

Abstract
Human amyloid deposits always contain the normal plasma protein serum amyloid P component (SAP), owing to its avid but reversible binding to all amyloid fibrils, including the amyloid β (Aβ) fibrils in the cerebral parenchyma plaques and cerebrovascular amyloid deposits of Alzheimer's disease (AD) and cerebral amyloid angiopathy (CAA). SAP promotes amyloid fibril formation in vitro, contributes to persistence of amyloid in vivo and is also itself directly toxic to cerebral neurons. We therefore developed (R)-1-[6-[(R)-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl]pyrrolidine-2-carboxylic acid (CPHPC), a drug that removes SAP from the blood, and thereby also from the cerebrospinal fluid (CSF), in patients with AD. Here we report that, after introduction of transgenic human SAP expression in the TASTPM double transgenic mouse model of AD, all the amyloid deposits contained human SAP. Depletion of circulating human SAP by CPHPC administration in these mice removed all detectable human SAP from both the intracerebral and cerebrovascular amyloid. The demonstration that removal of SAP from the blood and CSF also removes it from these amyloid deposits crucially validates the strategy of the forthcoming ‘Depletion of serum amyloid P component in Alzheimer's disease (DESPIAD)’ clinical trial of CPHPC. The results also strongly support clinical testing of CPHPC in patients with CAA.
Keywords: Alzheimer's disease, Aβ amyloid, serum amyloid P component, cerebral amyloid angiopathy, CPHPC
1. Background
The amyloid fibrils which are deposited as the pathognomonic intracerebral amyloid plaques and cerebrovascular amyloid deposits in Alzheimer's disease (AD) are composed of the Aβ peptide [1]. Aβ is derived by sequential proteolytic cleavage of the amyloid precursor protein (APP) first by β-secretase [2], and subsequently by the γ-secretase complex, of which the presenilin proteins (PSEN 1, PSEN 2) are the catalytic subunits [3]. Autosomal dominant hereditary AD is caused by mutations in, or duplication of, the APP gene and by mutations in the PSEN 1, PSEN 2 genes [4]. The APP–Aβ pathway thus must have a causal role in neurodegeneration but neither the actual pathogenic moiety nor the mechanisms underlying neuronal damage in AD are known and all attempts so far at therapeutic targeting of this pathway have failed through lack of efficacy and/or drug toxicity [5]. In contrast, the Aβ amyloid in sporadic cerebral amyloid angiopathy (CAA) clearly causes the structural disruption which results in vascular dysfunction and cerebral haemorrhage, just as the amyloid deposits in the tissues cause disease in systemic amyloidosis [6]. Indeed, CAA is by the far the most common and serious form of local amyloidosis.
Serum amyloid P component (SAP) has long been identified by our laboratory as a therapeutic target in all forms of amyloid deposition [7–9]. SAP is an invariant, normal, highly proteinase resistant, plasma glycoprotein which is always present in all human amyloid deposits [10], including the Aβ amyloid of AD and CAA [11–13]. SAP binds avidly but reversibly to all amyloid fibrils, including those which are the direct cause of disease in systemic amyloidosis [6] and in CAA [14], and those in AD where their role in pathogenesis, if any, is not known. Binding of SAP promotes amyloid fibrillogenesis in vitro [15,16] and stabilizes the fibrils, protecting them from proteolytic cleavage and acting as an anti-opsonin to prevent their destruction by phagocytic cells [8,17]. SAP thus is likely to contribute significantly to the unique persistence of amyloid deposits in vivo [18] that contrasts with the generally very efficient clearance of autologous debris from the extracellular space.
In addition, unrelated to its role in amyloid deposits, human SAP is directly neurotoxic to cerebral neurons [19–21]. It binds to them, is internalized, traffics to the nucleus [22] where it binds to chromatin [23], and leads to apoptotic cell death. SAP is synthesized and catabolized only by the liver [24] and human SAP is not expressed in the brain [25], which it enters only from the blood. It is normally present in the cerebrospinal fluid (CSF) at only about one-thousandth of the plasma concentration [26,27]. The binding of SAP to intracerebral Aβ amyloid deposits and plaques must increase the brain content of SAP [28]. The reversible non-covalent interaction with amyloid fibrils [10] must then also provide an increased local concentration of free SAP adjacent to amyloid deposits. Furthermore, other known risk factors for dementia, including advanced age, cerebral haemorrhage, traumatic brain injury (TBI) and severe or repeated non-penetrating head injury, all, certainly or probably, increase exposure of the brain to SAP, which could then contribute to neurodegeneration. In old age, the increase is simply the prolonged duration of exposure to normal CSF SAP concentrations but the other conditions, with compromised cerebral vascular integrity, enable plasma, with its 1000-fold higher SAP concentration, to enter the cerebral substance.
