinical trial to confirm this hypothesis is scheduled to open shortly.
ATP7A-related distal motor neuropathy
A novel ATP7A-related phenotype
A third clinical phenotype—distal motor neuropathy without overt copper metabolic abnormalities—was recently found to be associated with mutations in ATP7A in two large families with multiple affected males.51 Distal hereditary motor neuropathies (HMNs) comprise a clinically and genetically heterogeneous group of disorders that predominantly affect motor neurons in the PNS.106 Distal HMNs have been classified into seven main subtypes on the basis of mode of inheritance, age of onset, distribution of muscle weakness, and clinical progression.107 14 genetic loci associated with distal HMNs have been mapped, with 10 genes identified to date.51,108,109 These genes encode a functionally diverse array of proteins, including two cation transporters (ATP7A and transient receptor potential cation channel subfamily V member 4),51,109 a transfer RNA synthetase,110 two heat shock proteins,111,112 and a microtubule motor protein involved in axonal transport.113 ATP7A-related distal motor neuropathy is associated with unique missense mutations affecting amino acids within or near transmembrane segments of the protein (Figure 3). The resulting amino acid substitutions—Thr994Ile in transmembrane domain 6 and Pro1386Ser in the short extracellular loop between transmembrane domains 7 and 8 (Figure 3)—may contribute to the abnormal intra-cellular trafficking phenotype observed with these defects (Supplementary Figure 3 online),51 and may prove relevant to the underlying mechanism of this form of motor neuron disease.
Clinical features
As in CMT2,114 the newly described ATP7A-related phenotype features progressive distal motor neuropathy with less prominent sensory loss (Table 1). Symptoms begin with distal muscle weakness and atrophy of the lower extremities, followed by involvement of the upper limbs, reductions in tactile and vibratory sensation, and loss of deep tendon reflexes. Foot and hand deformities such as pes cavus (Figure 2c), hammer toes and curled fingers are typical.49 The age of onset varies from the first to sixth decade of life, with the majority of cases presenting between 10–35 years of age.51 Nerve conduction studies show reductions in compound motor amplitudes with generally normal (≥40 s) conduction velocities,49–51 indicative of an axonopathy rather than a demyelinating process. The phenomenon of ‘dying-back neuropathy’, in which degeneration begins in the distal portions of axons and slowly advances back towards the motor neuron cell body, is usually a result of a metabolic disturbance or toxin. The delayed-onset (often in adulthood) character of ATP7A-related distal motor neuropathy implies that the mutations associated with this disease have subtle effects that require years to provoke pathological consequences.
A novel mechanism of disease?
The mechanism underlying ATP7A-related distal motor neuropathy seems to be distinct from the pathophysiology of Menkes disease and OHS. Individuals with ATP7A-related distal motor neuropathy have no neurological problems other than motor neuron disease, and no clinical or biochemical findings similar to those observed in patients with Menkes or OHS (Table 1). Specifically, no patients with ATP7A-related distal motor neuropathy examined to date have shown hair, skin or joint abnormalities, low serum copper, abnormal plasma catecholamine levels, or renal tubular dysfunction,51 all of which are considered to be hallmarks of mutations at the ATP7A locus.10,18 Conversely, three patients with Menkes disease and four individuals with OHS examined recently showed no clinical or electro physiological evidence of motor neuron dysfunction (S. G. Kaler, unpublished work). The individuals with OHS included the first patient in whom the condition was molecularly defined,10 who is now 32 years old, and an unrelated individual with OHS, who is now 19 years old.43 At these ages, one would have expected ATP7A-related distal motor neuropathy to be clinically manifest, if a common pathogenetic mechanism were involved.
Expression of the Thr994Ile and Pro1386Ser ATP7A alleles tagged with Venus fluorescent protein in HEK 293 cells (Supplementary Figure 3 online) and in a motor neuron-enriched cell line (NSC-34) suggests prolonged retention of the mutant proteins at the plasma and axonal membranes, as well as at the trans-Golgi membrane (S. G. Kaler, unpublished work). In addition, the expressed mutant proteins show a slightly more diffuse intra cellular pattern than wild-type ATP7A. In conjunction with prior observations in fibroblasts from affected patients,51 these results confirm the existence of abnormal ATP7A trafficking in the distal motor neuropathy phenotype, although the meaning and implications of the specific abnormalities remain to be elucidated. These findings might, for example, reflect protein misfolding with oligomerization or aggregate formation, or impaired endocytic retrieval from the cell periphery. Abnormal aggregation115–117 and endocytic pathway errors118–120 are known to be associated with motor neuron disease involving other gene products.121–123 In the case of ATP7A, potential consequences of such events could include a chronic deficiency of one or more copper-dependent enzymes (Supplementary Table 1 online), oxidative damage, axonal ionic imbalances, or alterations in synaptic neurotransmission.
Delineation of the underlying mechanism in this form of motor neuropathy will help elucidate the normal role of ATP7A in peripheral nerve biology (Figure 6). Copper deficiency from various causes is known to induce transient sensory or motor neuropathy,124–130 and both ATP7A and ATP7B are expressed in mouse spinal cord neurons;131 however, the identification of ATP7A-related distal motor neuropathy51 is the first evidence that ATP7A has a direct role in motor neuron function.
Figure 6.

Proposed roles of ATP7A in motor neurons. Since various cuproenzymes (SOD1, CCO, ceruloplasmin and PAM), as well as copper transporters and chaperones (ATP7A, ATP7B, ATOX1, CCS and COX11), are expressed in mouse spinal cord,131 normal motor neuron function clearly seems to require copper. The discovery of ATP7A-related distal motor neuropathy,51 combined with case reports of peripheral neuropathy involving transient disturbances of copper metabolism, confirmed the importance of copper in these cells. Here, in addition to metallation of cuproenzymes, ATP7A is postulated to traffic down axons and mediate copper release from the axonal membrane of motor neurons and, possibly, at the neuromuscular junction. Golgi organelles and other translation machinery may reside within the axon itself, remote from the motor neuron cell body.163,164 Subtle but chronic metabolic insults produced by the Thr994Ile or Pro1386Ser mutations might engender the ‘dying back’ axonopathy that resembles Charcot–Marie–Tooth disease type 2. Abbreviations: ATOX1, copper transport protein ATOX1; ATP7A, copper-transporting ATPase 1; ATP7B, copper-transporting ATPase 2; CCO, cytochrome c oxidase; CCS, copper chaperone for SOD1; COX11, cytochrome c oxidase assembly protein COX11; COX17, cytochrome c oxidase copper chaperone; CTR1, copper transporter 1; DMT1, divalent metal transporter 1; PAM, peptidyl α-amidating monooxygenase; SCO1, protein SCO1 homolog; SOD1, copper–zinc superoxide dismutase.
Treatment
The five patients with ATP7A-related distal motor neuropathy for whom serum copper levels have been measured had levels in the range of 80–100 μg/dl (normal range 75–150 μg/dl). Thus, until more information is gathered regarding the pathogenetic mechanisms involved in this disease, exogenous copper replacement in affected individuals does not seem warranted. Nevertheless, the known relationship between acquired copper deficiency and peripheral neuropathy124–130 suggests that oral or parenteral copper supplementation might eventually emerge as a rational therapeutic approach for patients with ATP7A-related distal motor neuropathy. Indeed, these subtle ATP7A defects might lead to a chronic copper deficiency that, in turn, affects motor neurons and causes symptoms in patients with this condition. Copper replacement treatment seems especially relevant for pediatric patients with a family history of ATP7A-related distal motor neuropathy if such individuals are known to possess the mutant ATP7A allele but have not developed neurological symptoms.51 Ideally, responses to treatment should be tracked at regular intervals with objective measures of distal motor neuron structure and function (for example, diffusion tensor imaging and nerve conduction studies). Gene therapy that targets motor neurons132 might also be considered as an alternative to copper replacement or as part of a combinatorial treatment approach.
