

Abstract
Large-scale recovery of genomes from isolates, single cells, and metagenomic data has been made possible by advances in computational methods and substantial reductions in sequencing costs. Although this increasing breadth of draft genomes is providing key information regarding the evolutionary and functional diversity of microbial life, it has become impractical to finish all available reference genomes. Making robust biological inferences from draft genomes requires accurate estimates of their completeness and contamination. Current methods for assessing genome quality are ad hoc and generally make use of a limited number of “marker” genes conserved across all bacterial or archaeal genomes. Here we introduce CheckM, an automated method for assessing the quality of a genome using a broader set of marker genes specific to the position of a genome within a reference genome tree and information about the collocation of these genes. We demonstrate the effectiveness of CheckM using synthetic data and a wide range of isolate-, single-cell-, and metagenome-derived genomes. CheckM is shown to provide accurate estimates of genome completeness and contamination and to outperform existing approaches. Using CheckM, we identify a diverse range of errors currently impacting publicly available isolate genomes and demonstrate that genomes obtained from single cells and metagenomic data vary substantially in quality. In order to facilitate the use of draft genomes, we propose an objective measure of genome quality that can be used to select genomes suitable for specific gene- and genome-centric analyses of microbial communities.
Recent advances in high-throughput sequencing combined with improving computational methods are enabling the rapid, cost effective recovery of genomes from cultivated and uncultivated microorganisms across a wide range of host-associated and environmental samples. Large-scale initiatives, such as the Genomic Encyclopedia of Bacteria and Archaea (GEBA) (Wu et al. 2009), aim to provide reference genomes from isolated species across the Tree of Life, whereas targeted efforts such as the Human Microbiome Project (HMP) (Turnbaugh et al. 2007) and the GEBA-Root Nodulating Bacteria (GEBA-RNB; http://jgi.doe.gov/) initiatives are providing reference genomes necessary for understanding the role of microorganisms in specific habitats. These efforts are complemented by initiatives such as the GEBA-Microbial Dark Matter (GEBA-MDM) project, which used single-cell genomics to obtain genomes from uncultivated bacterial and archaeal lineages (Rinke et al. 2013). Several studies have also demonstrated the successful recovery of high-quality population genomes directly from metagenomic data (Tyson et al. 2004; Wrighton et al. 2012; Albertsen et al. 2013; Sharon et al. 2013). Together these initiatives have produced thousands of draft genomes and stand to provide tens of thousands more as sequencing technology and computational methodologies continue to improve. Although this rapid recovery of genomes stands to greatly improve our understanding of the microbial world, it is outpacing our ability to manually assess the quality of individual genomes.
In order to make robust inferences from the increasing availability of draft genomes, it is critical to distinguish between genomes of varying quality (Mardis et al. 2002; Chain et al. 2009). In particular, genomes recovered from single cells or metagenomic data require careful scrutiny due to the additional complications inherent in obtaining genomes using these approaches (Dick et al. 2010; Albertsen et al. 2013). The quality of isolate genomes has traditionally been evaluated using assembly statistics such as N50 (Salzberg et al. 2012; Gurevich et al. 2013), whereas single-cell and metagenomic studies have relied on the presence and absence of universal single-copy “marker” genes for estimating genome completeness (Wrighton et al. 2012; Haroon et al. 2013; Rinke et al. 2013; Sharon et al. 2013). However, the accuracy of this completeness estimate has not been evaluated, and the approach is likely to be limited by both the uneven distribution of universal marker genes across a genome and their low number, typically accounting for <10% of all genes (Sharon and Banfield 2013). These limitations have been partially addressed by identifying genes that are ubiquitous and single copy within a specific phylum, which increases the number of marker genes used in the estimate (Swan et al. 2013). Single-copy marker genes present multiple times within a recovered genome have also been used to estimate potential contamination (Albertsen et al. 2013; Soo et al. 2014; Sekiguchi et al. 2015).
Here we describe CheckM, an automated method for estimating the completeness and contamination of a genome using marker genes that are specific to a genome's inferred lineage within a reference genome tree. Using simulated genomes of varying degrees of quality, we demonstrate that lineage-specific marker genes provide refined estimates of genome completeness and contamination compared to the universal or domain-level marker genes commonly used. Marker genes that are consistently collocated within a lineage do not provide independent evidence of a genome's quality, so collocated marker genes were grouped into marker sets in order to further refine estimates of genome quality. We show that lineage-specific collocated marker sets provide robust estimates across all bacterial and archaeal lineages, with completeness and contamination estimates generally having a low absolute error even when genomes are relatively incomplete (70%) with medium contamination (10%). We also propose a fixed vocabulary for defining genome quality based on estimates of completeness and contamination that is suitable for automated screening of genomes from large-scale sequencing initiatives and for annotating genomes in reference databases. We envisage that CheckM will help identify problematic genomes before they are deposited in public databases. For single-cell genomes and population genomes recovered from metagenomic data, the improved quality estimates provided by CheckM allow biological inferences to be made in the context of genome quality and highlight genomes that would benefit from further refinement.
Results
Simulation models for evaluating the accuracy of quality estimates
Three independent models were used to generate simulated genomes suitable for evaluating the completeness and contamination estimates provided by CheckM. Under the “random fragment” model, 3324 high-quality draft genomes (spanning 39 classes and 20 phyla) obtained from IMG were fragmented into nonoverlapping windows of 5–50 kbp and randomly sampled in order to generate genomes with varying degrees of completeness and contamination (see Methods for more details). This model allows a large number of genomes to be simulated at varying degrees of quality and provides a baseline for assessing the accuracy of completeness and contamination estimates. In order to simulate genomes reflecting the characteristics of assembled contigs, the 2430 high-quality draft genomes (spanning 31 classes and 18 phyla) comprised of 20 or more contigs were used to simulate incomplete genomes contaminated with foreign DNA. Under this “random contig” model, incomplete genomes were generated by randomly removing contigs to achieve a desired level of completeness and contamination introduced by randomly adding contigs from a randomly selected draft genome.
The final model simulates genomes that reflect the limitations of metagenomic binning methods that rely on the statistical properties of assembled contigs (e.g., tetranucleotide signatures, coverages) to determine their source genome. Since the variance of genome statistics increase with decreasing contig length, binning methods are more likely to incorrectly bin shorter contigs (Dick et al. 2010; Albertsen et al. 2013; Imelfort et al. 2014). The “inverse length” model captures this limitation by generating incomplete and contaminated genomes in a manner similar to the random contig model, but with contigs removed or added with a probability inversely proportional to a contig's length.
For all three models, genomes were generated at 50%–100% completeness and 0%–20% contamination. Although genomes recovered using single-cell genomics are often <50% complete, we have focused on relatively complete genomes as these have a broader range of applications, and we believe the methodology of combining reads from multiple single cells belonging to the sample population, which often results in highly complete genomes, will become common practice. Similarly, we have focused on genomes with ≤20% contamination as there are limited applications for genomes with excessive levels of contamination.