Our drug, (R)-1-[6-[(R)-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl]pyrrolidine-2-carboxylic acid (CPHPC), was developed to abrogate the pathogenic contribution of human SAP to amyloid formation and persistence [9]. CPHPC is specifically bound by human SAP in the circulation to form stable complexes of pairs of native pentameric SAP molecules cross-linked by the drug [9]. These complexes are immediately cleared by the liver, leading safely and effectively to almost complete depletion of plasma SAP for as long as the drug is administered [9,29]. We have shown that depletion of plasma SAP by administration of CPHPC to patients with AD leads to complete disappearance of SAP from the CSF [30]. CPHPC also enters the CSF in AD patients [30] and is thus available to inhibit binding of SAP to amyloid fibrils. Hence the rationale for University College London's forthcoming ‘Depletion of serum amyloid P component in Alzheimer's disease (DESPIAD)’ clinical trial of CPHPC in AD is: (i) that removal of all SAP from the brain by CPHPC will abrogate the direct neurotoxicity of human SAP; and (ii) that stripping of SAP from the cerebral plaques and cerebrovascular Aβ amyloid deposits will promote amyloid clearance. The same rationale underpins further planned clinical studies of CPHPC in CAA and TBI.
We already know that the first goal of removing all SAP from the CSF will be achieved [30], but it is obviously not possible to directly demonstrate removal of SAP from amyloid deposits in living human subjects. Indeed, although CPHPC treatment very substantially reduces the amount of SAP in systemic amyloid deposits, the affinity of binding of SAP to CPHPC is not sufficient for the drug to strip all SAP from its avid multivalent binding to the very abundant amyloid deposits that are present in these patients [29]. In contrast, cerebral and cerebrovascular amyloid deposits are several orders of magnitude less abundant than the deposits in systemic amyloidosis. Also the intracerebral plaques are exposed to a 1000-fold lower SAP concentration than is present in the plasma. On purely thermodynamic grounds, therefore, CPHPC should completely remove SAP from cerebral Aβ amyloid plaques and CAA deposits in patients.
Mouse SAP binds about 30-fold more weakly than human SAP to all known ligands so that, although mouse SAP is present in murine systemic amyloid deposits, it is not found in cerebral Aβ amyloid in mouse models of human AD [31], and circulating mouse SAP is not depleted by CPHPC in vivo [9]. In order to investigate the capacity of CPHPC to remove bound SAP from amyloid deposits in the brain, we therefore introduced transgenic expression of human SAP into the TASTPM double transgenic mouse model of familial AD [32]. The TAS and TPM transgenes encode pathogenic variants of human APP and human presenilin-1, respectively, and the mice develop abundant intracerebral and cerebrovascular human Aβ amyloid deposits. Here we report that the triple transgenic mice showed the expected presence of human SAP in all their amyloid deposits and that it was completely removed by sustained depletion of circulating human SAP produced by long-term administration of CPHPC.
2. Material and methods
2.1. Human CAA tissue
Formalin fixed wax embedded blocks of cerebral cortex tissues from nine patients with histologically confirmed sporadic CAA were provided by the Queen Square Brain Bank for Neurological Disorders, UCL.
2.2. Transgenic mice
TASTPM transgenic mice [32], homozygous for both the TAS and TPM transgenes, were provided by GlaxoSmithKline. The human SAP gene was amplified by PCR using Phusion High-Fidelity DNA Polymerase (NEB) and cloned in a plasmid vector as a 2.6 kb HindIII fragment, comprising the 1 kb SAP gene with 0.6 and 1 kb of 5′ and 3′ flanking sequence, respectively. After verifying the exon sequences, the 2.6 kb HindIII insert was released from the vector, gel purified and microinjected into pronuclei of C57BL/6J mouse embryos to generate transgenic mice expressing human SAP on the inbred C57BL/6J background. Lines were established and maintained hemizygous by backcrossing to wild-type C57BL/6J partners. TASTPM, human SAP triple transgenic mice were generated by crossing TASTPM homozygous mice with human SAP transgenic mice. The progeny were thus either hemizygous for all three transgenes or were hemizygous for the TAS and TPM transgenes. Genotypes were assessed by PCR. Data shown are representative of findings with both males and females.
2.3. Immunoassay for SAP
SAP protein concentrations were assayed by electroimmunoassay or immunoradiometric assay, as described previously [27,33].
2.4. Immunohistochemical staining for Aβ and SAP
Immunoperoxidase histochemistry was performed on a Ventana Discovery XT staining platform using the Ventana DAB Map Kit. Wax sections of formalin fixed tissue were pretreated with formic acid (for Aβ staining) or with Ventana Protease 3 (for SAP staining) and blocked for 8 min using Superblock (Medite). A mouse monoclonal primary antibody was used for detection of Aβ (Dako, clone 6F-3D), and an in-house monospecific polyclonal rabbit antiserum was used to detect human SAP, and counterstained with haematoxylin. Immunofluorescence histochemistry was performed on 14 µm frozen sections of unfixed frozen mouse brains. Sections were dried at room temperature and stored at −20°C before use. Non-specific binding was blocked with 2% w/v BSA, 0.1% Triton X-100 in PBS, and SAP was then detected with the monospecific rabbit anti-human SAP antibody followed by Alexa-488 labelled anti-rabbit IgG antibody, followed by counterstaining with bisbenzimide.