Models of ATP7A-related disease
From yeast to fish
Several excellent model systems for ATP7A-related copper transport diseases currently exist. The Saccharomyces cerevisiae homolog of both ATP7A and ATP7B is named ccc2.133 The yeast deletion strain ccc2Δ is unable to transport copper into a post-Golgi compartment and requires increased copper concentrations for growth compared with wild-type strains.134 Transformation of ccc2Δ with ATP7A or ATP7B complements the copper transport abnormality, providing a convenient functional assay to determine whether any copper ATPase activity is generated from specific mutant alleles (Figure 4).24,43,51,83,84,90,91,135,136 For example, consistent with the subtle clinical effects of ATP7A-related distal motor neuropathy, yeast complementation assays indicated that copper pumping capacity was largely retained in the disease-associated ATP7A variants, with the activities of Pro1386Ser and Thr994Ile ATP7A estimated at 70%51 and 60% (S. G. Kaler, unpublished work), respectively, of wild-type ATP7A activity.
The Caenorhabditis elegans homolog of ATP7A and ATP7B, cua-1, also complements the yeast copper transport knockout.137 The phenotype of the C. elegans copper transport knockout has not yet been described.
Drosophila melanogaster harboring a loss-of-function allele of the ATP7A ortholog DmATP7 have been shown to be strikingly more lethargic than wild-type flies, and exhibit smaller mouthparts, markedly less pigmentation, and a shorter lifespan (typically dying within 36 h after hatching).138 These knock-out animals have been rescued by injecting affected embryos with the wild-type DmATP7 allele.
The lower vertebrate Danio rerio (zebrafish) has also emerged as a useful model of ATP7A-related disease. Presence of calamity—a mutant allele of the zebrafish ortholog of ATP7A—causes abnormalities in pigmentation and notochord development, as well as hindbrain degeneration.62,139,140 Normal copper metabolism in zebrafish harboring the calamity allele was restored by injecting zygotes with either synthetic RNA encoding human ATP7A,62 or antisense morpholinos crafted to restore proper splicing in a specific splice junction mutation (calamityvu69).139 In a related chemical genetic screening study, mutagenized zebrafish embryos were exposed to a copper chelator that reduced copper availability. Through this approach, two other mutant alleles conferring sensitivity to copper deficiency were identified, namely calamitygw71, a mild calamity allele associated with skeletal abnormalities, and catastropheQ136X, an allele comprising a nonsense mutation affecting the δ1 subunit of F-ATPase protein 6, a hydrogen ion- transporting ATPase.140 This last result is intriguing, as it implies that copper transport is linked with the exchange of other ions, and that a proton gradient may be necessary for normal copper transport, perhaps to balance charge transfers across membranes.
Yeast, worms, flies and fish have been and remain immensely important model systems for analysis of copper homeostasis; however, mammalian systems will probably manifest greater complexity owing to the presence of two copper-transporting ATPases rather than one.1–6
Mouse models
The so-called mottled mouse provides an excellent mammalian model for Menkes disease.34,35 The mottled (Atp7a) and human ATP7A loci are located in homologous regions of their respective X chromosomes. The Mouse Genome Informatics database of the Jackson Laboratory lists 88 Atp7a alleles, including 38 spontaneous or induced (through radiation and/or chemical mutagenesis) mutants, as well as 48 gene-trapped and two gene-targeted alleles.141 The molecular bases for 14 Atp7a allelic variants have been characterized (Supplementary Table 4 online).36,47,102,141–147
One of the best studied mouse mutants, the mottled brindled male hemizygote, shows tremor, a decrease in coat pigmentation, general inactivity, death at ≈14 days of age, an increase in intestinal copper levels with low levels of the metal in the liver and in the brain, and reductions in copper enzyme activities.35 Of interest has been the observation that normal viability can be restored in these mutant animals if a single copper injection is provided during the first 7–10 days of life, a response also characteristic of the mottled macular mouse, a biochemically similar model of Menkes disease discovered in Japan.37,147 Nevertheless, at least in mottled brindled animals, this treatment response seems to be dependent on modifier genes, as no response to therapy is shown in mottled brindled mice on a homogeneous C57BL/6J background, a strain that often maximizes expression of mutant phenotypes. In contrast to mottled brindled animals, mice harboring the mottled blotchy mutant allele are viable, even without treatment, although these latter mice show more pronounced connective tissue abnormalities than the former. Cultured fibroblasts from all Atp7a mutant mice tested have demonstrated increased copper accumulation compared with similar cells from wild-type animals.148
Investigation of the biochemical phenotype in mottled brindled and mottled blotchy mutant lines has been extensive,18,35,38–40,141 while recent cell biological studies have provided new insights into the effects of specific mottled (Atp7a) mutations.149,150 The mottled brindled and mottled blotchy mutant proteins do not traffic efficiently from the trans-Golgi to the plasma membrane in response to elevations in copper.149 The mottled viable brindled and mottled macular defects each shift the steady-state equilibrium of Atp7a to one where the protein is mainly localized to the plasma membrane under basal copper conditions, whereas mottled 11H, an embryonic lethal allele, encodes an isoform of Atp7a that is unable to exit the endoplasmic reticulum.150 In conjunction with earlier work,151 the findings in mottled viable brindled and mottled macular imply that Atp7a trafficking is delicately controlled by phosphorylation and dephosphorylation. Specifically, these mutations seem to stabilize Atp7a in a distinct conformation that leads to an increase in phosphorylation and alters intracellular localization.150,151
Embryonic gene replacement accomplished by crossing mottled female heterozygotes with male transgenic strains corrected lethality in mottled tohm (a large intragenic deletion of Atp7a) and mottled brindled mutant mice.146,152 To begin to assess the safety and efficacy of postnatal gene-based therapies that could be relevant to human newborns with Menkes disease, we designed a brain-directed treatment approach for mottled brindled C57BL/6J mice. A combination of gene therapy with AAV5 harboring a truncated (owing to AAV vector packaging limits) but functional version of ATP7A, plus copper chloride, both delivered by intra cerebroventricular injection, enhanced the survival of mottled brindled males in comparison with no treatment.97 AAV5 transduces the choroid plexuses of the cerebral ventricles,153 and these specialized polarized epithelia appear crucial for copper transport to the CNS.154
The mottled mouse mutants will continue to serve as useful agents for the study of Menkes disease and other ATP7A-related diseases, and for evaluation of potential new therapies. For example, mouse knock-in models of the Thr994Ile and Pro1386Ser ATP7A mutations will be used to evaluate the development of motor neuron disease, as well as potential treatments involving small or large molecules.
Conclusions and future perspectives
Spawned by identification of the genes responsible for Menkes disease and Wilson disease, the past two decades have witnessed a remarkable growth in our knowledge and understanding of eukaryotic copper metabolism. Appreciation of the basic pathways that guide cellular copper uptake, transport and export has reached a reason able level; however, we know considerably less about the precise mechanisms that underlie the neurological consequences of disturbed copper homeostasis, and the ideal remedies.