Assessment of universal- and domain-level marker gene sets
Within CheckM, a gene identified as single copy in ≥97% of genomes is considered to be a marker gene. The genome quality estimates provided by the marker sets used by CheckM were compared with three universal (Mende et al. 2013; Rinke et al. 2013; Darling et al. 2014), three bacterial (Dupont et al. 2012; Wu and Scott 2012; Rinke et al. 2013), and two archaeal (Wu and Scott 2012; Rinke et al. 2013) marker sets using genomes simulated under the random fragment model. The universal marker sets had similar performance with the exception of the SpecI set (Mende et al. 2013), which provided superior contamination estimates at the expense of substantially less accurate completeness estimates (Supplemental Table S1). Among the CheckM, GEBA-MDM (Rinke et al. 2013), and PhyloSift (Darling et al. 2014) sets, the mean absolute error in quality estimates provided by CheckM never deviated by more than 1.4% and was typically within 1% of the best performing universal marker set. The four bacterial-specific and three archaeal-specific marker sets provided far more accurate estimates than the universal sets with the exception of the poorly performing Amphora 2 set (Supplemental Table S1; Wu and Scott 2012). The mean absolute error in the completeness estimates provided by CheckM's domain-specific marker sets were always within 1% of the best performing marker set, whereas the contamination estimates were generally within 1% and always within 1.8%.
Organizing marker genes into collocated sets
Because marker genes are required to be present in nearly all genomes within a lineage (e.g., all bacteria or archaea), they often encode essential functions and are frequently organized into operons (Supplemental Fig. S1). Marker genes that are consistently collocated do not provide independent information regarding the completeness or contamination of a genome. To address this, we grouped marker genes that were consistently collocated within a lineage into marker sets and used this grouping structure to refine estimates of genome completeness and contamination. Collocated marker genes are common across all taxonomic groups with 36% of marker genes, on average, being grouped into a set with one or more other marker genes (Supplemental Table S2).
We evaluated the benefit of collocated marker sets for assessing genome quality by applying CheckM's domain-specific markers (bacteria: 104 markers organized into 58 sets; archaea: 150 markers organized into 108 sets) to genomes simulated under the random fragment model. Completeness and contamination estimates calculated with collocated marker sets were superior to estimates determined with individual marker genes regardless of the completeness or contamination of the simulated genomes (Fig. 1; Supplemental Table S3). The mean absolute error in completeness (contamination) estimates across all simulated genomes increased from 4.3% to 5.7% (3.8% to 4.7%) when using marker sets compared to 5.5% to 9.0% (4.7% to 6.8%) when using individual marker genes as the window size was increased from 5 to 50 kbp (Supplemental Table S3).
Figure 1.

Error in completeness and contamination estimates on simulated genomes with 50%, 70%, 80%, or 90% completeness (comp.) and 5%, 10%, or 15% contamination (cont.). Quality estimates were determined using domain-level marker genes treated as individual markers (IM) or organized into collocated marker sets (MS). Simulated genomes were generated under the random fragment model from 3324 draft genomes spanning 39 classes (20 phyla) with each draft genome being used to generate 20 simulated genomes. A systematic bias in the estimates results in completeness being overestimated on average (median value to the right of zero) and contamination being underestimated on average (median value to the left of zero). Results are summarized using box-and-whisker plots showing the 1st (99th), 5th (95th), 25th (75th), and 50th percentiles.
To further evaluate the benefits of using collocated marker sets, domain-specific markers were used to estimate the quality of genomes simulated under the random contig and inverse length models. Under the random contig model, the mean absolute error in the completeness and contamination estimates across all simulated genomes was reduced from 8.5% to 5.4% and 5.9% to 4.1%, respectively, when genome quality was estimated with collocated marker sets as opposed to individual marker genes (Supplemental Fig. S2; Supplemental Table S4). Similar improvements were obtained under the inverse length model, although estimates were less accurate for genomes generated under this model (completeness: 10.3%–6.6%; contamination: 8.2%–5.6%) (Supplemental Fig. S3; Supplemental Table S5).
Inference of reference genome tree
Estimates of completeness and contamination can be further refined by using lineage-specific, collocated marker sets determined from the placement of a query genome within a reference genome tree (Fig. 2). The reference tree used by CheckM was inferred from the concatenation of 43 conserved marker genes with largely congruent phylogenetic histories (Supplemental Tables S6, S7). It incorporates 2052 finished and 3604 draft genomes obtained from the Integrated Microbial Genomes (IMG) database (Markowitz et al. 2014) identified as being near complete with minimal contamination (see Methods). The inferred tree (Supplemental Fig. S4) shares features in common with recently published genome trees, including the class Clostridia being highly paraphyletic (Yutin and Galperin 2013) and the class Epsilonproteobacteria residing outside the Proteobacteria phylum (Rinke et al. 2013). These discrepancies between phylogeny and taxonomy will cause marker genes calculated from named lineages within the genome tree to deviate from those determined strictly from assigned taxonomy. More importantly, a reference tree allows lineage-specific marker genes to be established for any internal nodes and not just those representing a named taxonomic group.
Figure 2.
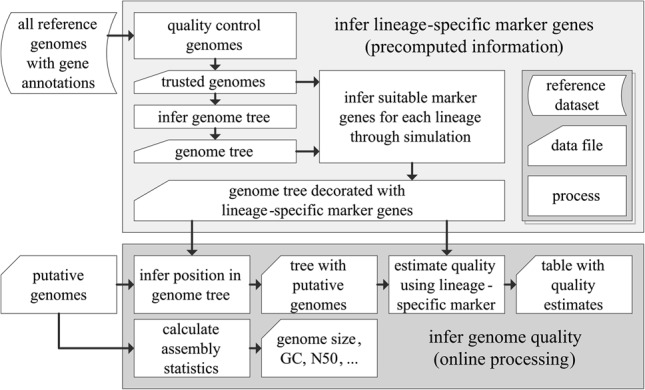
CheckM consists of a workflow for precomputing lineage-specific marker genes for each branch within a reference genome tree (top box) and an online workflow for inferring the quality of putative genomes (bottom box). Starting with a set of annotated reference genomes, the quality of these genomes is assessed in order to produce a set of near-complete genomes suitable for inferring marker genes. These genomes form the basis of a reference genome tree. A simulation framework is then used to associate each branch in the reference genome tree with a lineage-specific marker set suitable for robustly estimating the quality of genomes placed along a given branch (Fig. 3). To determine the quality of a putative genome, its position within the reference genome tree is inferred in order to establish the set of marker genes suitable for assessing its quality. These marker genes are identified within the putative genome and the presence/absence of these genes used to estimate its completeness and contamination.
Assessment of lineage-specific marker sets
Lineage-specific marker sets were determined for all nodes within the reference genome tree by identifying single-copy genes present in ≥97% of all descendant genomes. The quality of a genome can be estimated using the marker set defined at any parental node between the genome's position in the reference tree and the root. A simulation framework was used to establish the parental lineage with the most favorable set of markers for assessing the quality of genomes placed along any branch in the reference tree (Fig. 3). Briefly, finished genomes were used to simulate incomplete and contaminated genomes placed along a branch, and the parental lineage whose marker genes most accurately estimated the quality of these genomes was determined.
Figure 3.
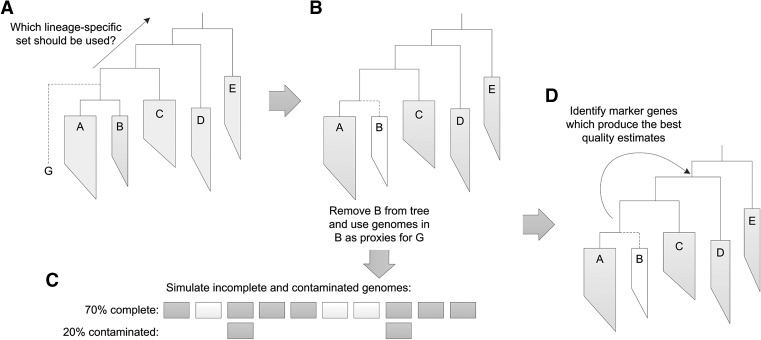
Overview of simulation framework for selecting lineage-specific marker genes. (A) To evaluate a genome, G, it is placed into a reference genome tree. Each parental node from the point of insertion to the root of the tree defines a lineage-specific marker set which can be used to estimate the completeness and contamination of this genome. (B) To select a suitable set of lineage-specific marker genes for evaluating G, the genomes in the child lineage of G with the fewest genomes were used as proxies for G. (C) Genomes at different levels of completeness and contamination were simulated from these proxy genomes by subsampling and duplicating fixed sized genomic fragments. (D) Each parental marker set was then used to estimate the completeness and contamination of these simulated genomes, and the marker set resulting in the best average performance over all simulated genomes was identified. This marker set is used to assess the quality of any genomes subsequently inserted along this branch.