2.5. Histochemical staining for amyloid
Amyloid deposits in histological sections were stained with Congo red [34] and identified by their pathognomonic green birefringence in high intensity cross polarized light.
3. Results
3.1. SAP in human cerebral and cerebrovascular Aβ amyloid deposits
The universal presence of human SAP in Aβ amyloid has been extensively confirmed since our original 1984 observations on sporadic and hereditary CAA [11]. However, in order to validate our methods for subsequent application to studies in human SAP transgenic mice, we examined brain sections from nine confirmed sporadic CAA patients using Congo red staining for amyloid and immunohistochemical staining of adjacent sections for SAP and Aβ. In each human case, SAP and Aβ staining were observed in congruent patterns in blood vessel walls (figure 1), and in eight of the nine cases, the SAP staining was strong. In seven of the cases, intracerebral Aβ amyloid plaques were also present. They stained with anti-SAP antibodies but much less intensely than the vascular amyloid in the same sections (figure 1e,f). Diffuse immunostaining for SAP was also seen in perivascular areas (figure 1g–j), presumably reflecting recent vascular leakage.
Figure 1.

Localization of SAP in human CAA. (a,b) Congo red stained section viewed with bright field illumination (a) and with intense crossed polarized light (b), showing abundant amyloid in blood vessel walls. The arrowhead in (b) shows the white birefringence of collagen often seen in normal blood vessels, in contrast to the green birefringence of Congo red stained amyloid. (c,d) Immunoperoxidase staining for Aβ (c) and SAP (d), showing strong immunoreactivity in vessel walls. In one vessel, the amyloid is mostly confined to one segment of the vessel wall (arrow in a–d). (e,f) Lower magnification images of the sections shown in (c,d), showing amyloid plaques which stain strongly for Aβ and very weakly for SAP. Higher magnification images of the arrowed plaque are inset. (g–j) Sections of an amyloid-containing blood vessel surrounded by blood. (g,h) Congo red stained section viewed with bright field illumination (g) and with intense cross polarized light (h). (i,j) Immunostaining for Aβ and SAP, respectively. The arrow in (j) shows diffuse staining in adjacent neural tissue, not associated with amyloid, likely due to a microbleed. Scale bars, (a–d), 250 µm; (e,f), 500 µm; (g–j), 250 µm.
3.2. Aβ amyloid deposition in TASTPM mice
Almost all patients with AD have CAA, although usually of lesser severity than in patients with typical sporadic CAA and predominantly affecting capillaries rather than arterioles. Similar cerebrovascular amyloid deposition is seen in mouse models of AD [31,35]. The double transgenic TASTPM mice have been reported to develop immunologically detectable human Aβ deposits from three months of age and amyloid plaques by six months [32]. Here we also observed extensive vascular amyloid deposition at six months of age in TASTPM mouse brains, with pathognomonic green birefringence in polarized light of both plaques and cerebral blood vessels stained with Congo red (figure 2).
Figure 2.
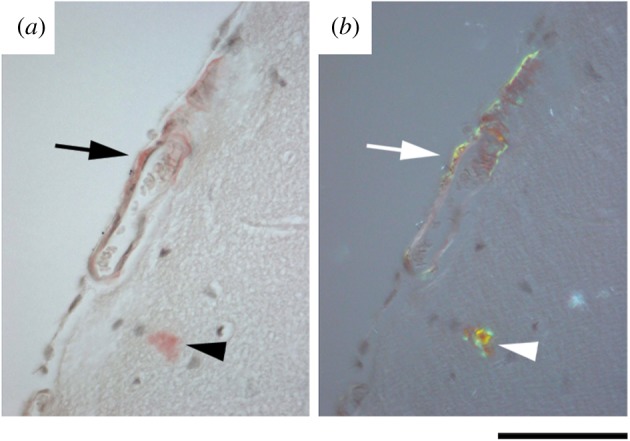
Cerebrovascular amyloid in TASTPM mice. Unfixed frozen brain sections from six-month-old female TASTPM mouse stained with Congo red showing cerebrovascular amyloid (arrow) and cerebral plaque (arrowhead). (a) Bright field illumination; (b) intense cross polarized light showing pathognomonic green birefringence of Congo red bound to amyloid. Scale bar, 150 µm.
3.3. Human SAP transgenic mice and CPHPC administration
Several human SAP transgenic lines were established, two of which were expanded for further study. The serum concentration of human SAP in line 9 transgenic mice was mean (s.d.) 29.2 (4.0) mg l−1 (n = 6), closely corresponding to the human reference range [27]. In contrast, the human SAP concentration in sera of line 38 transgenic mice was mean (s.d.) 216.9 (154.9) mg l−1 (n = 31) (figure 3a), with no statistically significant difference between males and females. Following administration to line 38 mice of CPHPC at 5 mg ml−1 in their drinking water, the serum concentration of human SAP rapidly fell to 1.33 (0.64) mg l−1 (n = 32) (figure 3a) and remained reduced by more than 97% for as long as drug exposure continued.