Specific issues ripe for further research include the complete crystal structure of ATP7A, dissection of the roles of ATP7A at the BBB and the blood–CSF barrier, and the specific functions of this transporter in glutamatergic, acetylcholinergic and other neurons. The role of ATP7A in axonal and synaptic physiology remains obscure, as does the question of whether the basis for ATP7A-related distal motor neuropathy involves a copper deficiency124–130 or a copper excess.155 Various clinical and translational research questions also remain to be addressed. Notably, can population-based newborn screening assays based on blood neurochemical levels or other biochemical or molecular methods be developed to test for ATP7A-related disorders? Are some or all ATP7A missense mutations that cause Menkes disease responsive to early copper treatment? In relation to therapy, will gene replacement approaches in mouse models be applicable to human patients with large ATP7A deletions or other severe loss-of-function mutations? Will l-DOPS correct symptoms of dysautonomia in individuals with Menkes disease or OHS that is caused by DBH deficiency, and can connective tissue abnormalities caused by lysyl oxidase deficiency in these diseases be treated more effectively? The answers to such clinical and translational research questions will have important implications for patients with ATP7A-related diseases, and their families.
The evaluation of ATP7A function represents an intersection of biochemistry, cell biology, genetics and gene regulation, neuroscience, and structural biology. Many uncertainties remain; however, the pace of discovery concerning this fascinating molecule across multiple disciplines has quickened noticeably in recent years, auguring well for substantial future advances relevant to the neurology of ATP7A-related copper transport disease.
Supplementary Material
MedscapeCMESM Continuing Medical Education online.
This activity has been planned and implemented in accordance with the Essential Areas and policies of the Accreditation Council for Continuing Medical Education through the joint sponsorship of Medscape, LLC and Nature Publishing Group. Medscape, LLC is accredited by the ACCME to provide continuing medical education for physicians.
Medscape, LLC designates this educational activity for a maximum of 1.0 AMA PRA Category 1 Credits™. Physicians should only claim credit commensurate with the extent of their participation in the activity. All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test and/or complete the evaluation at http://www.medscapecme.com/journal/nrneuro; and (4) view/print certificate.
Learning objectives
Upon completion of this activity, participants should be able to:
Describe the pathophysiology, clinical features, and management of Menkes disease.
Describe the pathophysiology, clinical features, and management of occipital horn syndrome (OHS).
Describe the pathophysiology, clinical features, and management of isolated distal motor neuropathy.
Key points.
The critical involvement of copper-transporting ATPase 1 (ATP7A) in axonal outgrowth, synapse integrity and neuronal activation underlines the fundamental role of copper metabolism in neurological function
Mutations in ATP7A yield three distinct clinical syndromes—Menkes disease, occipital horn syndrome (OHS) and isolated distal motor neuropathy—each of which has distinct neurological effects
Menkes disease results in lethal infantile neurodegeneration if left untreated, but normal neurodevelopmental outcomes are sometimes possible if therapy can be administered on the basis of neonatal diagnosis
In OHS, leaky splice junction or hypomorphic missense mutations in ATP7A allow considerable ATP7A-mediated copper transport, thereby sparing the CNS, but cuproenzyme deficiencies can cause dysautonomia and connective tissue problems
A newly discovered ATP7A phenotype, adult-onset distal motor neuropathy, shares no clinical or biochemical abnormalities with Menkes disease or OHS, and results from missense mutations that cause mistrafficking of ATP7A
A rich array of model organisms provides an opportunity for further exploration of human copper metabolism and evaluation of potential disease remedies, including gene therapy
Acknowledgments
Written consent was obtained for publication of Figure 2a from the patient's mother. The author is supported by the Intramural Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NIH). The author apologizes to any colleagues whose work was not cited in this Review as a result of length constraints.
L. Barclay, freelance writer and reviewer, is the author of and is solely responsible for the content of the learning objectives, questions and answers of the MedscapeCME-accredited continuing medical education activity associated with this article.
Footnotes
Competing interests: The author, the journal Chief Editor H. Wood and the CME questions author L. Barclay declare no competing interests.
Supplementary information: Supplementary information is linked to the online version of the paper at www.nature.com/nrneurol
References
- 1.Banci L, Bertini I, Cantini F, Ciofi-Baffoni S. Cellular copper distribution: a mechanistic systems biology approach. Cell Mol Life Sci. 2010;67:2563–2589. doi: 10.1007/s00018-010-0330-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lalioti V, Muruais G, Tsuchiya Y, Pulido D, Sandoval IV. Molecular mechanisms of copper homeostasis. Front Biosci. 2009;14:4878–4903. doi: 10.2741/3575. [DOI] [PubMed] [Google Scholar]
- 3.Kim BE, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol. 2008;4:176–185. doi: 10.1038/nchembio.72. [DOI] [PubMed] [Google Scholar]
- 4.Barry AN, Shinde U, Lutsenko S. Structural organization of human Cu-transporting ATPases: learning from building blocks. J Biol Inorg Chem. 2010;15:47–59. doi: 10.1007/s00775-009-0595-4. [DOI] [PubMed] [Google Scholar]
- 5.Veldhuis NA, Gaeth AP, Pearson RB, Gabriel K, Camakaris J. The multi-layered regulation of copper translocating P-type ATPases. Biometals. 2009;22:177–190. doi: 10.1007/s10534-008-9183-2. [DOI] [PubMed] [Google Scholar]
- 6.La Fontaine S, Mercer JF. Trafficking of the copper-ATPases, ATP7A and ATP7B: role in copper homeostasis. Arch Biochem Biophys. 2007;463:149–167. doi: 10.1016/j.abb.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 7.Vulpe CB, Levinson S, Whitney S, Packman S, Gitschier J. Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat Genet. 1993;3:7–13. doi: 10.1038/ng0193-7. [DOI] [PubMed] [Google Scholar]
- 8.Chelly J, et al. Isolation of a candidate gene for Menkes disease which encodes for a potential heavy metal binding protein. Nat Genet. 1993;3:14–19. doi: 10.1038/ng0193-14. [DOI] [PubMed] [Google Scholar]
- 9.Mercer JF, et al. Isolation of a partial candidate gene of Menkes disease by positional cloning. Nat Genet. 1993;3:20–25. doi: 10.1038/ng0193-20. [DOI] [PubMed] [Google Scholar]
- 10.Kaler SG, et al. Occipital horn syndrome and a mild Menkes phenotype associated with splice site mutations at the MNK locus. Nat Genet. 1994;8:195–202. doi: 10.1038/ng1094-195. [DOI] [PubMed] [Google Scholar]