We evaluated the effectiveness of the selected lineage-specific marker sets on genomes generated under all three simulation models. The quality of each simulated genome was estimated using collocated marker sets inferred from genomes within (1) the archaeal or bacterial lineage, (2) the lineage selected by the simulation framework, and (3) the parental lineage producing the most accurate estimates. Case 3 represents an idealized case in which the parental lineage is selected independently for each simulated genome, which is in contrast to Case 2, where the single parental lineage selected by the simulation framework must be used for all simulated genomes placed along a given branch. Under all three models, the selected lineage-specific marker sets provided more accurate completeness and contamination estimates than domain-specific marker sets and produced estimates nearly as accurate as the best performing lineage-specific marker sets (Fig. 4; Table 1; Supplemental Figs. S5, S6; Supplemental Tables S8–S10). The improvement in quality estimates can be substantial with the mean absolute error in completeness and contamination being reduced by 44.4% (5.4%–3.0%) and 19.5% (4.1%–3.3%), respectively, when using selected lineage-specific sets instead of the domain-specific sets to estimate the quality of genomes generated with the random contig model. In addition, the variances of the completeness and contamination estimates are substantially reduced. Summarizing results by the taxonomic group affiliated with each simulated genome indicated that the selected lineage-specific sets provided improved estimates across all 39 classes (20 phyla) considered in this study, with the exception of the poorly sampled Synergistetes lineage, in which the estimates were largely unchanged (Supplemental Fig. S7; Supplemental Tables S11–S13).
Figure 4.

Error in completeness and contamination estimates on simulated genomes with 50%, 70%, 80%, or 90% completeness and 5%, 10%, or 15% contamination. Quality estimates were determined using (1) domain: marker sets inferred across all archaeal or bacterial genomes; (2) selected: marker sets inferred from genomes within the lineage selected by CheckM; and (3) best: marker sets inferred from genomes within the lineage producing the most accurate estimates. Marker genes were organized into collocated marker sets in all cases. Simulated genomes were generated under the random contig model from 2430 draft genomes spanning 31 classes (18 phyla) with each draft genome being used to generate 20 simulated genomes.
Table 1.
Mean absolute error (±SD) in completeness (comp.) and contamination (cont.) estimates for (1) Domain: marker sets inferred across all archaeal or bacterial genomes; (2) Selected: marker sets inferred from genomes within the lineage selected by CheckM; and (3) Best: marker sets inferred from genomes within the parental lineage producing the most accurate estimates

Influence of taxonomic novelty
“Natural” taxonomic novelty within the test set of 3324 draft genomes was examined in order to explore the influence of novelty on genome quality estimates. As expected, increasing taxonomic novelty results in the preferential use of more broadly defined lineage-specific marker sets (Supplemental Table S14). For example, 69.7% of the 33 test genomes that are sole representatives of different taxonomic families within the reference genome tree were evaluated with a lineage-specific marker set at a rank of class or higher, whereas only 17.9% of the 1923 genomes with multiple strains within the reference genome tree were evaluated with such broad marker sets. The use of more broadly defined marker sets had a direct impact on the genome quality estimates for simulated genomes generated under the random fragment model (Supplemental Table S15). Although simulated genomes derived from the six draft genomes representing novel phyla or classes resulted in less accurate quality estimates then genomes from less novel lineages, the estimates were consistent with the performance expected for genomes evaluated with a domain-specific marker set (cf. Supplemental Tables S8, S15). This suggests that the novelty of a genome is less critical than the specificity of the marker set used to evaluate genome quality, and that even genomes representing novel phyla can be assessed at the error rates expected for domain-specific marker sets. Although genome quality estimates improved gradually as the taxonomic novelty of genomes decreases from order to species, the mean absolute error of the quality estimates across the test sets never deviated by more than 0.8%, suggesting that the accuracy of lineage-specific marker sets are relatively stable at this degree of novelty (Supplemental Table S15).
Bias in genome quality estimates
Quality estimates based on individual marker genes or collocated marker sets exhibit a bias resulting in completeness being overestimated and contamination being underestimated (Figs. 1, 4). This bias is the result of marker genes residing on foreign DNA that are otherwise absent in a genome being mistakenly interpreted as an indication of increased completeness as opposed to contamination. This bias approximately follows a binomial distribution, suggesting a potential avenue for bias correction (see Supplemental Methods). We have elected not to correct for this bias because confounding factors such as gene collocation make the correction approximate and the bias is small (<3%) when genomes are substantially complete (>70%) with medium contamination (≤10%) (Supplemental Fig. S8).
Assessment of isolate genomes
To benchmark CheckM on real-world data, we assessed the quality of 2281 isolate genomes from the GEBA, GEBA-KMG, GEBA-PCC, GEBA-RNB, and HMP data sets (Table 2). Using lineage-specific marker sets, 2190 (96%) of these genomes were estimated to be ≥95% complete with ≤5% contamination (Supplemental Table S16), making them excellent reference genomes for analyses such as assigning taxonomy to anonymous genomic fragments (Brady and Salzberg 2009; Parks et al. 2011) or characterizing metagenomic samples using marker genes (Darling et al. 2014). The remaining 91 (4%) genomes were found to be <95% complete or >5% contaminated making them less reliable reference genomes for some analyses. A small number of the genomes have an estimated completeness <90% (14 genomes) or an estimated contamination >10% (five genomes). These genomes suffer from a diverse range of problems, which we illustrate using three public genomes from the HMP available at the time of preparing this manuscript:
The Capnocytophaga sp. oral taxon 329 genome (HMP id: 9074; GenBank id: AFHP00000000; IMG id: 651324019) was estimated as 100% complete and 100% contaminated by CheckM. Investigation of the 157 contigs comprising this genome revealed a bimodal GC-distribution, suggesting the presence of two distinct genomes (Supplemental Fig. S9). We separated the contigs into two clusters by applying k-means clustering with k=2 to the tetranucleotide signatures of each contig. Placing the resulting clusters into a genome tree identified one cluster as a novel Capnocytophaga genome (99.0% complete, 0.2% contaminated) and the other cluster as closely related to Paraprevotella clara YIT 11840 (100% complete, 0.4% contaminated) (Supplemental Fig. S10).
The least complete HMP genome reported by CheckM was the gastrointestinal Clostridiales sp. SM4/1 genome (HMP id: 924; GenBank id: FP929060; IMG id: 2524023221) annotated as finished at IMG and GOLD, but estimated as only 56% complete. CheckM determined the coding density of this genome to be 66%, suggesting substantial assembly or gene calling errors. Further investigation revealed that 667 kbp (21.5%) of this 3.1 Mbp genome is comprised of ambiguous base pairs (N's).
