

Abstract
Nearly 8% of the human population carries an inactivating point mutation in the gene that encodes the cardioprotective enzyme aldehyde dehydrogenase 2 (ALDH2). This genetic polymorphism (ALDH2*2) is linked to more severe outcomes from ischemic heart damage and an increased risk of coronary artery disease (CAD), but the underlying molecular bases are unknown. We investigated the ALDH2*2 mechanisms in a human model system of induced pluripotent stem cell–derived cardiomyocytes (iPSC-CMs) generated from individuals carrying the most common heterozygous form of the ALDH2*2 genotype. We showed that the ALDH2*2 mutation gave rise to elevated amounts of reactive oxygen species and toxic aldehydes, thereby inducing cell cycle arrest and activation of apoptotic signaling pathways, especially during ischemic injury. We established that ALDH2 controls cell survival decisions by modulating oxidative stress levels and that this regulatory circuitry was dysfunctional in the loss-of-function ALDH2*2 genotype, causing up-regulation of apoptosis in cardiomyocytes after ischemic insult. These results reveal a new function for the metabolic enzyme ALDH2 in modulation of cell survival decisions. Insight into the molecular mechanisms that mediate ALDH2*2-related increased ischemic damage is important for the development of specific diagnostic methods and improved risk management of CAD and may lead to patient-specific cardiac therapies.
INTRODUCTION
Heart failure is a leading cause of death worldwide, with coronary artery disease (CAD) and stroke together killing more than 10 million people annually. The single-nucleotide polymorphism that generates a Glu-to-Lys change at position 487 (E487K) of the cardioprotective enzyme aldehyde dehydrogenase 2 (ALDH2*2) (1, 2) has been linked to both an increased risk of CAD and more severe outcomes (3-6), a higher incidence of hypertension and cancer, and an increase in complications from type 2 diabetes (7-9). The ALDH2*2 genotype is present in ~8% of the human population, predominantly in people of East Asian heritage.
CAD causes cardiomyocytes to experience ischemic challenge that results in altered metabolism and elevated oxidative stress. During myocardial infarction (MI), reactive oxygen species (ROS) cause oxidative damage, including lipid peroxidation and enhanced production of toxic aldehydes such as 4-hydroxynonenal (4HNE) (2, 10-12). Furthermore, dysregulation of ROS-mediated signaling events during ischemia and reperfusion is known to trigger apoptosis and necrosis in heart tissue (13). Mitochondrial ALDH2 fulfills a critical role in metabolic health as a detoxifying enzyme of reactive aldehydes that are generated by ROS, such as 4HNE (2, 12, 14), and as a beneficial factor in acetaldehyde-associated and aging-induced cardiovascular complications (15-18). Moreover, bioactivation of the vasodilator nitroglycerin is catalyzed by ALDH2 and is impaired in carriers of the ALDH2*2 polymorphism (19-21). Oxidative stress and nitroglycerin tolerance are caused by inactivation of cardioprotective ALDH2, which is associated with cardiac dysfunction and increased infarct size after MI (19, 22, 23). Specific activation of mitochondrial ALDH2 has been shown to overcome ischemic heart damage (2, 19, 23-25).
In heterozygous carriers of the dominant-negative ALDH2*2 genotype, the enzymatic activity of ALDH2 is reduced to less than 40% of wild type (26), thereby rendering ALDH2*2 carriers more susceptible to oxidative damage. In addition, previous studies have shown that toxic aldehydes such as 4HNE rapidly inactivate ALDH2 (2). 4HNE also acts as a signaling molecule that modulates transcriptional regulation (27), causing cell cycle arrest and activation of proapoptotic pathways, as well as inflammatory pain (27-30).
The mechanism by which the nonbenign ALDH2*2 genotype (7, 9, 31) causes more severe outcomes from ischemic heart damage has not been studied in human cardiomyocytes at the cellular level. Here, we hypothesize that increased levels of ROS and 4HNE in ALDH2*2 cells not only affect the metabolic balance (11, 16, 32) but also cause dysregulation of certain signaling events, especially after ischemia. To test this notion, we used two different cell types, human fibroblasts and human induced pluripotent stem cell–derived cardiomyocytes (iPSC-CMs), as correlating model systems. Our data recapitulated the phenotype of the ALDH2*2 polymorphism and revealed its underlying dysfunctional signaling mechanisms, which led to increased apoptotic cell death after ischemic challenge.
RESULTS
Correlation of ALDH2*2 polymorphism with elevated 4HNE and ROS
To characterize the phenotype of the ALDH2*2 mutation, we recruited a cohort of 10 age- and sex-matched East Asian individuals who carried either the common heterozygous mutation ALDH2*2/1 or the ALDH2 wild-type gene (Fig. 1A). We derived fibroblast cell lines from skin biopsies of these individuals. ALDH2 activity was significantly reduced in ALDH2*2/1 cell lysates relative to wild-type controls (Fig. 1B). Alda-1, a specific small-molecule activator of ALDH2 (2, 33), increased ALDH2 activity in both wild-type and ALDH2*2/1 cells. By contrast, the toxic aldehyde 4HNE, which inactivates ALDH2 (2), inhibited ALDH2 activity (Fig. 1B).
Fig. 1. Analysis of a human model system for the ALDH2*2/1 polymorphism reveals that reduced ALDH2 enzymatic activity correlates with increased levels of ROS and 4HNE in ALDH2*2/1 cells.

(A) Genotyping of skin biopsy–derived fibroblasts confirms the ALDH2 wild type (wt) or heterozygous 2*2/1 mutation. In ALDH2*2/1, the G peak corresponding to the single remaining wt allele is strongly reduced and overlaid by the A peak of the mutated allele. (B) Enzymatic activity of ALDH2 in lysates from ALDH2*2/1 human fibroblasts (n = 5) is strongly reduced compared to that in wt control (n = 4). ALDH2-specific small molecules were used as a control for specificity. The ALDH2 activator Alda-1 enhances enzymatic activity, whereas the ALDH2 inhibitor 4HNE has the opposite effect. (C) Total baseline ROS are quantified via cellular hydrogen peroxide (H2O2) levels in an Amplex Red–dependent fluorescent readout. ALDH2*2/1 fibroblasts show elevated ROS levels. Data represent n = 5 per group. (D) ALDH2 expression levels in wt and ALDH2*2/1 are comparable, as assessed by Western blot (n = 5 per group). (E) Quantification of expression levels shown in (D) for n = 5 per group. Differences between groups are not significant. (F) Significantly increased levels of 4HNE in ALDH2*2/1 human fibroblasts are confirmed by Western blot, normalizing via β-actin as loading control. (G) Quantification of Western blot analysis for n = 5 per group. Experiments were performed in the presence or absence of 20 μM Alda-1, 80 μM 4HNE, or control vehicle [dimethyl sulfoxide (DMSO)] as indicated. Data are expressed as means ± SEM. ns, not significant. *P < 0.05, calculated by Student’s t test.
To exclude contribution of other dehydrogenases, we measured ALDH2 activity in the presence of 4-methylpyrazole (4-MP), an alcohol dehydrogenase inhibitor (fig. S1, A and B), and daidzin, an ALDH2 inhibitor (fig. S1, A and B). We next quantified amounts of ROS in ALDH2*2/1 and wild-type human fibroblasts. In line with ALDH2’s function as a detoxifying enzyme reducing aldehydic load in the cell and increasing mitochondrial integrity (2, 34), we found a significant elevation in basal amounts of ROS in ALDH2*2/1 fibroblasts compared to wild-type controls (Fig. 1C). ROS levels were also higher in ALDH2*2/1 fibroblasts compared to wild-type controls after external challenge with 4HNE (fig. S2A). As additional control, we confirmed that an unrelated strong ROS scavenger, the enzyme catalase, also significantly reduced cellular ROS in ALDH2*2/1 and wild-type fibroblasts (4.4 ± 0.5–fold and 2.9 ± 0.2–fold, respectively; fig. S2B). Moreover, neither Alda-1 nor 4HNE interfered with the cellular ROS readout in an independent enzymatic reaction (fig. S3, A and B), and ALDH2 protein expression levels were comparable in ALDH2*2/1 versus wild type (Fig. 1, D and E).