- 11.Schlief ML, Craig AM, Gitlin JD. NMDA receptor activation mediates copper homeostasis in hippocampal neurons. J Neurosci. 2005;25:239–246. doi: 10.1523/JNEUROSCI.3699-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El Meskini R, et al. ATP7A (Menkes protein) functions in axonal targeting and synaptogenesis. Mol Cell Neurosci. 2007;34:409–421. doi: 10.1016/j.mcn.2006.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qin Z, et al. Role of Menkes ATPase in angiotensin II-induced hypertension: a key modulator for extracellular superoxide dismutase function. Hypertension. 2008;52:945–951. doi: 10.1161/HYPERTENSIONAHA.108.116467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rabik CA. Role of copper transporters in resistance to platinating agents. Cancer Chemother Pharmacol. 2009;64:133–142. doi: 10.1007/s00280-008-0860-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White C, Lee J, Kambe T, Fritsche K, Petris MJ. A role for the ATP7A copper-transporting ATPase in macrophage bactericidal activity. J Biol Chem. 2009;284:33949–33956. doi: 10.1074/jbc.M109.070201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim BE, et al. Cardiac copper deficiency activates a systemic signaling mechanism that communicates with the copper acquisition and storage organs. Cell Metab. 2010;11:353–363. doi: 10.1016/j.cmet.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Menkes JH, Alter M, Weakley D, Sung DH. A sex-linked recessive disorder with growth retardation, peculiar hair, and focal cerebral and cerebellar degeneration. Pediatrics. 1962;29:764–779. [PubMed] [Google Scholar]
- 18.Kaler SG. Menkes disease. Adv Pediatr. 1994;41:263–304. [PubMed] [Google Scholar]
- 19.Danks DM, Campbell PE, Stevens BJ, Mayne V, Cartwright E. Menkes' kinky hair syndrome: an inherited defect in copper absorption with widespread effects. Pediatrics. 1972;50:188–201. [PubMed] [Google Scholar]
- 20.Goka TJ, Stevenson RE, Hefferan PM, Howell RR. Menkes disease: a biochemical abnormality in cultured human fibroblasts. Proc Natl Acad Sci USA. 1976;73:604–606. doi: 10.1073/pnas.73.2.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beratis NG, Price P, Labadie G, Hirschhorn K. 64Cu metabolism in Menkes and normal cultured skin fibroblasts Pediatr Res. 1978;12:699–702. doi: 10.1203/00006450-197806000-00004. [DOI] [PubMed] [Google Scholar]
- 22.Kaler SG, Goldstein DS, Holmes C, Salerno JA, Gahl WA. Plasma and cerebrospinal fluid neurochemical pattern in Menkes disease. Ann Neurol. 1993;33:171–175. doi: 10.1002/ana.410330206. [DOI] [PubMed] [Google Scholar]
- 23.Bennetts HW, Chapman FE. Copper deficiency in sheep in western Australia: a preliminary account of the aetiology of enzootic ataxia of lambs and an anaemia of ewes. Aust Vet J. 1937;13:138–149. [Google Scholar]
- 24.Kaler SG, et al. Neonatal diagnosis and treatment of Menkes disease. N Engl J Med. 2008;358:605–614. doi: 10.1056/NEJMoa070613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaler SG. In: Small Molecule Therapy for Genetic Disease. Thoene J, editor. Cambridge University Press; New York: 2010. pp. 202–212. [Google Scholar]
- 26.Das S, et al. Diverse mutations in patients with Menkes disease often lead to exon skipping. Am J Hum Genet. 1994;55:883–889. [PMC free article] [PubMed] [Google Scholar]
- 27.Kaler SG. Molecular and metabolic bases of Menkes disease and occipital horn syndrome. Pediatr Dev Path. 1998;1:85–98. doi: 10.1007/s100249900011. [DOI] [PubMed] [Google Scholar]
- 28.Tümer Z, et al. Identification of point mutations in 41 unrelated patients affected with Menkes disease. Am J Hum Genet. 1997;60:63–71. [PMC free article] [PubMed] [Google Scholar]
- 29.Liu PC, McAndrew PE, Kaler SG. Rapid and robust screening of the Menkes disease/occipital horn syndrome gene. Genet Test. 2002;6:255–260. doi: 10.1089/10906570260471778. [DOI] [PubMed] [Google Scholar]
- 30.Moizard MP, et al. Twenty-five novel mutations including duplications in the ATP7A gene. Clin Genet. doi: 10.1111/j.1399-0004201001461.x. [DOI] [PubMed] [Google Scholar]
- 31.Kaler SG, et al. Early copper therapy in classic Menkes disease patients with a novel splicing mutation. Ann Neurol. 1995;38:921–928. doi: 10.1002/ana.410380613. [DOI] [PubMed] [Google Scholar]
- 32.Kaler SG. Menkes disease mutations and response to early copper histidine treatment. Nat Genet. 1996;13:21–22. doi: 10.1038/ng0596-21. [DOI] [PubMed] [Google Scholar]
- 33.Liu PC, et al. Downregulation of myelination, energy, and translational genes in Menkes disease brain. Mol Genet Metab. 2005;85:291–300. doi: 10.1016/j.ymgme.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 34.Fraser AS, Sobey S, Spicer CC. Mottled, a sex-modified lethal in the house mouse. J Genet. 1953;51:217–221. [Google Scholar]
- 35.Hunt DM. Primary defect in copper transport underlies mottled mutants in mouse. Nature. 1974;249:852–854. doi: 10.1038/249852a0. [DOI] [PubMed] [Google Scholar]
- 36.Levinson B, et al. The mottled gene is the mouse homologue of the Menkes disease gene. Nat Genet. 1994;6:369–373. doi: 10.1038/ng0494-369. [DOI] [PubMed] [Google Scholar]
- 37.Meguro Y, et al. Changes in copper level and cytochrome c oxidase activity in the macular mouse with age. Brain Dev. 1991;13:184–186. doi: 10.1016/s0387-7604(12)80027-1. [DOI] [PubMed] [Google Scholar]
- 38.Royce PM, Camakaris J, Mann JR, Danks DM. Copper metabolism in mottled mouse mutants. The effect of copper therapy on lysyl oxidase activity in brindled (Mobr) mice. Biochem J. 1982;202:369–371. doi: 10.1042/bj2020369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wenk G, Suzuki K. Congenital copper deficiency: copper therapy and dopamine-β-hydroxylase activity in the mottled (brindled) mouse. J Neurochem. 1983;41:1648–1652. doi: 10.1111/j.1471-4159.1983.tb00876.x. [DOI] [PubMed] [Google Scholar]
- 40.Fujii T, Ito M, Tsuda H, Mikawa H. Biochemical study on the critical period for treatment of the mottled brindled mouse. J Neurochem. 1990;55:885–889. doi: 10.1111/j.1471-4159.1990.tb04574.x. [DOI] [PubMed] [Google Scholar]
- 41.Lazoff SG, Rybak JJ, Parker BR, Luzzatti L. Skeletal dysplasia, occipital horns, diarrhea and obstructive uropathy—a new hereditary syndrome. Birth Defects. 1975;11:71–74. [PubMed] [Google Scholar]
- 42.Tsukahara M, Imaizumi K, Kawai S, Kajii T. Occipital horn syndrome: report of a patient and review of the literature. Clin Genet. 1994;45:32–35. doi: 10.1111/j.1399-0004.1994.tb03986.x. [DOI] [PubMed] [Google Scholar]
- 43.Tang J, Robertson SP, Lem KE, Godwin SC, Kaler SG. Functional copper transport explains neurologic sparing in occipital horn syndrome. Genet Med. 2006;8:711–718. doi: 10.1097/01.gim.0000245578.94312.1e. [DOI] [PubMed] [Google Scholar]
- 44.Siegel RC. Lysyl oxidase. Int Rev Connect Tissue Res. 1979;8:73–118. doi: 10.1016/b978-0-12-363708-6.50009-6. [DOI] [PubMed] [Google Scholar]
- 45.Robertson D, et al. Isolated failure of autonomic noradrenergic neurotransmission. N Eng J Med. 1986;314:1494–1497. doi: 10.1056/NEJM198606053142307. [DOI] [PubMed] [Google Scholar]
