

Abstract
Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis (FHHNC) is an autosomal-recessive renal tubular disorder characterized by excessive urinary losses of magnesium and calcium, bilateral nephrocalcinosis and progressive chronic renal failure. Presentation with FHHNC symptoms generally occurs early in childhood or before adolescence. At present, the only therapeutic option is supportive and consists of oral magnesium supplementation and thiazide diuretics. However, neither treatment seems to have a significant effect on the levels of serum magnesium or urine calcium or on the decline of renal function. In end-stage renal disease patients, renal transplantation is the only effective approach. This rare disease is caused by mutations in the CLDN16 or CLDN19 genes. Patients with mutations in CLDN19 also present severe ocular abnormalities such as myopia, nystagmus and macular colobamata. CLDN16 and CLDN19 encode the tight-junction proteins claudin-16 and claudin-19, respectively, which are expressed in the thick ascending limb of Henle's loop and form an essential complex for the paracellular reabsorption of magnesium and calcium. Claudin-19 is also expressed in retinal epithelium and peripheral neurons. Research studies using mouse and cell models have generated significant advances on the understanding of the pathophysiology of FHHNC. A recent finding has established that another member of the claudin family, claudin-14, plays a key regulatory role in paracellular cation reabsorption by inhibiting the claudin-16–claudin-19 complex. Furthermore, several studies on the molecular and cellular consequences of disease-causing CLDN16 and CLDN19 mutations have provided critical information for the development of potential therapeutic strategies.
Keywords: claudin-16, claudin-19, genetic disease, magnesium, renal tubulopathy
Introduction
Magnesium plays an important role in many cellular processes, including energy metabolism, DNA replication, RNA transcription and protein synthesis [1, 2]. Consequently magnesium homeostasis, which depends on magnesium uptake in the intestine, magnesium storage in bone and magnesium excretion in the kidney, needs to be strictly controlled. Several rare inherited diseases due to renal magnesium loss including autosomal dominant and autosomal recessive hypomagnesaemias have been described [2–4]. Hypomagnesaemia is usually defined as plasma magnesium levels <0.7 mM. Identification of the genes and their encoded proteins involved in the pathogenesis of these diseases has contributed greatly to our knowledge of epithelial magnesium transport in the kidney.
In 1972, Michelis et al. [5] reported clinical data from two siblings with hypomagnesaemia, decreased bicarbonate threshold, distal renal tubular acidosis, nephrocalcinosis, polyuria and convulsions. Subsequently, similar cases were described showing that the patients also had hypercalciuria, nephrocalcinosis, progressive decline of renal function and, sometimes, severe ocular defects [6–10]. In these patients, the hypomagnesaemia and hypercalciuria were resistant to magnesium oral supplementation and thiazide treatment, respectively. This renal tubulopathy was named ‘familial hypomagnesaemia with hypercalciuria and nephrocalcinosis’ (FHHNC) [10]. Rodríguez-Soriano et al. [8] were the first to suggest that FHHNC was caused by a defect in magnesium and calcium reabsorption in the thick ascending limb (TAL) of Henle's loop, the main nephron segment for paracellular reabsorption of these two cations [11, 12].
Using a positional cloning method, Simon et al. [13] analyzed 12 families with FHHNC and identified a gene, PCLN1 (now described as CLDN16), associated with the disorder. A few years later, Konrad et al. [14] studied a group of FHHNC patients, also affected with severe ocular defects and lacking mutations in CLDN16, and identified a second gene, CLDN19, linked to the disease. CLDN16 and CLDN19 encode the tight-junction (TJ) proteins claudin-16 and claudin-19, respectively, which are expressed in the TAL, interact with each other and play a key role in regulating the paracellular transport of magnesium and calcium [15]. During the last decade there have been significant advances in the pathophysiology of FHHNC. The current knowledge of clinical and molecular features of this disease is the focus of the present review.
Clinical characteristics
FHHNC [type 1, Online Mendelian Inheritance in Man (OMIM) #248250] is a rare autosomal recessive tubular disease characterized by excessive renal magnesium and calcium excretion [8, 10]. Patients develop hypomagnesaemia, hypercalciuria and bilateral nephrocalcinosis and their clinical course is often complicated by the development of chronic renal failure. Hypomagnesaemia may disappear with the deterioration of the glomerular filtration rate (GFR) due to a decline in filtered magnesium. Furthermore, patients usually present with recurrent urinary tract infections, nephrolithiasis, polyuria, polydipsia and/or failure to thrive [8, 10, 16–18]. Presentation with clinical traits of FHHNC usually occurs early in childhood or before adolescence. Many patients show a marked decline in GFR at the time of diagnosis, and about one-third of them progress to chronic renal failure during adolescence. Problems associated with acute hypomagnesaemia, such as convulsions and muscular tetany, are less common. These features differentiate FHHNC from other inherited types of renal hypomagnesaemia (Table 1). Other biochemical anomalies consist of elevated serum parathyroid hormone (PTH) levels before the onset of chronic renal failure, incomplete distal tubular acidosis, hyperuricaemia and hypocitraturia [17]. It should be emphasized that the high serum levels of PTH observed in FHHNC patients contrast with the normal or reduced PTH levels associated with hypomagnesaemia [2]. This fact seems counterintuitive since magnesium deficiency can suppress PTH secretion. Ocular abnormalities, mainly severe myopia, macular colobomata and nystagmus, have been reported for a subset of patients [10, 14, 19]. This disease form is known as FHHNC with severe ocular involvement (type 2, OMIM #24819).
Table 1.