The Lactobacillus gasseri MV-22 genome (HMP id: 515) available from IMG (id: 643886189) consists of 93 contigs comprising 1.89 Mbp with only 193 ambiguous bases. CheckM estimated the completeness of this genome as 90.9% when using the lineage-specific marker sets and 81.2% complete when using the bacterial marker set (Supplemental Table S17). Although these low completeness estimates could be the result of lineage-specific gene loss, the other three Lactobacillus gasseri genomes from HMP are all estimated to be ≥96% complete with only the Leucyl-tRNA synthetase protein family (PF13603) exhibiting lineage-specific gene loss across the bacterial marker genes (Supplemental Table S18). This suggests that Lactobacillus gasseri MV-22 is incomplete with ≥9% of its genome estimated to be missing. The incomplete state of this genome is not transparent from its genome size, as available Lactobacillus gasseri genomes are between 1.78 and 2.01 Mbp.
Table 2.
Completeness and contamination of genomes from large-scale sequencing projects

The issues exemplified above are not limited to the HMP or large-scale sequencing efforts. For example, the Paracoccus denitrificans SD1 genome (Siddavattam et al. 2011) at IMG (id: 2511231195) was estimated to be only 59% complete by CheckM (Supplemental Table S19). Comparing this genome to Paracoccus denitrificans PD1222 suggests that this species has two chromosomes and a plasmid, and that the SD1 genome is currently missing both a chromosome and its plasmid. CheckM also identified several submission errors as exemplified by the type strain Oligotropha carboxidovorans OM5 (IMG id: 650716069), which is reported as 99.7% complete and 100.9% contaminated as a result of both draft and finished versions of its chromosome and plasmid being contained in its genome sequence file.
Assessment of single-cell genomes
The GEBA-MDM initiative applied single-cell genomics to novel uncultivated bacterial and archaeal cells (Rinke et al. 2013). Although this is the largest single-cell sequencing initiative currently published, other large-scale initiatives are underway and have submitted initial genomes to IMG. To assess the quality of genomes recovered through single-cell genomics, we applied CheckM to (1) 201 genomes recovered from individual cells in the GEBA-MDM initiative; (2) 21 genomes coassembled from GEBA-MDM cells belonging to the same population; and (3) 410 additional genomes from unpublished studies annotated as “uncultured type” or “single cell” in IMG (Table 2).
Technical challenges in obtaining single-cell genomes such as low DNA yield and the associated need for genome amplification make it challenging to recover complete genomes. CheckM quality estimates indicate that only three of the 201 (1.5%) GEBA-MDM genomes and 17 of the 410 (4.4%) unpublished single-cell genomes have an estimated completeness ≥90%. Combining cells from the same population can substantially improve completeness with the 21 combined assemblies in GEBA-MDM having an average completeness of 64.9% ± 24.3% compared to 34.9% ± 20.6% for the 201 single-cell genomes (Supplemental Table S20). Although current techniques for recovering genomes from single cells result in highly incomplete genomes, these are still valuable reference genomes for analyses such as assigning taxonomy to anonymous genomic fragments and resolving phylogenetic relationships (Rinke et al. 2013). However, these reference genomes should be free from substantial contamination as this will be a source of inaccuracy in such analyses. CheckM estimates identified 62 of the 410 (15.1%) unpublished single-cell genomes have ≥5% contamination. All the GEBA-MDM genomes were found to have <5% contamination, except one Omnitrophica sp. with 5.3% contamination and two combined assemblies that were estimated to be 11.3% (Marinimicrobia sp.) and 21.5% (Cloacimonetes sp.) contaminated. Comparison of duplicate marker genes within these genomes suggests the contamination is the result of foreign DNA being amplified and not an assembly error.
Assessment of population genomes
Unlike genomes recovered from cultured isolates or single cells, genomes obtained from metagenomic data represent a consensus across a microbial population. CheckM was applied to 146 population genomes recovered from four metagenomic studies (Table 2). As expected, the estimated completeness and contamination of these genomes vary substantially (Fig. 5). Although population genomes are often incomplete (74 of 146 genomes are between 50% and 95% complete), they can be recovered with relatively little contamination (43 of the 74 partial genomes have ≤5% contamination) (Supplemental Table S21). In addition to this set of 74 partial genomes, an additional 16 (11%) population genomes were estimated to be ≥95% complete with <5% contamination. These estimates account for several of the population genomes recoding the opal stop codon as CheckM automatically identifies such recodings (see Supplemental Methods).
Figure 5.

Lineage-specific completeness and contamination estimates for 262 isolates annotated as finished in IMG (A), 2019 isolates annotated as draft in IMG (B), 632 genomes recovered using single-cell genomics (C), and 146 population genomes recovered from metagenomic data (D). Dashed lines indicate the criteria required for a genome to be considered a near-complete genome with low contamination. Insets give a more detailed view of the quality of the isolate genomes. The 2281 isolate genomes were obtained from IMG and sequenced as part of the GEBA, GEBA-KMG, GEBA-PCC, GEBA-RNB, or HMP initiatives.
Poor quality estimates are expected for genomic elements such as plasmids or phage as the marker genes used by CheckM are specific to bacterial and archaeal chromosomes. The 10 plasmids and 11 phage identified within the acetate-amended aquifer (Wrighton et al. 2012) and infant gut (Sharon et al. 2013) data sets were estimated to be 0% complete and 0% contaminated, with the exception of two plasmids (CARSEP1P, ACD71) and one phage (ACD33), which were estimated as 4.2%, 2.7%, and 0.15% complete, respectively (Supplemental Table S21). The completeness of reduced genomes without representation in the reference genome tree will also be underestimated when genome reduction has resulted in the loss of marker genes. This is illustrated by the four candidate phylum Saccharibacteria (TM7) genomes obtained from sludge bioreactor metagenomes, which were estimated to be 60%–70% complete by CheckM, although shown to be ≥85% complete after accounting for lineage-specific gene loss (Albertsen et al. 2013).
We compared the quality estimates obtained for the 90 putative population genomes recovered from the acetate-amended aquifer (Wrighton et al. 2012) community using domain-level and lineage-specific marker sets (Supplemental Table S22). Although the completeness and contamination of these population genomes is unknown, these results demonstrate the degree to which quality estimates can change under these two conditions. We have focused on the acetate-amended aquifer data set as it contains population genomes spanning a wide range of qualities, whereas other studies have focused exclusively on high-quality population genomes. On average, completeness changed by 13.0% and contamination by 5.1% when using lineage-specific instead of domain-specific marker sets. Estimates varied substantially for some genomes with completeness estimates changing by ≥15% for 36 genomes and contamination estimates changing by ≥10% for 12 genomes. Although completeness estimates with domain-level and lineage-specific marker sets are highly correlated (R2 = 0.84), domain-level estimates tend to overestimate the completeness of genomes relative to lineage-specific estimates (Supplemental Fig. S11). The correlation between contamination estimates is weaker (R2 = 0.69), and any global trend is less clear as the majority of population genomes exhibit <5% contamination (Supplemental Fig. S12).
Estimating strain heterogeneity
CheckM can distinguish between contamination resulting from the presence of genomic fragments from multiple strains and contamination resulting from the inclusion of genomic fragments from more divergent taxa. This is particularly useful for genomes recovered from metagenomic data as separating strains into individual genomes remains a challenging problem (Imelfort et al. 2014). These two types of contamination are differentiated automatically by CheckM by using the amino acid identity (AAI) between multicopy genes as a measure of phylogenetic relatedness (Konstantinidis and Tiedje 2005). Reanalysis of the methanotrophic ANME-1 genome recovered from metagenomic data by Meyerdierks et al. (2010) with CheckM illustrates that this population genome is a chimera of closely related strains. Of the 229 lineage-specific marker genes used to evaluate the quality of this genome, 42 were identified as being multicopy within the ANME-1 genome (38 present twice, two present three times; 82.3% completeness). Although this represents ∼21% contamination, 82.0% of the comparisons between multicopy genes have an AAI ≥90% (76.0% at ≥95% AAI) (Supplemental Fig. S13), revealing that the contamination is largely the result of incorporating genomic fragments from closely related taxa and that multiple ANME-1 strains are likely present within this environment.