- 46.Biaggioni I, Goldstein DS, Atkinson T, Robertson D. Dopamine-β-hydroxylase deficiency in humans. Neurology. 1990;40:370–373. doi: 10.1212/wnl.40.2.370. [DOI] [PubMed] [Google Scholar]
- 47.Das S, et al. Similar splicing mutations of the Menkes/mottled copper-transporting ATPase gene in occipital horn syndrome and the blotchy mouse. Am J Hum Genet. 1995;56:570–576. [PMC free article] [PubMed] [Google Scholar]
- 48.Andrews EJ, White WJ, Bullock LP. Spontaneous aortic aneurysms in blotchy mice. Am J Pathol. 1975;78:199–210. [PMC free article] [PubMed] [Google Scholar]
- 49.Takata RI, et al. A new locus for recessive distal spinal muscular atrophy at Xq13.1-q21. J Med Genet. 2004;41:224–229. doi: 10.1136/jmg.2003.013201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kennerson M, et al. X-linked distal hereditary motor neuropathy maps to the DSMAX locus on chromosome Xq13.1–q21. Neurology. 2009;72:246–252. doi: 10.1212/01.wnl.0000339483.86094.a5. [DOI] [PubMed] [Google Scholar]
- 51.Kennerson ML, et al. Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy. Am J Hum Genet. 2010;86:343–352. doi: 10.1016/j.ajhg.2010.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van den Berghe PV, Klomp LW. New developments in the regulation of intestinal copper absorption. Nutr Rev. 2009;67:658–672. doi: 10.1111/j.1753-4887.2009.00250.x. [DOI] [PubMed] [Google Scholar]
- 53.Liu N, et al. Transcuprein is a macroglobulin regulated by copper and iron availability. J Nutr Biochem. 2007;18:597–608. doi: 10.1016/j.jnutbio.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Petris MJ, et al. Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: a novel mechanism of regulated trafficking. EMBO J. 1996;15:6084–6095. [PMC free article] [PubMed] [Google Scholar]
- 55.Choi BS, Zheng W. Copper transport to the brain by the blood–brain barrier and blood–CSF barrier. Brain Res. 2009;1248:14–21. doi: 10.1016/j.brainres.2008.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aoki I, Wu YJ, Silva AC, Lynch RM, Koretsky AP. In vivo detection of neuroarchitecture in the rodent brain using manganese-enhanced MRI. Neuroimage. 2004;22:1046–1059. doi: 10.1016/j.neuroimage.2004.03.031. [DOI] [PubMed] [Google Scholar]
- 57.Kuo YM, Gitschier J, Packman S. Developmental expression of the mouse mottled and toxic milk genes suggests distinct functions for the Menkes and Wilson disease copper transporters. Hum Mol Genet. 1997;6:1043–1049. doi: 10.1093/hmg/6.7.1043. [DOI] [PubMed] [Google Scholar]
- 58.Niciu MJ, et al. Developmental changes in the expression of ATP7A during a critical period in postnatal neurodevelopment. Neuroscience. 2006;139:947–964. doi: 10.1016/j.neuroscience.2006.01.044. [DOI] [PubMed] [Google Scholar]
- 59.Quinton PM, Wright EM, Tormey JM. Localization of sodium pumps in the choroid plexus epithelium. J Cell Biol. 1973;58:724–730. doi: 10.1083/jcb.58.3.724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alper SL, Stuart-Tilley A, Simmons CF, Brown D, Drenckhahn D. The fodrin–ankyrin cytoskeleton of choroid plexus preferentially colocalizes with apical Na+K+-ATPase rather than with basolateral anion exchanger AE2. J Clin Invest. 1994;93:1430–1438. doi: 10.1172/JCI117120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robinson NJ, Winge DR. Copper metallochaperones. Annu Rev Biochem. 2010;79:537–562. doi: 10.1146/annurev-biochem-030409-143539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mendelsohn BA, et al. Atp7a determines a hierarchy of copper metabolism essential for notochord development. Cell Metab. 2006;4:155–162. doi: 10.1016/j.cmet.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 63.Bousquet-Moore D, et al. Interactions of peptide amidation and copper: novel biomarkers and mechanisms of neural dysfunction. Neurobiol Dis. 2010;37:130–140. doi: 10.1016/j.nbd.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.El Meskini R, Cline LB, Eipper BA, Ronnett GV. The developmentally regulated expression of Menkes protein ATP7A suggests a role in axon extension and synaptogenesis. Dev Neurosci. 2005;27:333–348. doi: 10.1159/000086713. [DOI] [PubMed] [Google Scholar]
- 65.Schlief ML, West T, Craig AM, Holtzman DM, Gitlin JD. Role of the Menkes copper-transporting ATPase in NMDA receptor-mediated neuronal toxicity. Proc Natl Acad Sci USA. 2006;103:14919–14924. doi: 10.1073/pnas.0605390103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Odermatt A, Suter H, Krapf R, Solioz M. Primary structure of two P-type ATPases involved in copper homeostais in Enterococcus hirae. J Biol Chem. 1993;268:12775–12779. [PubMed] [Google Scholar]
- 67.Pedersen PL, Carafoli E. Ion motive ATPases. I. Ubiquity, properties, and significance to cell function. Trends Biochem Sci. 1987;12:146–150. [Google Scholar]
- 68.Kaler SG, et al. Successful early copper therapy in Menkes disease associated with a mutant transcript containing a small in-frame deletion. Biochem Mol Med. 1996;57:37–46. doi: 10.1006/bmme.1996.0007. [DOI] [PubMed] [Google Scholar]
- 69.Friedman E, Harden A, Koivikko M, Pampiglione G. Menkes' disease: neurophysiological aspects. J Neurol Neurosurg Psychiatry. 1978;41:505–510. doi: 10.1136/jnnp.41.6.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bahi-Buisson N, et al. Epilepsy in Menkes disease: analysis of clinical stages. Epilepsia. 2006;47:380–386. doi: 10.1111/j.1528-1167.2006.00432.x. [DOI] [PubMed] [Google Scholar]
- 71.White SR, Reese K, Sato S, Kaler SG. Spectrum of EEG findings in Menkes disease. Electroencephalogr Clin Neurophysiol. 1993;87:57–61. doi: 10.1016/0013-4694(93)90175-u. [DOI] [PubMed] [Google Scholar]
- 72.Amador E, Domene R, Fuentes C, Carreño JC, Enríquez G. Long-term skeletal findings in Menkes disease. Pediatr Radiol. 2010;40:1426–1429. doi: 10.1007/s00247-010-1551-8. [DOI] [PubMed] [Google Scholar]
- 73.Kaler SG, et al. Gastrointestinal hemorrhage associated with gastric polyps in Menkes disease. J Pediatr. 1993;122:93–95. doi: 10.1016/s0022-3476(05)83496-1. [DOI] [PubMed] [Google Scholar]
- 74.Grange DK, et al. Severe bilateral panlobular emphysema and pulmonary arterial hypoplasia: unusual manifestations of Menkes disease. Am J Med Genet. 2005;139:151–155. doi: 10.1002/ajmg.a.31001. [DOI] [PubMed] [Google Scholar]
- 75.Godwin SC, Shawker T, Chang M, Kaler SG. Brachial artery aneurysms in Menkes disease. J Pediatr. 2006;149:412–415. doi: 10.1016/j.jpeds.2006.05.041. [DOI] [PubMed] [Google Scholar]
- 76.Price D, Ravindranath T, Kaler SG. Internal jugular phlebectasia in Menkes disease. Int J Pediatr Otorhinolarygol. 2007;71:1145–1148. doi: 10.1016/j.ijporl.2007.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Setty SR, et al. Cell-specific ATP7A transport sustains copper-dependent tyrosinase activity in melanosomes. Nature. 2008;454:1142–1146. doi: 10.1038/nature07163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Goldstein DS, Holmes CS, Kaler SG. Relative efficiencies of plasma catechol levels and ratios for neonatal diagnosis of Menkes disease. Neurochem Res. 2009;34:1464–1468. doi: 10.1007/s11064-009-9933-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Eipper BA, Stoffers DA, Mains RE. The biosynthesis of neuropeptides: peptide α-amidation. Annu Rev Neurosci. 1992;15:57–85. doi: 10.1146/annurev.ne.15.030192.000421. [DOI] [PubMed] [Google Scholar]