Characteristics of FHHNC and other inherited types of renal hypomagnesaemia
| Disease | Mutated gene (protein) | Pattern of inheritance | Serum Mg++ | Urine Mg++ | Serum Ca++ | Urine Ca++ | Renal failure | Other symptoms |
|---|---|---|---|---|---|---|---|---|
| FHHNC type 1 |
CLDN16 (claudin-16) |
AR | ↓ | ↑ | – | ↑ | +a | Nephrocalcinosis |
| FHHNC type 2 |
CLDN19 (claudin-19) |
AR | ↓ | ↑ | – | ↑ | +a | Nephrocalcinosis, severe ocular defectsb |
| Gitelman syndrome |
SLC12A3 (NCC) |
AR | ↓ | ↑ | – | ↓ | – | Muscle weakness, tetany, hypokalaemia, chondrocalcinosis |
| Hypomagnesaemia with secondary hypocalcaemia |
TRPM6 (TRPM6) |
AR | ↓ | ↑ | ↓ | – | – | Tetany, muscle spams, mental retardation |
| Isolated dominant hypomagnesaemia |
FXYD2 (Na+/K+-ATPase γ subunit) |
AD | ↓ | ↑ | – | ↓ | – | Convulsions |
| Isolated recessive hypomagnesaemia |
EGF (EGF) |
AR | ↓ | ↑ | – | – | – | Tetany, mental retardation |
ATPase, adenosine 5′-triphosphatase; AR, autosomal recessive; AD, autosomal dominant; NCC, Na+/Cl− cotransporter; TRPM6, transient receptor potential channel malastatin member 6; EGF, epidermal growth factor.
aChronic renal failure in infancy or early adolescence.
bMyopia, macular colobamata and nystagmus.
Genetics
Sixteen years ago Simon et al. [13] identified the CLDN16 gene, which is linked to chromosome 3q, and found the first pathogenic mutations in patients with FHHNC from several countries. CLDN16 encodes the TJ protein claudin-16. To date, >50 CLDN16 pathogenic mutations have been reported, including mainly missense mutations, and also nonsense mutations, splice site mutations and small deletions [13, 17–36]. The mutations are found in either a homozygous or compound heterozygous state and are spread over the five exons of the CLDN16 gene. In the claudin-16 protein, most missense mutations are located in or near the four transmembrane domains, in the two extracellular segments and in the carboxy-terminal cytoplasmic region. Patients from Germany or Eastern European countries show a common CLDN16 mutation, L151F, which is due to a widespread founder effect [17]. This mutation is located in the carboxy terminus of the first extracellular segment of claudin-16. In contrast, patients from North Africa frequently show another founder mutation, A139V, also affecting the first extracellular segment [19].
A second locus for FHHNC was mapped on chromosome 1p34.2 and three pathogenic mutations were identified in the CLDN19 gene of patients who lacked mutations in CLDN16 and showed severe ocular defects [14]. CLDN19 encodes claudin-19, another member of the claudin family. Up to now, 17 different CLDN19 pathogenic mutations have been identified [14, 19, 37–41]. These include mostly missense mutations located in exons 1–4 and a few nonsense mutations and large or small deletions. Missense mutations are found in the first extracellular segment or in the first three transmembrane domains of claudin-19. The majority of patients present homozygous mutations and a few are compound heterozygous. A founder effect has been indicated for the recurrent G20D mutation in Spanish and French families [14, 19, 37]. Recently a Mexican family of Spanish origin with three FHHNC-affected sisters homozygous for this mutation was reported, which suggests a remote common ancestor [42].
Konrad et al. [31] have established a phenotype–genotype correlation with regard to renal failure progression in FHHNC patients with CLDN16 mutations. They have studied a cohort of 71 affected individuals and found that patients with loss-of-function mutations in both alleles exhibit a younger age at the presentation of FHHNC symptoms and a faster decline in renal function when compared with those bearing at least one mutation with partial function. These data suggest that the presence of at least one CLDN16 allele with partial function is enough to predict a milder clinical phenotype. On the other hand, there are phenotypic differences between FHHNC patients with the same mutation. Most patients with CLDN19 mutations develop disease symptoms during the first 2 years of life, but some may remain asymptomatic until the second decade of life [37, 43]. Phenotypic variability has been shown even in families with affected siblings harbouring the same CLDN16 or CLDN19 homozygous mutation [34, 42]. These differences suggest the likely contribution of epigenetic factors or other unknown genetic modifiers [37, 43].
CLDN16 mutations that cause FHHNC can alter intracellular trafficking of claudin-16 or disturb its capacity to facilitate paracellular magnesium transport. Several claudin-16 mutants, such as R149L, G162V, L167P, G233D, S235P and S235F, are retained in the endoplasmic reticulum (ER) and are subjected to proteasomal degradation [25], while others accumulate in the Golgi complex (R149X, L151W and L151F) or are delivered to lysosomes (M71T, G191R and T303R) [22, 25, 31]. However, other mutants, including G88E, S110R, R114Q, H141D, L145P, L151W, G198A, R216C and G227R, are properly targeted to the TJ but show a complete or partial loss of function [25, 31]. In addition, Hou et al. [38] have shown that some CLDN16 mutations, comprising L145P, L151F, G191R, A209T and F232C, prevent claudin-16/claudin-19 interactions. CLDN19 mutations also have different consequences. Claudin-19 proteins encoded by mutations G20D and Q57E have trafficking defects [25, 38]; mutant G20D is confined to the ER, consistent with a defect of the normal signal peptide sequence of this protein, whereas mutant Q57E is found in the apical membrane with a diffuse pattern. Both mutations result in complete loss of claudin-19 function. Mutants L90P and G123R are localized in the TJ and cause partial loss of claudin-19 function [38]. These two mutations severely affect heteromeric interactions with claudin-16.
An understanding of the molecular mechanism by which CLDN16 and CLDN19 mutations cause FHHNC might provide novel alternatives for therapeutic intervention [44]. For instance, Müller et al. [23, 24] have shown that surface expression of several retained claudin-16 mutants could be improved by inhibiting endocytosis or using pharmacological chaperones. Moreover, the consequences of presumed missense mutations at the level of mRNA should be evaluated. We have found that two CLDN16 missense mutations, R149L and G198A, can alter pre-mRNA splicing, resulting in defective mRNAs [45].