Figure 3.
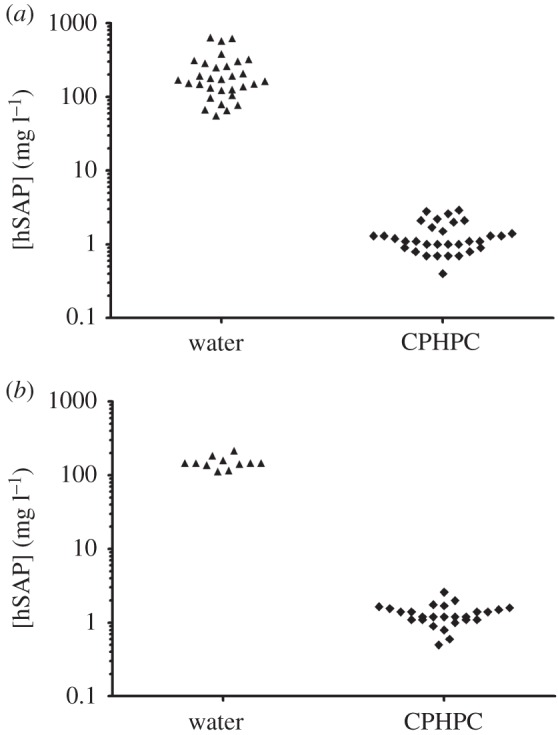
Human SAP expression in transgenic mice, and depletion by CPHPC. Human SAP concentrations in sera of individual transgenic mice, while on water alone or water containing 5 mg ml−1 CPHPC. (a) Line 38 human SAP transgenic C57BL/6 mice; (b) line 38 human SAP transgenic TASTPM mice. Results are from males and females with no apparent differences between sexes.
3.4. Human SAP transgenic TASTPM mice
After introduction of the human SAP transgene into TASTPM mice by crossing them with line 38, the circulating concentration of human SAP was in the same range as in the transgenic mice on the wild-type background (figure 3b). When examined at 20 months of age, both the vascular and plaque Aβ amyloid deposits were positive for human SAP by immunoperoxidase staining of sections of formalin fixed wax embedded brain (figure 4a–c), while control sections from age matched TASTPM mice without the human SAP transgene were entirely negative (figure 4f–h). This was confirmed by immunofluorescence staining of frozen sections of unfixed tissue from human SAP transgenic TASTPM mice (figure 4d,e) and control TASTPM animals (figure 4i,j).
Figure 4.

Depletion of human SAP from cerebrovascular amyloid by CPHPC treatment of transgenic mice. Sections of formalin fixed wax embedded cerebral cortex from 20-month-old mice stained with Congo red and viewed in bright field (a,f,k) or intense cross polarized illumination (b,g,l), and also immunoperoxidase (IP) stained with anti-human SAP antibodies (c,h,m). Unfixed frozen sections from the same tissues stained by immunofluorescence (IF) with anti-human SAP antibodies (green) and bisbenzimide counterstain (blue) (d,i,n) and also viewed in phase contrast (e,j,o). (a–e) Human SAP transgenic TASTPM mouse, (f–j) TASTPM control mouse, (k–o) CPHPC-treated human SAP transgenic TASTPM mouse. Amyloid-containing blood vessels are labelled with arrows, and example amyloid plaques are indicated by arrowheads. Tissues in (a–c), (f–h), (k–m) are from males and (d,e,i,j,n,o) from female mice.
3.5. Effects of CPHPC administration and SAP depletion
Administration to human SAP transgenic TASTPM mice of 5 mg ml−1 of CPHPC in drinking water, from age four or six months until age 20 months, had no discernible adverse effects. The body weights of CPHPC-treated mice were indistinguishable from those of controls (data not shown). The circulating human SAP concentration in CPHPC-treated animals fell to between 0.1 and 2.9 mg l−1 in samples taken during treatment and at termination (figure 3b). Furthermore, no immunoperoxidase staining for human SAP was observed in the amyloid deposits after CPHPC treatment (figure 4k–m), in marked contrast to the positive staining in untreated human SAP transgenic TASTPM mice (figure 4a–c). The immunoreactivity of human SAP is diminished by standard formalin fixation and wax embedding [36], and antigen retrieval techniques are required for its optimal detection by immunoperoxidase staining. The presence of human SAP was therefore also sought by immunofluorescence staining of cryosections of unfixed tissue. The results confirmed the absence of appreciable staining for human SAP (figure 4n,o) with appearances indistinguishable from those of tissues from control TASTPM mice without the human SAP transgene (figure 4i,j).
