

Abstract
Genes encoding orthologs of the vaccinia virus G1 protein are present in all poxviruses for which sequence information is available, yet neither the role of the protein nor its requirement for virus replication is known. G1 was predicted to be involved in the cleavage of core proteins, based on a transfection study and the presence of an HXXEH motif found in a subset of metallopeptidases. In the present study, we engineered a recombinant vaccinia virus containing a single copy of the G1L gene with a C-terminal epitope tag that is stringently regulated by the Escherichia coli lac repressor. In the absence of inducer, expression of G1 was repressed and virus replication was inhibited. Rescue of infectious virus was achieved by expression of wild-type G1 in trans, but not when the putative protease active site residues histidine-41, glutamate-44, or histidine-45 were mutated. Nevertheless, the synthesis and proteolytic processing of major core and membrane proteins appeared unaffected under nonpermissive conditions, distinguishing the phenotype of the G1L mutant from one in which the gene encoding the I7 protease was repressed. Noninfectious virus particles, assembled in the absence of inducer, did not attain the oval shape or characteristic core structure of mature virions. The polypeptide composition of these particles, however, closely resembled that of wild-type virus. Full-length and shorter forms of the G1 protein were found in the core fraction of virus particles assembled in the presence of inducer, suggesting that G1 is processed by self-cleavage or by another protease.
Vaccinia virus (VV), a large DNA virus that replicates entirely in the cytoplasm of infected cells, is predicted to encode nearly 200 proteins with roles in DNA replication, transcription, virus assembly, and host interactions (14). Of these open reading frames (ORFs), only about 50 are recognizable in all insect and vertebrate poxviruses (17). Surprisingly, little or nothing is known about the roles of the products of many of these highly conserved genes. The protein encoded by one such gene, ORF 078 (henceforth referred to as G1L) of VV strain WR (accession number AY243312), contains the sequence HXXEH (19), which is present in a subset of metallopeptidases and is an inversion of the classical HEXXH metallopeptidase motif (3, 15). Such short sequences, however, are found in numerous proteins that are not known to have proteolytic activity. Using a plasmid transfection assay, Whitehead and Hruby (19) reported that expression of G1 induced the cleavage of the VV core protein precursor L4 (also known as P25K), albeit at an apparently unnatural AG/S site, and that this activity was inhibited by mutations of the HXXEH sequence. However, it is not known whether G1 is required for protein cleavages during virus replication or even whether the gene is essential. The suggested role of G1 is clouded by recent reports indicating that I7, a protein with a cysteine protease domain, induces cleavage of L4 at the natural AG/A site as well as the AG/S site (1, 6). Furthermore, conditional lethal mutants that express a temperature-sensitive I7 protein (6, 12) or repress expression of I7 (1) have a block in morphogenesis and fail to cleave L4 and other core proteins.
To investigate the role of G1, we made a recombinant VV with an inducible G1L gene that exhibited a conditional lethal phenotype. Virus rescue could be achieved by transfection of a plasmid that expresses wild-type G1 but not by plasmids with mutations in the HXXEH motif, supporting its function as a metalloprotease. Nevertheless, cleavage of core and membrane proteins occurred under nonpermissive conditions, distinguishing it from the I7 protease. In the absence of inducer, aberrant, noninfectious particles containing the major virion proteins formed. That finding and the association of G1 with the cores of purified virions suggested a role at a late stage of morphogenesis. The presence of both a full-length and shorter form of G1 raised the possibility that the protein has self-cleaving activity or is cleaved by another protease.
MATERIALS AND METHODS
Cells and viruses.
BS-C-1 and HeLa S3 cells were grown in minimum essential medium with Earle's salts (EMEM) and Dulbecco's modified Eagle's medium, respectively, each obtained from Quality Biologicals, Inc. (Gaithersburg, Md.) and supplemented with 10% fetal bovine serum (FBS). VV strain WR and the recombinant VV vT7LacOI were propagated in HeLa cells as described previously (10). The recombinant vG1Li was replicated in HeLa cells in the presence of 50 μM isopropyl-β-d-thiogalactopyranoside (IPTG) and 2.5% FBS. Virus stocks were stored at −70°C.
Plasmids.
To construct pVOTE-G1L-HA, a copy of the G1L ORF flanked by BspMI and BamHI sites was generated from viral DNA by PCR using primers that added an influenza virus hemagglutinin (HA) epitope tag at the C terminus, cloned into pGEM-T (Promega), digested with BspMI and BamHI, and inserted into NcoI and BamHI sites in pVOTE.1 (18).
To produce pGFP.ΔG1L, we generated three separate DNA segments by PCR. The first segment comprised the entire G3L ORF, including 7 nucleotides that overlap with the 5′ region of the G2R ORF as well as the next 273 nucleotides of the latter. The second PCR product contained the ORF encoding enhanced green fluorescent protein (GFP) regulated by the VV late P11 promoter. The third DNA contained the last 673 nucleotides of the I8R ORF. The three PCR products were assembled so that the GFP-containing fragment was flanked by the G3L/G2R and I8R DNA at its 5′ and 3′ ends, respectively.
PSLP-G1L-HA was made by inserting a copy of the G1L ORF generated by PCR with primers containing SalI and BamHI sites and an influenza virus HA epitope tag at the C terminus of G1L into like sites of pUC19 (pSLP), which was modified to contain a VV strong late synthetic promoter upstream of the SalI site (8). Mutations in nucleotides that encode the putative catalytic residues of the G1 protein were made using the QuikChange mutagenesis kit (Stratagene, La Jolla, Calif.).
Construction of plasmid pSLP-L4R was described previously (1). Fluorescence dideoxy termination sequencing was used to confirm the sequences of relevant portions of all plasmids.
Construction of recombinant viruses.
vG1Li was constructed in two steps using the VOTE system essentially as described for vI7Li (1). First, plasmid pVOTE-G1L-HA was used to insert an IPTG-inducible copy of a C-terminal HA-tagged version of the G1L ORF into the HA locus (A56R ORF) of vT7LacOI by homologous recombination. This step generated an intermediate virus named vG1L/G1Li, with both the original and an inducible copy of the G1L gene. pGFP.ΔG1L, containing the G1L ORF replaced by a GFP reporter gene under the control of a VV late promoter, was then transfected into cells infected with vG1L/G1Li, and fluorescent plaques were picked. The resulting virus, named vG1Li, contained a single IPTG-inducible G1L gene.