Claudin-16 and claudin-19 proteins
Claudins are members of a family of 28 tetraspan membrane proteins that are found in TJs of epithelia and endothelia [15, 46, 47]. They form strands that mediate cell adhesion and function as both paracellular barriers and selective ion channels regulating the flow of ions and solutes between the luminal and basolateral spaces (Figure 1). Expression studies have indicated that claudin-16 functions predominantly as a pore-forming claudin increasing permeability to cations, whereas claudin-19 performs mainly as a barrier-forming claudin decreasing permeability to anions [48, 49]. Claudin proteins consist of four transmembrane domains (TM1–4), two extracellular segments (ECS1 and ECS2), amino- and carboxy-terminal cytoplasmic tails and a short cytoplasmic loop (Figure 2). Research studies on several claudins have suggested that ECS1 contains the ion selectivity filter while ECS2 is involved in claudin–claudin interactions [50–52]. Recently Suzuki et al. [53] reported the first crystal structure of a mammalian claudin (claudin-15), providing structural information on this family of TJ proteins. The structure showed that the four transmembrane segments form a tight four-helix package and that sections of the two extracellular segments form a domain with a beta-sheet structure. This palm-shaped domain is likely to contribute to trans-interactions between claudin molecules of adjacent cells, resulting in the formation of paracellular channels [54]. A disulphide bridge between two cysteine residues located in the first extracellular segment stabilizes the beta-sheet structure [53]. These cysteine residues are highly conserved among all members of the claudin family [15]. In addition, ECS1 contains a conserved glycine-leucine-tryptophan motif that also seems to contribute to stabilization of the appropriately folded ECS1 structure [55]. At the end of ECS1 there is a short extracellular helix (ECH) that links to TM2.
Fig. 1.

Schematic representation of tight junctions and putative location of claudin-16 and claudin-19. (A) TJs are the most apical intercellular junctions in epithelial cells of the nephron. They are composed of multiprotein complexes that include mainly claudins, occludin and the junctional adhesion molecules. (B) These protein components are arranged like beads on a string that span the adjacent membranes of each TJ. (C) Claudin-16 and claudin-19 are mainly found in TJs of epithelial cells of the TAL. They interact with each other, generating cation-selective pores.
Fig. 2.

Membrane model of claudins. The conserved structural features of claudins, four transmembrane domains (TM1–4), extracellular segments (ECS1 and ECS2) and ECH, are shown.
The intracellular amino terminus of claudins is usually very short, although claudin-16 could be an exception. The human CLDN16 mRNA has two potential in-frame start codons that may produce a short and a long protein isoform. The difference between them would be only within the intracellular amino termini (3 and 73 amino acids, respectively), and it is still a matter of discussion whether the long version exists under physiological conditions [48, 56]. Although the two isoforms could exist in kidney cells, some findings support the hypothesis that the second start codon is the real translation start site [17, 48]. The intracellular carboxy-terminal tail of claudins seems to be involved in exit from the ER and trafficking to the TJ [57]. It contains phosphorylation and palmitoylation sites that seem to regulate the localization of claudins and the barrier properties of TJs [58]. At the end of this carboxy terminus there is usually a PDZ domain-binding motif that can interact with PDZ domains of TJ scaffolding/adapter proteins including ZO-1, ZO-2 and ZO-3 [59, 60]. These adaptor proteins directly or indirectly bind actin and therefore attach the TJ within the cytoskeleton. Mutation T303R in the carboxy terminus of claudin-16 results in inactivation of the PDZ domain-binding motif [22]. The mutant protein fails to interact with ZO-1 and is misdirected to the lysosomes for degradation. Thus the association between claudin-16 and ZO-1 is required for proper localization of claudin-16 in the TJ.
The expression pattern of claudins is tissue specific and most tissues express many claudins, which can interact with other claudin molecules to form the TJ strands. Claudin-16 is mainly expressed in the kidney, and in the nephron it is selectively expressed in TJs of renal epithelial cells of the TAL [13]. Expression of claudin-19 has been detected in the kidney, nervous system and retina [13, 61, 62]. Along the nephron, claudin-19 is expressed together with claudin-16 mainly in the TAL [38, 63, 64]. In peripheral neurons, claudin-19 is found in TJ-like structures of Schwann cells [65]. Claudin-19-deficient mice present peripheral neuropathy that might be attributed to deficient electrophysiological sealing of Schwann cells [65].
Pathophysiology
The first inherited disorder of claudins to be identified was FHHNC [13]. It was then established that mutations in claudin-16 (then PCLN1) lead to renal loss of magnesium and calcium. These initial discoveries indicated that claudin-16 was involved in the formation of a cation-selective TJ pore for the reabsorption of magnesium and calcium in the TAL. This segment of the nephron is where ∼60% of the filtered magnesium and 20% of the filtered calcium are recovered [11, 12]. The selective transport of magnesium and calcium in the TAL occurs through a paracellular pathway that is dependent on the lumen-positive transepithelial voltage generated by the activity of both the Na+–K+–2Cl− co-transporter (NKCC2) and the renal outer medullary potassium (ROMK) channel at the apical membrane (Figure 3). Using claudin-16 pathogenic mutants and a cell model, Hou et al. [48] later found that claudin-16 just moderately enhanced magnesium permeability but greatly increased transepithelial Na+ permeability. This indicated that instead of acting as a magnesium channel, claudin-16 regulates the transepithelial voltage gradient, increasing paracellular Na+ permeability. The same group generated Cldn16 knockdown (KD) mice that presented hypomagnesaemia, hypercalciuria, hypermagnesuria and nephrocalcinosis, a comparable phenotype to that of FHHNC patients [66].
Fig. 3.
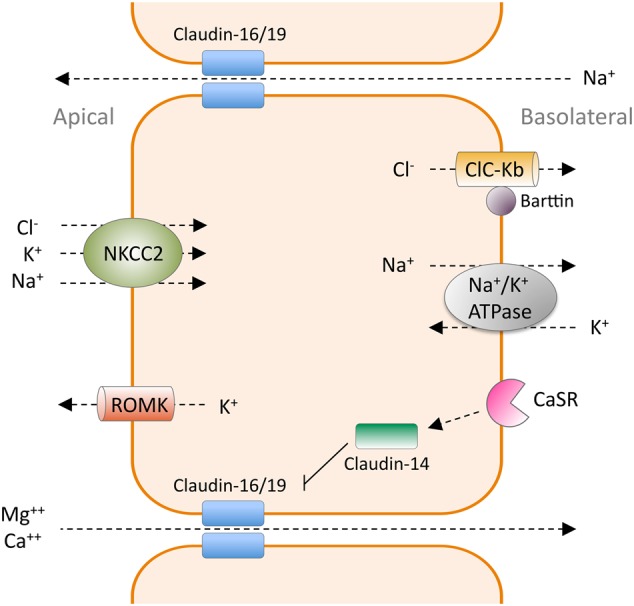
Paracellular reabsorption of magnesium and calcium in the TAL of Henle's loop. This transport depends on the lumen-positive electrical potential established by the transcellular reabsorption of other cations and anions. Reabsorption of Na+, K+ and Cl− through the apical membrane occurs via the NKCC2 co-transporter. Na+ and Cl− leave the epithelial cell through the Na+/K+-adenosine 5′-triphosphatase (ATPase) and the ClC-Kb channel at the basolateral membrane, respectively. K+ is excreted to the lumen by the ROMK channel. The backflow of Na+ through the paracellular channel, as a consequence of diminishing luminal Na+ concentrations, is an additional contributor to the lumen-positive voltage that forces magnesium and calcium reabsorption. Claudin-16 and claudin-19 facilitate the paracellular transport of magnesium and calcium. Activation of CaSR by extracellular calcium upregulates claudin-14, which in turn interacts with the claudin-16/claudin-19 complex and inhibits its cation permeability.