Having established specificity and efficacy in our experimental platform, we subsequently measured amounts of endogenous 4HNE in ALDH2*2/1 and wild-type fibroblast by quantification of 4HNE-adducted proteins, as previously reported (2). 4HNE amounts were found to be significantly increased (3.5 ± 0.8–fold; P = 0.01, as determined by Student’s t test) in ALDH2*2/1 cell lysates compared to wild-type controls (Fig. 1, F and G).
4HNE- and ROS-induced cell cycle arrest in ALDH2*2/1 cells
Elevated cellular levels of 4HNE trigger a variety of deleterious effects such as a disturbance in the cell’s metabolic balance (11, 16, 32, 34) and inhibition of cell proliferation (35, 36). We also observed slower growth rates in isolated cultures of ALDH2*2/1 fibroblasts compared to wild-type control fibroblasts by measuring cellular growth and viability 48 hours after plating equal numbers of cells per group. Cell numbers and viability levels for ALDH2*2/1 fibroblasts were significantly reduced compared to wild-type controls (42.5 ± 16.9%; P < 0.01; Fig. 2A; and 54.6 ± 18.6%; P < 0.01, as determined by Student’s t test; Fig. 2B). Activation of ALDH2 in ALDH2*2/1 fibroblasts with Alda-1 restored cell growth and viability to wild-type control levels, indicating that the cellular deficiencies were directly dependent on the ALDH2*2/1 genotype. By contrast, a challenge with 80 μM 4HNE had no apparent effect on wild-type controls but did decrease proliferation of ALDH2*2/1 fibroblasts by 1.7-fold (P < 0.05, as determined by Student’s t test; Fig. 2A) and cellular viability by 2.2-fold (P < 0.001, as determined by Student’s t test; Fig. 2B), indicating a greater sensitivity of ALDH2*2/1 cells to toxic aldehydic load.
Fig. 2. ALDH2*2/1 fibroblasts show reduced cell proliferation and viability due to cell cycle arrest.

Cell proliferation and viability were determined in a 96-well plate, high-content readout format. (A) Quantification of cell proliferation 48 hours after plating using a fluorescent, membrane permeable nuclear dye, Syto-60. Experiments were performed in triplicates and n = 5 per group. (B) Viability in ALDH2*2/1 fibroblasts was quantified via spectrophotometric measurement of XTT [2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide] absorbance. Experiments were performed in triplicates and n = 5 per group. (C) Representative confocal images show cell cycle arrest in ALDH2*2/1 fibroblasts analyzed via BrdU incorporation and costaining with both DAPI as a nuclear marker and the mitochondrial structural protein TOM20. Scale bar, 100 μM. (D) Quantification and statistical analysis (n = 5 cell lines per group) analyzing a total of 42 images for wt and 44 images for ALDH2*2/1. Experiments were performed in the presence or absence of 20 μM Alda-1, 80 μM 4HNE, or control vehicle (DMSO) as indicated. Data are expressed as means ± SEM. *P < 0.05 and **P < 0.01, calculated by Student’s t test.
To determine whether diminished cell growth rates of ALDH2*2/1 cells were caused by a cell cycle defect, we next cultured wild-type control and ALDH2*2/1 fibroblasts for 24 hours in the presence of bromodeoxyuridine (BrdU) to label DNA in actively dividing nuclei. Strikingly, we found that the ratio of BrdU (dividing nuclei) to 4′,6-diamidino-2-phenylindole (DAPI) (total number of nuclei) was significantly reduced in ALDH2*2/1 fibroblasts relative to wild type (Fig. 2, C and D).
Together, these data establish that the ALDH2*2 genotype is associated with increased cellular levels of 4HNE and concomitantly increased ROS, which directly correlate with reduced cell viability and proliferation. We suggest that 4HNE- and ROS-induced cell cycle arrest in ALDH2*2/1 cells is a possible underlying mechanism.
Recapitulation of increased susceptibility to ischemia in ALDH2*2 iPSC-CMs
To further elucidate the underlying principles in a human cardiomyocyte model system, we moved to human iPSC-CMs. We reprogrammed ALDH2*2/1 and wild-type fibroblasts to bona fide iPSCs (figs. S4 and S5, and table S1) and then differentiated them into beating iPSC-CMs as described previously (figs. S6 and S7, tables S2 and S3, and videos S1 and S2) (37, 38). We also confirmed equal expression of ALDH2 in ALDH2*2/1 and wild-type iPSC-CMs (fig. S8). Our previous findings established increased ROS levels as well as a higher sensitivity in response to 4HNE challenge in ALDH2*2/1 fibroblasts (Fig. 2, A and B, and fig. S2A). Therefore, we hypothesized that reduced viability caused by the loss-of-function mutation in the gene encoding mitochondrial ALDH2 might be linked to increased ROS production, due to elevated aldehydic load and mitochondrial dysfunction. Analysis of ROS levels in wild-type and ALDH2*2/1 iPSC-CMs indeed revealed a boost of ROS in the latter cells, especially after in vitro ischemic challenge using an ischemia-mimetic solution (39) combined with hypoxia (Fig. 3, A and B). An elevation in ROS was dependent on reduced ALDH2 function in ALDH2*2/1 iPSC-CMs, because Alda-1–mediated activation of ALDH2 significantly reduced excess ROS in post-ischemic ALDH2*2/1 iPSC-CMs (23.0 ± 2.1%; P = 0.015, as determined by Student’s t test; Fig. 3B). As further confirmation of our assay system, we also demonstrated that elevated ROS in ALDH2*2/1 iPSC-CMs, especially after ischemia, could be reduced by the addition of catalase, an independent ROS scavenger, to the cultures (fig. S9).
Fig. 3. Increased ischemic damage in ALDH2*2/1 iPSC-CMs is caused by enhanced apoptosis.

(A and B) Total ROS in ALDH2*2/1 iPSC-CMs are quantified via cellular H2O2 in an Amplex Red–dependent fluorescent readout. ALDH2*2/1 iPSC-CMs show elevated ROS levels, especially post-ischemia, which can be ameliorated by Alda-1–mediated activation of ALDH2 before ischemic challenge. Data represent n = 5 per group. (C and D) Viability of ALDH2*2/1 iPSC-CMs is significantly reduced compared to wt iPSC-CMs after ischemia. Alda-1–mediated activation of ALDH2 can rescue this phenotype. Absorbance of XTT was measured ina 96-well plate under a high-content readout format (n = 3 per group performed in duplicate). (E to G) Ischemic damage in ALDH2*2/1 iPSC-CMs is due to apoptosis, as shown by TUNEL staining. After ischemia, ALDH2*2/1 iPSC-CMs express significantly increased numbers of TUNEL-positive nuclei, compared to wt control. (E) Representative confocal images showing TUNEL and DAPI staining, as well as costaining for cardiac troponin T (TNNT2). Scale bar, 50 μM. (F) Quantification of TUNEL/DAPI ratio for wt control (n = 7 images), wt ischemia (n = 8), ALDH2*2/1 control (n = 9), and ALDH2*2/1 ischemia (n = 11). (G) Post-ischemic ALDH2*2/1 iPSC-CMs show fragmented DNA. All data are expressed as means ± SEM. ns, not significant. *P < 0.05 and **P < 0.01, calculated by Student’s t test.
To determine whether elevated amounts of ROS in ALDH2*2/1 iPSC-CMs impaired the cellular mitochondria-dependent metabolic balance as a consequence, we next assessed the mitochondrial membrane potential (MMP). The eletrochemical gradient and potential difference across the mitochondrial membrane represents a key indicator for cellular metabolism and health. We quantified the 590:525-nm fluorescence ratio of the cationic-potential sensor dye JC1 in ALDH2*2/1 and wild-type iPSC-CMs and found that simulated ischemia in vitro as described above did not affect the MMP of wild-type control iPSC-CMs but significantly reduced the MMP of ALDH2*2/1 iPSC-CMs (59.4 ± 15.3%; P < 0.05, as determined by Student’s t test; fig. S10). Together, our data recapitulate the phenotype of the ALDH2*2 mutation in iPSC-CMs under ischemic conditions. No differences were noted under control conditions when comparing ALDH2*2/1 to wild-type iPSC-CMs, except for a reduced metabolic rate in ALDH2*2/1 iPSC-CMs, reflected by diminished oxygen consumption (fig. S11). However, ALDH2*2/1 iPSC-CMs were found to be overall significantly more affected by ischemic challenge in vitro than wild-type controls.