3.6. Human SAP content of intracerebral plaques and cerebrovascular amyloid deposits
In brain sections from human SAP transgenic TASTPM mice, anti-SAP antibodies consistently stained the cerebrovascular amyloid deposits much more intensely than the parenchymal amyloid plaques (figure 5), in both immunoperoxidase staining of formalin fixed wax embedded tissue (figure 5a,b) and immunofluorescence staining of cryosections of unfixed tissue (figure 5e,f). The SAP content of cerebrovascular amyloid is thus considerably greater than that of plaque amyloid, consistent with in vivo exposure of the plaques only to the SAP concentration of CSF fluid which, in humans and presumably also in mice, is about 1000-fold lower than the plasma and extracerebral interstitial fluid concentration. Using up to 10-fold longer exposures, even trace immunofluorescent staining with anti-SAP antibody could be sensitively detected (figure 5e,f) and the abundance of SAP in the deposits semi-quantitatively assessed. This confirmed that after CPHPC treatment both the cerebrovascular deposits and the cerebral plaques had been stripped of SAP (figure 5g,h), with staining intensity not different from the background autofluorescence of plaques in TASTPM controls with no human SAP transgene (figure 5c,d).
Figure 5.

Depletion of human SAP from cerebral plaques by CPHPC treatment of transgenic mice. (a,b) Sections of formalin fixed wax embedded cerebral cortex from 20-month-old human SAP transgenic male TASTPM mice, immunoperoxidase stained with anti-human SAP antibodies, showing intense staining of vascular amyloid (arrows) and very weak staining of intracerebral plaques (arrowheads). (c–h) Amyloid plaques (outlined) in unfixed frozen sections of cerebral cortex from 20-month-old female mice stained with fluorescent labelled anti-human SAP antibodies (green). (c,d) TASTPM control; (e,f) human SAP transgenic TASTPM; (g,h) CPHPC-treated human SAP transgenic TASTPM. The green channel exposures used for panels (c–h) were 10 times longer than for figure 4d,i,n. At the exposure that demonstrated SAP in the cerebrovascular amyloid in figure 4, there was no signal from the intracerebral plaques. Scale bars, (a,b), 100 µm; (c–h), 150 µm.
4. Discussion
We have previously identified human SAP as a therapeutic target in both systemic amyloidosis and in neurodegenerative conditions associated with cerebral and cerebrovascular Aβ amyloid deposition [7–9]. We have also shown that administration of the SAP depleting drug, CPHPC, produces sustained depletion of plasma SAP for as long as the drug is administered [29] and that depletion of circulating SAP leads to complete disappearance of SAP from the CSF in patients with AD [30]. Since SAP is not expressed in the human brain [25], the SAP depletion effected by CPHPC should abrogate the direct neurotoxicity of this plasma protein. However, for depletion of free soluble SAP to promote the desired enhanced clearance of amyloid deposits, it should ideally lead to essentially complete removal of SAP from its binding to amyloid fibrils. This does not happen in systemic amyloidosis because, despite the ability to safely administer high doses of this non-toxic, non-metabolized drug, the CPHPC concentrations which are attainable in vivo are not sufficient to reverse the avid multivalent binding of the pentameric SAP molecule to the very abundant amyloid fibrils. Even after months of daily dosing with CPHPC, some SAP, albeit a much reduced amount, remains associated with visceral amyloid deposits [29]. Although no regression of amyloid then ensues, observations in CPHPC-treated systemic amyloidosis patients suggest that the treatment nonetheless may reduce new amyloid accumulation and stabilize the function of amyloidotic organs [29].
The situation regarding cerebral and cerebrovascular Aβ amyloid is radically different. The total mass of Aβ amyloid is extremely small in comparison with systemic amyloid deposits. We therefore expected that plasma SAP depletion by CPHPC would successfully remove all SAP bound to cerebral parenchymal and vascular Aβ amyloid deposits, and this has been confirmed by the present observations. Furthermore, parenchymal plaques are exposed to about 1000-fold lower ambient SAP concentrations in the CSF [26,30] than the cerebrovascular deposits bathed in plasma-derived extracellular fluid. The relative quantities of bound SAP differ accordingly, as we have confirmed here immunohistochemically, and this explains why, over 30 years ago, we [11] and others [37] did not initially detect SAP on the cerebral parenchymal plaques using much less sensitive immunostaining procedures.
It is important to note that the present experiments were not designed to show an effect of CPHPC treatment and human SAP depletion on Aβ amyloid load. TASTPM mice strongly overexpress two different familial AD genes under the mouse Thy-1 gene promoter [32] and develop extensive cerebral amyloid plaques and CAA in the absence of human SAP expression. It is therefore unlikely that their Aβ amyloid deposition will be detectably modulated by the presence or depletion of human SAP.