Proposed genome quality classification scheme
Genomes recovered from isolates, single cells, or metagenomic data vary substantially in their quality (Fig. 5). To make full use of these genomes, their quality must be reported in reference databases along with other essential genome information (Field et al. 2008). A qualitative vocabulary for discussing genomes of varying quality was proposed by Chain et al. (2009); here, we supplement this effort by proposing a vocabulary based on a quantitative threshold that augments existing schemes for describing draft genome quality (Table 3). The status of finished is reserved for genomes assembled into a single contiguous sequence containing no gaps or ambiguities, where extensive efforts have been made to identify errors (Mardis et al. 2002; Chain et al. 2009). Genomes assembled into multiple sequences as a result of repetitive regions, but otherwise of a finished quality, may be classified as noncontiguous finished (Chain et al. 2009). We propose that all other genomes be designated as draft, and the quality of genomes qualified based on both established vocabularies for describing genome quality and estimates of genome completeness and contamination.
Table 3.
Controlled vocabulary of draft genome quality based on estimated genome completeness and contamination

The proposed quantitative vocabulary permits automated assignments of completeness and contamination estimates, which are critical for quality control in large-scale genome sequencing initiatives and for updating genome databases as new genomes are added or techniques for estimating genome quality improve. Of the 3059 genomes (2281 isolates, 632 single cell, 146 metagenomic) considered in this study, 2292 (74.9%) were classified as being near complete with either no detectable (833 genomes; 27.2%) or low (1461 genomes; 47.8%) contamination. These genomes are strong candidates for being classified as finished or noncontiguous finished, but this designation should only be applied after extensive additional verification. The wide range of quality within the remaining 767 (25.1%) genomes illustrates the need for a verbose vocabulary when discussing draft genomes, e.g., 84 (2.7%) were classified as substantially complete drafts with 22 (0.7%) exhibiting no contamination, 55 (1.8%) having low contamination, and 5 (0.2%) having medium contamination. The presence of metagenomic and single-cell genomes was also transparent as 125 (4.1%) of the genomes were classified as moderately complete drafts, and 521 (17.0%) were classified as partial draft genomes.
Discussion
Here, we introduce CheckM, a new tool developed to estimate the completeness and contamination of genomes derived from isolates, single cells, and metagenomes using lineage-specific marker genes. To evaluate the robustness of genome quality estimates, we simulated genomes under three distinct models: (1) a random fragment model in which genomic fragments were removed or added uniformly across the genome; (2) a random contig model that accounts for the characteristics of assembled contigs; and (3) an inverse length model reflecting the limitations of metagenomic binning methods. Our results on simulated genomes demonstrate that when lineage-specific marker genes are organized into collocated sets, they are sufficiently spaced throughout a genome to provide accurate estimates of genome quality. For substantially complete genomes (≥70%–90%) with medium contamination (5% to ≤10%), our results suggest that completeness and contamination estimates generally have an absolute error of ≤6%, and the error in the quality estimates tends to decrease as the quality of a genome improves (Fig. 4; Table 1; Supplemental Figs. S5, S6; Supplemental Tables S8–S10).
The robust estimates of genome quality provided by CheckM allow for automated quality screening of bacterial and archaeal genomes. Using CheckM, we were able to identify isolate genomes exhibiting a wide range of problems. Incorporation of these low-quality genomes into reference data sets will diminish the accuracy of inferences made in many studies. For example, a study of horizontal gene transfer might incorrectly predict a large number of transfers between Capnocytophaga and Paraprevotella genomes due to the Capnocytophaga sp. oral taxon 329 genome erroneously containing genes from both of these genera. Similarly, a comparative genomics study including the Clostridiales sp. SM4/1 genome identified as 56% complete may incorrectly report the number of core genes among Clostridiales genomes or the ubiquity of key metabolic pathways. Comparison of the incomplete Lactobacillus gasseri MV-22 genome considered in this study to its GenBank (id: GL531761) counterpart revealed that this issue was localized to the IMG repository, illustrating the benefit of independently verifying the quality of genomes at different repositories.
Many of the erroneous genomes reported in this study were brought to the attention of IMG and have subsequently been removed from their database. The Capnocytophaga sp. oral taxon 329 has also been retracted from NCBI. Although removal of contaminated or incomplete genomes is warranted, the statistics provided by CheckM can help identify the problems associated with these genomes. In the case of Capnocytophaga sp. oral taxon 329, the CheckM statistics directly suggested the presence of two distinct populations, which allowed for the recovery of two near-complete genomes with low contamination.
Incomplete draft genomes are valuable references for many genomic analyses, and their use is likely to increase as partial genomes of novel species are recovered from single cells and metagenomic data. Although methodologies for handling genomes of varying qualities are currently in their infancy, it is clear many analyses will benefit from accurate estimates of completeness and contamination. The benefit of using partial genomes with low contamination for assigning taxonomy to anonymous genomic fragments and resolving phylogenetic relationships has already been demonstrated (Rinke et al. 2013). Other analyses such as comparing the metabolic capability of different groups of genomes will likely benefit from restricting the analyses to only near-complete genomes in order to ensure confident predictions can be made regarding differences in their metabolic capabilities. Because the quality of a genome is essential for determining its suitability for different analyses, we recommend public genome repositories and new genome announcements include completeness and contamination estimates (Table 3).
The limitations of the proposed approach must be considered when interpreting CheckM quality estimates. Eukaryotic or phage genomes will be reported as highly incomplete because we have focused on marker sets suitable for evaluating bacterial and archaeal genomes. The quality of plasmids must also be assessed independently of CheckM. When recovering genomes from metagenomic data, the additional genome statistics reported by CheckM (e.g., coding density, coverage) can be used along with the quality estimates to help distinguish putative genomes representing fragments of an archaeal or bacterial chromosome from phage, plasmids, or eukaryotic genomes. However, incorporation of CheckM into a complete quality control and annotation pipeline where plasmids (Jørgensen et al. 2014), genomic islands (Langille et al. 2010), phage (Akhter et al 2012), and other genomic elements are automatically identified and screened will likely improve the quality and characterization of recovered genomes. Use of taxonomic assignment methods (Patil et al. 2011; Dröge et al. 2014) would also be of benefit within a complete quality control pipeline in order to identify genomes exhibiting clear chimerism and sources of contamination within single-cell and isolate genomes. The estimates for highly incomplete or highly contaminated genomes must be interpreted with regard to the observed systematic bias, which arises from marker genes from foreign genomes being misinterpreted as an indication of additional completeness (Supplemental Fig. S8). The novelty of a genome will also influence the accuracy of CheckM estimates. Estimates for bacterial and archaeal genomes from deep basal lineages with few reference genomes are necessarily determined using domain-level marker sets instead of lineage-specific markers that generally provide superior estimates. This limitation is most evident for novel lineages undergoing genome reduction as demonstrated by our reanalysis of the candidate phylum Saccaribacteria (TM7) genomes. Although CheckM can provide refined estimates for reduced genomes in well-characterized lineages (see Supplemental Results), a manual assessment of gene loss or duplication is required to improve quality estimates for reduced genomes recovered from novel lineage (Albertsen et al. 2013). CheckM provides outputs to aid in performing this refinement.