Apoptosis-induced increase of ischemic damage in ALDH2*2/1 cardiomyocytes
Overall, these cellular profiling experiments have characterized the deleterious phenotypic and functional effects of the ALDH2*2 mutation in cardiomyocytes, especially with regard to ischemic challenge. Our data revealed that the great reduction of ALDH2 function in ALDH2*2/1 iPSC-CMs is connected to elevated ROS levels as well as a disturbed mitochondrial integrity and metabolic balance, resulting in reduced cellular viability.
On the basis of these findings, we decided to establish whether the higher sensitivity to ischemic challenge noted in ALDH2*2/1 iPSC-CMs also affects cellular viability. Using a high-content spectrophotometric approach, we found cellular viability in post-ischemic ALDH2*2/1 iPSC-CMs to be 2.3-fold lower, compared to equally ischemia-treated wild-type controls (Fig. 3, C and D). Activation of ALDH2 has been shown to partially overcome ischemic damage in cardiomyocytes (2). Accordingly, Alda-1–mediated ALDH2 activation before ischemic challenge improved the viability of ALDH2*2/1 iPSC-CMs after ischemia by 52.1 ± 8.0% (P < 0.05, as determined by Student’s t test; Fig. 3D).
We next performed TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling) staining to investigate whether 4HNE- and ROS-associated loss of viability and increased ischemic damage in ALDH2*2/1 iPSC-CMs were associated with apoptosis. After ischemic challenge, TUNEL-positive nuclei were 4.4 ± 0.4–fold enriched in ALDH2*2/1 iPSC-CMs but not in wild-type controls (Fig. 3, E and F). ALDH2*2/1 iPSC-CMs also showed fragmented DNA after ischemia (Fig. 3G). Together, these experiments establish a proapoptotic phenotype in post-ischemic ALDH2*2/1 iPSC-CMs compared to wild-type control iPSC-CMs.
Identification of signaling mechanisms underlying increased ischemic damage in ALDH2*2/1 cardiomyocytes
To further analyze the molecular basis of increased apoptosis after ischemia, we next performed genome-wide RNA sequencing in ALDH2*2/1 and wild-type control iPSC-CMs by comparing control and post-ischemic conditions. On the basis of changes in expression levels of 26,000 genes, Ingenuity Pathway Analysis (IPA) revealed significant up-regulation of mitogen-activated protein kinase (MAPK), apoptosis, redox, and ROS metabolism pathways in ALDH2*2/1 iPSC-CMs after ischemia, whereas cell cycle signaling was strongly reduced (Fig. 4, A and B, and tables S4 and S5). These data are in line with our previous findings of increased cell cycle arrest in ALDH2*2/1 fibroblasts (Fig. 2, C and D) and altered metabolism in ALDH2*2/1 iPSC-CMs (figs. S10 and S11). IPA-based mapping of significantly altered genes involved in these pathways (Fig. 4B) demonstrated drastic changes in apoptotic and cell cycle signaling, as well as metabolism expression profiles in ALDH2*2/1 iPSC-CMs, especially after ischemia.
Fig. 4. Transcriptome analysis identifies signaling mechanisms underlying the proapoptotic phenotype of ALDH2*2/1 iPSC-CMs after ischemia.
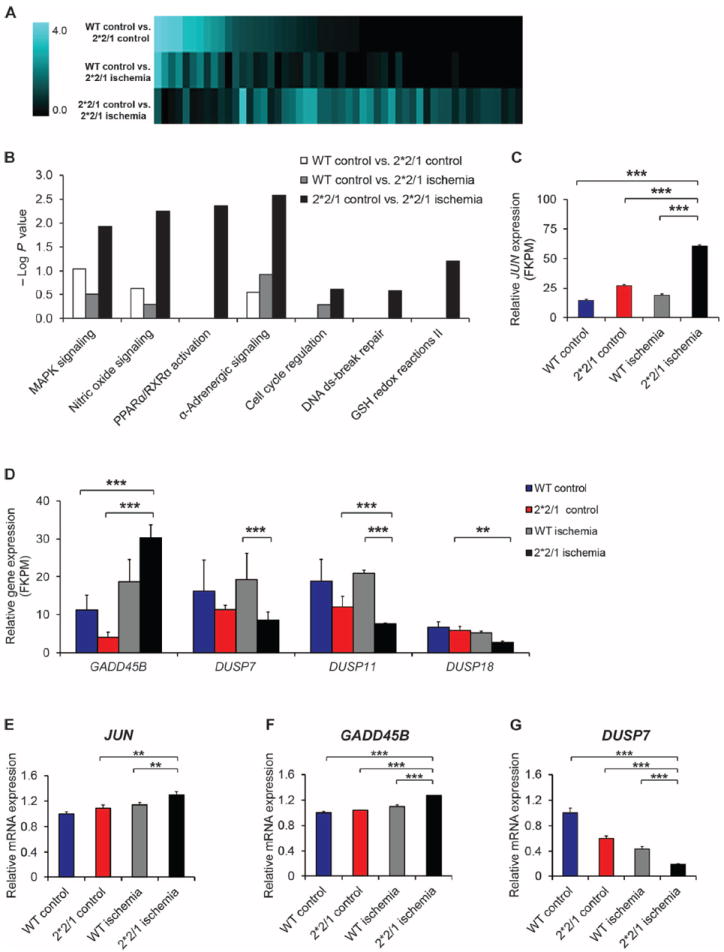
(A) Changes in the transcriptome of ALDH2*2/1 and wt iPSC-CMs after ischemia, as identified by whole-genome RNA sequencing and subsequent IPA. (B) IPA mapping for significantly altered pathways [−log (P value) >1.5] indicates marked profile changes comparing the baseline control (wt control versus ALDH2*2/1 control) iPSC-CMs to post-ischemia. MAPK, mitogen-activated protein kinase; PPARα/RXRα, peroxisome proliferator-activated receptor α and retinoid X receptor α; ds, double-stranded; GSH, glutathione. (C) Whole-genome RNA sequencing reveals that highly enhanced JUN expression was associated with the ALDH2*2/1 genotype, especially after ischemia. FKPM, fragments per kilobase of exon per million. (D) Changes in the expression levels of GADD45B, DUSP7, DUSP11, and DUSP18 as determined by whole-genome RNA sequencing. FKPM, fragments per kilobase of exon per million. (E) Validation by quantitative real-time polymerase chain reaction (qRT-PCR) confirms significantly increased JUN expression in ALDH2*2/1 iPSC-CMs after ischemia. Experiments performed in triplicate and n = 5 per group. (F and G) qRT-PCR–based validation also confirmed up-regulation of GADD45B as well as down-regulation of DUSP7 in post-ischemic ALDH2*2/1 iPSC-CMs. Experiments were performed in triplicate and n = 2 per group. Relative mRNA expression levels of each gene were determined by 2−ΔΔC, and expression was normalized to human 18S expression. Data are expressed as means ± SEM. **P < 0.01 and ***P < 0.001, as calculated by Student’s t test.
JUN expression was highly up-regulated in ALDH2*2/1 iPSC-CMs, again especially after ischemia (Fig. 4C). Previously, increased expression of JUN has been described to inhibit proliferation and induce apoptosis (40, 41). The JUN-encoded c-Jun protein exerts control of gene transcription via the activating protein 1 complex and can induce apoptotic and necrotic cell death (41, 42).
c-Jun is regulated by the Jun N-terminal kinase (JNK), which is activated by increased levels of ROS (43, 44). Therefore, we reasoned that ROS-mediated, prolonged activation of JNK in post-ischemic ALDH2*2/1 iPSC-CMs might lead to c-Jun–mediated downstream apoptotic signaling. Consistent with this notion, mRNA transcripts that encode the positive JNK regulator GADD45B (45) were up-regulated in post-ischemic ALDH2*2/1 iPSC-CMs. On the other hand, DUSP7, DUSP11, and DUSP18 transcripts, which encode the JNK-inactivating phosphatases DUSP7, DUSP11, and DUSP18, respectively (46), were significantly down-regulated (Fig. 4D) compared to control. Relative mRNA expression patterns were confirmed by qRT-PCR analysis (Fig. 4, E to G, and fig. S12).