On the other hand, CLDN19 mutations severely reduced Cl− permeability, indicating that claudin-19 acts as a barrier in the TJ [38, 49]. Cldn19 KD mice also developed FHHNC features, including hypomagnesaemia, hypercalciuria and hypermagnesuria [63]. siRNA knockdown of Cldn19 expression causes a loss of claudin-16 from TJs in the TAL, while siRNA knockdown of Cldn16 produces an analogous result on claudin-19. Both claudin-16- and claudin-19-depleted TJs show normal barrier function but defective ion selectivity [63]. These data suggested that a heteromeric caludin-16–claudin-19 interaction is needed for assembling of the complex into the TJ structure and generating the cation-selective paracellular pathway [38]. On the other hand, some CLDN16 and CLDN19 mutations disturb this interaction, implying a function in the TAL for claudin interaction in the development of FHHNC [38]. However, in retinal pigment epithelium, claudin-19 is expressed and functional in the absence of claudin-16 [67].
Alterations in extracellular calcium seem to affect the transport of calcium, magnesium and other ions in the TAL. The calcium/magnesium-sensing receptor (CaSR), highly expressed in the TAL, has been known to play an essential role in regulating divalent cation reabsorption in this segment of the nephron [68]. However, the precise role of CaSR is not entirely comprehended. Stimulation of CaSR by high concentrations of extracellular calcium may result in inhibition of the electrolyte transport handled by NKCC2 and ROMK, reduction in the lumen-positive electrical potential and, consequently, increased urinary excretion of magnesium and calcium. Nevertheless, recent evidence indicates that the principal task of CaSR in the TAL is to control paracellular permeability via regulation of claudin-14. This protein, encoded by CLDN14, is found mainly in the TAL and therefore it is also considered to be involved in divalent cation transport [69]. A genome-wide association analysis recognized CLDN14 as a main risk factor for hypercalciuric nephrolithiasis [70]. Pathogenic mutations in this gene cause autosomal recessive deafness in children [71]. Hou et al. [69] have elegantly shown that claudin-14 interacts with claudin-16, repressing the cation selectivity of the heteromeric claudin-16/claudin-19 complex. Accordingly, under high calcium dietary conditions, Cldn14 knockout (KO) mice progress to hypermagnesaemia, hypomagnesuria and hypocalciuria, the opposed phenotype to that of Cldn16 KD mice [69]. On the other hand, transgenic mice overexpressing claudin-14 selectively in the TAL epithelium of the kidney present the FHHNC phenotype [72]. The same authors determined that the mechanism by which calcium regulates claudin-14 is through microRNA-based repression of CLDN14 expression. Two kidney-specific microRNAs (miR-9 and miR-374) bind to the 3′-untranslated region of the CLDN14 mRNA, repress its translation and induce its decay [69]. Extracellular calcium, through activation of CaSR, reduces the expression levels of these microRNAs, thus stimulating claudin-14 and preventing claudin-16/claudin-19-facilitated divalent cation reabsorption (Figure 3), which results in hypercalciuria [72–74]. These findings have encouraged the pursuit of therapeutic alternatives to reduce urinary calcium excretion. Recently, Gong et al. [75] showed that treatment with histone deacetylase (HDAC) inhibitors stimulates transcription of the two claudin-14 targeting microRNAs, repressing claudin-14 expression and reducing renal calcium excretion specifically in the TAL.
As mentioned before, FHHNC patients usually develop chronic renal failure, which is correlated with progressive tubulointerstitial nephritis; however, its pathophysiology is uncertain. Although it has been suggested that renal failure in FHHNC could be associated with the hypercalciuria and nephrocalcinosis present in these patients [10], a proper correlation has not been found. Recent findings have revealed a previously unidentified mechanism for crystal nephropathies and progression of chronic kidney disease (CKD) [76, 77]. This mechanism involves activation of the inflammasome, a high-molecular-weight enzymatic complex that triggers pro-inflammatory cytokine production in response to infection and tissue injury [78]. The most exhaustively characterized inflammasome is the NLRP3 (or NALP3) inflammasome, which seems to be involved in the pathogenesis of many renal diseases and their complications. NLRP3-mediated inflammasome activation could also initiative CKD in hereditary forms of crystal nephropathies like FHHNC. Although studies with several animal models have indicated that inhibition of the NLRP3 inflammasome reduces the progression of renal injury, additional research will be needed to establish if this could represent a therapeutic possibility for treating FHHNC or other renal tubulopathies. Moreover, it has been hypothesized that claudin-16 could also be implicated in tubular cell proliferation and differentiation [79]. This is supported by the phenotype of Japanese Black cattle with homozygous claudin-16 gene deletions, which shows renal failure due to interstitial nephritis [80, 81]. Nevertheless, another study argues that renal dysplasia may be unrelated to the absence of claudin-16 in cattle [82].
Diagnosis
Several hereditary magnesium transport diseases have been described and their associated genes have been identified [3, 4]. Their clinical characteristics, biochemical data and mode of inheritance can in many cases be used to distinguish them [3, 4]. In FHHNC, these characteristics include hypomagnesaemia, hypercalciuria, normocalcemia, hypermagnesuria, bilateral nephrocalcinosis, renal failure, autosomal recessive mode of inheritance and, in FHHNC type 2, ocular abnormalities such as severe myopia, macular colobomata and nystagmus (Table 1). As with other traits of FHHNC, the ocular defects are generally present at diagnosis early in childhood or before adolescence [19, 37, 43]. It should be noted that normal serum levels of magnesium do not exclude FHHNC [37, 83]. In case of normomagnesaemia, the fractional excretion values of magnesium can be valuable for diagnosis [83]. Because of the autosomal recessive mode of inheritance, many FHHNC patients originate from populations with a high frequency of consanguineous marriages.