We anticipate several improvements that will further refine the estimates produced by CheckM. The most substantial impact is likely to be the inclusion of additional reference genomes from lineages that are currently poorly represented. This will mitigate the number of genomes that are evaluated using broad, less accurate marker sets and improve refinements for lineage-specific gene loss and duplication. Incorporation of eukaryotic genomes into the reference tree would also be a substantive benefit when assessing population genomes recovered from environmental samples where fungi and other microbial Eukaryotes may be present. Further exploration of the parameter space of CheckM may also result in improved estimates. For instance, the 97% ubiquity criteria used to delineate marker genes is likely not optimal, and the use of a probabilistic model for assessing the presence/absence of a gene across all genomes in a lineage may improve the inferred marker sets (Segata et al. 2013). Ultimately, we expect to adopt a strategy that will allow optimal values for key parameters to be determined independently for each lineage.
CheckM is the first automated tool for estimating the completeness and contamination of isolate, single-cell, and population genomes. The need for accurate estimates of genome quality will only grow in importance as we continue to fill out the microbial tree of life and are better able to utilize draft genomes to inform gene- and genome-centric analyses of microbial communities.
Methods
Inference of reference genome tree
A genome tree incorporating 5656 trusted reference genomes (see Supplemental Methods) was inferred from a set of 43 genes with largely congruent phylogenetic histories. An initial set of 66 universal marker genes was established by taking the intersection between bacterial and archaeal genes determined to be single copy in >90% of genomes. From this initial gene set, 18 multicopy genes with divergent phylogenetic histories in >1% of the reference genomes were removed. A multicopy gene within a genome was only deemed to have a congruent phylogenetic history if all copies of the gene were situated within a single conspecific clade (i.e., all copies were contained in a clade from a single named species) within its gene tree. Genes were aligned with HMMER v3.1b1 (http://hmmer.janelia.org), and gene trees inferred with FastTree v2.1.3 (Price et al. 2009) under the WAG (Whelan and Goldman 2001) and GAMMA (Yang 1994) models. Trees were then modified with DendroPy v3.12.0 (Sukumaran and Holder 2010) in order to root the trees between archaea and bacteria unless these groups were not monophyletic, in which case midpoint rooting was used. A further five genes found to be incongruent with the IMG taxonomy were also removed as these genes may be subject to lateral transfer. Testing of taxonomic congruency was performed as described in Soo et al. (2014). The final set of 43 phylogenetically informative marker genes (Supplemental Table S6) consists primarily of ribosomal proteins and RNA polymerase domains and is similar to the universal marker set used by PhyloSift (Supplemental Table S7; Darling et al. 2014). A reference genome tree was inferred from the concatenated alignment of 6988 columns with FastTree v2.1.3 under the WAG+GAMMA model and rooted between bacteria and archaea. Internal nodes were assigned taxonomic labels using tax2tree (McDonald et al. 2012).
Identification of marker genes in putative genomes
Genes were predicted using Prodigal v2.60 (Hyatt et al. 2012) and Pfam (Finn et al. 2014), and TIGRFAMs (Haft et al. 2003) protein families were identified using HMMER v3.1b1 (http://hmmer.janelia.org) with model specific cutoff values for both the Pfam (-cut_gc) and TIGRFAMs (-cut_nc) HMMs. Pfam annotations are assigned using the same methodology as the Sanger Institute and IMG, which accounts for homologous relationships between Pfam clans (see pfam_scan.pl on the Sanger Institute FTP site, https://www.sanger.ac.uk/). Gene calling errors occasionally occur due to ambiguous bases in a contig that can result in adjacent, erroneous genes being called that are assigned to the same marker gene. These errors are resolved by checking if adjacent marker genes have a best match to adjacent, nonoverlapping portions of a marker gene's HMM.
Determination of lineage-specific marker genes
Single-copy Pfam and TIGRFAMs genes were identified within reference genomes using the annotations provided by IMG. A gene was defined as a lineage-specific marker gene if it occurs only once in >97% of the genomes within a lineage. Lineage-specific marker genes were inferred for all internal nodes within the reference genome tree. Pfam and TIGRFAMs families were considered redundant if they matched the same genes in >90% of the finished IMG genomes, in which case only the Pfam gene was used as a marker.
Organization of marker genes into collocated marker sets
A pair of marker genes were considered to be collocated within a lineage if they occurred within 5 kbp of each other in >95% of genomes within a lineage. Sets of collocated markers were formed from collocated gene pairs by clustering together all pairs with a shared gene (e.g., if genes A and B, and genes B and C are collocated, then they are clustered into the collocated set ABC).
Estimation of completeness, contamination, and strain heterogeneity
Genome completeness is estimated as the number of marker sets present in a genome taking into account that only a portion of a marker set may be identified:
| (1) |
where s is a set of collocated marker genes; M is the set of all collocated marker sets s; and GM is the set of marker genes identified in a genome. Genome contamination is estimated from the number of multicopy marker genes identified in each marker set:
| (2) |
where Cg is N − 1 for a gene g identified N ≥ 1 times, and 0 for a missing gene. CheckM also supports estimating completeness and contamination without arranging marker genes into collocated sets. Equations 1 and 2 can be applied to this case by assigning each marker gene to its own set (i.e., ).
Contamination resulting from multiple strains or closely related species being binned into a single putative genome is identified by examining the AAI between multicopy marker genes. Specifically, a strain heterogeneity index is calculated as the fraction of multicopy gene pairs above a specified AAI threshold:
| (3) |
where g = {g1,g2,…,gN} is the set of hits to a marker gene; G is the set of all marker genes; and aai is 1 if the AAI between gi and gj is greater than t (default = 0.9) and 0 otherwise.
Placement of genomes into the reference genome tree
Identification of the 43 phylogenetically informative marker genes within a putative genome are identified using HMMs as described in the preceding section Identification of Marker Genes in Putative Genomes. Identified genes are aligned with HMMER and the concatenated alignment used to place a genome into the reference genome tree using pplacer v2.6.32 (Matsen et al. 2010). Putative genomes consisting of an insufficient number of unique phylogenetic marker genes (default = 10) to be robustly placed within the reference genome tree are evaluated using the universal marker set.
Selection of lineage-specific marker genes
Marker genes can be inferred for all internal nodes in the reference tree along the path from the putative genome to the root (Fig. 3A). The most suitable set of marker genes for assessing a genome depends on a number of factors, including the novelty of the putative genome relative to the surrounding reference genomes and the breadth of diversity covered by these genomes. A simulation framework was used to establish the parent node producing the most suitable marker set for estimating the completeness and contamination of a genome placed on a given branch within the reference genome tree.