Restoration of regular ROS levels and cell cycle progression by JNK inhibition
We next hypothesized that in ALDH2*2/1 iPSC-CMs, elevated levels of ROS and 4HNE might result in enhanced JNK activation and a subsequent increase in c-Jun–mediated apoptotic signaling and cell death. Therefore, to test whether inhibition of JNK signaling can reverse increased ischemic damage in ALDH2*2/1 iPSC-CMs, we used a JNK-specific inhibitor, JNK-interacting protein 1(JIP) (47). JNK inhibition significantly reduced endogenous ROS levels in post-ischemic ALDH2*2/1 iPSC-CMs (27.6 ± 1.4%; P = 0.01, as determined by Student’s t test; Fig. 5A). To evaluate the effects of ROS-enhanced JNK up-regulation in post-ischemic ALDH2*2/1 iPSC-CMs, we also challenged wild-type and ALDH2*2/1 iPSC-CMs with H2O2 before ischemia (Fig. 5B). Although wild-type iPSC-CMs were not significantly affected, ALDH2*2/1 iPSC-CMs were highly sensitive to external ROS challenge with H2O2. Moreover, JNK inhibition with JIP significantly reduced ROS susceptibility in post-ischemic ALDH2*2/1 iPSC-CMs even in the presence of H2O2 (34.4 ± 4.9%; P = 0.002, as determined by Student’s t test; Fig. 5B).
Fig. 5. JNK inhibition recovers ROS scavenging in ALDH2*2/1 iPSC-CMs and restores cell cycle progression in ALDH2*2/1 fibroblasts.

(A) Selective inhibition of JNK, the upstream regulator of c-Jun, by JIP decreases ROS levels in ALDH2*2/1 iPSC-CMs after ischemia (n = 3 per group). (B) ALDH2*2/1 and wt iPSC-CMs were challenged with H2O2 before ischemia in the presence or absence of control vehicle (DMSO) or JIP as indicated. In contrast to the wt control, ALDH2*2/1 iPSC-CMs cannot scavenge increased endogenous ROS during ischemia. This effect is enhanced if an external ROS challenge, H2O2, is applied to ALDH2*2/1 iPSC-CMs before ischemia. JIP-mediated JNK inhibition during H2O2 challenge can restore ROS scavenging in post-ischemic ALDH2* 2/1 iPSC-CMs. (C) Western blot analysis confirms reduced levels of phosphorylated JNK (p-JNK) upon JIP treatment in post-ischemic ALDH2*2/1 iPSC-CMs, compared to wt. Activation of ALDH2 and hence lower ROS levels also reduce p-JNK levels. Data are normalized for β-actin as loading control. (D) Quantification of Western blot analysis for seven experiments performed in ALDH2*2/1 or wt iPSC-CMs (n = 2 per group). See also table S6. (E) Viability ofALDH2*2/1 fibroblasts is rescued by inhibition of JNK via the chemical inhibitors SP600125 and JIP. Experiments were performed as n = 5 per group in triplicate. (F) siRNA-mediated knockdown of JUN confirms the rescue of cell viability in ALDH2*2/1 fibroblasts. Experiments were performed as n = 5 per group in triplicate. (G and H) Cell proliferation and cell cycle arrest can be overcome by JNK inhibition. (G) Representative confocal images show BrdU staining and costaining for total number of nuclei with DAPI, as well as the mitochondrial marker TOM20. Scale bar, 100 μM. (H) Quantification and statistical analysis. Experiments were performed in three independent cell lines per group, quantifying 10 images each. All data are expressed as means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001, as calculated by Student’s t test.
To confirm enhanced activation of JNK in post-ischemic ALDH2*2/1 iPSC-CMs, we analyzed lysates from post-ischemic ALDH2*2/1 and wild-type iPSC-CMs by immunoblotting and found increased phospho-JNK levels in ALDH2*2/1 iPSC-CMs (Fig. 5, C and D, and table S6). In line with our hypothesis, treatment of ALDH2*2/1 iPSC-CMs with JIP before ischemic challenge resulted in reduced levels of phospho-JNK (Fig. 5, C and D, and table S6). Activation of ALDH2 via Alda-1 before ischemic challenge showed comparable results, verifying that ALDH2-mediated ROS regulation directly affects JNK activity, with important consequences for cell survival after ischemia.
To further corroborate these findings, we established that application of JNK inhibitors (SP600125 and JIP) to ALDH2*2/1 fibroblasts can recover viability in these cells (Fig. 5E). Rescue of viability in ALDH2*2/1 fibroblasts can also be achieved by small interfering RNA (siRNA)–mediated knockdown of JUN (Fig. 5F). Finally, JNK inhibition with JIP also reversed cell cycle arrest in ALDH2*2/1 fibroblasts (Fig. 5, G and H). Together, these data imply that in post-ischemic cells with the ALDH2*2/1 genotype, proapoptotic signaling events as confirmed by transcriptome analysis (Fig. 4) are mediated via JNK and triggered through excess ROS, caused by greatly reduced ALDH2 function in ALDH2*2/1 cells.
ALDH2 as modulator in a cell survival–regulating cascade
These findings indicate a role of ALDH2 in the regulation of cell survival decisions via modulation of cellular ROS levels, which affects JNK-mediated downstream signaling and, eventually, c-Jun–dependent transcription of proapoptotic genes. To further support this hypothesis, we tested whether JNK inhibition before ischemic challenge can restore viability of ALDH2*2/1 iPSC-CMs. Using two independent JNK inhibitors (SP600125 and JIP), we found that the viability of post-ischemic ALDH2*2/1 iPSC-CMs was restored to levels found in nonischemic, untreated ALDH2*2/1 iPSC-CMs (Fig. 6, A and B). Quantification of TUNEL-positive nuclei (Fig. 6, C and D) also revealed reduced apoptosis in JIP-treated ALDH2*2/1 iPSC-CMs subjected to ischemia, implying a rescue from ischemia-associated damage. Collectively, our data indicate that cells with the ALDH2*2 loss-of-function mutation cannot efficiently control ROS levels, a deficiency that is linked to subsequent JNK-mediated downstream signaling, which leads to defective cell cycle signaling and, especially after ischemic challenge, c-Jun–mediated cell death signaling (Fig. 6E).
Fig. 6. JNK inhibition rescues the proapoptotic phenotype in ALDH2*2/1 iPSC-CMs.

(A and B) Inhibition of JNK via two selective inhibitors, SP600125 and JIP, increases viability in ALDH2*2/1 iPSC-CMs and wt iPSC-CMs after ischemia and reverses the proapoptotic phenotype of post-ischemic ALDH2*2/1 iPSC-CMs (n = 3 per group). (C and D) Inhibition of JNK significantly reduces apoptotic damage in ALDH2*2/1 iPSC-CMs after ischemia. (C) Representative confocal images showing TUNEL and DAPI staining, as well as costaining for TNNT2. Scale bar, 50 μM. (D) Quantification of TUNEL/DAPI ratio for n = 10 images per group. (E) Mechanistic schema of the cell survival cascade regulated by ALDH2. Loss of ALDH2 activity, such as that occurring in the ALDH2*2 genotype, leads to elevated 4HNE and increased ROS, which activates JNK and leads to downstream proapoptotic signaling events. These events are enhanced during ischemic challenge, which triggers additional ROS. In ischemic ALDH2*2/1 iPSC-CMs, accumulating 4HNE and unscavenged ROS build up over time, causing higher levels of JNK activity and downstream c-Jun–dependent activation of apoptotic genes. ALDH2 activation via Alda-1 as well as JNK inhibition via JIP can rescue the proapoptotic phenotype in post-ischemic ALDH2*2/1 iPSC-CMs, whereas 4HNE inhibits ALDH2. All data are expressed as means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001, calculated by Student’s t test.
DISCUSSION
Here, we used a human iPSC-CM platform to investigate the molecular mechanisms underlying the cardiac disease phenotypes observed in carriers of the dominant-negative ALDH2*2 genetic polymorphism.
