

Abstract
Background
Whole-exome sequencing (WES) has led to an exponential increase in identification of causative variants in mitochondrial disorders (MD).
Methods
We performed WES in 113 MD suspected patients from Polish paediatric reference centre, in whom routine testing failed to identify a molecular defect. WES was performed using TruSeqExome enrichment, followed by variant prioritization, validation by Sanger sequencing, and segregation with the disease phenotype in the family.
Results
Likely causative mutations were identified in 67 (59.3 %) patients; these included variants in mtDNA (6 patients) and nDNA: X-linked (9 patients), autosomal dominant (5 patients), and autosomal recessive (47 patients, 11 homozygotes). Novel variants accounted for 50.5 % (50/99) of all detected changes. In 47 patients, changes in 31 MD-related genes (ACAD9, ADCK3, AIFM1, CLPB, COX10, DLD, EARS2, FBXL4, MTATP6, MTFMT, MTND1, MTND3, MTND5, NAXE, NDUFS6, NDUFS7, NDUFV1, OPA1, PARS2, PC, PDHA1, POLG, RARS2, RRM2B, SCO2, SERAC1, SLC19A3, SLC25A12, TAZ, TMEM126B, VARS2) were identified. The ACAD9, CLPB, FBXL4, PDHA1 genes recurred more than twice suggesting higher general/ethnic prevalence. In 19 cases, variants in 18 non-MD related genes (ADAR, CACNA1A, CDKL5, CLN3, CPS1, DMD, DYSF, GBE1, GFAP, HSD17B4, MECP2, MYBPC3, PEX5, PGAP2, PIGN, PRF1, SBDS, SCN2A) were found. The percentage of positive WES results rose gradually with increasing probability of MD according to the Mitochondrial Disease Criteria (MDC) scale (from 36 to 90 % for low and high probability, respectively). The percentage of detected MD-related genes compared with non MD-related genes also grew with the increasing MD likelihood (from 20 to 97 %). Molecular diagnosis was established in 30/47 (63.8 %) neonates and in 17/28 (60.7 %) patients with basal ganglia involvement. Mutations in CLPB, SERAC1, TAZ genes were identified in neonates with 3-methylglutaconic aciduria (3-MGA) as a discriminative feature. New MD-related candidate gene (NDUFB8) is under verification.
Conclusions
We suggest WES rather than targeted NGS as the method of choice in diagnostics of MD in children, including neonates with 3-MGA aciduria, who died without determination of disease cause and with limited availability of laboratory data. There is a strong correlation between the degree of MD diagnosis by WES and MD likelihood expressed by the MDC scale.
Electronic supplementary material
The online version of this article (doi:10.1186/s12967-016-0930-9) contains supplementary material, which is available to authorized users.
Keywords: Whole-exome sequencing, Mitochondrial disorders, Mitochondrial disease criteria scale, Neonates, Basal ganglia involvement, Leigh syndrome, 3-methylglutaconic aciduria, Novel mutation, Candidate gene
Background
The diagnostics of mitochondrial disorders (MD) remains a challenge due to clinical heterogeneity [1] and the constantly expanding amount of gene candidates [2] as well as new phenotypes of these conditions [3]. There are eight published studies evaluating diagnostic utility of next generation sequencing (NGS) in mitochondrial patient cohorts, selected either based on particular biochemical signatures of disease [4–8] or centre/cohort-based studies [9–11]. However, of these only four used whole exome sequencing (WES) [7–10].
A particular challenge is the diagnosis of MD in neonates below 3 months of age as these patients may account for up to 30 % of all MD cases [12, 13]. However, so far, this group has not been specifically focused on in terms of diagnostic effectiveness of WES. The prevailing majority (96.5 %) of cases with a molecular diagnosis of MD established at our national reference centre until 2013 included children older than 3 months, indicating considerable under-diagnosis rates in the youngest infants in the Polish population. We have achieved some improvement in neonatal MD detection by performing targeted DNA sequencing (frequently post mortem) in cases of neonates with lactic aciduria (LA-uria) found in selective GC–MS screening, including over 90 % of SCO2 [14] and DGUOK [15] deficiencies, and ~ 50 % of SURF1 deficiency [16].
The purpose of our study was to evaluate WES as a tool for diagnosis of MD depending on the disease probability assessed according to mitochondrial disease criteria (MDC) [17]. We considered both patients with full-range mitochondrial diagnostics (Leigh syndrome features in MRI and/or muscle biopsy evaluation) and those in whom only fragmentary clinical data e.g. abnormal result of GC–MS screening indicating the presence LA-uria and/or 3-methylglutaconic aciduria (3-MGA-uria) were available.
Methods
Patients
WES was performed in patients with probable or possible MD, in whom a molecular defect had not been identified within the analysed period. In the retrospective subgroup (88/113 patients) the lag time was 2–25 years (mean 7.5 +/5.9 years). Since 2013 WES has been considered in consecutive patients (25/113). To undergo WES, a patient had to fulfil at least one of the following criteria: 1/neonatal onset; 2/basal ganglia involvement (Leigh syndrome—LS, nonspecific basal ganglia involvement); 3/increased 3-MGA in urine (patients recruited from a group of >250 cases of 3-MGA aciduria identified by national selective GC–MS screening for metabolic disorders since 2000), and 4/genetic counselling demands. Access to biological material and informed consent of parents were sine qua non conditions for participation in the study. Details of criteria for patient selection and their clinical characteristics are shown in Table 1 and Additional file 1: Table S1.
Table 1.
Characteristics of 113 MD suspected patients; inclusion criteria
| ID patient | Sex | Date of birth (year) | Neonatal onset | 3-MGA in urine | Basal ganglia involvement | Death | MDC score | Muscle biopsy | Period from onset to WES (year) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 2009 | + | + | 5 | 5 | |||
| 2 | F | 2013 | + | + | 4 | Autopsy | 0 | ||
| 3 | M | 2012 | + | 5 | + | 2 | |||
| 4 | F | 2007 | 4 | + | 0 | ||||
| 5 | F | 2013 | + | + | + | 5 | 0 | ||
| 6 | F | 2011 | 4 | 0 | |||||
| 7 | M | 2006 | + | 5 | + | 7 | |||
| 8 | M | 2008 | + | 2 | 0 | ||||
| 9 | M | 2011 | + | 6 | + | 2 | |||
| 10 | M | 2004 | + | 5 | + | 7 | |||
| 11 | M | 2005 | 2 | 2 | |||||
| 12 | M | 2005 | + | 3 | Autopsy | 7 | |||
| 13 | M | 2014 | + | + | + | 4 | Autopsy | 0 | |
| 14 | F | 2006 | + | 3 | + Autopsy | 7 | |||
| 15 | F | 2008 | + | + | 4 | + | 5 | ||
| 16 | M | 2012 | + | 3 | 0 | ||||
| 17 | F | 1992 | + | 3 | + | 21 | |||
| 18 | F | 2003 | + | 3 | + | 7 | |||
| 19 | M | 2009 | 5 | + | 3 | ||||