To confirm the clinical diagnosis in FHHNC patients it is necessary to identify a pathogenic mutation in both alleles of the CLDN16 or CLDN19 genes. Mutational analysis is carried out by sequencing the coding exons and flanking intronic regions of these two genes [19, 37, 84]. Quantitative multiplex polymerase chain reaction of short fragments can be used to detect deletions of exons. Taking into account the presence or absence of severe ocular defects, depending on the origin of the patient, it might be reasonable to begin the mutational analysis with a study of frequent disease-causing mutations. As mentioned earlier, CLDN16 mutations L151F and A139V are recurrent in patients from Germany, Poland and the Czech Republic and North Africa, respectively [17–19, 31, 83], and CLDN19 mutation G20D is very frequent among Spanish patients or patients with Spanish ancestors [14, 19, 37, 85]. Therefore the first genetic screening in patients of these populations should be testing for the presence of the respective recurrent mutations. Specific mutations can be analyzed by SNaPshot or by sequencing the corresponding exon.
It should be noted that FHHNC types 1 and 2 are very rare disorders and their population prevalence is unknown. Only ∼70 apparently unrelated families (∼100 patients) with a CLDN16 causative mutation and 60 families (∼70 patients) with a CLDN19 causative mutation have been described in the literature [84]. Nevertheless, this disease is probably underdiagnosed due to a lack of awareness. FHHNC families with pathogenic mutations in either gene should seek genetic counselling for carrier status, disease members or prenatal diagnosis.
Treatment
Like with other inherited rare disorders, modification of the primary genetic defects is not yet realistic. There is no specific treatment for this disease and there have been no clinical trials that may guide us toward a therapeutic option. Therapeutic approaches to regulate the claudin genes involved in renal calcium and magnesium transport have been lacking. Therefore the current therapy is basically supportive. FHHNC patients are given oral magnesium supplementation and thiazide diuretics to prevent the progression of nephrocalcinosis or nephrolithiasis, but neither treatment has had a considerable effect on the levels of urinary calcium or serum magnesium or on the deterioration of renal function [10, 17, 19, 37]. However, a recent study of 25 FHHNC patients in Poland argues that treatment with thiazides effectively reduced hypercalciuria in most cases [83]. Vitamin D deficit is common in patients with CKD and is a risk factor for secondary hyperparathyroidism. Therefore supplementation with active vitamin D is sometimes included in the treatment of FHHNC patients. In addition, a recent study indicates that maintaining normal vitamin D levels helps preserve kidney function in children with CKD [86]. However, vitamin D supplementation is occasionally a controversial aspect of treatment because it increases intestinal calcium absorption and urinary calcium excretion, which might enhance calcium-stone formation. Consequently some physicians may be reluctant to prescribe vitamin D therapy to patients with hypercalciuria. The only alternative for treatment of renal dysfunction in FHHNC is renal replacement therapy in end-stage renal disease patients. Calcium and magnesium excretion is normalized after transplantation and there is no recurrence of the disease. Severe myopia can be corrected with glasses, contact lenses, laser surgery or implanting artificial lenses. At present, there is no therapy for nystagmus or macular colobomata.
Conclusions and perspectives
Since the identification of the CLDN16 gene and its association with FHHNC in 1999, there have been important advances in our knowledge of the pathogenesis of this disease. However, although the essential role of claudin-16, claudin-19 and claudin-14 in paracellular transport of magnesium and calcium in the TAL has been elucidated, much needs to be done in order to fully comprehend the pathophysiology of FHHNC. Undoubtedly the recent elucidation of the claudin-15 crystal structure will improve the molecular comprehension of paracellular barriers between epithelial cells.
Claudin-19 is expressed at high levels in the retina, but why mutations in this claudin cause ocular defects is still unknown. Claudin-19 is also expressed in peripheral neurons, and in fact Cldn19 KO mice present peripheral neuropathy [65]. However, the function of claudin-19 in this part of the nervous system has not been determined. On the other hand, the possible contribution of epigenetic factors or genetic modifiers on the phenotypic variability observed in some patients with FHHNC has been suggested, but there are no studies addressing this question. We need to learn more in relation to the interactions of claudin-16, claudin-19 and claudin-14 with other factors and their potential signalling functions. In addition, further evaluation of the consequences of specific CLDN16 and CLDN19 mutations remains an area for further investigation that could deliver new therapeutic strategies.
For many years, the only available therapy for hypercalciuria has been through the use of diuretics such as thiazide and amiloride. Therapeutic approaches to regulate claudin function in renal calcium transport have been lacking. The recent discovery that claudin-14 is an inhibitory subunit of the claudin-16/claudin-19 complex has prompted the search for new therapeutic alternatives to reduce urinary calcium excretion relying on this pathway. This has led to the identification for a new approach based on HDAC inhibitors and microRNA regulation that seems promising for the treatment of hypercalciuric diseases.
Acknowledgements
This work was supported by grant PI14/00760 from Instituto de Salud Carlos III, Spain, and co-financed by the European Regional Development Fund, ‘A way to build Europe’.
Conflicts of interest statement
None declared.