Analysis and characterization of human ALDH2*2/1 iPSC-CMs revealed no significant differences compared to wild-type control iPSC-CMs under normoxic conditions; however, relative to wild type, ALDH2*2/1 iPSC-CMs were highly sensitive to ischemia simulated in vitro. These findings make functional sense in light of the higher incidence of MI and CAD in ALDH2*2 carriers reported previously (3, 4, 31). Independent genome-wide association and meta-analysis studies on the determining factors associated with the genetic basis of CAD have revealed the ALDH2*2 genotype as a risk factor for CAD (5, 48, 49). Carriers of the ALDH2*2 polymorphism in Chinese, Korean, and Japanese populations were found to display an increased susceptibility to both MI and CAD. In addition, studies in animal models have shown that the ALDH2*2 genotype is more susceptible to oxidative damage than wild-type animals and that oxidative stress–mediated inhibition of ALDH2 causes cardiac dysfunction (16). Oxidative stress and ROS have been reported to play major roles in CAD pathogenesis (50). During MI, ROS not only directly cause cell injury but also induce signaling events that contribute to cellular dysfunction, including damage of membranes and degradation of extracellular matrix components. The resulting structural changes induce left ventricular (LV) dilatation and, consequently, LV remodeling after MI (50, 51).
We show here that under ischemic conditions, ALDH2*2/1 iPSC-CMs displayed significantly elevated levels of ROS and apoptosis. Transcriptome profiling identified differences in ROS, cell cycle–related, and apoptosis signaling pathways and strong up-regulation of JUN and related transcripts. Thus, we reasoned that in ALDH2*2/1 iPSC-CMs, especially after ischemia, c-Jun–related apoptotic signaling might be caused by enhanced ROS-mediated activation of JNK. This is in line with previous evidence that JNK/c-Jun can negatively regulate cell proliferation (40).
Indeed, JNK inhibition reversed ROS sensitivity of ALDH2*2/1 iPSC-CMs after ischemia and restored viability to wild-type control levels. Our findings also demonstrated that in ALDH2*2/1 cells, an increase in 4HNE load and subsequent ROS accumulation induced downstream signaling events that led to reduced viability and cell death—a process that is further enhanced during ischemic stress—and that these events functioned via JNK and its downstream effecter c-Jun, enhancing transcription of proapoptotic genes. Mitochondrial ALDH2 metabolizes and inactivates the ROS enhancer 4HNE. Hence, impaired ALDH2 function in the ALDH2*2 genotype causes elevated levels of ROS and 4HNE, which support sustained JNK activation and c-Jun–mediated induction of apoptosis. Consequently, the lack of ALDH2-dependent ROS scavenging renders ALDH2*2/1 iPSC-CMs more susceptible to ischemic damage. Both activation of ALDH2 and inhibition of JNK can partially reverse these deleterious effects in ALDH2*2/1 cells.
We propose that the ALDH2*2 genotype is associated with defective regulation of ALDH2-dependent signaling events, which induces apoptosis and results in increased damage of ALDH2*2 iPSC-CMs during ischemia. These findings reveal a newly identified regulatory loop that provides insight into novel functions of ALDH2. We have discovered a direct link between ALDH2 activity and cell survival decisions by describing new aspects of mitochondrial ALDH2’s cardioprotective function.
Pinpointing the molecular basis underlying ALDH2-mediated cardioprotection has potential implications for the treatment of ischemic heart disease and regenerative medicine therapy in human carriers of the ALDH2*2 genetic polymorphism.
MATERIALS AND METHODS
Please see Supplementary Methods for full details.
Study design
To investigate the molecular mechanisms underlying increased ischemic damage in the ALDH2*2/1 genotype, a cohort of 10 consenting, 21- to 22-year-old male individuals of East Asian origin and with no family relations were recruited. Skin punch biopsies were digested using Collagenase IV and were kept in GlutaMAX-containing Dulbecco’s modified Eagle’s medium (Life Technologies) supplied with 10% fetal bovine serum (Gibco) under sterile culture conditions at 37°C. After 1 week, outgrowing fibroblast cell lines were subjected to genotyping. On the basis of the genotyping results, cell lines were grouped into control (wild-type ALDH2 genotype, n = 5) or heterozygous ALDH2 mutant (ALDH2*2/1, n = 5).
Genotyping
The ALDH2*2/1 genotype was determined by isolating genomic DNA from ~5 × 106 human fibroblasts per each line using a DNeasy Blood & Tissue kit (Qiagen) according to the manufacturer’s instructions. A ~300–base pair (bp) stretch of DNA upstream and downstream of the point mutation E487K was amplified via PCR using the following primers: ALDH2 E487K: forward, 5′gtcaactgctatgatgtgtttggagccc3′; reverse, 5′caagcatgagggagaggccaaaagg3′. PCR products were run on a 2% agarose gel, and DNA was extracted using a QIAquick Gel Extraction Kit (Qiagen) and sequenced at Quintara Biosciences (San Francisco) using the primers ALDH2 E487K–forward and ALDH2 E487K–reverse, respectively.
Cardiac differentiation of iPSCs
Human iPSCs were grown to 90% confluence (52-54), after which iPSCs were differentiated into beating cardiomyocytes using a small molecule–based monolayer method (37) as described in detail previously (38). After differentiation, human iPSC-CMs were cultured in RPMI medium plus B-27 Supplement (Life Technologies). iPSC-CMs expressed typical cardiac markers such as cardiac troponin T (TNNT2), sarcomeric α-actinin, and myosin light chain 2a and showed electrophysiological profiles effective of atrial-, nodal-, and ventricular-like cells as well as normal calcium transients. About 30 days after cardiac differentiation, beating iPSC-CM monolayers were dissociated using TripleE (10 min) or Accutase (30 min), washed, and plated in the respective assay format.
Measurement of ALDH2 activity
Cell lysates (200 μg) were added into a cuvette containing activity assay buffer and substrate [50 mM sodium pyrophosphate buffer at pH 9.0, 2.5 mM NAD+ (nicotinamide adenine dinucleotide), 10 mM acetaldehyde, and 460 μl of H2O] with a final volume of 2 ml. Optical density at A340 nm was measured at 25°C for increase of NADH (reduced form of NAD+) for 5 min. Blank control was no acetaldehyde. To show ALDH2 sensitivity of this assay, the ALDH2-specific small-molecule activator Alda-1 (20-μM final concentration) or the ALDH2-specific inhibitor daidzin (50 μM final concentration) was added to the lysates. When indicated, 4-MP, an alcohol dehydrogenase inhibitor, was preincubated with the lysates at 200-μM final concentration for 1 hour before measurements.
Measurement of ROS
Fibroblasts or iPSC-CMs cultured on 96-well plates were treated with small molecules or vehicle control as indicated below. Subsequently, basal cellular levels of ROS were determined using an Amplex Red assay kit (Life Technologies) according to the manufacturer’s instructions. The Amplex Red assay is a measure of cellular ROS by specific detection of cellular H2O2 resulting from degrading intracellular free radicals, such as superoxide. Cellular H2O2 and horseradish peroxidase present in the Amplex Red assay convert Amplex Red reagent to its fluorescent product, resorufin. Fluorescence emitted by this reaction was measured at 562 nm using a GloMax spectrophotometer plate reader (Promega). Other than as indicated in the figure, no additional external ROS challenge was added to the cells. An H2O2 standard curve was used for confirming the assay’s sensitivity and background signal. Specificity of this reaction was further confirmed by pretreating fibroblasts or iPSC-CMs, where indicated, for 4 hours with an unrelated ROS scavenger, catalase. To exclude interference of Alda-1 and 4HNE with the Amplex Red experimental readout, a Xanthine/Xanthine Oxidase Amplex Red–based assay kit (Life Technologies) was used according to the manufacturer’s instructions. Alternatively, cellular ROS were detected by addition of the fluorogenic dye 5-(and-6)-carboxy-2′,7′-dichlorofluorescein diacetate (DCFDA, Life Technologies) according to the manufacturer’s instructions.
RNA sequencing and data analysis
RNA was isolated from ALDH2*2/1 and wild-type control iPSC-CMs (n = 2 per group) using a Qiagen microRNA kit, and 100 ng of total RNA was converted to complementary DNA (cDNA) and amplified using NuGEN V2 RNA-Seq kit (NuGEN). cDNA was sonicated to an average fragment size of 300 bp, and Illumina sequencing adapters were ligated to 500 ng of cDNA using NEBNext mRNA Library Prep Reagent Set for Illumina (New England Biolabs). PCR was performed on the adapter-ligated cDNA under the following conditions: denaturation at 98°C for 30 s, followed by eight cycles of denaturation at 98°C for 10 s, annealing at 65°C for 30 s, and extension at 72°C for 30 s, ending with an additional extension at 72°C for 5 min. Sequencing was performed using Illumina’s HiSeq2000 platform using paired in reads at an average length of 100 bp (2 × 100). Reads were filtered to produce an average of 60 million reads per sample and were aligned via the hg19 reference genome using TopHat version 2.0.8b (PMID 19289445). Differentially regulated transcripts and gene expression levels were ascertained using Cufflinks v2.1.1 (55) algorithms.