| 20 | M | 2009 | 4 | 2 | |||||
| 21 | F | 2006 | + | 6 | + | 8 | |||
| 22 | M | 2010 | + | + | 8 | + | 2 | ||
| 23 | M | 2011 | + | + | 4 | + | 3 | ||
| 24 | F | 2008 | + | 6 | + | 4 | |||
| 25 | M | 2010 | + | + | 7 | + | 3 | ||
| 26 | M | 2011 | + | + | 8 | + | 2 | ||
| 27 | M | 2008 | + | + | + | 5 | 6 | ||
| 28 | M | 2004 | + | + | + | 3 | + | 11 | |
| 29 | F | 2007 | + | 5 | + | 7 | |||
| 30 | F | 2002 | + | + | 2 | + | 13 | ||
| 31 | F | 2005 | + | 6 | + | 9 | |||
| 32 | M | 2002 | + | + | 5 | Autopsy | 3 | ||
| 33 | F | 2006 | 3 | 2 | |||||
| 34 | M | 2006 | + | 6 | + | 4 | |||
| 35 | M | 2012 | + | 6 | + | 2 | |||
| 36 | M | 2006 | + | + | 5 | + | 6 | ||
| 37 | M | 2003 | + | + | + | 7 | + | 12 | |
| 38 | M | 1985 | 3 | + | 12 | ||||
| 39 | M | 1996 | 3 | + | 11 | ||||
| 40 | M | 2010 | + | + | + | 5 | Autopsy | 4 | |
| 41 | F | 2011 | + | 4 | + | 3 | |||
| 42 | F | 2013 | 2 | 0 | |||||
| 43 | M | 1967 | + | 2 | 10 | ||||
| 44 | F | 1956 | 4 | 3 | |||||
| 45 | F | 1995 | 2 | + | 11 | ||||
| 46 | M | 2009 | 3 | + | 4 | ||||
| 47 | M | 2013 | 2 | 0 | |||||
| 48 | F | 2007 | 2 | 4 | |||||
| 49 | M | 2012 | + | + | 6 | Autopsy | 2 | ||
| 50 | M | 2009 | + | + | 2 | Autopsy | 5 | ||
| 51 | M | 2003 | + | + | + | 5 | + | 12 | |
| 52 | F | 2011 | + | 5 | 3 | ||||
| 53 | M | 2007 | 6 | + | 7 | ||||
| 54 | M | 1990 | + | 6 | + | 25 | |||
| 55 | F | 1981 | 4 | + | 21 | ||||
| 56 | F | 2012 | 4 | 0 | |||||
| 57 | M | 2010 | + | 6 | + | 0 | |||
| 58 | M | 2012 | + | 6 | + | 0 | |||
| 59 | F | 2010 | + | 6 | + | 4 | |||
| 60 | M | 2003 | + | + | + | 6 | + | 10 | |
| 61 | M | 1989 | + | 8 | + | 23 | |||
| 62 | M | 1997 | + | + | 6 | + | 18 | ||
| 63 | F | 1989 | 4 | + | 16 | ||||
| 64 | F | 2012 | + | + | 6 | + | 2 | ||
| 65 | M | 1991 | + | + | 4 | + | 23 | ||
| 66 | F | 2012 | + | 5 | + | 2 | |||
| 67 | F | 2014 | + | + | 4 | 0 | |||
| 68 | M | 2012 | + | 4 | + | 0 | |||
| 69 | M | 2013 | 3 | 0 | |||||
| 70 | F | 2004 | + | 5 | + | 11 | |||
| 71 | M | 2001 | + | + | 5 | + | 14 | ||
| 72 | M | 2011 | + | + | 4 | Autopsy | 3 | ||
| 73 | F | 2002 | + | 3 | + | 11 | |||
| 74 | F | 1989 | 4 | + | 12 | ||||
| 75 | M | 2008 | + | + | 5 | + | 6 | ||
| 76 | F | 2003 | + | 4 | + | 6 | |||
| 77 | F | 2011 | + | + | + | 6 | + | 3 | |
| 78 | M | 1994 | + | 3 | 17 | ||||
| 79 | M | 2004 | 3 | + | 6 | ||||
| 80 | F | 2012 | + | + | 2 | 0 | |||
| 81 | F | 1990 | + | + | 4 | + Autopsy | 21 | ||
| 82 | F | 2000 | + | 3 | 2 | ||||
| 83 | F | 2003 | + | + | 4 | + | 12 | ||
| 84 | M | 2010 | + | 3 | + | 4 | |||
| 85 | F | 2013 | + | + | 3 | 0 | |||
| 86 | M | 2008 | + | 2 | 5 | ||||
| 87 | M | 2010 | 3 | 0 | |||||
| 88 | M | 1997 | 2 | 0 | |||||
| 89 | F | 2004 | + | + | + | 4 | + | 11 | |
| 90 | M | 2002 | + | 4 | + | 13 | |||
| 91 | M | 2009 | + | 6 | + | 5 | |||
| 92 | M | 1995 | + | 2 | 5 | ||||
| 93 | M | 2011 | + | + | 3 | Autopsy | 3 | ||
| 94 | F | 2010 | + | + | 4 | 3 | |||
| 95 | F | 2011 | + | 4 | + | 3 | |||
| 96 | M | 2011 | + | 2 | 2 | ||||
| 97 | M | 2005 | + | ND | 4 | + | 10 | ||
| 98 | F | 2012 | + | + | + | 2 | Autopsy | 2 | |
| 99 | F | 1974 | 2 | 0 | |||||
| 100 | M | 2009 | + | 3 | + | 5 | |||
| 101 | M | 2012 | + | 4 | 0 | ||||
| 102 | F | 2006 | + | 3 | 0 | ||||
| 103 | F | 2008 | + | + | + | 3 | Autopsy | 4 | |
| 104 | F | 1988 | + | 4 | + | 18 | |||
| 105 | F | 2014 | + | + | 5 | 0 | |||
| 106 | M | 2011 | + | 3 | + | 2 | |||
| 107 | M | 2006 | 4 | + | 8 | ||||
| 108 | M | 2012 | + | 3 | + | 2 | |||
| 109 | M | 1997 | + | + | 4 | Autopsy | 18 | ||
| 110 | M | 2010 | 2 | + | 4 | ||||
| 111 | F | 2014 | + | + | 4 | 0 | |||
| 112 | F | 2010 | + | + | 3 | Autopsy | 4 | ||
| 113 | M | 2013 | + | 4 | + | 0 |
F female, M male
The study included cases with a high probability of MD and those in whom MD was considered possible. The level of probability was assessed according to the MDC score proposed by the Nijmegen mitochondrial team as follows: 2–4 points: MD possible; 5–8 points: MD probable [17]. The MDC scoring for this study did not include the results of muscle biopsy (panels A+B, without C). The mean MD score in the study group was 4.1 ± 1.5 (range 2–8). Muscle biopsy with subsequent OXPHOS evaluation was performed in 67 cases, and autopsy in 15 cases. The family history was positive in 26 cases and three couples were consanguineous.
In the retrospective group, DNA was isolated from fibroblast cultures or frozen tissue samples obtained by muscle/liver biopsy or by autopsy. Whenever possible, skeletal muscle was preferred. In the remaining cases, DNA was isolated from blood. Throughout the paper the genes were classified as MD-related if they had a connection with mitochondrial disorders documented in the literature [9] or non MD-related when this was not the case.
Parents of the patients gave informed consent for the WES analysis. The study protocol was in agreement with the Helsinki Convention and the study was approved by the Ethics Committee of The Children’s Memorial Health Institute.
Whole-exome sequencing
WES was performed using TruSeqExome Enrichment Kits according to the manufacturer’s instructions (Illumina). The samples were run on 1/4 of a lane on HiSeq 1500 using 2 × 100 bp paired-end reads. Bioinformatics analysis was performed as previously described [18]. Briefly, after initial processing with CASAVA, the sequencing reads were aligned to the hg19 reference genome with the Burrows-Wheeler Alignment Tool and further processed by Genome Analysis Toolkit [19]. Base quality score recalibration, indel realignment, duplicate removal, and SNP/INDEL calling were done as described [20]. The detected variants were annotated using Annovar and converted to MS Access format for final manual analyses. Alignments were viewed with Integrative Genomics Viewer [21, 22]. The complete results of WES, including VCF and/or FASTQ files, are available on demand to qualified researchers. All samples were sequenced so that min. 80 % of target was covered 20× or more.
The presence of the variants identified by WES was confirmed by Sanger sequencing.
Results
Among 67 probands, we found 99 variants in 49 different genes with a Known disease link (Table 2). They were variants in mtDNA (6 patients) and nuclear DNA (nDNA): X-linked (9 patients), autosomal dominant (5 patients), and autosomal recessive (47 patients), including 11 homozygotes. In 50.5 % (50/99) the detected variants were novel (Table 3). Sixty-six of the variants found in the study group occurred in MD-related genes, whereas 31 were found in non MD-related loci. In addition, deleterious variants in a gene not previously linked to disease in humans were identified in one proband (Table 2).
Table 2.