The simulation framework was restricted to the 2052 finished reference genomes in IMG, as draft genomes were used for evaluating the performance of CheckM. For each branch, the descendant lineage with the fewest genomes was removed from the reference tree (Fig. 3B). These genomes were used as proxies to simulate genomes placed on this branch. Each genome was fragmented into 10-kbp windows and used to simulate 100 independent genomes with completeness randomly selected between 50% and 100% and contamination randomly selected between 0% and 20% (Fig. 3C). Marker genes were then inferred for each parent node using the 97% single-copy criterion and used to assess the completeness and contamination of the simulated genomes. Marker genes were not formed into marker sets in order to reduce computational complexity and to allow a fair assessment of how this feature influences genome assessment. The parental node whose inferred marker genes minimize the error in the estimated completeness and contamination over all simulated genomes was assigned to the branch (Fig. 3D):
| (4) |
where m is a set of marker genes; M is the set of marker genes for each parent node; compest(gi,m) is the estimated completeness of simulated genome gi using m; compt(gi) is the true completeness of gi; contest(gi,m) and contt(gi) are analogous functions for contamination; and N is the number of simulated genomes derived from g. Marker genes associated with each internal node were calculated de novo during the simulation to reflect removing the test genomes and then recalculated afterward using all available reference genomes in order to produce refined sets of marker genes. This simulation framework is computationally expensive, but only needs to be performed once, and the results are distributed with the CheckM software.
Simulation of incomplete and contaminated isolate and population genomes
Simulated genomes were generated from an initial set of 3604 draft genomes within IMG identified as being of high quality (see Supplemental Methods). To help alleviate bias toward well-sampled lineages, 280 of the 3604 high-quality draft genomes with identical phylogenetic marker genes were not used during the generation of simulated genomes. Simulated genomes were generated at varying degrees of completeness and contamination using three distinct random sampling models. Under the random fragment model, each contig comprising a genome was fragmented into nonoverlapping windows of a fixed size between 5 and 50 kbp. This size range was selected because it approximates the contig lengths of genomes recovered from metagenomic data or single-cell genomics: The mean N50 of the GEBA-MDM single-cell genomes, Wrighton acetate-amended aquifer population genomes, and Sharon infant gut population genomes is ∼28 kbp, ∼17 kbp, and ∼ 12 kbp, respectively. In order to generate genomes at a desired level of completeness and contamination, fragments were sampled without or with replacement, respectively. Windows were sampled until a simulated genome had completeness and contamination equal to or just greater than the target values. Generation of simulated genomes was limited to draft genomes as finished genomes were used to determine appropriate lineage-specific marker sets suitable for evaluating genomes (Fig. 3).
The 2430 draft reference genomes comprised of 20 or more contigs were used to simulate partial and contaminated genomes reflecting the characteristics of assembled contigs. Under this random contig model, genomes were generated by randomly removing contigs until the simulated genome reached or fell below a target completeness level. Contamination was introduced by randomly adding contigs with replacement from a single randomly selected genome until the desired level of contamination was reached or exceeded. These 2430 draft genomes were also used to generate genomes reflecting the limitations of metagenomic binning methods that rely on the statistical properties of contigs (e.g., tetranucleotide signature, coverage) to establish putative population genomes. To simulate this, partial genomes were generated by randomly removing contigs with a probability inversely proportional to their length until the simulated genome reached or fell below a target completeness level. Contamination was introduced by randomly selecting another draft reference genome and adding contigs from this genome with a probability inversely proportional to length until the desired level of contamination was reached or exceeded.
Evaluation using simulated genomes
Evaluation of CheckM was performed using simulated genomes generated at all combinations of 50%, 70%, 80%, 90%, 95%, and 100% completeness with 0, 5%, 10%, 15%, or 20% contamination. Marker genes and marker sets were inferred with the test genome removed from the set of reference genomes (i.e., leave-one-out testing), and their performance was evaluated by considering the error in completeness and contamination estimates. To evaluate the performance of the lineage-specific markers selected by the simulation framework (Fig. 3), results were compared to the lineage-specific markers resulting in the best performance as determined by applying Equation 4 independently to each set of simulated genomes generated from a test genome at a specific level of completeness and contamination. This represents a highly idealized case, as it assumes a method capable of selecting different optimal lineage-specific markers for the same genome under varying levels of completeness and contamination.
Evaluation of universal- and domain-level marker gene sets
Several universal- and domain-level marker gene sets were obtained either directly from the authors or from available software packages (Supplemental Table S1). PhyloSift (Darling et al. 2014) originally used the 40 marker genes suggested by Wu et al. (2013), but was reduced to 37 markers in version 1.0.1 of the PhyloSift software. Marker genes were identified using the precalculated “gathering” and “noise” cutoffs for the Pfam and TIGRFAMs models, respectively. Marker genes of COG or custom protein families were identified using an E-value cutoff of 1 × 10−5, 1 × 10−10, 1 × 10−15, and 1 × 10−20 and results reported for the cutoff providing the best mean performance. Results were highly similar across this range of E-values.
Evaluation of taxonomic novelty
The influence of taxonomic novelty on genome quality estimates was evaluated by identifying “natural” novelty within the test set of high-quality draft genomes used to generate simulated genomes. A test genome was classified as having taxonomic novelty at a given rank (e.g., genus) if (1) it was the only genome within the reference genome tree from the named group (e.g., the only genome from genus Ruminobacter); and (2) the parent group (e.g., the family Succinivibrionacea) contains at least two named groups at the rank of interest (e.g., Ruminobacter and at least one other Succinivibrionacea genus). The taxonomic rank of a lineage-specific marker set is defined as the most specific taxonomic rank containing the internal node from which the marker set was inferred (e.g., a marker set defined at an internal node between a named order and class was assigned to the broader rank of class).
Genome data sets
Population genomes from the Wrighton et al. (2012) and Sharon et al. (2013) studies were downloaded from ggKbase (http://ggkbase.berkeley.edu/) on March 31, 2014. Tyson et al. (2004) and Meyerdierks et al. (2010) population genomes were obtained from NCBI. The population genomes from the Albertsen et al. (2013) study can be obtained from http://ecogenomic.org/checkm/public-data. Reference genomes at NCBI and IMG are occasionally removed or modified. For posterity, the reference genomes analyzed in this paper have been archived at http://ecogenomic.org/checkm/public-data. The GEBA, GEBA-KMG, GEBA-PCC, GEBA-RNB, GEBA-MDM, and HMP genomes comprise part of the data downloaded from IMG on April 4, 2014.
Software availability
CheckM is open source software available at http://ecogenomics.github.io/CheckM. CheckM v0.9.4 was used during the preparation of this manuscript and is available as Supplemental Material.
Acknowledgments
Many of the genomes considered in this manuscript were produced by the US Department of Energy Joint Genome Institute (http://www.jgi.doe.gov) in collaboration with the user community. D.H.P. is supported by the Natural Sciences and Engineering Research Council of Canada. M.I. is supported by a Great Barrier Reef Foundation Postdoctoral Research Fellowship through the ReFuGe2020 Consortium. C.T.S. was supported by an Australian Postgraduate Award from the Australian Research Council. G.W.T. and P.H. are supported by a Discovery Outstanding Researcher Award (DORA) and Queen Elizabeth II Fellowship from the Australian Research Council, grants DP120103498 and DP1093175, respectively.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.186072.114.
Freely available online through the Genome Research Open Access option.