Chemical inhibitors, activators, and siRNA-mediated knockdown
Fibroblasts or iPSC-CMs were treated with small-molecule activators or inhibitors, H2O2, or control vehicle as indicated below for 48 hours. Lyophilized Alda-1 was dissolved in DMSO and used at a final concentration of 20 μM. SP600125 (Cell Signaling Technology) was used at a final concentration of 20 uM. TAT-TI-JIP (EMD Millipore) was used at a final concentration of 1 μM. Vehicle control indicates the application of an equal concentration of solvent (DMSO). Challenge with H2O2 was performed at a final concentration of 200 μM. siRNA-mediated knockdown of JUN was performed with a Silencer Select siRNA, s7659 (Applied Biosystems), at 50-nM final concentration, as well as Silencer Negative Control #1 siRNA (Applied Biosystems).
Statistical analysis
An unpaired Student’s t test was used to calculate significances between two groups. If the data were normally distributed, a one-way analysis of variance (ANOVA) was used, and multiple comparison correction analysis was performed. A P value of <0.05 was considered statistically significant.
Supplementary Material
Methods
Fig. S1. Effect of 4-MP and daidzin on ALDH2 activity in cell lysates from ALDH2*2/1 and wild-type fibroblasts.
Fig. S2. ROS are enhanced upon external challenge with 4HNE in ALDH2*2/1 fibroblasts, but not in wild-type control.
Fig. S3. Alda-1 and 4HNE do not interfere with Amplex Red–based analysis of xanthine oxidase–catalyzed H2O2 formation.
Fig. S4. Characterization of a human iPSC model for the ALDH2*2/1 genetic polymorphism.
Fig. S5. ALDH2*2/1 and wild-type iPSCs form teratoma and present a regular karyotype.
Fig. S6. Profiling of ALDH2*2/1 and wild-type control iPSC-CMs.
Fig. S7. Electrophysiological analysis of ALDH2*2/1 and wild-type iPSC-CMs reveals typical profiles.
Fig. S8. ALDH2*2/1 iPSC-CMs express equal levels of ALDH2 compared to wild-type control.
Fig. S9. Effect of an unrelated ROS scavenger, catalase, on total cellular ROS levels in post-ischemic ALDH2*2/1 iPSC-CMs, compared to wild-type control.
Fig. S10. Analysis of differences in mitochondrial-based metabolism comparing ALDH2*2/1 iPSC-CMs versus wild-type control iPSC-CMs.
Fig. S11. Metabolic profiling in ALDH2*2/1 and wild-type iPSC-CMs.
Fig. S12. Validation of relative transcriptome changes for selected target genes in ALDH2*2/1 and wild-type iPSC-CMs, comparing control (normoxia) and post-ischemia.
Table S1. Overview on profiling assays performed for characterization of iPSCs.
Table S2. Overview on profiling assays performed for characterization of iPSC-CMs.
Table S3. Quantification and detailed parameters of experiments shown in fig. S4.
Table S4. Canonical pathways and corresponding fold changes [−log (P value)] displayed in Fig. 4A.
Table S5. Canonical pathways and corresponding fold changes [−log (P value)] displayed in Fig. 4B.
Table S6. Increased levels of p-JNK in post-ischemic ALDH2*2/1 iPSC-CMs are reduced by JNK inhibition (JIP) and ALDH2 activation (Alda-1).
Video S1. Contracting monolayer of control wild-type iPSC-CMs.
Video S2. Contracting monolayer of ALDH2*2/1 iPSC-CMs.
Acknowledgments
We thank B. Wu for the critical reading of the manuscript. We are grateful for the support by the shared resource facilities at Stanford, including the Neuroscience Microscopy Service supported by NIH grant NS069375, the FACS Core at the Institute for Stem Cell Biology and Regenerative Medicine, and the Stanford Shared FACS Facility. Funding: This work is supported by research grants from the American Heart Association Established Investigator Award, NIH grant R01 HL113006, NIH grant R01 HL123968, NIH grant U01 099776, NIH grant R24 HL117756, NIH grant P01 GM099130, and Fondation Leducq grant 11CDV02 (J.C.W.), NIH grant AA11147 (D.M.-R.), and Deutsche Forschungsgemeinschaft (German Research Foundation) EB480/1-1 (A.D.E.).
Footnotes
Author contributions: A.D.E., J.C.W., and D.M.-R. designed the study and participated in data analysis as well as manuscript writing. J.D.G. participated in manuscript editing. A.D.E. participated in all experimental work. K.K. performed RNA sequencing validation. H.W. performed calcium imaging. P.L. performed electrophysiology. J.C. performed the Illumina library preparation. J.L., B.C.H., J.R., P.d.A., P.W.B., S.D., and F.L. participated in the acquisition and/or analysis of the data.
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/6/255/255ra130/DC1
Competing interests: J.C.W. is a cofounder of Stem Cell Theranostics. D.M.-R. is the founder of ALDEA Pharmaceuticals. However, she has no role in the company, and the research in her laboratory is supported only by the NIH and is not disclosed to the company.
Data and materials availability: The data for this study have been deposited in the database Gene Expression Omnibus, accession no. GSE59100.
REFERENCES AND NOTES
- 1.Larson HN, Weiner H, Hurley TD. Disruption of the coenzyme binding site and dimer interface revealed in the crystal structure of mitochondrial aldehyde dehydrogenase “Asian” variant. J Biol Chem. 2005;280:30550–30556. doi: 10.1074/jbc.M502345200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–1495. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guo YJ, Chen L, Bai YP, Li L, Sun J, Zhang GG, Yang TL, Xia J, Li YJ, Chen XP. The ALDH2 Glu504Lys polymorphism is associated with coronary artery disease in Han Chinese: Relation with endothelial ADMA levels. Atherosclerosis. 2010;211:545–550. doi: 10.1016/j.atherosclerosis.2010.03.030. [DOI] [PubMed] [Google Scholar]
- 4.Takagi S, Iwai N, Yamauchi R, Kojima S, Yasuno S, Baba T, Terashima M, Tsutsumi Y, Suzuki S, Morii I, Hanai S, Ono K, Baba S, Tomoike H, Kawamura A, Miyazaki S, Nonogi H, Goto Y. Aldehyde dehydrogenase 2 gene is a risk factor for myocardial infarction in Japanese men. Hypertens Res. 2002;25:677–681. doi: 10.1291/hypres.25.677. [DOI] [PubMed] [Google Scholar]
- 5.Takeuchi F, Yokota M, Yamamoto K, Nakashima E, Katsuya T, Asano H, Isono M, Nabika T, Sugiyama T, Fujioka A, Awata N, Ohnaka K, Nakatochi M, Kitajima H, Rakugi H, Nakamura J, Ohkubo T, Imai Y, Shimamoto K, Yamori Y, Yamaguchi S, Kobayashi S, Takayanagi R, Ogihara T, Kato N. Genome-wide association study of coronary artery disease in the Japanese. Eur J Hum Genet. 2012;20:333–340. doi: 10.1038/ejhg.2011.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang H, Gong DX, Zhang YJ, Li SJ, Hu S. Effect of mitochondrial aldehyde dehydrogenase-2 genotype on cardioprotection in patients with congenital heart disease. Eur Heart J. 2012;33:1606–1614. doi: 10.1093/eurheartj/ehs061. [DOI] [PubMed] [Google Scholar]
- 7.Xu F, Chen Y, Lv R, Zhang H, Tian H, Bian Y, Feng J, Sun Y, Li R, Wang R, Zhang Y. ALDH2 genetic polymorphism and the risk of type II diabetes mellitus in CAD patients. ALDH2 genetic polymorphism and the risk of T2DM in CAD patients. Hypertens Res. 2010;33:49–55. doi: 10.1038/hr.2009.178. [DOI] [PubMed] [Google Scholar]