Antibodies.
Rabbit antisera to the N-terminal peptide of the mature A17 protein (4) and to a C-terminal peptide of the G7 protein (16) were previously described. Antiserum to the C-terminal peptide QYISARHITELF of the A3 protein was made in rabbits. Mouse monoclonal antibodies (MAbs) against an influenza virus HA peptide were purchased from Covance Research Products (Berkeley, Calif.).
Western blotting.
Cells were lysed in buffer containing 0.06 M Tris-HCl (pH 6.8), 3% sodium dodecyl sulfate (SDS), 10% (vol/vol) glycerol, and 0.002% bromophenol blue. After addition of β-mercaptoethanol to a final concentration of 5%, the lysates were heated at 100°C and the proteins were resolved by SDS-polyacrylamide gel electrophoresis (SDS-PAGE). The proteins were transferred to a nitrocellulose membrane, which was blocked overnight with 1% nonfat dried milk in 0.05% Tween 20 in phosphate-buffered saline. Membranes were incubated with a 1:1,000 dilution of antiserum to A17, A3L, or G7 followed by horseradish peroxidase-conjugated donkey anti-rabbit immunoglobulin G. MAb HA.11 was also diluted 1:1,000 but was followed by peroxidase-conjugated anti-mouse immunoglobulin G (Amersham Biosciences, Piscataway, N.J.). Bound antibodies were detected by chemiluminescence with West Pico reagents (Pierce Biotechnology, Rockford, Ill.).
Plaque assay and one-step virus growth.
BS-C-1 cell monolayers, in six-well tissue culture plates, were infected with 10-fold serial dilutions of virus. After 1 h, the inocula were removed and replaced with complete EMEM containing 2.5% FBS and 0.5% methylcellulose, with or without 50 μM IPTG. The infected cells were incubated at 37°C for 2 days and stained with crystal violet, and the plaques were counted.
For one-step virus growth determinations, BS-C-1 cell monolayers in six-well tissue culture plates were infected with 5 PFU of virus per cell. After 1 h, the inocula were removed and the cell monolayers were washed twice with EMEM containing 2.5% FBS. The cells were then incubated in EMEM containing 2.5% FBS with or without 50 μM IPTG and harvested at various times after infection. The infected cells were subjected to three freeze-thaw cycles, sonicated, and stored at −70°C. Virus titers were determined by plaque assay in the presence of 50 μM IPTG.
Electron microscopy.
BS-C-1 cells were infected with vG1Li at a multiplicity of infection of 5 in the presence or absence of 50 μM IPTG. After 24 h, the cells were fixed with 2% glutaraldehyde, embedded in Epon resin, thin sectioned, and viewed on a Philips CM100 electron microscope. Sucrose-gradient-purified vG1Li grown in the presence or absence of 50 μM IPTG was deposited on a grid, negatively stained, and viewed with the electron microscope.
Pulse-chase analysis of viral late proteins.
BS-C-1 cells were infected at a multiplicity of infection of 10 with vT7LacOI in the presence or absence of 100 μg of rifampin per ml or with vG1Li in the presence or absence of 50 μM IPTG. At 8 h after infection, cells were washed with methionine- and cysteine-deficient medium and incubated for 45 min in the same medium supplemented with a mixture of 35S-labeled methionine and cysteine. Cells were either harvested immediately or washed and incubated with medium containing unlabeled methionine and cysteine for an additional 16 h and harvested. Cells were lysed in SDS sample buffer with mercaptoethanol and analyzed by SDS-PAGE and autoradiography.
Complementation assays.
BS-C-1 cells were infected with vI7Li at a multiplicity of infection of 3 in the absence of IPTG. The cells were washed and overlaid with Opti-MEM I reduced serum medium (Invitrogen Corp., Carlsbad, Calif.) in the presence or absence of IPTG and transfected with 1 μg of plasmid DNA(s) in Lipofectamine 2000. After 5 h, the medium was removed and replaced with EMEM containing 2.5% FBS (with or without IPTG). At 24 h after infection, the cells were harvested, frozen and thawed three times, and stored at −70°C. Experiments were done in triplicate, and virus yields were determined by plaque assay.
Purification of virus particles.
Fourteen T-150 flasks of BS-C-1 cells were infected with vG1Li at a multiplicity of infection of 3 in the presence or absence of 50 μM IPTG. After 48 h, cells were harvested and disrupted by Dounce homogenization. Virus particles were purified by sedimentation through a sucrose cushion and a sucrose gradient as described elsewhere (10). Absorbance of virus samples was measured at 260 nm to determine their concentration.
Virion fractionation.
Sucrose gradient-purified vG1Li virions grown in the presence of 50 μM IPTG were incubated in 50 mM Tris-HCl, pH 7.5, alone or with 0.5% NP-40 or with 0.5% NP-40 plus 50 mM dithiothreitol for 1 h at 37°C. Samples were centrifuged for 30 min at 4°C. Pellet and supernatant fractions were collected and analyzed by Western blotting.
Multiple sequence alignment.
The seed alignment for the M16 family of metalloproteases was obtained from the Protein Families Database of Alignments and Hidden Markov Models (PF00675). Using the hidden Markov model software (HMMER) version 2.3.2, a profile hidden Markov model was generated for local alignments with respect to the profile. The profile was calibrated and used to align a subgroup of the M16 family of proteins with G1L orthologs from representatives of poxvirus genera. Manual editing was required to include the more distantly related representative of the Entomopoxvirinae subfamily.
RESULTS
Construction of a conditional lethal VV mutant with an inducible G1L gene.