Molecular variants identified in 67 individuals of the study group
| Gene | Chromosome:RefSeq | Variant 1 | Variant 2 | Zygosity status | Mode | ID patient | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Type | Status | Origin | Type | Status | Origin | |||||
| Mitochondrial disease gene | ||||||||||
| ACAD9 | chr3:NM_014049.4 | c.514G>A/p.Gly172Arg | Novel | mat | c.803C>T/p.Ser268Phe | Novel | pat | comphtz | AR | 15 |
| ACAD9 | chr3:NM_014049.4 | c.1552C>T/p.Arg518Cys | Known | mat | c.1553G>A/p.Arg518His | Known | pat | comphtz | AR | 23 |
| ACAD9 | chr3:NM_014049.4 | c.728C>G/p.Thr243Arg | Novel | ND | c.1552C>T/p.Arg518Cys | Known | mat | comphtz | AR | 53 |
| ADCK3 | chr1:NM_020247.4 | c.827A>G/p.Lys276Arg | Novel | mat | c.1702delG/p.Gly568Argfs | Novel | pat | comp htz | AR | 61 |
| AIFM1 | chrX:NM_004208.3 | c.1474T>C/p.Tyr492His | Novel | mat | – | – | hemi | XLR | 25 | |
| CLPB | chr11:NM_030813.4 | c.2045T>A/p.Ile682Asn | Known | pat | c.1937_1938insG/p.645Gly_646Cysfs | Known | mat | comphtz | AR | 5 |
| CLPB | chr11:NM_030813.4 | c.1249C>T/p.Arg417a | Known | pat | c.748C>T/p.Arg250a | Known | mat | comphtz | AR | 27 |
| CLPB | chr11:NM_030813.4 | c.1249C>T/p.Arg417a | Known | pat | c.1222A>G/p.Arg408Gly | Known | mat | comphtz | AR | 67 |
| COX10 | chr17:NM_001303.3 | c.1030A>G/p.Met344Val | Novel | pat | c.1270dupC/p.Leu424Profs | Novel | mat | comphtz | AR | 9 |
| COX10 | chr17:NM_001303 | c.674C>T/p.Pro225Leu | Known | mat | c.674C>T/p.Pro225Leu | Known | pat | hom | AR | 36 |
| DLD | chr7:NM_000108.4 | c.1123G>A/p.Glu375Lys | Known | mat | c.1123G>A/p.Glu375Lys | Known | pat | hom | AR | 31 |
| EARS2 | chr16:NM_001083614.1 | c.164G>A/p.Arg55His | Known | mat | c.325G>C/p.Gly109Arg | Novel | pat | comphtz | AR | 7 |
| EARS2 | chr16:NM_001083614.1 | c.164G>A/p.Arg55His | Known | pat | c.1256C>T/p.Pro419Leu | Novel | mat | comphtz | AR | 70 |
| FBXL4 | chr6:NM_012160.4 | c.858+1G>T/p.? | Novel | pat | c.585+5G>C/p.? | Novel | mat | comphtz | AR | 3 |
| FBXL4 | chr6:NM_012160.4 | c.1303C>T/p.Arg435a | Known | ND | c.64C>T/p.Arg22a | Novel | mat | comphtz | AR | 52 |
| FBXL4 | chr6:NM_012160.4 | c.64C>T/p.Arg22a | Novel | mat | c.64C>T/p.Arg22a | Novel | pat | hom | AR | 55 |
| MTATP6 | chrM:NC_012920.1 | m.9185T>C/p.Leo220Pro | Known | mat | – | – | hompl | M | 32 | |
| MTFMT | chr15:NM_139242.3 | c.994C>T/p.Arg332a | Known | ND | c.626C>T/p.Ser209Leu | Known | ND | comphtz | AR | 91 |
| MTND1 | chrM:NC_012920.1 | m.3902_3908invACCTTGC/p.? | Known | de novo | – | – | hetpl | M | 22 | |
| MTND1 | chrM:NC_012920.1 | m.3688G>A/p.Ala128Thr | Known | ND | – | – | hompl | M | 64 | |
| MTND3 | chrM:NC_012920.1 | m.10254G>A/p.Asp66Asn | Known | de novo | – | – | hetpl | M | 57 | |
| MTND5 | chrM:NC_012920.1 | m.12706T>C/p.Phe124Leu | Known | de novo | – | – | hetpl | M | 34 | |
| MTND5 | chrM:NC_012920.1 | m.13513G>A/p.Asp393Asn | Known | de novo | – | – | hetpl | M | 35 | |
| NAXE | chr1:NM_144772.2 | c.653A>T/p.Asp218Val | Known | mat | c.743_744delC/p.247Ala_248Thrfs | Known | pat | comphtz | AR | 12 |
| NDUFS6 | chr5:NM_004553.4 | c.313_315delAAAG/p.104Lys_106Thrfs | Novel | pat | c.334_359del26ins13/p.Glu112 fs | Novel | mat | comphtz | AR | 1 |
| NDUFS7 | chr19:NM_024407.4 | c.376C>T/p.Leu126Phe | Novel | ND | c.504G>C/p.Arg168Ser | Novel | ND | het | AR | 75 |
| NDUFV1 | chr11:NM_007103.3 | c.733G>A/p.Val245Met | Novel | pat | c.383G>T/p.Arg128Leu | Novel | mat | comphtz | AR | 10 |
| OPA1 | chr3:NM_015560.2 | c.1146A>G/p.Ile382Met | Known | mat | – | – | htz | AD | 33 | |
| PARS2 | chr1:NM_152268.3 | c.1091C>G/p.Pro364Arg | Novel | mat | c.239T>C/p.Ile80Thr | Novel | pat | comphtz | AR | 60 |
| PC | chr11:NM_000920.3 | c.808C>T/p.Arg270Trp | Known | pat | c.2381_2383delTGG/p.Val794del | Novel | mat | comphtz | AR | 29 |
| PC | chr11:NM_000920.3 | c.1487G>A/p.Arg496Gln | Novel | ND | c.584C>T/p.Ala195Val | Novel | ND | comphtz | AR | 71 |
| PDHA1 | chr X:NM_000284.3 | c.262C>T/p.Arg88Cys | Known | mat | – | – | hemi | XLD | 19 | |
| PDHA1 | chrX:NM_000284.3 | c.856_859dupACTT/p. Arg288Leufs | Novel | de novo | – | – | htz | XLD | 56 | |
| PDHA1 | chrX:NM_000284.3 | c.933_935del/p.Arg311del l | Known | de novo | – | – | htz | XLD | 66 | |
| PDHA1 | chrX:NM_000284.3 | c.291G>A/p.? | Novel | de novo | – | – | hemi, mosaic | XLD | 68 | |
| POLG | chr15:NM_001126131.1 | c.2639C>A/p.Ala880Asp | Novel | pat | c.2243G>C/p.Trp748Ser | Known | mat | comphtz | AR | 113 |
| RARS2 | chr6:NM_020320.3 | c.1026G>A/p.Met342Ile | Novel | mat | c.622C>T/p.Gln208a | Novel | pat | comphtz | AR | 41 |
| RRM2B | chr8:NM_015713.4 | c.414_415delCA/p.Tyr138a | Novel | mat | c.414_415delCA/p.Tyr138a | Novel | ND | hom | AR | 21 |
| RRM2B | chr8:NM_015713.4 | c.686G>T/p.Gly229Val | Known | mat | c.686G>T/p.Gly229Val | Known | pat | hom | AR | 51 |
| SCO2 | chr22:NM_005138.2 | c.418G>A/p.Glu140Lys | Known | ND | c.418G>A/p.Glu140Lys | Known | ND | hom | AR | 54 |
| SERAC1 | chr6:NM_032861.3 | c.1822_1828+10delinsACCAACAGG | Known | ND | c.1822_1828+10delinsACCAACAGG | Known | ND | hom | AR | 37 |
| SLC19A3 | chr2:NM_025243.3 | c.68G>T/p.Gly23Val | Known | Pending | c.68G>T/p.Gly23Val | Known | Pending | hom | AR | 58 |
| SLC19A3 | chr2:NM_025243.3 | c.74dupT/p.Ser26Leufs | Known | ND | c.74dupT/p.Ser26Leufs | Known | ND | hom | AR | 109 |