- 8.Yokoyama A, Mizukami T, Matsui T, Yokoyama T, Kimura M, Matsushita S, Higuchi S, Maruyama K. Genetic polymorphisms of alcohol dehydrogenase-1B and aldehyde dehydrogenase-2 and liver cirrhosis, chronic calcific pancreatitis, diabetes mellitus, and hypertension among Japanese alcoholic men. Alcohol Clin Exp Res. 2013;37:1391–1401. doi: 10.1111/acer.12108. [DOI] [PubMed] [Google Scholar]
- 9.Wu C, Kraft P, Zhai K, Chang J, Wang Z, Li Y, Hu Z, He Z, Jia W, Abnet CC, Liang L, Hu N, Miao X, Zhou Y, Liu Z, Zhan Q, Liu Y, Qiao Y, Zhou Y, Jin G, Guo C, Lu C, Yang H, Fu J, Yu D, Freedman ND, Ding T, Tan W, Goldstein AM, Wu T, Shen H, Ke Y, Zeng Y, Chanock SJ, Taylor PR, Lin D. Genome-wide association analyses of esophageal squamous cell carcinoma in Chinese identify multiple susceptibility loci and geneenvironment interactions. Nat Genet. 2012;44:1090–1097. doi: 10.1038/ng.2411. [DOI] [PubMed] [Google Scholar]
- 10.Dickinson BC, Chang CJ. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat Chem Biol. 2011;7:504–511. doi: 10.1038/nchembio.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lucas DT, Szweda LI. Cardiac reperfusion injury: Aging, lipid peroxidation, and mitochondrial dysfunction. Proc Natl Acad Sci U S A. 1998;95:510–514. doi: 10.1073/pnas.95.2.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eaton P, Li JM, Hearse DJ, Shattock MJ. Formation of 4-hydroxy-2-nonenal-modified proteins in ischemic rat heart. Am J Physiol. 1999;276(Pt 2):H935–H943. doi: 10.1152/ajpheart.1999.276.3.H935. [DOI] [PubMed] [Google Scholar]
- 13.Madamanchi NR, Runge MS. Redox signaling in cardiovascular health and disease. Free Radic Biol Med. 2013;61C:473–501. doi: 10.1016/j.freeradbiomed.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohta S, Ohsawa I, Kamino K, Ando F, Shimokata H. Mitochondrial ALDH2 deficiency as an oxidative stress. Ann N Y Acad Sci. 2004;1011:36–44. doi: 10.1007/978-3-662-41088-2_4. [DOI] [PubMed] [Google Scholar]
- 15.Chen CH, Sun L, Mochly-Rosen D. Mitochondrial aldehyde dehydrogenase and cardiac diseases. Cardiovasc Res. 2010;88:51–57. doi: 10.1093/cvr/cvq192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J, Wang H, Hao P, Xue L, Wei S, Zhang Y, Chen Y. Inhibition of aldehyde dehydrogenase 2 by oxidative stress is associated with cardiac dysfunction in diabetic rats. Mol Med. 2011;17:172–179. doi: 10.2119/molmed.2010.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee SH, Oe T, Blair IA. Vitamin C–induced decomposition of lipid hydroperoxides to endogenous genotoxins. Science. 2001;292:2083–2086. doi: 10.1126/science.1059501. [DOI] [PubMed] [Google Scholar]
- 18.Li SY, Du M, Dolence EK, Fang CX, Mayer GE, Ceylan-Isik AF, LaCour KH, Yang X, Wilbert CJ, Sreejayan N, Ren J. Aging induces cardiac diastolic dysfunction, oxidative stress, accumulation of advanced glycation endproducts and protein modification. Aging Cell. 2005;4:57–64. doi: 10.1111/j.1474-9728.2005.00146.x. [DOI] [PubMed] [Google Scholar]
- 19.Beretta M, Gorren AC, Wenzl MV, Weis R, Russwurm M, Koesling D, Schmidt K, Mayer B. Characterization of the East Asian variant of aldehyde dehydrogenase-2: Bioactivation of nitroglycerin and effects of Alda-1. J Biol Chem. 2010;285:943–952. doi: 10.1074/jbc.M109.014548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Z, Zhang J, Stamler JS. Identification of the enzymatic mechanism of nitroglycerin bioactivation. Proc Natl Acad Sci U S A. 2002;99:8306–8311. doi: 10.1073/pnas.122225199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Z, Stamler JS. Bioactivation of nitroglycerin by the mitochondrial aldehyde dehydrogenase. Trends Cardiovasc Med. 2006;16:259–265. doi: 10.1016/j.tcm.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 22.Ferreira JC, Mochly-Rosen D. Nitroglycerin use in myocardial infarction patients. Circ J. 2012;76:15–21. doi: 10.1253/circj.cj-11-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun L, Ferreira JC, Mochly-Rosen D. ALDH2 activator inhibits increased myocardial infarction injury by nitroglycerin tolerance. Sci Transl Med. 2011;3:107ra111. doi: 10.1126/scitranslmed.3002067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun A, Ren J. ALDH2, a novel protector against stroke? Cell Res. 2013;23:874–875. doi: 10.1038/cr.2013.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Budas GR, Disatnik MH, Chen CH, Mochly-Rosen D. Activation of aldehyde dehydrogenase 2 (ALDH2) confers cardioprotection in protein kinase C epsilon (PKCe) knockout mice. J Mol Cell Cardiol. 2010;48:757–764. doi: 10.1016/j.yjmcc.2009.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Larson HN, Zhou J, Chen Z, Stamler JS, Weiner H, Hurley TD. Structural and functional consequences of coenzyme binding to the inactive Asian variant of mitochondrial aldehyde dehydrogenase: Roles of residues 475 and 487. J Biol Chem. 2007;282:12940–12950. doi: 10.1074/jbc.M607959200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barrera G, Pizzimenti S, Dianzani MU. 4-Hydroxynonenal and regulation of cell cycle: Effects on the pRb/E2F pathway. Free Radic Biol Med. 2004;37:597–606. doi: 10.1016/j.freeradbiomed.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 28.Chaudhary P, Sharma R, Sharma A, Vatsyayan R, Yadav S, Singhal SS, Rauniyar N, Prokai L, Awasthi S, Awasthi YC. Mechanisms of 4-hydroxy-2-nonenal induced pro- and anti-apoptotic signaling. Biochemistry. 2010;49:6263–6275. doi: 10.1021/bi100517x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang Y, Sharma R, Sharma A, Awasthi S, Awasthi YC. Lipid peroxidation and cell cycle signaling: 4-Hydroxynonenal, a key molecule in stress mediated signaling. Acta Biochim Pol. 2003;50:319–336. [PubMed] [Google Scholar]
- 30.Zambelli VO, Gross ER, Chen CH, Gutierrez VP, Cury Y, Mochly-Rosen D. Aldehyde dehydrogenase-2 regulates nociception in rodent models of acute inflammatory pain. Sci Transl Med. 2014;6:251ra118. doi: 10.1126/scitranslmed.3009539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu F, Chen YG, Xue L, Li RJ, Zhang H, Bian Y, Zhang C, Lv RJ, Feng JB, Zhang Y. Role of aldehyde dehydrogenase 2 Glu504lys polymorphism in acute coronary syndrome. J Cell Mol Med. 2011;15:1955–1962. doi: 10.1111/j.1582-4934.2010.01181.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen J, Henderson GI, Freeman GL. Role of 4-hydroxynonenal in modification of cytochrome c oxidase in ischemia/reperfused rat heart. J Mol Cell Cardiol. 2001;33:1919–1927. doi: 10.1006/jmcc.2001.1454. [DOI] [PubMed] [Google Scholar]
- 33.Perez-Miller S, Younus H, Vanam R, Chen CH, Mochly-Rosen D, Hurley TD. Alda-1 is an agonist and chemical chaperone for the common human aldehyde dehydrogenase 2 variant. Nat Struct Mol Biol. 2010;17:159–164. doi: 10.1038/nsmb.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gomes KM, Campos JC, Bechara LR, Queliconi B, Lima VM, Disatnik MH, Magno P, Chen CH, Brum PC, Kowaltowski AJ, Mochly-Rosen D, Ferreira JC. Aldehyde dehydrogenase 2 activation in heart failure restores mitochondrial function and improves ventricular function and remodelling. Cardiovasc Res. 2014;103:498–508. doi: 10.1093/cvr/cvu125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moneypenny CG, Gallagher EP. 4-Hydroxynonenal inhibits cell proliferation and alters differentiation pathways in human fetal liver hematopoietic stem cells. Biochem Pharmacol. 2005;69:105–112. doi: 10.1016/j.bcp.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 36.Fazio VM, Rinaldi M, Ciafrè S, Barrera G, Farace MG. Control of neoplastic cell proliferation and differentiation by restoration of 4-hydroxynonenal physiological concentrations. Mol Aspects Med. 1993;14:217–228. doi: 10.1016/0098-2997(93)90008-2. [DOI] [PubMed] [Google Scholar]