To investigate the role of G1 in the VV replication cycle, we engineered a recombinant VV with an inducible G1L gene. The previously constructed virus vT7LacOI, which encodes a continuously expressed Escherichia coli lac repressor and an IPTG-inducible bacteriophage T7 RNA polymerase, was used as the starting virus (18). A copy of the G1L ORF, with an influenza virus HA epitope tag at the C terminus and regulated by the bacteriophage T7 promoter and E. coli lac operator, was inserted by homologous recombination into the VV HA locus (A56R ORF) of vT7LacOI. The resulting virus, vG1L/G1Li, had both the original and inducible copies of the G1L gene. In the second recombination step, a GFP reporter gene under the control of a strong VV late promoter replaced the original G1L ORF, except for the four nucleotides at the N terminus that overlapped with the G3L ORF. In addition, since the predicted G1L promoter and its start codon were within the neighboring G3L ORF, we made silent mutations to disrupt them and avoid synthesis of a spurious polypeptide. PCR and DNA sequencing were used to confirm that both homologous recombination steps occurred as planned, and the final virus was named vG1Li (Fig. 1A).
FIG. 1.
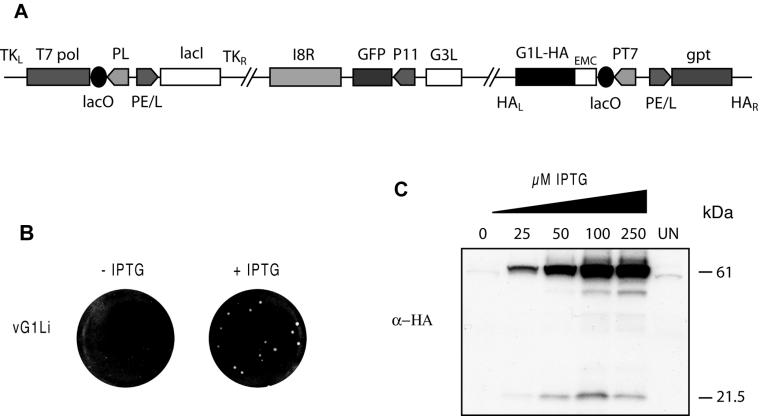
Construction and initial characterization of an inducible G1L mutant. (A) Representation of the genome of vG1Li. Important features include the presence of the bacteriophage T7 RNA polymerase gene (T7 pol) regulated by the VV late P11 promoter (PL) and the E. coli lac operator (lacO); the E. coli lac repressor (lacI) regulated by the VV early-late P7.5 promoter (PE/L); the GFP gene regulated by the late P11 promoter and replacing the original G1L gene; an inducible copy of the G1L gene (G1L-HA) encoding an influenza virus HA epitope tag at the C-terminal end of the protein and regulated by the bacteriophage T7 promoter (PT7); the lacO; and the encephalomyocarditis (EMC) leader. TKL, TKR, HAL, and HAR represent left and right flanking sequences of the VV TK and HA genes. gpt, E. coli guanine phosphoribosyltransferase gene. (B) Effect of IPTG on plaque formation. BS-C-1 cell monolayers were infected with vG1Li in the presence or absence of 50 μM IPTG. After 48 h, plaques were stained and visualized with crystal violet. (C) Effect of IPTG on the expression of G1. Uninfected cells (UN) and cells infected with vG1Li at a multiplicity of infection of 5 in the presence of 0 to 250 μM IPTG were harvested at 18 h after infection. Cell extracts were separated by SDS-PAGE and detected by Western blotting with a MAb to the influenza virus HA epitope tag (α-HA). The masses of marker proteins in kilodaltons are indicated on the right.
The conditional lethal phenotype of vG1Li was demonstrated by plaque assay. Plaques of vG1Li formed only in the presence of the inducer (Fig. 1B), whereas the inducer had no effect on either the size or number of plaques formed by the parental virus, vT7LacOI, or the intermediate virus, vG1L/G1Li (data not shown). Plaques made by vG1Li in the presence of 50 μM IPTG were similar in size to plaques made by the parental virus, indicating that the addition of the epitope tag at the C terminus of the G1 protein had no deleterious effect on in its function.
We next investigated whether the expression of G1 was dependent on the presence of the inducer. To do so, we infected BS-C-1 cells with vG1Li in the presence of increasing concentrations of IPTG and carried out Western blotting using a MAb to the C-terminal HA epitope tag. A major band, estimated to have a mass of 61 kDa relative to the mobility of marker proteins, was induced in a concentration-dependent manner by IPTG and was not detected in the absence of inducer (Fig. 1C). Two additional bands with more rapid mobilities were also induced by IPTG. Since the predicted mass of G1 is 68 kDa, we believe that the major band corresponds to the full-length protein and the others to C-terminal fragments, perhaps formed by cleavage. Corresponding N-terminal fragments would not have been detected with the HA MAb.
Effect of IPTG on virus replication.
IPTG-dependent plaque formation of vG1Li could result from either a defect in virus replication or spread. To distinguish between these possibilities, we investigated the effect of IPTG under one-step virus growth conditions. Cells were infected with vG1Li in the presence of a range of IPTG concentrations and harvested 24 h later. An incremental increase in infectious virus yield was observed as the IPTG concentration was raised from 2.5 to 25 μM (Fig. 2A). At 50 μM, there was a 2-log increase in virus production. Higher IPTG concentrations, however, slightly decreased the yield of infectious virus (Fig. 2A). With the intermediate virus vG1L/G1Li, which carries two copies of the G1L gene, the decrease in virus yield occurred at lower IPTG concentrations than with vG1Li. High concentrations of IPTG did not reduce the yield of vT7lacOI, indicating that the effects on vG1Li and vG1L/G1Li were probably due to overexpression of G1.
FIG. 2.
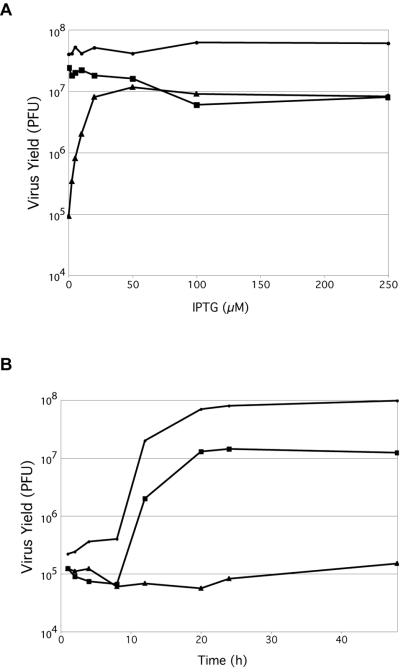
Effect of IPTG on the production of infectious vG1Li. (A) BS-C-1 cells were infected with vT7LacOI (•), vG1L/G1Li (▪), or vG1Li (▴) at a multiplicity of infection of 5 and incubated in the presence of 0 to 250 μM IPTG. At 24 h after infection, virus titers were determined by plaque assay in the presence of 50 μM IPTG. (B) BS-C-1 cells were infected with vT7LacOI (•) or vG1Li in the presence (▪) or absence (▴) of 50 μM IPTG. Cells were harvested at the indicated times after infection, and virus titers were determined as described for panel A.