| SLC25A12 | chr2:NM_003705.4 | c.1335C>A/p.Asn445Lys | Novel | mat | c.1335C>A/p.Asn445Lys | Novel | pat | hom | AR | 24 |
| TAZ | chrX:NM_000116.3 | c.684_685insC/p.227Phe_228Profs | Novel | ND | – | – | hemi | XLR | 28 | |
| TMEM126B a | chr11:NM_018480.4 | c.635G>T/p.Gly212Val | Known | mat | c.635G>T/p.Gly212Val | Known | pat | hom | AR | 59 |
| VARS2 | chr6:NM_001167734.1.5 | c.1100C>T/p.Thr367Ile | Known | Pending | c.1490G>A/p.Arg497His | Novel | Pending | comphtz | AR | 97 |
| Non mitochondrial disease gene | ||||||||||
| ADAR | chr1:NM_001111.4 | c.3202+1G>A/p.? | Novel | ND | c.577C>G/p.Pro193Ala | Known | ND | comphtz | AR | 18 |
| CACNA1A | chr19:NM_001127221.1 | c.1997C>T/p.Thr666Met | Known | mat | – | – | htz | AD | 39 | |
| CDKL5 | chrX:NM_003159.2 | c.1942C>T/p.Gln648a | Novel | mat | – | – | hemi | XLD | 65 | |
| CLN3 | chr16:NM_001042432.1 | c.954_962+18del27/p.Leu313_Trp321del | Known | pat | c.461-280_677+382del966 | Known | Pending | comphtz | AR | 88 |
| CPS1 | chr2:NM_001875.4 | c.1837-8A>G/p.? | Known | mat | c.3691G>C/p.Ala1231Pro | Novel | Paternal | comphtz | AR | 13 |
| CPS1 | chr2:NM_001875.4 | c.1289C>G/p.Ser430a | Novel | mat | c.3971_3972delT/p.1323Ile_1324Leufs | Novel | pat | comphtz | AR | 40 |
| DMD | chr X:NM_004006 | c.31+1G>A/p.? | Novel | mat | – | – | hemi | XLR | 38 | |
| DYSF | chr2:NM_003494.3 | c.1180+5G>A/p.? | Known | ND | c.6124C>T/p.Arg2042Cys | Known | ND | comphtz | AR | 45 |
| GBE1 | chr3:NM_000158.3 | c.1621A>T/p.Asn541Tyr | Novel | mat | c.263G>A/p.Cys88Tyr | Novel | pat | comphtz | AR | 14 |
| GFAP | chr17:NM_002055.4 | c.1100G>C/p.Arg367Thr | Novel | de novo | – | – | htz | AD | 42 | |
| HSD17B4 | chr5:NM_000414.3 | c.46G>A/p.Gly16Ser | Known | ND | c.367C>T/p.His123Tyr | Novel | ND | comphtz | AR | 30 |
| MECP2 | chrX:NM_004992.3 | c.89delA/p.Lys30Argfs | Novel | de novo | – | – | hemi | XLD | 106 | |
| MYBPC3 | chr11:NM_000256.3 | c.1351+1G>A/p.? | Known | pat | – | – | htz | AD | 8 | |
| PEX5 | chr12:NM_001131025.1 | c.1669C>T/p.Arg557Trp | Known | mat | c.1799C>T/p.Ser600Leu | Novel | pat | comphtz | AR | 20 |
| PGAP2 | chr11:NM_001256240.1 | c.2T>G/p.Met1? | Known | mat | c.221G>A/p.Arg74His | Known | pat | comphtz | AR | 73 |
| PIGN | chr18:NM_176787.4 | c.932T>G/p.Leu311Trp | Known | mat | c.790G>A/p.Gly264Arg | Known | pat | comphtz | AR | 6 |
| PRF1 | chr10:NM_001083116.1 | c.808_812delGGCAG/p.Gly270 fs | Novel | mat | c.658G>A/p.Gly220Ser | Known | pat | comphtz | AR | 2 |
| SBDS | chr7:NM_016038.2 | c.258+2T>C/p.? | Known | pat | c.184A>T/p.Lys62a | Novel | mat | comphtz | AR | 95 |
| SCN2A | chr2:NM_021007.2 | c.2948T>G/p.Leu983Trp | Novel | de novo | – | – | htz | AD | 47 | |
| New candidate gene for mitochondrial disease | ||||||||||
| NDUFB8 | chr10:NM_005004.3 | c.432C>G/p.Cys144Trp | Novel | mat | c.227C>A/p.Pro76Gln | Novel | pat | comphtz | AR | 26 |
mat maternal, pat paternal, ND not determined (DNA not available), hom homozygote, htz heterozygote, comp htz compound heterozygote, hemi hemizygote, hompl homoplasmic, hetpl heteroplasmic, AR autosomal recessive inheritance, AD autosomal dominant inheritance, XLR X-linked recessive inheritance, XLD X-linked dominant inheritance, M mitochondrial inheritance
aData published on ESHG 2016 by Alston et al.
Table 3.
Novel molecular variants identified in the study; pathogenicity status
| Gene | Variant | MAF | Pathogenicity statusa | Genotype–Phenotype correlationb | Parental results status | Family history | ID patient | |
|---|---|---|---|---|---|---|---|---|
| 1000 G | POL 400 | |||||||
| ACAD9 | c.514G>A/p.Gly172Arg | 0 | 0 | Pathogenic | Moderate | in-trans | Negative | 15 |
| ACAD9 | c.803C>T/p.Ser268Phe | 0 | 0 | Pathogenic | Moderate | in-trans | Negative | 15 |
| ACAD9 | c.728C>G/p.Thr243Arg | 0 | 0 | Pathogenic | Low | in-trans | Negative | 53 |
| ADAR | c.3202+1G>A/p.? | 0 | 0.0014 | Pathogenic | Moderate | ND | Affected brother | 18 |
| ADCK3 | c.827A>G/p.Lys276Arg | 0 | 0 | Pathogenic | High | in-trans | Negative | 61 |
| ADCK3 | c.1702delG/p.Gly568Argfs | 0 | 0 | Pathogenic | High | in-trans | Negative | 61 |
| AIFM1 | c.1474T>C/p.Tyr492His | 0 | 0 | Pathogenic | Moderate | X-linked | Negative | 25 |
| CDKL5 | c.1942C>T/p.Gln648a | 0 | 0 | Pathogenic | Moderate | X-linked | Negative | 65 |
| COX10 | c.1030A>G/p.Met344Val | 0 | 0.0007 | Pathogenic | Moderate | in-trans | Negative | 9 |
| COX10 | c.1270dupC/p.Leu424Profs | 0 | 0 | Pathogenic | Moderate | in-trans | Negative | 9 |
| CPS1 | c.3691G>C/p.Ala1231Pro | 0 | 0.0014 | Pathogenic | Low | In-trans | Affected sister | 13 |
| CPS1 | c.1289C>G/p.Ser430a | 0 | 0.0014 | Pathogenic | Moderate | in-trans | Affected brother | 40 |
| CPS1 | c.3971_3972delT/p.1323Ile_1324Leufs | 0 | 0.0014 | Pathogenic | Moderate | in-trans | Affected brother | 40 |
| DMD | c.31+1G>A/p.? | 0 | 0 | Pathogenic | Low | X-linked | Affected many males | 38 |
| EARS2 | c.325G>C/p.Gly109Arg | 0 | 0.0014 | Likely pathogenic | High | in-trans | Negative | 7 |
| EARS2 | c.1256C>T/p.Pro419Leu | 0 | 0 | Likely pathogenic | Moderate | in-trans | Negative | 70 |
| FBXL4 | c.858+1G>T/p.? | 0 | 0 | Pathogenic | High | in-trans | Miscarriage | 3 |
| FBXL4 | c.585+5G>C/p.? | 0 | 0 | Pathogenic | High | in-trans | Miscarriage | 3 |
| FBXL4 | c.64C>T/p.Arg22a | 0 | 0 | Pathogenic | Moderate | in-trans | Empty ovum | 52 |