References
- 1.Jahnen-Dechent W, Ketteler M. Magnesium basics. Clin Kidney 2012; 5(Suppl 1): i3–i14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Baaij JH, Hoenderop JG, Bindels RJ. Magnesium in man: implications for health and disease. Physiol Rev 2015; 95: 1–46 [DOI] [PubMed] [Google Scholar]
- 3.de Baaij JH, Hoenderop JG, Bindels RJ. Regulation of magnesium balance: lessons learned from human genetic diseases. Clin Kidney J 2012; 5(Suppl 1): i15–i24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Konrad M, Schlingmann KP. Inherited disorders of renal hypomagnesaemia. Nephrol Dial Transplant 2014; 29(Suppl 4): iv63–iv71 [DOI] [PubMed] [Google Scholar]
- 5.Michelis MF, Drash AL, Linarelli LG, et al. Decreased bicarbonate threshold and renal magnesium wasting in a sibship with distal renal tubular acidosis. (Evaluation of the pathophysiological role of parathyroid hormone). Metabolism 1972; 21: 905–920 [DOI] [PubMed] [Google Scholar]
- 6.Manz F, Schärer K, Janka P, et al. Renal magnesium wasting, incomplete tubular acidosis, hypercalciuria and nephrocalcinosis in siblings. Eur J Pediatr 1978; 128: 67–79 [DOI] [PubMed] [Google Scholar]
- 7.Ulmann A, Hadj S, Lacour B, et al. Renal magnesium and phosphate wastage in a patient with hypercalciuria and nephrocalcinosis: effect of oral phosphorus and magnesium supplements. Nephron 1985; 40: 83–87 [DOI] [PubMed] [Google Scholar]
- 8.Rodríguez-Soriano J, Vallo A, García-Fuentes M. Hypomagnesaemia of hereditary renal origin. Pediatr Nephrol 1987; 1: 465–472 [DOI] [PubMed] [Google Scholar]
- 9.Nicholson JC, Jones CL, Powell HR, et al. Familial hypomagnesaemia—hypercalciuria leading to end-stage renal failure. Pediatr Nephrol 1995; 9: 74–76 [DOI] [PubMed] [Google Scholar]
- 10.Praga M, Vara J, González-Parra E, et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis. Kidney Int 1995; 47: 1419–1425 [DOI] [PubMed] [Google Scholar]
- 11.Cole DE, Quamme GA. Inherited disorders of renal magnesium handling. J Am Soc Nephrol 2000; 11: 1937–1947. [DOI] [PubMed] [Google Scholar]
- 12.Mount DB. Thick ascending limb of the loop of Henle. Clin J Am Soc Nephrol 2014; 9: 1974–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simon DB, Lu Y, Choate KA, et al. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science 1999; 285: 103–106 [DOI] [PubMed] [Google Scholar]
- 14.Konrad M, Schaller A, Seelow D, et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet 2006; 79: 949–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Günzel D, Yu AS. Claudins and the modulation of tight junction permeability. Physiol Rev 2013; 93: 525–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benigno V, Canonica CS, Bettinelli A, et al. Hypomagnesaemia-hypercalciuria-nephrocalcinosis: a report of nine cases and a review. Nephrol Dial Transplant 2000; 15: 605–610 [DOI] [PubMed] [Google Scholar]
- 17.Weber S, Schneider L, Peters M, et al. Novel paracellin-1 mutations in 25 families with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J Am Soc Nephrol 2001; 12: 1872–1881 [DOI] [PubMed] [Google Scholar]
- 18.Blanchard A, Jeunemaitre X, Coudol P, et al. Paracellin-1 is critical for magnesium and calcium reabsorption in the human thick ascending limb of Henle. Kidney Int 2001; 59: 2206–2215 [DOI] [PubMed] [Google Scholar]
- 19.Godron A, Harambat J, Boccio V, et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: phenotype-genotype correlation and outcome in 32 patients with CLDN16 or CLDN19 mutations. Clin J Am Soc Nephrol 2012; 7: 801–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weber S, Hoffmann K, Jeck N, et al. Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis maps to chromosome 3q27 and is associated with mutations in the PCLN-1 gene. Eur J Hum Genet 2000; 8: 414–422 [DOI] [PubMed] [Google Scholar]
- 21.Tajima T, Nakae J, Fujieda K. Two heterozygous mutations of CLDN16 in a Japanese patient with FHHNC. Pediatr Nephrol 2003; 18: 1280–1282 [DOI] [PubMed] [Google Scholar]
- 22.Müller D, Kausalya PJ, Claverie-Martin F, et al. A novel claudin 16 mutation associated with childhood hypercalciuria abolishes binding to ZO-1 and results in lysosomal mistargeting. Am J Hum Genet 2003; 73: 1293–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Müller D, Kausalya PJ, Bockenhauer D, et al. Unusual clinical presentation and possible rescue of a novel claudin-16 mutation. J Clin Endocrinol Metab 2006; 91: 3076–3079 [DOI] [PubMed] [Google Scholar]
- 24.Müller D, Kausalya PJ, Meij IC, et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: blocking endocytosis restores surface expression of a novel Claudin-16 mutant that lacks the entire C-terminal cytosolic tail. Hum Mol Genet 2006; 15: 1049–1058 [DOI] [PubMed] [Google Scholar]
- 25.Kausalya PJ, Amasheh S, Günzel D, et al. Disease-associated mutations affect intracellular traffic and paracellular Mg2+ transport function of Claudin-16. J Clin Invest 2006; 116: 878–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang JH, Choi HJ, Cho HY, et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis associated with CLDN16 mutations. Pediatr Nephrol 2005; 20: 1490–1493 [DOI] [PubMed] [Google Scholar]
- 27.Kutluturk F, Temel B, Uslu B, et al. An unusual patient with hypercalciuria, recurrent nephrolithiasis, hypomagnesemia and G227R mutation of Paracellin-1. An unusual patient with hypercalciuria and hypomagnesemia unresponsive to thiazide diuretics. Horm Res 2006; 66: 175–181 [DOI] [PubMed] [Google Scholar]
- 28.Sanjad SA, Hariri A, Habbal ZM, et al. A novel PCLN-1 gene mutation in familial hypomagnesemia with hypercalciuria and atypical phenotype. Pediatr Nephrol 2007; 22: 503–508 [DOI] [PubMed] [Google Scholar]
- 29.Staiger K, Staiger H, Haas C, et al. Hypomagnesemia and nephrocalcinosis in a patient with two heterozygous mutations in the CLDN16 gene. J Nephrol 2007; 20: 107–110 [PubMed] [Google Scholar]