The kinetics of vG1Li production in the presence of 50 μM IPTG was similar to that of the parental virus, although the yields were lower at all times (Fig. 2B). In the absence of IPTG, a slight increase in infectious vG1Li was detected at 48 h, probably due to accumulation of small amounts of G1.
Putative protease catalytic site residues of G1 are required for trans complementation.
The G1L ORF of VV strain WR is predicted to encode a protein of 591 amino acids that is conserved in every poxvirus whose genome sequence is known (17). The orthopoxvirus G1 orthologs are more than 98% identical to each other, whereas the G1 ortholog of the distantly related entomopoxvirus Amsacta moorei is 21% identical (39% similar) to vaccinia virus G1. At the time that Whitehead and Hruby (19) noted HXXEH in the VV G1L ORF, other poxvirus genome sequences were unavailable for comparison. Presently, there are complete genome sequences for 30 poxviruses, and the G1L orthologs of each one contains the HXXEH motif. Figure 3 contains an alignment of the HXXEH and adjacent sequences of representative poxviruses and of prokaryotic and eukaryotic metalloproteinases of the ME group (http://www.sanger.ac.uk/Software/Pfam/). The sequence in the top line is of G1. Previous mutagenesis studies of pitrilysin, an E. coli metalloproteinase, demonstrated that both histidine residues and the glutamate of the HXXEH motif are required for catalytic activity (3). Glutamate 169 of pitrilysin was also required for zinc binding (2), but it aligned poorly with other members of the ME group, including G1 and its poxvirus orthologs, and therefore was not included in the alignment. The second and third positions within the HXXEH motif are generally hydrophobic residues, such as leucine, valine, or phenylalanine. G1 orthologs from the various poxvirus families have two leucines in the X positions; the only known exception is in entomopoxviruses, which have isoleucine for the second X. Flanking the active site motif are a number of highly conserved residues, such as glycine 28 of VV strain WR, which is in all the aligned proteins (Fig. 3) and likely contributes to the structure of the catalytic site. Glycine 38 is also highly conserved in G1 orthologs as well as in nonpoxvirus homologs. In addition, residue 32 is almost always acidic, even in the distantly related Amsecta moorei entomopoxvirus (Fig. 3). However, in Melanoplus sanguinipes entomopoxvirus, this position is occupied by the polar but uncharged asparagine residue (data not shown). Except for sequences around the HXXEH motif, the cellular and poxvirus metalloproteinases appeared unrelated to each other.
FIG. 3.

Multiple sequence alignment of poxvirus G1L orthologs and HXXEH-containing metallopeptidases. Invariant amino acid residues are shaded gray. The HXXEH motif is indicated by asterisks at the top. The alignment includes two orthopoxvirus sequences, one representative sequence from the other chordopoxvirus genera, and one entomopoxvirus sequence. Numbers indicate first and last amino acids in the alignment. Virus protein names consist of an abbreviation of the virus and gene name as listed in the University of Victoria database (http://www.poxvirus.org/pocs.asp). Nonviral entries consist of abbreviations of gene and organism names. Virus name abbreviations are as follows: VAR-I, variola major virus strain India (GI 420468); VV-WR, Western Reserve strain of VV (GI 335671); FPV-V, fowlpox virus (GI 7271579); LSDV-L, lumpy skin disease virus Neethling vaccine LW 1959 (GI 15149061); MYX-L, myxoma virus strain Lausanne (GI 6523900); SPV-N, sheeppox virus strain NISKHI (GI 21492503); MCV-1, molluscum contagiosum virus subtype 1 (GI 7515386); YLDV, Yaba-like disease virus (GI 12056209); AmEPV, Amsacta moorei entomopoxvirus (GI 9944779). Organism and gene name abbreviations are as follows: PTRA-ECOLI, pitrilysin from E. coli (GI 131573); IDE-DROME, insulin-degrading enzyme from Drosophila melanogaster (GI 124156); IDE-HUMAN, insulin-degrading enzyme from humans (GI 124157); MPPA-HUMAN, mitochondrial processing peptidase alpha subunit from humans (GI 29840846); MPPB-YEAST, mitochondrial processing peptidase beta subunit from Saccharomyces cerevisiae (GI 127290); PQQF-KLEPN, coenzyme pyrroloquinoline quinone biosynthesis protein f from Klebsiella pneumoniae (GI 130803); PQQL-ECOLI, putative coenzyme pyrroloquinoline quinone biosynthesis protein from E. coli (GI 2507259); SDP-EIMBO, sporozoite developmental protein from Eimeria bovis (GI 1173411); YMT1-CAEEL, hypothetical zinc protease f56d2.1 from Caenorhabditis elegans (GI 2507260); YMXG-BACSU, hypothetical zinc protease ymxg from Bacillus subtilis (GI 1176567); YQA4-CAEEL, hypothetical zinc protease c28f5.4 from C. elegans (GI 1730966).
trans complementation experiments were carried out to confirm that the replication defect of vG1Li was solely due to the lack of expression of G1 and to determine whether the HXXEH motif was essential. Cells were infected with vG1Li in the absence of IPTG and transfected with a plasmid containing an HA-tagged G1L gene under the control of a VV late promoter. G1 expressed from the wild-type plasmid increased the virus yield by more than 10-fold compared to that of vector alone but did not reach the yield of vG1Li in the presence of IPTG (Fig. 4A). Unlike the result obtained with the wild-type G1L gene, there was no significant increase in virus titer when cells were transfected with a plasmid containing the G1L ORF with histidine-41 or histidine-45 changed to arginine or with glutamate-44 changed to glutamine (Fig. 4A). This result was not due to low expression or instability of the G1 protein, as Western blotting indicated equivalent amounts of wild-type and mutated forms (Fig. 4B).