The pathogenic role, if any, of the cerebral parenchymal Aβ amyloid plaques in AD and that appear soon after TBI [38] is unknown, but an intervention which leads to clearance and/or prevention of the deposits would be very informative. This is one of the aims of our existing DESPIAD clinical trial of CPHPC in patients with AD, scheduled to start in 2016. In contrast to AD, the Aβ amyloid deposits in the walls of cerebral blood vessels in CAA are unequivocally the cause of vascular dysfunction and cerebral haemorrhage in this very important and prevalent disease [14]. Progressive deposition of amyloid disrupts blood vessel structure with loss of smooth muscle cells, thickened vessel walls and eventually detachment and delamination of the outer part of the tunica media. CAA is present at autopsy in 20–40% of non-demented and 50–60% of demented elderly populations, and in at least 90% of individuals with Alzheimer's disease (AD) [14]. CAA causes microbleeds and lobar intracerebral haemorrhages, comprising up to 20% of haemorrhagic strokes [14]. CAA is also associated with ischaemic brain injury, including small areas of infarction [39] and white matter abnormalities [40], and it impairs normal vascular function [41,42]. The very high, approximately 10%, annual rate of recurrent intracerebral haemorrhage in CAA increases the long-term risk of dementia [43]. The spectrum of vascular brain injury and dysfunction in CAA is also likely to cause cognitive impairment, independently of cerebral haemorrhage [44,45]. CAA is thus a particularly attractive indication both for an intervention designed to promote clearance of amyloid and for abrogation of the direct neurotoxicity of blood-derived SAP entering the brain.
Acknowledgements
TASTPM mice were provided by GlaxoSmithKline. We thank Beth Jones for formatting the manuscript.
Ethics
Animal studies were ethically reviewed and approved by the UCL Royal Free Campus Ethics and Welfare Committee and the UK Home Office, and complied fully with European Directive 86/609/EEC. Use of human brain tissues from the Queen Square Brain Bank for Neurological Disorders, UCL, was approved by the Research Ethics Committee of the National Hospital for Neurology and Neurosurgery.
Authors' contributions
The study was initiated and directed by M.B.P. The experiments were designed by J.P.S., R.A.-S., S.B. and M.B.P. and were conducted by J.P.S., R.A.-S., G.A.T., D.J.M. and A.R.-L. The paper was written by J.P.S. and M.B.P. with review and contributions from all authors.
Competing interests
M.B.P. is the sole inventor on the patents covering SAP depletion by CPHPC. These are owned by Pentraxin Therapeutics Ltd, a UCL spinout company that he founded and in which he owns shares; the company also owns other patents on CPHPC. The other authors declare no competing financial interests.
Funding
The Wolfson Drug Discovery Unit receives core support from the UK National Institute for Health Research Biomedical Research Centre and Unit Funding Scheme. Support from the Wolfson Foundation and the UCL Amyloidosis Research Fund is also gratefully acknowledged.
References
- 1.Glenner GG, Wong CW. 1984. Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 122, 1131–1135. (doi:10.1016/0006-291X(84)91209-9) [DOI] [PubMed] [Google Scholar]
- 2.De Strooper B, König G. 1999. Alzheimer's disease. A firm base for drug development. Nature 402, 471–472. (doi:10.1038/44973) [DOI] [PubMed] [Google Scholar]
- 3.Brunkan AL, Goate AM. 2005. Presenilin function and γ-secretase activity. J. Neurochem. 93, 769–792. (doi:10.1111/j.1471-4159.2005.03099.x) [DOI] [PubMed] [Google Scholar]
- 4.Bohm C, Chen F, Sevalle J, Qamar S, Dodd R, Li Y, Schmitt-Ulms G, Fraser PE, St George-Hyslop PH. 2015. Current and future implications of basic and translational research on amyloid-β peptide production and removal pathways. Mol. Cell. Neurosci. 66, 3–11. (doi:10.1016/j.mcn.2015.02.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schneider LS, et al. 2014. Clinical trials and late-stage drug development for Alzheimer's disease: an appraisal from 1984 to 2014. J. Intern. Med. 275, 251–283. (doi:10.1111/joim.12191) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pepys MB. 2006. Amyloidosis. Annu. Rev. Med. 57, 223–241. (doi:10.1146/annurev.med.57.121304.131243) [DOI] [PubMed] [Google Scholar]
- 7.Hind CRK, Collins PM, Caspi D, Baltz ML, Pepys MB. 1984. Specific chemical dissociation of fibrillar and non-fibrillar components of amyloid deposits. Lancet ii, 376–378. (doi:10.1016/S0140-6736(84)90544-0) [DOI] [PubMed] [Google Scholar]