- 30.Hampson G, Konrad MA, Scoble J. Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis (FHHNC): compound heterozygous mutation in the claudin 16 (CLDN16) gene. BMC Nephrol 2008; 9: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Konrad M, Hou J, Weber S, et al. CLDN16 genotype predicts renal decline in familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J Am Soc Nephrol 2008; 19: 171–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guran T, Akcay T, Bereket A, et al. Clinical and molecular characterization of Turkish patients with familial hypomagnesaemia: novel mutations in TRPM6 and CLDN16 genes. Nephrol Dial Transplant 2012; 27: 667–673 [DOI] [PubMed] [Google Scholar]
- 33.Kasapkara CS, Tumer L, Okur I, et al. A novel mutation of the claudin 16 gene in familial hypomagnesemia with hypercalciuria and nephrocalcinosis mimicking rickets. Genet Couns 2011; 22: 187–192 [PubMed] [Google Scholar]
- 34.Seeley HH, Loomba-Albrecht LA, Nagel M, et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis in three siblings having the same genetic lesion but different clinical presentations. World J Pediatr 2012; 8: 177–180 [DOI] [PubMed] [Google Scholar]
- 35.Deeb A, Abood SA, Simon J, et al. A novel CLDN16 mutation in a large family with familial hypomagnesaemia with hypercalciuria and nephrocalcinosis. BMC Res Notes 2013; 6: 527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nadarajah L, Khosravi M, Dumitriu S, et al. A novel claudin-16 mutation, severe bone disease, and nephrocalcinosis. Lancet 2014; 383: 98. [DOI] [PubMed] [Google Scholar]
- 37.Claverie-Martin F, Garcia-Nieto V, Loris C, et al. Claudin-19 mutations and clinical phenotype in Spanish patients with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. PLoS One 2013; 8: e53151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hou J, Renigunta A, Konrad M, et al. Claudin-16 and claudin-19 interact and form a cation-selective tight junction complex. J Clin Invest 2008; 118: 619–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naeem M, Hussain S, Akhtar N. Mutation in the tight-junction gene claudin 19 (CLDN19) and familial hypomagnesemia, hypercalciuria, nephrocalcinosis (FHHNC) and severe ocular disease. Am J Nephrol 2011; 34: 241–248 [DOI] [PubMed] [Google Scholar]
- 40.Ekinci Z, Karabaş L, Konrad M. Hypomagnesemia-hypercalciuria-nephrocalcinosis and ocular findings: a new claudin-19 mutation. Turk J Pediatr 2012; 54: 168–170 [PubMed] [Google Scholar]
- 41.Yuan T, Pang Q, Xing X, et al. First report of a novel missense CLDN19 mutations causing familial hypomagnesemia with hypercalciuria and nephrocalcinosis in a Chinese family. Calcif Tissue Int 2015; 96: 265–273 [DOI] [PubMed] [Google Scholar]
- 42.Arteaga ME, Hunziker W, Teo AS, et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: variable phenotypic expression in three affected sisters from Mexican ancestry. Ren Fail 2015; 37: 180–183 [DOI] [PubMed] [Google Scholar]
- 43.Faguer S, Chauveau D, Cintas P, et al. Renal, ocular, and neuromuscular involvements in patients with CLDN19 mutations. Clin J Am Soc Nephrol 2011; 6: 355–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schaeffer C, Creatore A, Rampoldi L. Protein trafficking defects in inherited kidney diseases. Nephrol Dial Transplant 2014; 29(Suppl 4): iv33–iv44 [DOI] [PubMed] [Google Scholar]
- 45.Claverie-Martin F, Acosta-Herrera M, González-Paredes FJ, et al. Missense and synonymous mutations in kidney disease-associated genes can cause mRNA defects. Pediatr Nephrol 2011; 26: 1573 [Google Scholar]
- 46.Furuse M. Molecular basis of the core structure of tight junctions. Cold Spring Harb Perspect Biol 2010; 2: a002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mineta K, Yamamoto Y, Yamazaki Y, et al. Predicted expansion of the claudin multigene family. FEBS Lett 2011; 585: 606–612 [DOI] [PubMed] [Google Scholar]
- 48.Hou J, Paul DL, Goodenough DA. Paracellin-1 and the modulation of ion selectivity of tight junctions. J Cell Sci 2005; 118: 5109–5118 [DOI] [PubMed] [Google Scholar]
- 49.Angelow S, El-Husseini R, Kanzawa SA, et al. Renal localization and function of the tight junction protein claudin-19. Am J Physiol Renal Physiol 2007; 293: F166–F177 [DOI] [PubMed] [Google Scholar]
- 50.Colegio OR, Van Itallie C, Rahner C, et al. Claudin extracellular domains determine paracellular charge selectivity and resistance but not tight junction fibril architecture. Am J Physiol Cell Physiol 2003; 284: C1346–C1354 [DOI] [PubMed] [Google Scholar]
- 51.Piontek J, Winkler L, Wolburg H, et al. Formation of tight junction: determinants of homophilic interaction between classic claudins. FASEB J 2008; 22: 146–158 [DOI] [PubMed] [Google Scholar]
- 52.Piehl C, Piontek J, Cording J, et al. Participation of the second extracellular loop of claudin-5 in paracellular tightening against ions, small and large molecules. Cell Mol Life Sci 2010; 67: 2131–2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suzuki H, Nishizawa T, Tani K, et al. Crystal structure of a claudin provides insight into the architecture of tight junctions. Science 2014; 344: 304–307 [DOI] [PubMed] [Google Scholar]
- 54.Suzuki H, Tani K, Tamura A, et al. Model for the architecture of claudin-based paracellular ion channels through tight junctions. J Mol Biol 2015; 427: 291–297 [DOI] [PubMed] [Google Scholar]
- 55.Van Itallie CM, Mitic LL, Anderson JM. Claudin-2 forms homodimers and is a component of a high molecular weight protein complex. J Biol Chem 2011; 286: 3442–3450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Günzel D, Amasheh S, Pfaffenbach S, et al. Claudin-16 affects transcellular Cl− secretion in MDCK cells. J Physiol 2009; 587: 3777–3793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Van Itallie CM, Colegio OR, Anderson JM. The cytoplasmic tails of claudins can influence tight junction barrier properties through effects on protein stability. J Membr Biol 2004; 199: 29–38 [DOI] [PubMed] [Google Scholar]
- 58.Ikari A, Matsumoto S, Harada H, et al. Dysfunction of paracellin-1 by dephosphorylation in Dahl salt-sensitive hypertensive rats. J Physiol Sci 2006; 56: 379–383 [DOI] [PubMed] [Google Scholar]