FIG. 4.
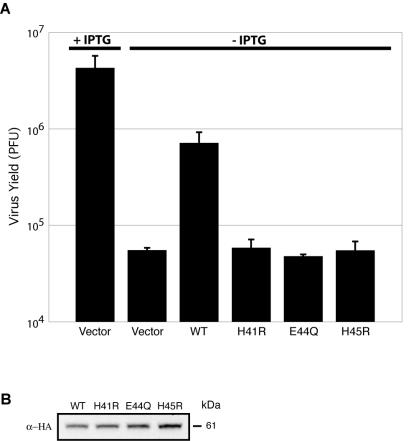
Complementation of vG1Li with plasmids expressing wild-type G1 but not those expressing mutated G1. (A) BS-C-1 cells were infected with vG1Li at a multiplicity of infection of 3 in the presence or absence of 50 μM IPTG as indicated. After 1 h, cells infected in the absence of IPTG were transfected with 1 μg of vector plasmid or plasmid expressing wild-type (WT) or mutated G1 proteins containing a C-terminal influenza virus HA epitope tag. H41R, histidine-41 mutated to arginine; E44Q, glutamate-44 mutated to glutamine; H45R, histidine-45 mutated to arginine. (B) Lysates from cells infected with vG1Li in the absence of IPTG and transfected with plasmids expressing wild-type or mutated G1-HA were analyzed by Western blotting with MAb to the influenza virus HA epitope (α-HA). The apparent molecular mass in kilodaltons is indicated on the right.
Synthesis and processing of viral late proteins in cells infected with vG1Li.
The previous report of a role for G1 in the processing of the L4 protein (19) and the present data showing that the putative catalytic site of G1 is required for trans-complementation of infectivity made it important to analyze the synthesis and processing of proteins expressed by vG1Li in the presence and absence of IPTG. At late times during VV infection, host protein synthesis is greatly inhibited, allowing the detection of viral proteins by metabolic labeling of infected cells followed by SDS-PAGE and autoradiography. Similar patterns of viral proteins were observed after pulse-labeling of cells at 8 h after infection with vG1Li in the presence or absence of IPTG or with vT7lacOI (Fig. 5A), indicating that G1 did not regulate gene expression directly or indirectly. To determine whether proteolytic processing of core proteins occurred, the pulse-labeled cells infected with vG1Li in the presence or absence of IPTG were chased for 16 h and the proteins were analyzed. As a control, cells infected with vT7LacOI in the presence of rifampin, which prevents virus assembly and cleavage of core proteins (13), were also pulsed and chased. Proteolytic processing of the major VV structural proteins was unaffected when G1 was repressed, as the gel patterns were similar to those made with extracts of cells infected with either vG1Li in the presence of IPTG or vT7LacOI in the absence of rifampin (Fig. 5A). In contrast, the major core proteins were not processed in cells infected with wild-type virus and treated with rifampin (Fig. 5A). The band marked with an asterisk (Fig. 5A), from cells infected with vG1Li in the presence or absence of IPTG but not from cells infected with vT7LacOI, is probably GFP, which has a predicted mass of 27 kDa (7).
FIG. 5.
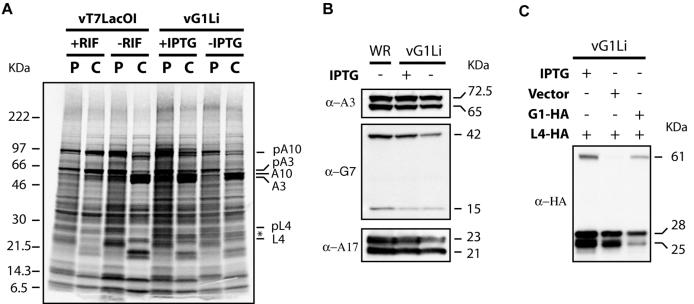
Synthesis and processing of viral late proteins. (A) BS-C-1 cells were infected with vT7LacOI in the presence or absence of 100 μg of rifampin (RIF) per ml or vG1Li in the presence or absence of 50 μM IPTG. At 8 h after infection, cells were pulse-labeled with a mixture of [35S]methionine and [35S]cysteine for 45 min and either harvested immediately (P) or chased for an additional 16 h with medium containing unlabeled methionine and cysteine (C). Cells were analyzed by SDS-PAGE under reducing conditions and by autoradiography. The protein bands that correspond to the proteolytically processed viral major structural proteins (A3, A10, and L4) and their uncleaved precursors (pA3, pA10, and pL4) are indicated on the right. The asterisk corresponds to the band believed to be GFP. (B) BS-C-1 cells were infected with wild-type VV (WR) or vG1Li in the presence (+) or absence (−) of 50 μM IPTG. At 18 h after infection, cells were harvested and analyzed by Western blotting with antibody to the A3 (α-A3), G7 (α-G7), or A17 (α-A17) protein. Apparent molecular masses in kilodaltons are indicated on the right. (C) BS-C-1 cells were infected with vG1Li in the presence (+) or absence (−) of 50 μM IPTG and transfected with vector plasmid or plasmids expressing G1 (G1-HA) or L4 (L4-HA) protein with an influenza virus HA epitope tag at the C terminus under the control of a VV synthetic strong late promoter. At 18 h after infection, cells were harvested and analyzed by Western blotting with MAb to the influenza virus HA epitope tag (α-HA). Apparent molecular masses in kilodaltons are indicated on the right.
We also used Western blotting to examine three viral proteins that normally undergo proteolytic processing. The proteins chosen were the two core proteins A3 (also known as P4b) and G7 and the membrane protein was A17. The relative amounts of uncleaved and cleaved proteins were similar from cells infected with vI7Li in the presence and absence of IPTG (Fig. 5B), indicating that G1 is not involved in processing of these proteins. Furthermore, by using a transfection assay, we found that the processing of a C-terminal HA-tagged version of L4 was not dependent on G1 expression (Fig. 5C). Importantly, we previously showed that the same HA-tagged L4 protein required I7 expression for cleavage at both AG/S and AG/A sites (1). Thus, these data were inconsistent with the previous report of an involvement of G1 in the processing of L4 and provided no evidence for such a role in the processing of P4a, P4b, G7, or A17. Thus, the putative substrates of G1 need to be identified.