| FBXL4 | c.64C>T/p.Arg22a | 0 | 0 | Pathogenic | Moderate | in-trans | Negative | 55 |
| GBE1 | c.1621A>T/p.Asn541Tyr | 0 | 0 | Pathogenic | Moderate | in-trans | Negative | 14 |
| GBE1 | c.263G>A/p.Cys88Tyr | 0 | 0 | Possibly pathogenic | Moderate | in-trans | Negative | 14 |
| GFAP | c.1100G>C/p.Arg367Thr | 0 | 0 | Pathogenic | Moderate | de novo | Negative | 42 |
| HSD17B4 | c.367C>T/p.His123Tyr | 0 | 0.0014 | Pathogenic | Moderate | ND | Affected brother | 30 |
| MECP2 | c.89delA/p.Lys30Argfs | 0 | 0.0 | Pathogenic | High | de novo | Negative | 106 |
| NDUFB8 | c.432C>G/p.Cys144Trp | 0 | 0.0014 | Possibly pathogenic | Moderate | in-trans | Negative | 26 |
| NDUFB8 | c.227C>A/p.Pro76Gln | 0 | 0 | Pathogenic | Moderate | in-trans | Negative | 26 |
| NDUFS6 | c.313_315delAAAG/p.104Lys_106Thrfs | 0 | 0 | Pathogenic | Moderate | in-trans | Affected brother | 1 |
| NDUFS6 | c.334_359del26ins13/p.Glu112 fs | 0 | 0 | Pathogenic | Moderate | in-trans | Affected brother | 1 |
| NDUFS7 | c.376C>T/p.Leu126Phe | 0 | 0 | Pathogenic | Moderate | ND | Similar symptoms in brother | 75 |
| NDUFS7 | c.504G>C/p.Arg168Ser | 0 | 0 | Likely Pathogenic | Moderate | ND | Similar symptoms in brother | 75 |
| NDUFV1 | c.733G>A/p.Val245Met | 0.0005 | 0 | Pathogenic | High | in-trans | Negative | 10 |
| NDUFV1 | c.383G>T/p.Arg128Leu | 0 | 0 | Pathogenic | High | in-trans | Negative | 10 |
| PARS2 | c.1091C>G/p.Pro364Arg | 0.0014 | 0.003 | Pathogenic | Moderate | in trans | Affected sibs | 60 |
| PARS2 | c.239T>C/p.Ile80Thr | 0 | 0 | Pathogenic | Moderate | in trans | Affected sibs | 60 |
| PC | c.2381_2383delTGG/p.Val794del | 0 | 0 | uncertain Pathogenic | High | in-trans | Affected brother | 29 |
| PC | c.1487G>A/p.Arg496Gln | 0 | 0 | Pathogenic | High | ND | Negative | 71 |
| PC | c.584C>T/p.Ala195Val | 0 | 0 | Pathogenic | High | ND | Negative | 71 |
| PDHA1 | c.856_859dupACTT/p. Arg288Leufs | 0 | 0 | Pathogenic | High | de novo | Negative | 56 |
| PDHA1 | c.291G>A/p.? | 0 | 0.0000 | Uncertain pathogenic | Moderate | de novo | Negative | 68 |
| PEX5 | c.1799C>T/p.Ser600Leu | 0 | 0 | Pathogenic | Low | in-trans | Negative | 20 |
| POLG | c.2639C>A/p.Ala880Asp | 0 | 0 | Pathogenic | Moderate | in-trans | Negative | 113 |
| PRF1 | c.808_812delGGCAG/p.Gly270 fs | 0 | 0.0000 | Pathogenic | Low | in trans | Negative | 2 |
| RARS2 | c.1026G>A/p.Met342Ile | 0 | 0 | Likely pathogenic | Moderate | in-trans | Affected brother | 41 |
| RARS2 | c.622C>T/p.Gln208a | 0 | 0.0014 | Pathogenic | Moderate | in-trans | Affected brother | 41 |
| RRM2B | c.414_415delCA/p.Tyr138a | 0 | 0.0014 | Pathogenic | High | ND | Negative | 21 |
| SBDS | c.184A>T/p.Lys62a | 0 | 0.002 | Pathogenic | Low | in-trans | PI neural tube defect | 95 |
| SCN2A | c.2948T>G/p.Leu983Trp | 0 | 0.0013 | Pathogenic | High | de novo | Negative | 47 |
| SLC25A12 | c.1335C>A/p.Asn445Lys | 0 | 0 | Pathogenic | Moderate | in-trans | Negative | 24 |
| TAZ | c.684_685insC/p.227Phe_228Profs | 0 | 0.0012 | Pathogenic | Low | ND | Negative | 28 |
| VARS2 | c.1490G>A/p.Arg497His | 0 | 0 | Pathogenic | Low | ND | Similar disease in sibs | 97 |
ND not determined due to lack of clinical data or DNA not available
aPathogenicity status evaluated according to in silico prediction algorithms (CADD, MetaSVM, Polyphen2 HDIV, Polyphen HVAR, mutation assessor, LRT, MetaLR, SIFT, mutationtaster) and classified as: pathogenic—nonsense, frameshift, splicesite and missense variants with pathogenic status at least in 7 of used algorithms; likely pathogenic - missense variants with pathogenic status in 4–6 of used algorithms; possibly pathogenic—missense variants with pathogenic status <4 of used algorithms
bGenotype-Phenotypecorrelationassessed by two independent specialists in clinical genetics and metabolic medicine
Mutations in MD-related genes were found in 47 probands. Identified pathogenic variants in 31 different genes included 27 located in nDNA and 4 in mtDNA (Table 2). Eleven genes were found defective more than once (PDHA1-4x, ACAD9, CLPB, and FBXL4-3x, COX10, EARS2, MTND1, MTND5, PC, RRM2B, SLC19A3-2x). The majority of these genes were not previously screened for in our mitochondrial diagnostic centre, with the exceptions of TAZ, PDHA1 [23], SCO2, and the genes encoding MTND and MTATP subunits. Below we present the results that were analysed according to selected phenotypic features (neonatal onset, basal ganglia involvement, 3-MGA) and MD likelihood.
Subgroup of neonates
WES yielded conclusive results in 63.9 % (30/47) of neonates studied (Fig. 1a). We found mutations in 23 different genes, including 16 MD-related (ACAD9, AIFM1, CLPB, FBXL4, NDUFS6, NDUFS7, PARS2, PC, PDHA1 [23], RRM2B, SERAC1, SLC19A3, SLC25A12, TAZ, TMEM126B, VARS2) and 7 non MD-related (CDKL5, CPS1, HSD17B4, MECP2, PGAP2, PRF1, SBDS). The majority of the neonates with positive WES results came from the first pregnancy of healthy unrelated parents. Twenty-nine neonates died before establishing a diagnosis; half in the early neonatal period. In 28 cases the mitochondrial testing was completed, including MR imaging and spectroscopy, muscle biopsy and fibroblast culture collection. In the remaining cases, mitochondrial diagnostics were absent or limited only to selective GC–MS screening showing increased excretion of lactate, Krebs cycle metabolites, 3-MGA and/or ketone bodies.
Fig. 1.