References
- Akhter S, Aziz RK, Edwards RA. 2012. PhiSpy: a novel algorithm for finding prophages in bacterial genomes that combines similarity- and composition-based strategies. Nucleic Acids Res 40: e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH. 2013. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31: 533–538. [DOI] [PubMed] [Google Scholar]
- Brady A, Salzberg SL. 2009. Phymm and PhymmBL: metagenomic phylogenetic classification with interpolated Markov models. Nat Methods 6: 673–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chain PS, Grafham DV, Fulton RS, Fitzgerald MG, Hostetler J, Muzny D, Ali J, Birren B, Bruce DC, Buhay C, et al. 2009. Genome project standard in a new era of sequencing. Science 326: 236–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AE, Jospin G, Lowe E, Matsen FA IV, Bik HM, Eisen JA. 2014. PhyloSift: phylogenetic analysis of genomes and metagenomes. PeerJ 2: e243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick GJ, Andersson AF, Baker BJ, Simmons SL, Thomas BC, Yelton AP, Banfield JF. 2010. Community-wide analysis of microbial genome sequences signatures. Genome Biol 10: R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dröge J, Gregor I, McHardy AC. 2014. Taxator-tk: precise taxonomic assignment of metagenomes by fast approximation of evolutionary neighborhoods. Bioinformatics 31: 817–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont CL, Rusch DB, Yooseph S, Lombardo MJ, Richter RA, Valas R, Novotny M, Yee-Greenbaum J, Selengut JD, Haft DH, et al. 2012. Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J 6: 1186–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. 2008. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 26: 541–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, et al. 2014. Pfam: the protein families database. Nucleic Acids Res 42: D222–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, Tesler G. 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics 15: 1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft DH, Selengut JD, White O. 2003. The TIGRFAMs database of protein families. Nucleic Acids Res 31: 371–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haroon MF, Hu S, Shi Y, Imelfort M, Keller J, Hugenholtz P, Yuan Z, Tyson GW. 2013. Anaerobic oxidation of methane coupled to nitrate reduction in a novel archaeal lineage. Nature 500: 567–570. [DOI] [PubMed] [Google Scholar]
- Hyatt D, Locascio PF, Hauser LJ, Uberbacher EC. 2012. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 28: 2223–2230. [DOI] [PubMed] [Google Scholar]
- Imelfort M, Parks DH, Woodcroft BJ, Dennis P, Hugenholtz P, Tyson GW. 2014. GroopM: an automated tool for the recovery of population genomes from related metagenomes. PeerJ 2: e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen TS, Xu Z, Hansen MA, Sørensen SJ, Hansen LH. 2014. Hundreds of circular novel plasmids and DNA elements identified in a rat cecum metamobilome. PLoS One 9: e87924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidis KT, Tiedje JM. 2005. Towards a genome-based taxonomy for prokaryotes. J Bacteriol 187: 6258–6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langille MGI, Hsiao WWL, Brinkman FSL. 2010. Detecting genomic islands using bioinformatics approaches. Nat Rev Microbiol 8: 373–382. [DOI] [PubMed] [Google Scholar]
- Mardis E, McPherson J, Martienssen R, Wilson RK, McCombie WR. 2002. What is finished, and why does it matter. Genome Res 12: 669–671. [DOI] [PubMed] [Google Scholar]
- Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Pillay M, Ratner A, Huang J, Woyke T, Huntemann M, et al. 2014. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res 42: D560–D567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsen FA, Kodner RB, Armbrust EV. 2010. pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics 11: 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. 2012. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6: 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mende DR, Sunagawa S, Zeller G, Bork P. 2013. Accurate and universal delineation of prokaryotic species. Nat Methods 10: 881–884. [DOI] [PubMed] [Google Scholar]
- Meyerdierks A, Kube M, Kostadinov I, Teeling H, Glöckner FO, Reinhardt R, Amann R. 2010. Metagenome and mRNA expression analyses of anaerobic methanotrophic archaea of the ANME-1 group. Environ Microbiol 12: 422–439. [DOI] [PubMed] [Google Scholar]
- Parks DH, MacDonald N, Beiko R. 2011. Classifying short genomic fragments from novel lineages using composition and homology. BMC Bioinformatics 12: 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil KR, Haider P, Pope PB, Turnbaugh PJ, Morrison M, Scheffer T, McHardy AC. 2011. Taxonomic metagenome sequence assignment with structured output models. Nat Methods 8: 191–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP. 2009. FastTree: computing large minimum-evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26: 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng JF, Darling A, Malfatti S, Swan BK, Gies EA, et al. 2013. Insights into the phylogeny and coding potential of microbial dark matter. Nature 499: 431–437. [DOI] [PubMed] [Google Scholar]
- Salzberg SL, Phillippy AM, Zimin A, Puiu D, Magoc T, Koren S, Treangen TJ, Schatz MC, Delcher AL, Roberts M, et al. 2012. GAGE: a critical evaluation of genome assemblies and assembly algorithms. Genome Res 22: 557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Börnigen D, Morgan XC, Huttenhower C. 2013. PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat Commun 4: 2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi Y, Ohashi A, Parks DH, Yamauchi T, Tyson GW, Hugenholtz P. 2015. First genomic insights into members of a candidate bacterial phylum responsible for wastewater bulking. PeerJ 3: e740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon I, Banfield JF. 2013. Genome from metagenomics. Science 342: 1057–1058. [DOI] [PubMed] [Google Scholar]
- Sharon I, Morowitz MJ, Thomas BC, Costello EK, Relman DA, Banfield JF. 2013. Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization. Genome Res 23: 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih PM, Wu D, Latifi A, Axen SD, Fewer DP, Talla E, Calteau A, Cai F, Tandeau de Marsac N, Rippka R, et al. 2013. Improving the coverage of the cyanobacterial phylum using diversity-driven genome sequencing. Proc Natl Acad Sci 110: 1053–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddavattam D, Karegoudar TN, Modde SK, Kumar N, Baddam R, Avasthi TS, Ahmed N. 2011. Genome of a novel isolate of Paracoccus denitrificans capable of degrading N,N-dimethylformamide. J Bacteriol 193: 5598–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soo RM, Skennerton CT, Sekiguchi Y, Imelfort M, Paech SJ, Dennis PG, Steen JA, Parks DH, Tyson GW, Hugenholtz P. 2014. An expanded genomic representation of the phylum Cyanobacteria. Genome Biol Evol 6: 1031–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumaran J, Holder MT. 2010. DendroPy: a Python library for phylogenetic computing. Bioinformatics 26: 1569–1571. [DOI] [PubMed] [Google Scholar]
- Swan BK, Tupper B, Sczyrba A, Lauro FM, Martinez-Garcia M, González JM, Luo H, Wright JJ, Landry ZC, Hanson NW, et al. 2013. Prevalent genome streamlining and latitudinal divergence of planktonic bacteria in the surface ocean. Proc Natl Acad Sci 110: 11463–11468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Hamady M, Frader-Liggett CM, Knight R, Gordon JI. 2007. The human microbiome project. Nature 449: 804–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM, Solovyev VV, Rubin EM, Rokhsar DS, Banfield JF. 2004. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 428: 37–43. [DOI] [PubMed] [Google Scholar]
- Whelan S, Goldman N. 2001. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol 18: 691–699. [DOI] [PubMed] [Google Scholar]
- Wrighton KC, Thomas BC, Sharon I, Miller CS, Castelle CJ, VerBerkmoes NC, Wilkins MJ, Hettich RL, Lipton MS, Williams KH, et al. 2012. Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337: 1661–1665. [DOI] [PubMed] [Google Scholar]
- Wu M, Scott AJ. 2012. Phylogenomic analysis of bacterial and archaeal sequences with AMPHORA2. Bioinformatics 28: 1033–1034. [DOI] [PubMed] [Google Scholar]
- Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, et al. 2009. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 462: 1056–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Doroud L, Eisen JA. 2013. TreeOTU: operational taxonomic unit classification based on phylogenetic trees. arXiv 1308.6333v1. [Google Scholar]
- Yang Z. 1994. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J Mol Evol 39: 306–314. [DOI] [PubMed] [Google Scholar]
- Yutin N, Galperin MY. 2013. A genomic update on clostridial phylogeny: gram-negative spore formers and other misplaced clostridia. Environ Microbiol 15: 2631–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]