Morphogenesis of vG1Li under nonpermissive conditions.
To determine the effect of repression of G1 on VV morphogenesis, cells were infected with vG1Li in the presence or absence of IPTG and examined by electron microscopy. At 24 h after infection in the presence of IPTG, immature and mature forms indistinguishable from those of wild-type virus were observed (data not shown). In the absence of IPTG, the crescents and immature forms, including some with nucleoids, appeared normal (Fig. 6A). There were also many dense particles that mostly appeared circular in cross section and had poorly developed core structures (Fig. 6B) but no typical intracellular mature virions (IMV). Despite these morphological defects, some aberrant particles progressed to the next stage of wrapping, which usually occurs when IMV are converted to intracellular enveloped virions (IEV) (Fig. 6C). For comparison, wild-type oval or brick-shaped IMV with distinctive cores are shown in Fig. 6D.
FIG. 6.
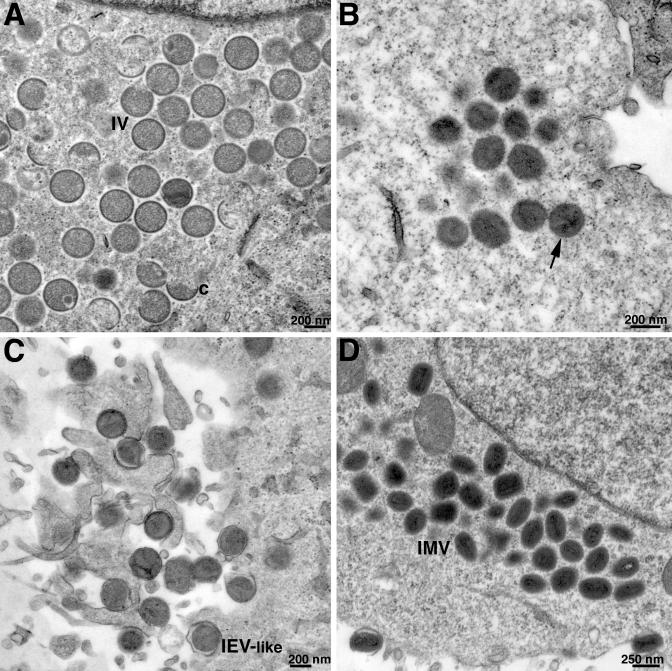
Electron microscopy of cells infected with vG1Li under nonpermissive conditions. BS-C-1 cells were infected with vG1Li at a multiplicity of infection of 5 in the absence of IPTG (A to C) or with wild-type VV (D). At 24 h after infection, cells were fixed and embedded in Epon, and ultrathin sections were prepared for transmission electron microscopy. (A) Crescents (c) and immature virions (IV), some with nucleoids. (B) Virus particles (arrow) that have progressed beyond the typical IV stage and resemble IMV but are mostly circular with poorly formed cores. (C) IEV-like particles that are wrapped with additional membranes. The images in panels A, B, and C were obtained from different cells in the same thin section. (D) Typical IMV in cells infected with wild-type VV. The intracellular clustering of particles at similar stages of morphogenesis, as seen in each of the panels, is typical.
Comparisons of purified virions formed in the absence or presence of IPTG.
We considered that a comparison of the protein compositions of purified virions grown under permissive and nonpermissive conditions might provide clues regarding the defect in morphogenesis and the putative substrate of G1. Virions were purified by sucrose gradient sedimentation from extracts of cells that had been infected with vG1Li in the presence or absence of IPTG. In each case, distinct opalescent bands were recovered, and the virions were examined by electron microscopy after negative staining. The virions made under nonpermissive conditions (Fig. 7A) were larger and more spherical than those made under permissive conditions (Fig. 7B). Similar amounts of viral DNA were associated with the virions isolated in the presence and absence of IPTG, as determined by DNA dot blot hybridization (data not shown).
FIG. 7.
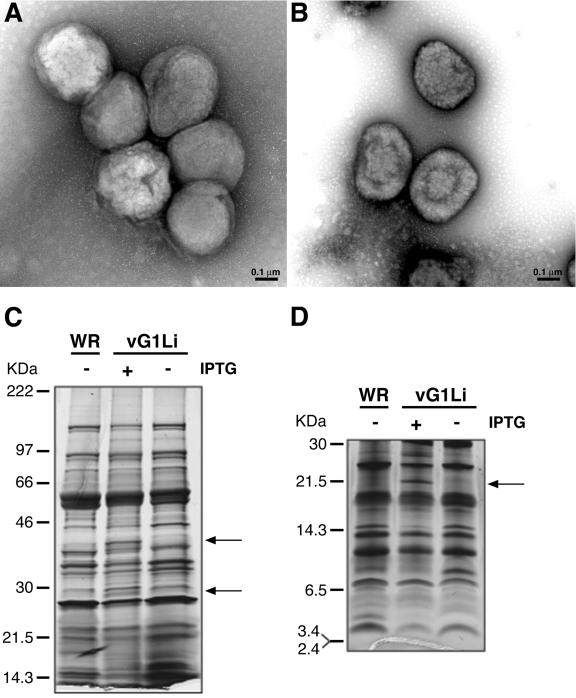
Morphology and protein composition of purified vG1Li formed in the presence and absence of IPTG. (A) Electron micrograph of negatively stained virus particles from cells infected with vG1Li in the absence of IPTG and purified by sedimentation through a 36% sucrose cushion followed by a 25-to-40% sucrose gradient. (B) Same experiment as shown in panel A, except that cells were infected with vG1Li in the presence of IPTG. (C) Purified wild-type VV (WR) and vG1Li grown in the presence (+) or absence (−) of IPTG were dissociated with SDS and mercaptoethanol and analyzed by electrophoresis in a 4-to-20% polyacrylamide gel in Tris-glycine buffer. A photograph of a silver-stained gel is shown. Distinctive vG1Li (+ IPTG) bands are indicated by arrows. The molecular masses of the marker proteins are on the left. (D) Same experiment as shown in panel C, but samples were analyzed on a 16% polyacrylamide gel in Tricine buffer.