- 8.Tennent GA, Lovat LB, Pepys MB. 1995. Serum amyloid P component prevents proteolysis of the amyloid fibrils of Alzheimer's disease and systemic amyloidosis. Proc. Natl Acad. Sci. USA 92, 4299–4303. (doi:10.1073/pnas.92.10.4299) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pepys MB, et al. 2002. Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nature 417, 254–259. (doi:10.1038/417254a) [DOI] [PubMed] [Google Scholar]
- 10.Pepys MB, Dyck RF, de Beer FC, Skinner M, Cohen AS. 1979. Binding of serum amyloid P component (SAP) by amyloid fibrils. Clin. Exp. Immunol. 38, 284–293. [PMC free article] [PubMed] [Google Scholar]
- 11.Rowe IF, Jensson O, Lewis PD, Candy J, Tennent GA, Pepys MB. 1984. Immunohistochemical demonstration of amyloid P component in cerebro-vascular amyloidosis. Neuropathol. Appl. Neurobiol. 10, 53–61. (doi:10.1111/j.1365-2990.1984.tb00340.x) [DOI] [PubMed] [Google Scholar]
- 12.Coria F, Castano E, Prelli F, Larrondo-Lillo M, van Duinen S, Shelanski ML, Frangione B. 1988. Isolation and characterization of amyloid P component from Alzheimer's disease and other types of cerebral amyloidosis. Lab. Invest. 58, 454–458. [PubMed] [Google Scholar]
- 13.Duong T, Pommier EC, Scheibel AB. 1989. Immunodetection of the amyloid P component in Alzheimer's disease. Acta Neuropathol. 78, 429–437. (doi:10.1007/BF00688180) [DOI] [PubMed] [Google Scholar]
- 14.Charidimou A, Gang Q, Werring DJ. 2012. Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. J. Neurol. Neurosurg. Psychiatry 83, 124–137. (doi:10.1136/jnnp-2011-301308) [DOI] [PubMed] [Google Scholar]
- 15.Hamazaki H. 1995. Amyloid P component promotes aggregation of Alzheimer's β-amyloid peptide. Biochem. Biophys. Res. Commun. 211, 349–353. (doi:10.1006/bbrc.1995.1819) [DOI] [PubMed] [Google Scholar]
- 16.Mold M, Shrive AK, Exley C. 2012. Serum amyloid P component accelerates the formation and enhances the stability of amyloid fibrils in a physiologically significant under-saturated solution of amyloid-β42. J. Alzheimers Dis. 29, 875–881. (doi:10.3233/jad-2012-120076) [DOI] [PubMed] [Google Scholar]
- 17.Noursadeghi M, Bickerstaff MCM, Gallimore JR, Herbert J, Cohen J, Pepys MB. 2000. Role of serum amyloid P component in bacterial infection: protection of the host or protection of the pathogen. Proc. Natl Acad. Sci. USA 97, 14 584–14 589. (doi:10.1073/pnas.97.26.14584) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Botto M et al. 1997. Amyloid deposition is delayed in mice with targeted deletion of the serum amyloid P component gene. Nat. Med. 3, 855–859. (doi:10.1038/nm0897-855) [DOI] [PubMed] [Google Scholar]
- 19.Urbányi Z, Lakics V, Erdó SL. 1994. Serum amyloid P component-induced cell death in primary cultures of rat cerebral cortex. Eur. J. Pharmacol. 270, 375–388. (doi:10.1016/0926-6917(94)90016-7) [DOI] [PubMed] [Google Scholar]
- 20.Duong T, Acton PJ, Johnson RA. 1998. The in vitro neuronal toxicity of pentraxins associated with Alzheimer's disease brain lesions. Brain Res. 813, 303–312. (doi:10.1016/S0006-8993(98)00966-4) [DOI] [PubMed] [Google Scholar]
- 21.Pisalyaput K, Tenner AJ. 2008. Complement component C1q inhibits β-amyloid- and serum amyloid P-induced neurotoxicity via caspase- and calpain-independent mechanisms. J. Neurochem. 104, 696–707. (doi:10.1111/j.1471-4159.2007.05012.x) [DOI] [PubMed] [Google Scholar]
- 22.Du Clos TW, Mold C, Stump RF. 1990. Identification of a polypeptide sequence that mediates nuclear localization of the acute phase protein C-reactive protein. J. Immunol. 145, 3869–3875. [PubMed] [Google Scholar]
- 23.Pepys MB, Booth SE, Tennent GA, Butler PJG, Williams DG. 1994. Binding of pentraxins to different nuclear structures: C-reactive protein binds to small nuclear ribonucleoprotein particles, serum amyloid P component binds to chromatin and nucleoli. Clin. Exp. Immunol. 97, 152–157. (doi:10.1111/j.1365-2249.1994.tb06594.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pepys MB, Booth DR, Hutchinson WL, Gallimore JR, Collins PM, Hohenester E. 1997. Amyloid P component. A critical review. Amyloid: Int. J. Exp. Clin. Invest. 4, 274–295. (doi:10.3109/13506129709003838) [Google Scholar]
- 25.Hawrylycz MJ, et al. 2012. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489, 391–399. (doi:10.1038/nature11405) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hawkins PN, Rossor MN, Gallimore JR, Miller B, Moore EG, Pepys MB. 1994. Concentration of serum amyloid P component in the CSF as a possible marker of cerebral amyloid deposits in Alzheimer's disease. Biochem. Biophys. Res. Commun. 201, 722–726. (doi:10.1006/bbrc.1994.1760) [DOI] [PubMed] [Google Scholar]