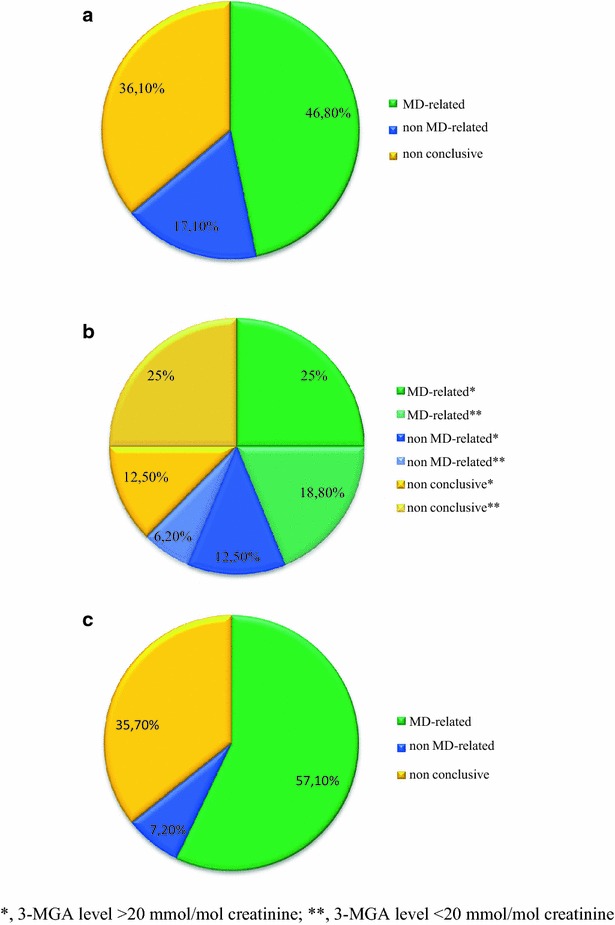
The percentage of detected MD-related genes, non MD-related genes and non-conclusive WES results in (a) neonates (n = 47), b patients with 3-MGA-uria (n = 16) and c patients with basal ganglia involvement (n = 28)
Subgroup with 3-methylglutaonic aciduria
Positive WES results were obtained in seven of 16 patients with persisting 3-MGA (Fig. 1b). In two subjects [P28 and P37] we found mutations in TAZ and SERAC1 genes known to cause mitochondrial diseases with 3-MGA as a discriminative feature [24]. Ex post it was apparent that earlier some important clinical features, including hearing impairment in the patient with SERAC1 mutations and increased excretion of 3-MGA in the terminal stage in the boy with the TAZ mutation, had been overlooked.
In three unrelated 3-MGA neonates included in this study, we identified mutations in the CLPB gene, whose link to human disease was subsequently established [25]. Two of them [P5 and P27] have already been reported in the first disease description [25].
Additionally, in two 3-MGA patients [P13, P40] we found molecular variants in the CPS1, a non MD-related gene linked to urea cycle disorder. In remaining patients in whom the reason for inclusion in the study group was a single GC–MS assessment (ACAD9 and MYBPC3 patients [P15, P8]), increased excretion of 3-MGA has been apparently transient or it was within normal limits after quantitative verification (Additional file 1: Table S1). Since traces of 3-MGA excretion were also found in a number of healthy siblings and parents of the patients the transient or mild increase in patients was most likely without a causal relationship.
Basal ganglia involvement (Leigh syndrome, Leigh-like, others)
In 15 of 28 patients from this group (Fig. 1c), molecular variants in LS-associated genes, including genes responsible for deficiency of complex I (MTND1, MTND3, MTND5, NDUFV1), complex IV (COX10), complex V (MTATP6), combined OXPHOS defect (EARS2, PARS2, RARS2, RRM2B, SERAC1, SLC19A3), and pyruvate dehydrogenase complex deficiency (DLD, PDHA1) [23] were identified. In the remaining 13 patients with LS or other basal ganglia involvement WES did not reveal variants in MD-related genes as listed by Neveling [9].
In three patients with basal ganglia involvement one MD-related candidate (NDUFB8) and two known non MD-related genes (ADAR, CDKL5) were identified.
Defects in non MD-related genes
In 19 patients who were included in the study because of a possible (low probability) mitochondrial disease, mutations in various non MD-related genes (ADAR, CACNA1A, CDKL5, CLN3, CPS1, DMD, DYSF, GBE1, GFAP, HSD17B4, MECP2, MYBPC3, PEX5, PGAP2, PIGN, PRF1, SBDS, SCN2A) were identified (Table 2; Additional file 1: Table S1).
New MD-related disorders
While our project was ongoing new candidate genes found by us including PARS2 [26] and CLPB have been described by other research teams [25]. The causal role of another two of our candidates has been recognized even more recently. The NAXE gene (APOA1BP according to old nomenclature), a susceptibility locus for migraine [27], in which likely pathogenic variants were found by us in two brothers with a fatal encephalitis-like disorder [P12], has been described in April 2016 as the cause of lethal infantile leukoencephalopathy in a large consanguineous family [28]. A homozygous variant in the TMEM126B gene encoding a subunit required for mitochondrial complex I assembly [29, 30], found by us in a complex I deficient girl with extra-neurological presentation [P59], has been discovered and verified functionally as a cause of the disease in a subset of other patients (ESHG 2016, Alston et al.).
The interesting remaining candidate for a novel disease gene identified in our study is NDUFB8. Compound heterozygosity for two variants in NDUFB8 was found in a boy with a typical course of LS and complex I deficiency in muscle homogenate [P26] (Additional file 1: Table S1). NDUFB8 [31] encodes a known subunit of complex I, but, to the extent of our knowledge, its association with complex I deficiency and LS in humans has not been published so far.
Mitochondrial disease criteria score
In the studied cohort there were 40 patients with high probability of MD, i.e., with an MDC score above 4 (5–8, criteria A+B, without C). Positive WES results were obtained in 36 of them (90 %). In this group, pathogenic variants were found mainly in MD-related genes (CPS1 being the exception). WES failed in four patients [P49, P62, P77, P105] with an MDC score above 4. Some of them were found to carry a deleterious variant in one of the known MD-related genes only on one allele. The definite diagnosis still remains open in these cases. Bioinformatics tools for identification of structural variants using NGS have not been applied to our data so it is possible that in some cases the disease may be caused by large deletions/duplications. The complete lists of variants detected in the subjects without fully conclusive results and/or the respective FASTQ files are available on demand to qualified researchers.
Intermediate probability of MD (MDC = 4) was associated with the occurrence of variants in both MD-related and non MD-related genes, in ten (10/31) and six (6/31) patients, respectively. MD-related genes were represented in this subset twice by ACAD9 [P15, P23] and PDHA1 [P56, P68], and in single cases by CLPB [P67], FBLX4 [P55], POLG [P113], RARS2 [P41], SLC19A3 [P109], and VARS2 [P97].
In the subgroup with low probability of MD, i.e., a MDC score of 2–3 points, positive WES results were obtained in 15 of 42 cases (36 %). Three MD-related genes (7 %) including: OPA1 [P33], TAZ [P28] and NAXE [P12] were found. Non MD-related genes were identified in 12 of 42 cases (29 %).
The percentage of positive results rose gradually as the likelihood of MD increased, as shown by the MDC score (Fig. 2). In the subset of high probability of MD (MDC above 4), the detection percentage reached 90 %. There was a broad range of MD-related genes (Table 2). Only one non MD-related gene (CPS1) was found in a neonate with a MDC score of 5.
Fig. 2.
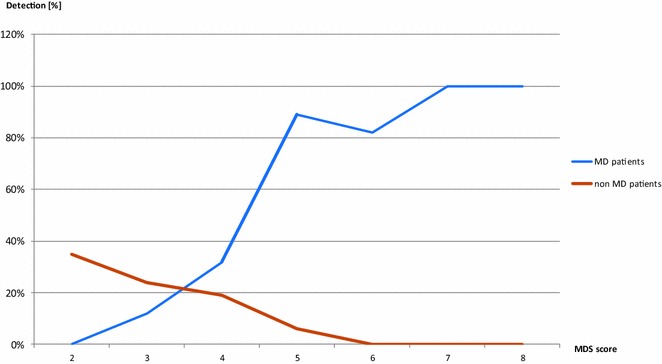
Efficacy of WES in 113 patients with possible or probable mitochondrial pathology depending on the level of probability expressed by MDC Nijmegen score
The participation of detected MD-related genes as compared with non MD-related genes also grew as the likelihood of MD probability increased (from 20 to 97 %, data not shown).
WES diagnostics of current cases vs. archival DNA samples
Characteristics of the patients stratified by the waiting period between disease onset and WES qualification into archival material and current diagnostics subset is shown in Table 4. WES efficacy assessed as percentage of molecularly confirmed diagnoses was comparable being higher than 50 % in both subsets. Contribution of MD-related genes expressed by the ratio of MD-related/non MD-related genes was higher in the archival than current subset (3.4 vs. 1.0, respectively) indicating that this subset contained more patients with non-mitochondrial genetic disorders and that our current qualification for WES became less demanding.