The protein constituents of the purified virions were analyzed by SDS-PAGE using two different gel systems in order to resolve species of high and low mass. Purified wild-type VV was analyzed in parallel. Overall, the silver-stained patterns were very similar (Fig. 7C and D). The most obvious difference was additional bands of approximately 40, 28, and 22 kDa in the preparation from cells infected with vG1L in the presence of IPTG. Their presence at lower intensity or absence from proteins of wild-type virus suggested that they may be caused by overexpression of G1 in the presence of 50 μM IPTG. The bands of 40, 28, and 22 kDa could represent processed forms of G1, whereas the full-length form would be obscured by major core proteins with electrophoretic mobilities similar to that of G1. There were no previous reports, however, regarding the association of G1 with virions.
Association of the G1 protein with virus particles.
The inducible copy of G1 expressed by vG1Li contained a C-terminal influenza virus HA epitope tag, allowing us to detect its presence with a specific MAb. The proteins resolved by SDS-PAGE of virions purified from cells infected with vG1Li in the presence of IPTG (Fig. 7B) were transferred to a membrane and probed with the MAb to the epitope tag. Bands of approximately 61 and 22 kDa were detected (Fig. 8, upper panel, lane 2), similar to those shown to be present in lysates of cells infected with vG1Li in the presence of IPTG (Fig. 1C). As suggested in a previous section, the 61-kDa band represents the full-length G1 protein, whereas the 22-kDa band may represent a C-terminal fragment. The latter is similar to one of the three unique bands detected by silver staining in Fig. 7D. We suspect, therefore, that the 40- and 28-kDa bands in Fig. 7C are N-terminal cleavage products of G1, which would not be detected by the MAb to the C-terminal epitope tag.
FIG. 8.
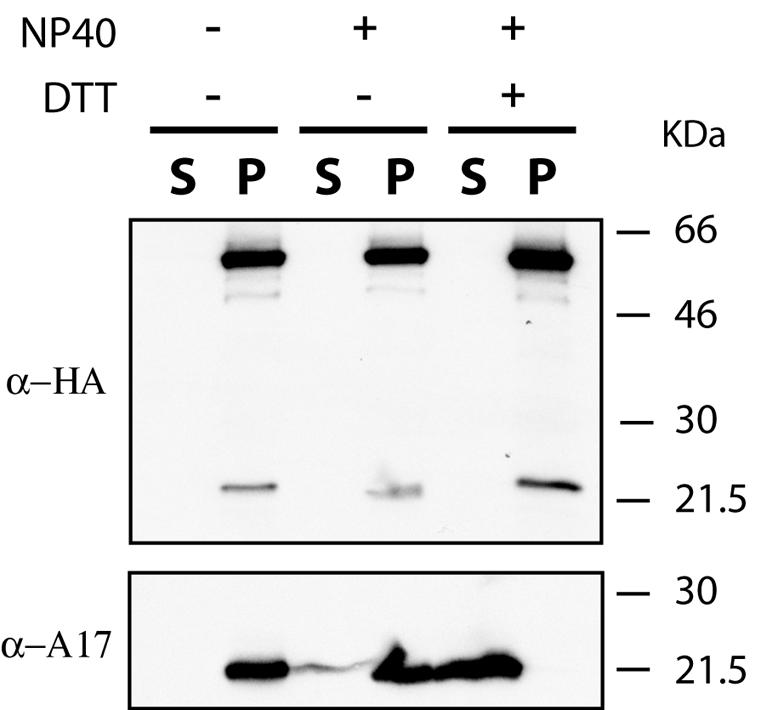
Localization of G1 in the virus particle. Sucrose gradient-purified vG1Li, from the preparation shown in Fig. 7B, was incubated in 50 mM Tris-HCl buffer (pH 7.5) alone or with 1% NP-40 or 1% NP-40 plus 50 mM dithiothreitol (DTT). Soluble and insoluble fractions were separated by centrifugation and analyzed by Western blotting with a MAb to the influenza virus HA epitope (α-HA) or polyclonal antibodies to an N-terminal peptide of A17 (α-A17). The molecular masses of the marker proteins are indicated on the right.
The purified virions were treated with NP-40 detergent in the presence or absence of reducing agent and submitted to high-speed centrifugation in order to separate the detergent-soluble membrane fraction from the particulate cores. The samples were then analyzed by SDS-PAGE followed by Western blotting with MAb to the epitope tag. Both the 61- and 22-kDa G1 proteins were associated with the cores (Fig. 8, upper panel, lanes 3 to 6). As a control, the Western blot membrane was stripped and reprobed with antibody against the membrane protein A17, which was found exclusively in the soluble fraction in the presence of NP-40 and dithiothreitol (Fig. 8, lower panel). We concluded that full-length G1 and the putative processed form are packaged in virus cores.
DISCUSSION
The conservation of the VV G1 protein in all poxviruses suggested that it has an essential role in the virus life cycle. We demonstrated this experimentally by constructing a recombinant virus in which expression of G1 was dependent on IPTG. In the absence of G1, the virus was unable to form plaques and replication was inhibited. The block occurred late in the virus replication cycle, consistent with the late promoter consensus sequence (9) preceding the G1L ORF. Viral protein synthesis appeared normal, and proteolytic processing of major core and membrane proteins occurred under nonpermissive conditions. Nevertheless, a defect in virus morphogenesis was demonstrated by electron microscopy. Virus particles formed, but they did not acquire the typical brick shape and the cores lacked the characteristic internal structure, although viral DNA was detected. Interestingly, the aberrant particles were wrapped with trans-Golgi or endosomal membranes to form structures that resemble IEV, suggesting that the membrane proteins were unaffected. Biochemical studies indicated that under permissive conditions, the G1 protein is incorporated into the core structure of mature virions. Therefore, the interruption in morphogenesis and consequent lack of infectivity may be associated with the absence of G1.