- 80.Hansel DE, May V, Eipper BA, Ronnett GV. Pituitary adenylyl cyclase-activating peptides and α-amidation in olfactory neurogenesis and neuronal survival in vitro. J Neurosci. 2001;21:4625–4636. doi: 10.1523/JNEUROSCI.21-13-04625.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ambrosini L, Mercer JF. Defective copper-induced trafficking and localization of the Menkes protein in patients with mild and copper-treated classical Menkes disease. Hum Mol Genet. 1999;8:1547–1555. doi: 10.1093/hmg/8.8.1547. [DOI] [PubMed] [Google Scholar]
- 82.Kim BE, Smith K, Petris MJ. A copper treatable Menkes disease mutation associated with defective trafficking of a functional Menkes copper ATPase. J Med Genet. 2003;40:290–295. doi: 10.1136/jmg.40.4.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.de Bie P, Muller P, Wijmenga C, Klomp LW. Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes. J Med Genet. 2007;44:673–688. doi: 10.1136/jmg.2007.052746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tang J, Donsante A, Desai V, Patronas N, Kaler SG. Clinical outcomes in Menkes disease patients with a copper-responsive ATP7A mutation, G727R. Mol Genet Metab. 2008;95:174–181. doi: 10.1016/j.ymgme.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kaler SG, et al. Molecular correlates of epilepsy in early diagnosed and treated Menkes disease. J Inher Metab Dis. 2010;33:583–589. doi: 10.1007/s10545-010-9118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gu YH, et al. ATP7A gene mutations in 16 patients with Menkes disease and a patient with occipital horn syndrome. Am J Med Genet. 2001;99:217–222. doi: 10.1002/1096-8628(2001)9999:9999<::aid-ajmg1167>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 87.Tümer Z, Birk Møller L, Horn N. Screening of 383 unrelated patients affected with Menkes disease and finding of 57 gross deletions in ATP7A. Hum Mutat. 2003;22:457–464. doi: 10.1002/humu.10287. [DOI] [PubMed] [Google Scholar]
- 88.Møller LB, et al. Identification and analysis of 21 novel disease-causing amino acid substitutions in the conserved part of ATP7A. Hum Mutat. 2005;26:84–93. doi: 10.1002/humu.20190. [DOI] [PubMed] [Google Scholar]
- 89.Møller LB, Mogensen M, Horn N. Molecular diagnosis of Menkes disease: genotype–phenotype correlation. Biochimie. 2009;91:1273–1277. doi: 10.1016/j.biochi.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 90.Donsante A, et al. Differences in ATP7A gene expression underlie intra-familial variability in Menkes disease/occipital horn syndrome. J Med Genet. 2007;44:492–497. doi: 10.1136/jmg.2007.050013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kaler SG, Tang JR, Donsante A, Kaneski C. Translational read-through of a nonsense mutation in ATP7A. Ann Neurol. 2009;65:108–113. doi: 10.1002/ana.21576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kaler SG, Tümer Z. Prenatal diagnosis of Menkes disease. Prenat Diagn. 1998;18:287–289. [PubMed] [Google Scholar]
- 93.Tümer Z, et al. Early copper-histidine treatment for Menkes disease. Nat Genet. 1996;12:11–13. doi: 10.1038/ng0196-11. [DOI] [PubMed] [Google Scholar]
- 94.Paulsen M, et al. Evidence that translation reinitiation leads to a partially functional Menkes protein containing two copper-binding sites. Am J Hum Genet. 2006;79:214–229. doi: 10.1086/505407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sheela SR, Manoj L, Liu PC, Lem KE, Kaler SG. Copper replacement treatment for symptomatic Menkes disease: ethical considerations. Clin Genet. 2005;68:278–283. doi: 10.1111/j.1399-0004.2005.00496.x. [DOI] [PubMed] [Google Scholar]
- 96.Brunetti-Pierri N, Auricchio A. Gene therapy of human inherited diseases. The Online Metabolic and Molecular Basis of Inherited Disease. 2010 [online], http://www.ommbid.com//OMMBID/the_online_metabolic_and_molecular_bases_of_inherited_disease/b/abstract/part2/ch5.2.
- 97.Donsante A, Kaler SG. Patterns of motor function recovery in a murine model of severe Menkes disease rescued by brain-directed AAV5 gene therapy plus copper. Molec Ther. 2010;18(Suppl. 1):S10. [Google Scholar]
- 98.Gacheru S, et al. Expression and accumulation of lysyl oxidase, elastin, and type I procollagen in human Menkes and mottled mouse fibroblasts. Arch Biochem Biophys. 1993;301:325–329. doi: 10.1006/abbi.1993.1151. [DOI] [PubMed] [Google Scholar]
- 99.Ronce N, et al. A C2055T transition in exon 8 of the ATP7A gene is associated with exon skipping in an occipital horn syndrome family. Am J Hum Genet. 1997;61:233–238. doi: 10.1016/S0002-9297(07)64297-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Qi M, Byers PH. Constitutive skipping of alternatively spliced exon 10 in the ATP7A gene abolishes Golgi localization of the menkes protein and produces the occipital horn syndrome. Hum Mol Genet. 1998;7:465–469. doi: 10.1093/hmg/7.3.465. [DOI] [PubMed] [Google Scholar]
- 101.Borm B, et al. Variable clinical expression of an identical mutation in the ATP7A gene for Menkes disease/occipital horn syndrome in three affected males in a single family. J Pediatr. 2004;145:119–121. doi: 10.1016/j.jpeds.2004.04.033. [DOI] [PubMed] [Google Scholar]
- 102.Mercer JF, et al. Mutations in the murine homologue of the Menkes gene in dappled and blotchy mice. Nat Genet. 1994;6:374–378. doi: 10.1038/ng0494-374. [DOI] [PubMed] [Google Scholar]
- 103.Levinson B, et al. A repeated element in the regulatory region of the MNK gene and its deletion in a patient with occipital horn syndrome. Hum Mol Genet. 1996;11:1737–1742. doi: 10.1093/hmg/5.11.1737. [DOI] [PubMed] [Google Scholar]
- 104.Dagenais SL, Adam AN, Innis JW, Glover TW. A novel frameshift mutation in exon 23 of ATP7A (MNK) results in occipital horn syndrome and not in Menkes disease. Am J Hum Genet. 2001;69:420–427. doi: 10.1086/321290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Goldstein DS. l-Dihydroxyphenylserine (l-DOPS): a norepinephrine prodrug. Cardiovasc Drug Rev. 2006;24:189–203. doi: 10.1111/j.1527-3466.2006.00189.x. [DOI] [PubMed] [Google Scholar]
- 106.De Jonghe P, Timmerman V, Van Broeckhoven C. 2nd workshop of the European CMT consortium: 53rd ENMC international workshop on classification and diagnostic guidelines for Charcot–Marie–Tooth type 2 (CMT2–HMSN II) and distal hereditary motor neuropathy (distal HMN–Spinal CMT) 26–28 September 1997, Naarden, The Netherlands. Neuromuscul Disord. 1998;8:426–431. [PubMed] [Google Scholar]
- 107.Harding AE. In: Peripheral Neuropathy. 3rd. Dyck PJ, Thomas PK, Griffin JW, Low PA, Poduslo JF, editors. WB Saunders; Philadelphia: 1993. pp. 1051–1064. [Google Scholar]
- 108.Irobi-Devolder J. A molecular genetic update of inherited distal motor neuropathies. Verh K Acad Geneeskd Belg. 2008;70:25–46. [PubMed] [Google Scholar]