- 37.Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, Raval KK, Zhang J, Kamp TJ, Palecek SP. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci U S A. 2012;109:E1848–E1857. doi: 10.1073/pnas.1200250109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu S, Wilson KD, Ghosh Z, Han L, Wang Y, Lan F, Ransohoff KJ, Burridge P, Wu JC. MicroRNA-302 increases reprogramming efficiency via repression of NR2F2. Stem Cells. 2013;31:259–268. doi: 10.1002/stem.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281:29776–29787. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- 40.Yeap YY, Ng IH, Badrian B, Nguyen TV, Yip YY, Dhillon AS, Mutsaers SE, Silke J, Bogoyevitch MA, Ng DC. c-Jun N-terminal kinase/c-Jun inhibits fibroblast proliferation by negatively regulating the levels of stathmin/oncoprotein 18. Biochem J. 2010;430:345–354. doi: 10.1042/BJ20100425. [DOI] [PubMed] [Google Scholar]
- 41.Podar K, Raab MS, Tonon G, Sattler M, Barilà D, Zhang J, Tai YT, Yasui H, Raje N, DePinho RA, Hideshima T, Chauhan D, Anderson KC. Up-regulation of c-Jun inhibits proliferation and induces apoptosis via caspase-triggered c-Abl cleavage in human multiple myeloma. Cancer Res. 2007;67:1680–1688. doi: 10.1158/0008-5472.CAN-06-1863. [DOI] [PubMed] [Google Scholar]
- 42.Wisdom R, Johnson RS, Moore C. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J. 1999;18:188–197. doi: 10.1093/emboj/18.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seki E, Brenner DA, Karin M. A liver full of JNK: Signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology. 2012;143:307–320. doi: 10.1053/j.gastro.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen YR, Wang X, Templeton D, Davis RJ, Tan TH. The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and g radiation. Duration of JNK activation may determine cell death and proliferation. J Biol Chem. 1996;271:31929–31936. doi: 10.1074/jbc.271.50.31929. [DOI] [PubMed] [Google Scholar]
- 45.Liebermann DA, Tront JS, Sha X, Mukherjee K, Mohamed-Hadley A, Hoffman B. Gadd45 stress sensors in malignancy and leukemia. Crit Rev Oncog. 2011;16:129–140. doi: 10.1615/critrevoncog.v16.i1-2.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 2008;27:253–261. doi: 10.1007/s10555-008-9123-1. [DOI] [PubMed] [Google Scholar]
- 47.Barr RK, Kendrick TS, Bogoyevitch MA. Identification of the critical features of a small peptide inhibitor of JNK activity. J Biol Chem. 2002;277:10987–10997. doi: 10.1074/jbc.M107565200. [DOI] [PubMed] [Google Scholar]
- 48.Wang Q, Zhou S, Wang L, Lei M, Wang Y, Miao C, Jin Y. ALDH2 rs671 polymorphism and coronary heart disease risk among Asian populations: A meta-analysis and meta-regression. DNA Cell Biol. 2013;32:393–399. doi: 10.1089/dna.2013.1995. [DOI] [PubMed] [Google Scholar]
- 49.Gu JY, Li LW. ALDH2 Glu504Lys polymorphism and susceptibility to coronary artery disease and myocardial infarction in East Asians: A meta-analysis. Arch Med Res. 2014;45:76–83. doi: 10.1016/j.arcmed.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 50.Hori M, Nishida K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc Res. 2009;81:457–464. doi: 10.1093/cvr/cvn335. [DOI] [PubMed] [Google Scholar]
- 51.Rohde LE, Ducharme A, Arroyo LH, Aikawa M, Sukhova GH, Lopez-Anaya A, McClure KF, Mitchell PG, Libby P, Lee RT. Matrix metalloproteinase inhibition attenuates early left ventricular enlargement after experimental myocardial infarction in mice. Circulation. 1999;99:3063–3070. doi: 10.1161/01.cir.99.23.3063. [DOI] [PubMed] [Google Scholar]
- 52.Chen G, Gulbranson DR, Hou Z, Bolin JM, Ruotti V, Probasco MD, Smuga-Otto K, Howden SE, Diol NR, Propson NE, Wagner R, Lee GO, Antosiewicz-Bourget J, Teng JM, Thomson JA. Chemically defined conditions for human iPSC derivation and culture. Nat Methods. 2011;8:424–429. doi: 10.1038/nmeth.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun N, Panetta NJ, Gupta DM, Wilson KD, Lee A, Jia F, Hu S, Cherry AM, Robbins RC, Longaker MT, Wu JC. Feeder-free derivation of induced pluripotent stem cells from adult human adipose stem cells. Proc Natl Acad Sci U S A. 2009;106:15720–15725. doi: 10.1073/pnas.0908450106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lan F, Lee AS, Liang P, Sanchez-Freire V, Nguyen PK, Wang L, Han L, Yen M, Wang Y, Sun N, Abilez OJ, Hu S, Ebert AD, Navarrete EG, Simmons CS, Wheeler M, Pruitt B, Lewis R, Yamaguchi Y, Ashley EA, Bers DM, Robbins RC, Longaker MT, Wu JC. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell. 2013;12:101–113. doi: 10.1016/j.stem.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Methods
Fig. S1. Effect of 4-MP and daidzin on ALDH2 activity in cell lysates from ALDH2*2/1 and wild-type fibroblasts.
Fig. S2. ROS are enhanced upon external challenge with 4HNE in ALDH2*2/1 fibroblasts, but not in wild-type control.
Fig. S3. Alda-1 and 4HNE do not interfere with Amplex Red–based analysis of xanthine oxidase–catalyzed H2O2 formation.
Fig. S4. Characterization of a human iPSC model for the ALDH2*2/1 genetic polymorphism.
Fig. S5. ALDH2*2/1 and wild-type iPSCs form teratoma and present a regular karyotype.
Fig. S6. Profiling of ALDH2*2/1 and wild-type control iPSC-CMs.
Fig. S7. Electrophysiological analysis of ALDH2*2/1 and wild-type iPSC-CMs reveals typical profiles.
Fig. S8. ALDH2*2/1 iPSC-CMs express equal levels of ALDH2 compared to wild-type control.
Fig. S9. Effect of an unrelated ROS scavenger, catalase, on total cellular ROS levels in post-ischemic ALDH2*2/1 iPSC-CMs, compared to wild-type control.
Fig. S10. Analysis of differences in mitochondrial-based metabolism comparing ALDH2*2/1 iPSC-CMs versus wild-type control iPSC-CMs.
Fig. S11. Metabolic profiling in ALDH2*2/1 and wild-type iPSC-CMs.
Fig. S12. Validation of relative transcriptome changes for selected target genes in ALDH2*2/1 and wild-type iPSC-CMs, comparing control (normoxia) and post-ischemia.
Table S1. Overview on profiling assays performed for characterization of iPSCs.
Table S2. Overview on profiling assays performed for characterization of iPSC-CMs.
Table S3. Quantification and detailed parameters of experiments shown in fig. S4.
Table S4. Canonical pathways and corresponding fold changes [−log (P value)] displayed in Fig. 4A.
Table S5. Canonical pathways and corresponding fold changes [−log (P value)] displayed in Fig. 4B.
Table S6. Increased levels of p-JNK in post-ischemic ALDH2*2/1 iPSC-CMs are reduced by JNK inhibition (JIP) and ALDH2 activation (Alda-1).
Video S1. Contracting monolayer of control wild-type iPSC-CMs.
Video S2. Contracting monolayer of ALDH2*2/1 iPSC-CMs.