The involvement of G1 in proteolytic processing of the viral core precursor protein L4 was suggested on the basis of transfection experiments and the presence of an HXXEH metalloprotease motif (19). We found that trans-expression of wild-type G1 rescued virus infectivity when cells were infected with vG1Li in the absence of IPTG. In contrast, rescue was not achieved when the transfected gene contained mutations in either of the two conserved histidines or glutamate. These data support the idea that G1 is a metalloprotease. The substrates of the putative protease, however, remain to be determined. As mentioned above, cleavage of core and membrane proteins occurred under nonpermissive conditions. In addition, we found that G1 was not required for cleavage of L4 expressed by transfection. Importantly, using the same L4 expression plasmid, we showed that I7, a cysteine protease, was required for cleavage (1). This result and other recent data (1, 5, 6) indicate that I7 is likely to be the sole protease responsible for cleavage of the major core and membrane proteins. In addition, it seems unlikely that a metalloprotease would have a substrate specificity similar to that of a cysteine protease, as had been suggested previously (19).
Analysis of the proteins in lysates and virions purified from cells infected with vG1Li in the presence of IPTG suggested that G1 has self-cleaving activity or is a substrate of another protease, as we found both a full-length and a shorter 22-kDa form. The latter was detected because of the presence of a C-terminal epitope tag. Other bands, detected by silver staining, may represent N-terminal fragments, but additional immunological reagents are needed to confirm this. If the cleaved G1 has a specific function in morphogenesis, then the protease activity may regulate this role. Vaccinia virus is thought to contain up to 100 polypeptides (11), and two-dimensional gels or a combination of gel electrophoresis and mass spectroscopy may be needed to detect additional substrates of G1.
Acknowledgments
We thank Norman Cooper for cells, Andrea Weisberg for electron microscopy, Wolfgang Resch for the multiple sequence alignment, and Tania Senkevich for assistance with protocols.
C.A.-S. received partial support from the Special Program for Microbiology of the Brazilian Council for Scientific Technological Development (CNPq).
REFERENCES
- 1.Ansarah-Sobrinho, C., and B. Moss. 2004. Role of the I7 protein in proteolytic processing of vaccinia virus membrane and core components. J. Virol. 78:6335-6343. [DOI] [PMC free article] [PubMed]
- 2.Becker, A. B., and R. A. Roth. 1993. Identification of glutamate-169 as the third zinc-binding residue in proteinase III, a member of the family of insulin-degrading enzymes. Biochem. J. 292:137-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Becker, A. B., and R. A. Roth. 1992. An unusual active site identified in a family of zinc metalloendopeptidases. Proc. Natl. Acad. Sci. USA 89:3835-3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Betakova, T., E. J. Wolffe, and B. Moss. 1999. Regulation of vaccinia virus morphogenesis: phosphorylation of the A14L and A17L membrane proteins and C-terminal truncation of the A17L protein are dependent on the F10L protein kinase. J. Virol. 73:3534-3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byrd, C. M., T. C. Bolken, and D. E. Hruby. 2003. Molecular dissection of the vaccinia virus I7L core protein proteinase. J. Virol. 77:11279-11283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byrd, C. M., T. C. Bolken, and D. E. Hruby. 2002. The vaccinia virus I7L gene product is the core protein proteinase. J. Virol. 76:8973-8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chalfie, M., Y. Tu, G. Euskirchen, W. W. Ward, and D. C. Prasher. 1994. Green fluorescent protein as a marker for gene expression. Science 263:802-805. [DOI] [PubMed] [Google Scholar]
- 8.Davison, A. J., and B. Moss. 1990. New vaccinia virus recombination plasmids incorporating a synthetic late promoter for high level expression of foreign proteins. Nucleic Acids Res. 18:4285-4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davison, A. J., and B. Moss. 1989. The structure of vaccinia virus late promoters. J. Mol. Biol. 210:771-784. [DOI] [PubMed] [Google Scholar]
- 10.Earl, P. L., B. Moss, L. S. Wyatt, and M. W. Carroll. 1998. Generation of recombinant vaccinia viruses, p. 16.17.1-16.17.19. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology, vol. 2. Greene Publishing Associates & Wiley Interscience, New York, N.Y.
- 11.Essani, K., and S. Dales. 1979. Biogenesis of vaccinia: evidence for more than 100 polypeptides in the virion. Virology 95:385-394. [DOI] [PubMed] [Google Scholar]
- 12.Kane, E. M., and S. Shuman. 1993. Vaccinia virus morphogenesis is blocked by a temperature-sensitive mutation in the I7 gene that encodes a virion component. J. Virol. 67:2689-2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katz, E., and B. Moss. 1970. Formation of a vaccinia virus structural polypeptide from a higher molecular weight precursor: inhibition by rifampicin. Proc. Natl. Acad. Sci. USA 6:677-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moss, B. 2001. Poxviridae: the viruses and their replication, p. 2849-2883. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 15.Rawlings, N. D., and A. J. Barrett. 1995. Evolutionary families of metallopeptidases. Methods Enzymol. 248:183-228. [DOI] [PubMed] [Google Scholar]
- 16.Szajner, P., H. Jaffe, A. S. Weisberg, and B. Moss. 2003. Vaccinia virus G7L protein interacts with the A30L protein and is required for association of viral membranes with dense viroplasm to form immature virions. J. Virol. 77:3418-3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Upton, C., S. Slack, A. L. Hunter, A. Ehlers, and R. L. Roper. 2003. Poxvirus orthologous clusters: toward defining the minimum essential poxvirus genome. J. Virol. 77:7590-7600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ward, G. A., C. K. Stover, B. Moss, and T. R. Fuerst. 1995. Stringent chemical and thermal regulation of recombinant gene expression by vaccinia virus vectors in mammalian cells. Proc. Natl. Acad. Sci. USA 92:6773-6777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whitehead, S. S., and D. E. Hruby. 1994. A transcriptionally controlled trans-processing assay: putative identification of a vaccinia virus-encoded proteinase which cleaves precursor protein P25K. J. Virol. 68:7603-7608. [DOI] [PMC free article] [PubMed] [Google Scholar]