- 27.Nelson SR, Tennent GA, Sethi D, Gower PE, Ballardie FW, Amatayakul-Chantler S, Pepys MB. 1991. Serum amyloid P component in chronic renal failure and dialysis. Clin. Chim. Acta 200, 191–200. (doi:10.1016/0009-8981(91)90090-Y) [DOI] [PubMed] [Google Scholar]
- 28.Crawford JR, Bjorklund NL, Taglialatela G, Gomer RH. 2012. Brain serum amyloid P levels are reduced in individuals that lack dementia while having Alzheimer's disease neuropathology. Neurochem. Res. 37, 795–801. (doi:10.1007/s11064-011-0674-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gillmore JD, et al. 2010. Sustained pharmacological depletion of serum amyloid P component in patients with systemic amyloidosis. Br. J. Haematol. 148, 760–767. (doi:10.1111/j.1365-2141.2009.08036.x) [DOI] [PubMed] [Google Scholar]
- 30.Kolstoe SE, et al. 2009. Molecular dissection of Alzheimer's disease neuropathology by depletion of serum amyloid P component. Proc. Natl Acad. Sci. USA 106, 7619–7623. (doi:10.1073/pnas.0902640106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winkler DT, Bondolfi L, Herzig MC, Jann L, Calhoun ME, Wiederhold K-H, Tolnay M, Staufenbiel M, Jucker M. 2001. Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. J. Neurosci. 21, 1619–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Howlett DR, Richardson JC, Austin A, Parsons AA, Bate ST, Davies DC, Gonzalez MI. 2004. Cognitive correlates of Aβ deposition in male and female mice bearing amyloid precursor protein and presenilin-1 mutant transgenes. Brain Res. 1017, 130–136. (doi:10.1016/j.brainres.2004.05.029) [DOI] [PubMed] [Google Scholar]
- 33.Millar DJ, Hutchinson WL, Pepys MB. 2011. Immunoradiometric assay for human serum amyloid P component. J. Immunol. Methods 371, 18–24. (doi:10.1016/j.jim.2011.06.010) [DOI] [PubMed] [Google Scholar]
- 34.Puchtler H, Waldrop FS, Meloan SN. 1985. A review of light, polarization and fluorescence microscopic methods for amyloid. Appl. Pathol. 3, 5–17. [PubMed] [Google Scholar]
- 35.Domnitz SB, Robbins EM, Hoang AW, Garcia-Alloza M, Hyman BT, Rebeck GW, Greenberg SM, Bacskai BJ, Frosch MP. 2005. Progression of cerebral amyloid angiopathy in transgenic mouse models of Alzheimer disease. J. Neuropathol. Exp. Neurol. 64, 588–594. (doi:10.1097/01.jnen.0000171644.00180.fc) [DOI] [PubMed] [Google Scholar]
- 36.Breathnach SM, Melrose SM, Bhogal B, de Beer FC, Dyck RF, Tennent G, Black MM, Pepys MB. 1981. Amyloid P component is located on elastic fibre microfibrils of normal human tissues. Nature 293, 652–654. (doi:10.1038/293652a0) [DOI] [PubMed] [Google Scholar]
- 37.Westermark P, Shirahama T, Skinner M, Brun A, Cameron R, Cohen AS. 1982. Immunohistochemical evidence for the lack of amyloid P component in some intracerebral amyloids. Lab. Invest. 46, 457–460. [PubMed] [Google Scholar]
- 38.Hong YT, et al. 2014. Amyloid imaging with carbon 11-labeled Pittsburgh compound B for traumatic brain injury. JAMA Neurol. 71, 23–31. (doi:10.1001/jamaneurol.2013.4847) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gregoire SM, et al. 2011. Acute ischaemic brain lesions in intracerebral haemorrhage: multicentre cross-sectional magnetic resonance imaging study. Brain 134, 2376–2386. (doi:10.1093/brain/awr172) [DOI] [PubMed] [Google Scholar]
- 40.Gurol ME, et al. 2012. Predicting sites of new hemorrhage with amyloid imaging in cerebral amyloid angiopathy. Neurology 79, 320–326. (doi:10.1212/WNL.0b013e31826043a9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dumas A, et al. 2012. Functional magnetic resonance imaging detection of vascular reactivity in cerebral amyloid angiopathy. Ann. Neurol. 72, 76–81. (doi:10.1002/ana.23566) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peca S, et al. 2013. Neurovascular decoupling is associated with severity of cerebral amyloid angiopathy. Neurology 81, 1659–1665. (doi:10.1212/01.wnl.0000435291.49598.54) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pendlebury ST, Rothwell PM. 2009. Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: a systematic review and meta-analysis. Lancet Neurol. 8, 1006–1018. (doi:10.1016/s1474-4422(09)70236-4) [DOI] [PubMed] [Google Scholar]
- 44.Arvanitakis Z, Leurgans SE, Wang Z, Wilson RS, Bennett DA, Schneider JA. 2011. Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann. Neurol. 69, 320–327. (doi:10.1002/ana.22112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poels MMF, Ikram MA, van der Lugt A, Hofman A, Niessen WJ, Krestin GP, Breteler MMB, Vernooij MW. 2012. Cerebral microbleeds are associated with worse cognitive function: the Rotterdam Scan Study. Neurology 78, 326–333. (doi:10.1212/WNL.0b013e3182452928) [DOI] [PubMed] [Google Scholar]