Table 4.
WES results related to the origin of the qualified material and to the specific inclusion criteria
| Subgroups of patients | MD or non-MD genes loci of variants | Diagnostics based on archival material | Current diagnostics | Total |
|---|---|---|---|---|
| Disease onset (year) | 1996–2012 | 2013–2014 | 1996–2014 | |
| Number of patients | 88 (5.5/year) | 25 (12.5/year) | 113 | |
| Period from onset to WES qualification (years) | 2–25 (mean 5.5 ± 5.9 ) | 0 | 0–25 | |
| MDC scale (A+B, without C) | 4.2 ± 1.5 (2–8) | 3.6 ± 1.2 (2–6) | 4.1 ± 1.5 | |
| Ratio of MD-related/non MD related genes | 3.4 | 1.0 | 2.4 | |
| Patients deceased | Total no. | 41 | 8 | 44 % |
| MD | 51.2 % (21) | 2 | 47 % (23) | |
| non MD | (3) | 2 | (5) | |
| Patients with neonatal onset | Total no. | 41 | 6 | 42 % |
| MD | 53.7 % (22) | 2 | 51 % (24) | |
| non MD | (5) | 2 | (7) | |
| Patients with LS or other basal ganglia involvement | Total no. | 21 | 7 | 25 % |
| MD | 61.9 % (13) | 3 | 57 % (16) | |
| non MD | (2) | 0 | (2) | |
| 3-methylglutaconic aciduria | Total no. | 13 | 3 | 14 % |
| MD | 53.8 % (7) | 2 | 53 % (9) | |
| non MD | 0 | 1 | (1) | |
| Muscle biopsy | Total no. | 62 | 5 | 67/113 |
| MD | 56.4 % (35) | (4) | 58 % (39) | |
| non MD | (10) | (0) | (10) | |
| Percentage of muscle biopsy | 70 % | 20 % | 59 % | |
aItalics in brackets indicates the number of patients in the given subset
LS Leigh syndrome, MD mitochondrial disorder, MD/non MD MD-related/non MD-related genes wherein variants were identified
Muscle biopsy findings
OXPHOS assessment available for 67 muscle homogenates showed isolated complex I deficiency in 16 cases, complex IV deficiency in 6 cases and combined OXPHOS defect in 10 cases. There were unspecific changes in 22 bioptates and normal OXPHOS activity in 10. The results were not conclusive in three cases due to technical problems (too small muscle specimen, low protein concentration, low citric synthase activity).
Complex I deficiency was found in 11 patients with molecular variants in MD-related genes (ACAD9 [P15, P23, P53], NDUFV1 [P10], NDUFS7 [P75], MTND1 [P64], MTND3 [P57], EARS2 [P7], SLC19A3 [P58], TMEM126B [P59]) and in one candidate (NDUFB8 [P26]. In one patient [P95] a defect in non MD-related gene (SBDS) was found. In 4 patients WES results were not conclusive.
In the subset with complex IV deficiency molecular defects were confirmed in three patients including COX10 [P9, P36] and EARS2 [P70]) while three WES analyses were not conclusive.
Combined OXPHOS defect occurred in 8 patients with variants identified in MD-related genes (FBXL4 [P3], ADCK3 [P61], RRM2B [P21, P51], AIFM1 [P25], TAZ [P28], PC [P71], MTND5 [P34]). In two cases WES results were not conclusive.
Histological and histochemical data of the patients with positive WES showed presence of ragged red fibers in four cases (ADCK3 [P61], ACAD9 [P15, P23, P53]), “lipid storage myopathy” in four (PC [P71, P29], MTND5 [P35], PDHA1 [P66]) and SMA-like pattern in three (AIFM1 [P25], SCO2 [P54], RRM2B [P51]).
Depletion of mitochondrial DNA (<30 % of reference value) was revealed in tissues of 8 patients. Molecular defect was established by WES in four of them (COX10 [P9], FBXL4 [3], RRM2B [P21, P51]).
Verification of mitochondrial genome variants
Interestingly, in six patients with typical MD phenotype the search for pathogenic variants in MD-related nuclear genes by WES was negative yet pathogenic variants were found in mtDNA. Each mtDNA variant identified by WES, was subsequently verified by Sanger sequencing using specific primers for mitochondrial genome. All detected changes are known and have been repeatedly reported. Examination of different tissues in probands and maternally related family members showed varying levels of heteroplasmy (Fig. 3).
Fig. 3.
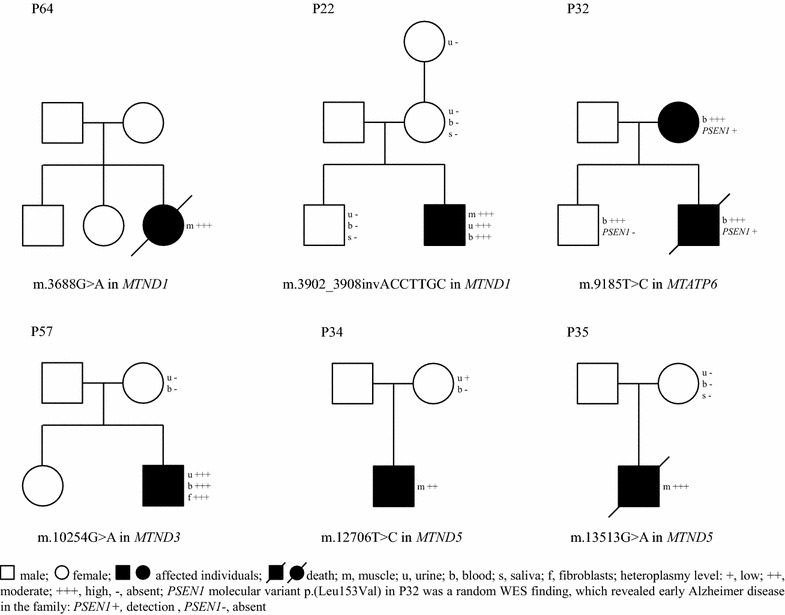
Family study in six probands with mtDNA known mutations
Discussion
Our results confirm that the implementation of WES led to a significant breakthrough in the diagnostics of MD in children [32]. This is expressed by both the increased number of identified genes and faster establishment of final diagnosis. The total number of genes with likely causative defects found in the present work was 47, a very satisfactory diagnostic yield when compared with 8 genes identified by us by single-gene Sanger sequencing before the introduction of WES (203 such diagnoses per ~1200 patients studied in the period from 1996 to 2013).
In our study we observed a pronounced upward trend in the detection of the molecular background of mitochondrial diseases that was associated with increased MD probability (Fig. 2). According to the MDC scale that we used, a final genetic diagnosis was achieved in over 90 % of patients with the highest MDC scores (5–8 points). In all such cases (with one exception for a neonate with CPS1 mutation), variants were found exclusively in MD-related genes. The diagnostic yield was the lowest (36 %) in the patients with low MD suspicion (MDC score 2–3), and most of the variants in this group were present in non MD-related genes.
A similar correlation between detection rate and the level of MD probability was described recently in a similar patient group studied by WES at the Nijmegen Mitochondrial Centre [10]. However, our results differed from that study in terms of the scope of detected defects. In our cohort, mutations in MTO1, TK2, C12orf65, COA6, TUFM, GFM1 were absent and the defects in nuclear encoded complex I subunits are different. This may be a result of random patient selection, but we should also take into account ethnic differences among European populations, e.g., the Slavonic vs. north-western European populations.
In addition, we identified six rare mtDNA pathogenic variants, not included in the common mutations screening i.e. m.9185T>C in MTATP6 [33–35] and in mitochondrial DNA genes encoding complex I subunits, MTND1 [36–38], MTND3 and MTND5 [39–42].