- 59.Ikari A, Hirai N, Shiroma M, et al. Association of paracellin-1 with ZO-1 augments the reabsorption of divalent cations in renal epithelial cells. J Biol Chem 2004; 279: 54826–54832 [DOI] [PubMed] [Google Scholar]
- 60.Itoh M, Furuse M, Morita K, et al. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J Cell Biol 1999; 147: 1351–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee NP, Tong MK, Leung PP, et al. Kidney claudin-19: localization in distal tubules and collecting ducts and dysregulation in polycystic renal disease. FEBS Lett 2006; 580: 923–931 [DOI] [PubMed] [Google Scholar]
- 62.Peng S, Adelman RA, Rizzolo LJ. Minimal effects of VEGF and anti-VEGF drugs on the permeability or selectivity of RPE tight junctions. Invest Ophthalmol Vis Sci 2010; 51: 3216–3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hou J, Renigunta A, Gomes AS, et al. Claudin-16 and claudin-19 interaction is required for their assembly into tight junctions and for renal reabsorption of magnesium. Proc Natl Acad Sci USA 2009; 106: 15350–15355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yu AS. Claudins and the kidney. J Am Soc Nephrol 2015; 26: 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miyamoto T, Morita K, Takemoto D, et al. Tight junctions in Schwann cells of peripheral myelinated axons: a lesson from claudin-19-deficient mice. J Cell Biol 2005; 169: 527–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hou J, Shan Q, Wang T, et al. Transgenic RNAi depletion of claudin-16 and the renal handling of magnesium. J Biol Chem 2007; 282: 17114–17122 [DOI] [PubMed] [Google Scholar]
- 67.Peng S, Rao VS, Adelman RA, et al. Claudin-19 and the barrier properties of the human retinal pigment epithelium. Invest Ophthalmol Vis Sci 2011; 52: 1392–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Riccardi D, Kemp PJ. The calcium-sensing receptor beyond extracellular calcium homeostasis: conception, development, adult physiology, and disease. Annu Rev Physiol 2012; 74: 271–297 [DOI] [PubMed] [Google Scholar]
- 69.Gong Y, Renigunta V, Himmerkus N, et al. Claudin-14 regulates renal Ca++ transport in response to CaSR signaling via a novel microRNA pathway. EMBO J 2012; 31: 1999–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thorleifsson G, Holm H, Edvardsson V, et al. Sequence variants in the CLDN14 gene associate with kidney stones and bone mineral density. Nat Genet 2009; 41: 926–930 [DOI] [PubMed] [Google Scholar]
- 71.Wilcox ER, Burton QL, Naz S, et al. Mutations in the gene encoding tight junction claudin-14 cause autosomal recessive deafness DFNB29. Cell 2001; 104: 165–172 [DOI] [PubMed] [Google Scholar]
- 72.Gong Y, Hou J. Claudin-14 underlies Ca++-sensing receptor-mediated Ca++ metabolism via NFAT-microRNA-based mechanisms. J Am Soc Nephrol 2014; 25: 745–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Toka HR, Al-Romaih K, Koshy JM, et al. Deficiency of the calcium-sensing receptor in the kidney causes parathyroid hormone-independent hypocalciuria. J Am Soc Nephrol 2012; 23: 1879–1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dimke H, Desai P, Borovac J, et al. Activation of the Ca(2+)-sensing receptor increases renal claudin-14 expression and urinary Ca(2+) excretion. Am J Physiol Renal Physiol 2013; 304: F761–F769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gong Y, Himmerkus N, Plain A, et al. Epigenetic regulation of microRNAs controlling CLDN14 expression as a mechanism for renal calcium handling. J Am Soc Nephrol 2015; 26: 663–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mulay SR, Kulkarni OP, Rupanagudi KV, et al. Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1β secretion. J Clin Invest 2013; 123: 236–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Knauf F, Asplin JR, Granja I, et al. NALP3-mediated inflammation is a principal cause of progressive renal failure in oxalate nephropathy. Kidney Int 2013; 84: 895–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chang A, Ko K, Clark MR. The emerging role of the inflammasome in kidney diseases. Curr Opin Nephrol Hypertens 2014; 23: 204–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee DB, Huang E, Ward HJ. Tight junction biology and kidney dysfunction. Am J Physiol Renal Physiol 2006; 290: F20–F34 [DOI] [PubMed] [Google Scholar]
- 80.Hirano T, Kobayashi N, Itoh T, et al. Null mutation of PCLN-1/claudin-16 results in bovine chronic interstitial nephritis. Genome Res 2000; 10: 659–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ohba Y, Kitagawa H, Kitoh K, et al. A deletion of the paracellin-1 gene is responsible for renal tubular dysplasia in cattle. Genomics 2000; 68: 229–236 [DOI] [PubMed] [Google Scholar]
- 82.Sugiyama A, Ozaki K, Miyazaki, et al. Renal dysplasia unrelated to claudin-16 deficiency in Japanese Black cattle. J Comp Pathol 2007; 137: 71–77 [DOI] [PubMed] [Google Scholar]
- 83.Sikora P, Zaniew M, Haisch L, et al. Retrospective cohort study of familial hypomagnesaemia with hypercalciuria and nephrocalcinosis due to CLDN16 mutations. Nephrol Dial Transplant 2015; 30: 636–644 [DOI] [PubMed] [Google Scholar]
- 84.Claverie-Martín F, Vargas-Poussou R, Müller D, et al. Clinical utility gene card for: familial hypomagnesemia with hypercalciuria and nephrocalcinosis with/without severe ocular involvement. Eur J Hum Genet 2015; 23: doi:10.1038/ejhg.2014.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Martin-Nuñez E, Cordoba-Lanus E, Gonzalez-Acosta H, et al. Haplotype analysis of CLDN19 single nucleotide polymorphisms in Spanish patients with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. World J Pediatr 2015; 11: 272–275 [DOI] [PubMed] [Google Scholar]
- 86.Shroff R, Aitkenhead H, Costa N, et al. Normal 25-hydroxyvitamin D Levels are associated with less proteinuria and attenuate renal failure progression in children with CKD. J Am Soc Nephrol 2015; doi:10.1681/ASN.2014090947 [DOI] [PMC free article] [PubMed] [Google Scholar]