- 109.Auer-Grumbach M, et al. Alterations in the ankyrin domain of TRPV4 cause congenital distal SMA, scapuloperoneal SMA and HMSN2C. Nat Genet. 2010;42:160–164. doi: 10.1038/ng.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Antonellis A, et al. Glycyl tRNA synthetase mutations in Charcot–Marie–Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet. 2003;72:1293–1299. doi: 10.1086/375039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Evgrafov OV, et al. Mutant small heat-shock protein 27 causes axonal Charcot–Marie–Tooth disease and distal hereditary motor neuropathy. Nat Genet. 2004;36:602–606. doi: 10.1038/ng1354. [DOI] [PubMed] [Google Scholar]
- 112.Irobi J, et al. Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat Genet. 2004;36:597–601. doi: 10.1038/ng1328. [DOI] [PubMed] [Google Scholar]
- 113.Puls I, et al. Distal spinal and bulbar muscular atrophy caused by dynactin mutation. Ann Neurol. 2005;57:687–694. doi: 10.1002/ana.20468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Züchner S, Vance JM. Molecular genetics of autosomal-dominant axonal Charcot–Marie–Tooth disease. Neuromolecular Med. 2006;8:63–74. doi: 10.1385/nmm:8:1-2:63. [DOI] [PubMed] [Google Scholar]
- 115.Windpassinger C, et al. Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat Genet. 2004;36:271–276. doi: 10.1038/ng1313. [DOI] [PubMed] [Google Scholar]
- 116.Levy JR, et al. A motor neuron disease-associated mutation in p150Glued perturbs dynactin function and induces protein aggregation. J Cell Biol. 2006;172:733–745. doi: 10.1083/jcb.200511068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Maystadt I, et al. The nuclear factor κB-activator gene PLEKHG5 is mutated in a form of autosomal recessive lower motor neuron disease with childhood onset. Am J Hum Genet. 2007;81:67–76. doi: 10.1086/518900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lupo V, et al. Missense mutations in the SH3TC2 protein causing Charcot–Marie–Tooth disease type 4C affect its localization in the plasma membrane and endocytic pathway. Hum Mol Genet. 2009;18:4603–4614. doi: 10.1093/hmg/ddp427. [DOI] [PubMed] [Google Scholar]
- 119.Roberts RC, et al. Mistargeting of SH3TC2 away from the recycling endosome causes Charcot–Marie–Tooth disease type 4C. Hum Mol Genet. 2010;19:1009–1018. doi: 10.1093/hmg/ddp565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cogli L, Piro F, Bucci C. Rab7 and the CMT2B disease. Biochem Soc Trans. 2009;37:1027–1031. doi: 10.1042/BST0371027. [DOI] [PubMed] [Google Scholar]
- 121.Niemann A, Berger P, Suter U. Pathomechanisms of mutant proteins in Charcot–Marie–Tooth disease. Neuromolecular Med. 2006;8:217–242. doi: 10.1385/nmm:8:1-2:217. [DOI] [PubMed] [Google Scholar]
- 122.Scherer SS, Wrabetz L. Molecular mechanisms of inherited demyelinating neuropathies. Glia. 2008;56:1578–1589. doi: 10.1002/glia.20751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dion PA, Daoud H, Rouleau GA. Genetics of motor neuron disorders: new insights into pathogenic mechanisms. Nat Rev Genet. 2009;10:769–782. doi: 10.1038/nrg2680. [DOI] [PubMed] [Google Scholar]
- 124.Goodman BP, et al. Clinical and electrodiagnostic findings in copper deficiency myeloneuropathy. J Neurol Neurosurg Psychiatry. 2009;80:524–527. doi: 10.1136/jnnp.2008.144683. [DOI] [PubMed] [Google Scholar]
- 125.Kelkar P, Chang S, Muley SA. Response to oral supplementation in copper deficiency myeloneuropathy. J Clin Neuromuscul Dis. 2008;10:1–3. doi: 10.1097/CND.0b013e3181828cf7. [DOI] [PubMed] [Google Scholar]
- 126.Kumar N, Ahlskog JE, Klein CJ, Port JD. Imaging features of copper deficiency myelopathy: a study of 25 cases. Neuroradiology. 2006;48:78–83. doi: 10.1007/s00234-005-0016-5. [DOI] [PubMed] [Google Scholar]
- 127.Spain RI, Leist TP, De Sousa EA. When metals compete: a case of copper-deficiency myeloneuropathy and anemia. Nat Clin Pract Neurol. 2009;5:106–111. doi: 10.1038/ncpneuro1008. [DOI] [PubMed] [Google Scholar]
- 128.Zara G, Grassivaro F, Brocadello F, Manara R, Pesenti FF. Case of sensory ataxic ganglionopathy-myelopathy in copper deficiency. J Neurol Sci. 2009;277:184–186. doi: 10.1016/j.jns.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 129.Weihl CC, Lopate G. Motor neuron disease associated with copper deficiency. Muscle Nerve. 2006;34:789–793. doi: 10.1002/mus.20631. [DOI] [PubMed] [Google Scholar]
- 130.Foubert-Samier A, et al. Axonal sensory motor neuropathy in copper-deficient Wilson's disease. Muscle Nerve. 2009;40:294–296. doi: 10.1002/mus.21425. [DOI] [PubMed] [Google Scholar]
- 131.Allen Institute for Brain Science. Allen Spinal Cord Atlas. 2010 [online], http://mousespinal.brain-map.org/
- 132.Federici T, Boulis N. Gene therapy for peripheral nervous system diseases. Curr Gene Ther. 2007;7:239–248. doi: 10.2174/156652307781369083. [DOI] [PubMed] [Google Scholar]
- 133.Fu D, Beeler TJ, Dunn TM. Sequence, mapping and disruption of CCC2, a gene that cross-complements the Ca2+-sensitive phenotype of csg1 mutants and encodes a P-type ATPase belonging to the Cu2+-ATPase subfamily. Yeast. 1995;11:283–292. doi: 10.1002/yea.320110310. [DOI] [PubMed] [Google Scholar]
- 134.Yuan DS, Dancis A, Klausner RD. Restriction of copper export in Saccharomyces cerevisiae to a late Golgi or post-Golgi compartment in the secretory pathway. J Biol Chem. 1997;272:25787–25793. doi: 10.1074/jbc.272.41.25787. [DOI] [PubMed] [Google Scholar]
- 135.Hung IH, et al. Biochemical characterization of the Wilson disease protein and functional expression in the yeast Saccharomyces cerevisiae. J Biol Chem. 1997;272:21461–21466. doi: 10.1074/jbc.272.34.21461. [DOI] [PubMed] [Google Scholar]
- 136.Hsi G, et al. Sequence variation in the ATP-binding domain of the Wilson disease transporter, ATP7B, affects copper transport in a yeast model system. Hum Mutat. 2008;29:491–501. doi: 10.1002/humu.20674. [DOI] [PubMed] [Google Scholar]
- 137.Sambongi Y, et al. Caenorhabditis elegans cDNA for a Menkes/Wilson disease gene homologue and its function in a yeast CCC2 gene deletion mutant. J Biochem. 1997;121:1169–1175. doi: 10.1093/oxfordjournals.jbchem.a021711. [DOI] [PubMed] [Google Scholar]
- 138.Norgate M, et al. Essential roles in development and pigmentation for the Drosophila copper transporter DmATP7. Mol Biol Cell. 2006;17:475–484. doi: 10.1091/mbc.E05-06-0492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Madsen EC, Morcos PA, Mendelsohn BA, Gitlin JD. In vivo correction of a Menkes disease model using antisense oligonucleotides. Proc Natl Acad Sci USA. 2008;105:3909–3914. doi: 10.1073/pnas.0710865105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Madsen EC, Gitlin JD. Zebrafish mutants calamity and catastrophe define critical pathways of gene–nutrient interactions in developmental copper metabolism. PLoS Genet. 2008;4:e1000261. doi: 10.1371/journal.pgen.1000261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.The Jackson Laboratory. Mouse Genome Informatics. 2010 [online], http://www.informatics.jax.org/searches/allele_report.cgi?_Marker_key=15197.
- 142.Grimes A, Hearn CJ, Lockhart P, Newgreen DF, Mercer JF. Molecular basis of the brindled mouse mutant (Mobr): a murine model of Menkes disease. Hum Mol Genet. 1997;6:1037–1042. doi: 10.1093/hmg/6.7.1037. [DOI] [PubMed] [Google Scholar]
- 143.Cecchi C, Biasotto M, Tosi M, Avner P. The mottled mouse as a model for human Menkes disease: identification of mutations in the Atp7a gene. Hum Mol Genet. 1997;6:425–433. doi: 10.1093/hmg/6.3.425. [DOI] [PubMed] [Google Scholar]
- 144.Reed V, Boyd Y. Mutation analysis provides additional proof that mottled is the mouse homologue of Menkes' disease. Hum Mol Genet. 1997;6:417–423. doi: 10.1093/hmg/6.3.417. [DOI] [PubMed] [Google Scholar]
- 145.Cunliffe P, Reed V, Boyd Y. Intragenic deletions at Atp7a in mouse models for Menkes disease. Genomics. 2001;74:155–162. doi: 10.1006/geno.2001.6529. [DOI] [PubMed] [Google Scholar]
- 146.Mototani Y, et al. Phenotypic and genetic characterization of the Atp7aMo–Tohm mottled mouse: a new murine model of Menkes disease. Genomics. 2006;87:191–199. doi: 10.1016/j.ygeno.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 147.Mori M, Nishimura M. A serine-to-proline mutation in the copper-transporting P-type ATPase gene of the macular mouse. Mamm Genome. 1997;8:407–410. doi: 10.1007/s003359900457. [DOI] [PubMed] [Google Scholar]
- 148.Masson W, Hughes H, Papworth D, Boyd Y, Horn N. Abnormalities of copper accumulation in cell lines established from nine different alleles of mottled are the same as those found in Menkes disease. J Med Genet. 1997;34:729–732. doi: 10.1136/jmg.34.9.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.La Fontaine S, et al. Intracellular localization and loss of copper responsiveness of Mnk, the murine homologue of the Menkes protein, in cells from blotchy (Moblo) and brindled (Mobr) mouse mutants. Hum Mol Genet. 1999;8:1069–1075. doi: 10.1093/hmg/8.6.1069. [DOI] [PubMed] [Google Scholar]
- 150.Kim BE, Petris MJ. Phenotypic diversity of Menkes disease in mottled mice is associated with defects in localisation and trafficking of the ATP7A protein. J Med Genet. 2007;44:641–646. doi: 10.1136/jmg.2007.049627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Petris MJ, et al. Copper-regulated trafficking of the Menkes disease copper ATPase is associated with formation of a phosphorylated catalytic intermediate. J Biol Chem. 2002;277:46736–46742. doi: 10.1074/jbc.M208864200. [DOI] [PubMed] [Google Scholar]
- 152.Llanos RM, et al. Correction of a mouse model of Menkes disease by the human Menkes gene. Biochim Biophys Acta. 2006;1762:485–493. doi: 10.1016/j.bbadis.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 153.Watson DJ, Passini MA, Wolfe JH. Transduction of the choroid plexus and ependyma in neonatal mouse brain by vesicular stomatitis virus glycoprotein-pseudotyped lentivirus and adeno-associated virus type 5 vectors. Hum Gene Ther. 2005;16:49–56. doi: 10.1089/hum.2005.16.49. [DOI] [PubMed] [Google Scholar]
- 154.Donsante A, Johnson P, Jansen LA, Kaler SG. Somatic mosaicism in Menkes disease suggests choroid plexus-mediated copper transport to the developing brain. Am J Med Genet. 2010;152A:2529–2534. doi: 10.1002/ajmg.a.33632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Jung KH, Ahn TB, Jeon BS. Wilson disease with an initial manifestation of polyneuropathy. Arch Neurol. 2005;62:1628–1631. doi: 10.1001/archneur.62.10.1628. [DOI] [PubMed] [Google Scholar]
- 156.Francis MJ, et al. A Golgi localization signal identified in the Menkes recombinant protein. Hum Mol Genet. 1998;7:1245–1252. doi: 10.1093/hmg/7.8.1245. [DOI] [PubMed] [Google Scholar]
- 157.Goodyer ID, Jones EE, Monaco AP, Francis MJ. Characterization of the Menkes protein copper-binding domains and their role in copper-induced protein relocalization. Hum Mol Genet. 1999;8:1473–1478. doi: 10.1093/hmg/8.8.1473. [DOI] [PubMed] [Google Scholar]
- 158.Strausak D, et al. The role of GMXCXXC metal binding sites in the copper-induced redistribution of the Menkes protein. J Biol Chem. 1999;274:11170–11177. doi: 10.1074/jbc.274.16.11170. [DOI] [PubMed] [Google Scholar]
- 159.Voskoboinik I, et al. Functional analysis of the N-terminal CXXC metal-binding motifs in the human Menkes copper-transporting P-type ATPase expressed in cultured mammalian cells. J Biol Chem. 1999;274:22008–22012. doi: 10.1074/jbc.274.31.22008. [DOI] [PubMed] [Google Scholar]
- 160.Petris MJ, Camakaris J, Greenough M, LaFontaine S, Mercer JF. A C-terminal di-leucine is required for localization of the Menkes protein in the trans-Golgi network. Hum Mol Genet. 1998;7:2063–2071. doi: 10.1093/hmg/7.13.2063. [DOI] [PubMed] [Google Scholar]
- 161.Francis MJ, et al. Identification of a di-leucine motif within the C terminus domain of the Menkes disease protein that mediates endocytosis from the plasma membrane. J Cell Sci. 1999;112:1721–1732. doi: 10.1242/jcs.112.11.1721. [DOI] [PubMed] [Google Scholar]
- 162.Greenough M, et al. Signals regulating trafficking of Menkes (MNK; ATP7A) copper-translocating P-type ATPase in polarized MDCK cells. Am J Physiol Cell Physiol. 2004;287:C1463–C1471. doi: 10.1152/ajpcell.00179.2004. [DOI] [PubMed] [Google Scholar]
- 163.Piper M, Holt C. RNA translation in axons. Annu Rev Cell Dev Biol. 2004;20:505–523. doi: 10.1146/annurev.cellbio.20.010403.111746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Merianda TT, et al. A functional equivalent of endoplasmic reticulum and Golgi in axons for secretion of locally synthesized proteins. Mol Cell Neurosci. 2009;40:128–142. doi: 10.1016/j.mcn.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.