One-third (15/47) of the identified gene defects were discovered during last 10 years and relatively poorly characterized in terms of phenotype. These included PGAP2 [43, 44], ACAD9 [45, 46], EARS2 [47], SERAC1 [48], SLC19A3 [49, 50], MTFMT [51], SLC25A12 [52] as well as VARS2 [53], AIFM1 [54], RARS2 [55], RRM2B [56], PIGN [44, 57], ADCK3 [58, 59] which were described in just individual cases. Notably, most of these genes are generally absent from commercial NGS panels available at present.
It is worth emphasizing that in some cases WES allowed for a diagnosis in statu nascendi, that is, at the time of the first publication of the new gene. This concerned, for example, mutations in CLPB [25, 60], PARS2 [26], FBXL4 [61, 62] and recently added TMEM126B (data published on ESHG 2016 by Alston et al.), and NAXE [28] In one of the patients with the MD phenotype we identified potentially pathogenic variants in candidate NDUFB8 which role in human pathology is under verification [Piekutowska-Abramczuk et al. submitted to SSIEM 2016].
According to published literature, every third paediatric MD case (approximately 30 % of all MD diagnoses in this age group) manifests clinically shortly after birth [12, 13]. The fatal outcome in such cases precludes transport to a reference centre and proper mitochondrial diagnostics. We have previously shown significantly reduced (up to ten times, about 3 % of all diagnoses) recognition of MD in this age group in Poland [16]. Therefore, neonates with suspected MD intentionally constituted a significant proportion of patients (47/113) undergoing WES in the present study.
Surprisingly, in the neonatal subgroup WES proved to be particularly useful, allowing identification of pathogenic variants in 24 various genes in 63.8 % of patients, including those without muscle biopsy or even autopsy. Our results extend the list recommended by Honzik [13] for neonatal MD diagnostics by at least 15 genes (MD-related: RRM2B, CLPB, ACAD9, FBXL4, PC, AIFM1, SLC25A12, MTND5, NDUFS6 and non MD-related: CPS1, PGAP2 and more).
In the LS subgroup WES expanded the set of patients from our centre diagnosed with complex I deficiency by three known genes: NDUFS6 [63, 64], NDUFV1 [65, 66], NDUFS7 [67], a new candidate NDUFB8 [68] and five MTNDs mentioned above. Despite this, complex I deficiency continues to be underrepresented in our cohort in relation to complex IV deficiency because of the high carriage rate of SURF1 mutations in Poland [69]. In a number of cases with basal ganglia brain changes, WES failed to show mutations in known LS-associated genes. This was especially the case in patients without lactic acidaemia and MDC scores below 5 (MD possible but not likely). We speculate that other, still unknown, genes or non-genetic factors might influence the occurrence of LS-brain changes.
Taken together, our results indicate that WES rather than targeted NGS should be the method of choice for MD testing, at least until all MD-associated genes are identified. Furthermore, the rationale for choosing WES in MD-suspected neonates is the non-specificity of symptoms and overlapping results of biochemical tests with non-mitochondrial errors of metabolism.
In 50.5 % the molecular variants were novel (Table 3). However, a number of recurrent rare pathogenic variants found in some recently discovered MD genes (p.Arg22* in FBLX4, p.Arg518Cys in ACAD9, p.Arg417* in CLPB and c.1822_1828+10delinsACCAACAGG in SERAC1) may extend the ethnic specificity of MD in the Polish population reported earlier by us for variants p.Glu140Lys in SCO2 [14] and c.845_846delCT in SURF1 genes [69]. Confirmation of these findings could facilitate in-house diagnostics in selected suspected cases.
Conclusions
In a nationwide reference centre, WES provided positive results in >90 % of children with high likelihood of MD (MDC score above 4);
WES should be recommended for diagnostics of mitochondrial pathology considering remarkable representation of non MD-related genes among causal factors in patients with lower likelihood of MD, as well as a possibility to discover new mitochondrial genes;
WES significantly improves recognition of MD in newborns, even in the case of limited availability of appropriate diagnostic procedures;
Despite being a sine qua non for certain diagnoses 3-MGA is not a universal marker of mitochondrial dysfunction;
Recurrent variants recognized in some relatively new MD genes (FBLX4, ACAD9, and CLPB) may extend the known ethnic specificity of MD in the Polish population reported earlier for SCO2 and SURF1 variants.
Authors’ contributions
Conception and design: EP, RP, EC, DPA, JT, DR. Analysis and interpretation of data: EP, DR, DPA, EC, JT, AKW, MPa, EJ, JK, AP, MR, MPr. Coordination and drafting the article: DPA, EC, JT, PH, EP, AP. Bioinformatic analysis: PS, RP. Revising article critically for important intellectual content EP, MPr, MKW, RP. All authors read and approved the final manuscript.
Acknowledgements
We thank all of the physicians who referred affected children to our mitochondrial centre, especially Hanna Mierzewska, Jacek Pilch, Ewa Jamroz, Jolanta Wierzba, Maria Giżewska, and others.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
The study protocol was in agreement with the Helsinki Convention and the study was approved by the Ethics Committee of The Children’s Memorial Health Institute. Parents of the patients gave informed consent for the WES analysis.
Funding
The study was supported by CMHI projects no. S136/13, no. 126/12, no. 216/12, no. 217/12, no. S134/13, no. S211/10, and by grants from the National Science Centre, 2012/05/B/NZ2/01627, 1154/B/P01/2011/40, 2857/B/P01/2010/39, and EU Structural Funds Project POIG.02.01.00-14-059/09.
Abbreviations
- MD
mitochondrial disorders
- WES
whole-exome sequencing
- MDC
mitochondrial disease criteria
- NGS
next generation sequencing
- LA-uria
lactic aciduria
- 3-MGA-uria
3-methylglutaconic aciduria
- nDNA
nuclear DNA
Additional file
10.1186/s12967-016-0930-9 Characteristics of 113 patients with probable/possible mitochondrial disease recruited for the study.
Footnotes
Dorota Piekutowska-Abramczuk, Elżbieta Ciara and Joanna Trubicka contributed equally to this work
Contributor Information
Ewa Pronicka, Email: e.pronicka@ipczd.pl.
Dorota Piekutowska-Abramczuk, Email: d.abramczuk@ipczd.pl.
Elżbieta Ciara, Email: e.ciara@ipczd.pl.
Joanna Trubicka, Email: j.trubicka@ipczd.pl.
Dariusz Rokicki, Email: d.rokicki@ipczd.pl.
Agnieszka Karkucińska-Więckowska, Email: A.Karkucinska-Wieckowska@IPCZD.pl.
Magdalena Pajdowska, Email: m.pajdowska@czd.pl.
Elżbieta Jurkiewicz, Email: e.jurkiewicz@ipczd.pl.
Paulina Halat, Email: p.halat@ipczd.pl.
Joanna Kosińska, Email: samocko@wp.pl.
Agnieszka Pollak, Email: a.pollak@ifps.org.pl.
Małgorzata Rydzanicz, Email: mrydzanicz@wum.edu.pl.
Piotr Stawinski, Email: stawinski@g.pl.
Maciej Pronicki, Email: m.pronicki@ipczd.pl.
Małgorzata Krajewska-Walasek, Email: M.Krajewska-Walasek@IPCZD.PL.
Rafał Płoski, Email: rploski@wp.pl.
References
- 1.DaRe JT, Vasta V, Penn J, Tran NT, Hahn SH. Targeted exome sequencing for mitochondrial disorders reveals high genetic heterogeneity. BMC Med Genet. 2013;14:118. doi: 10.1186/1471-2350-14-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vasta V, Ng SB, Turner EH, Shendure J, Hahn SH. Next generation sequence analysis for mitochondrial disorders. Genome Med. 2009;110:100. doi: 10.1186/gm100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koene S, Smeitink J. Mitochondrial medicine. J Inherit Metab Dis. 2011;342:247–248. doi: 10.1007/s10545-011-9292-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shamseldin HE, Alshammari M, Al-Sheddi T, Salih MA, Alkhalidi H, Kentab A, Repetto GM, Hashem M, Alkuraya FS. Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes. J Med Genet. 2012;494:234–241. doi: 10.1136/jmedgenet-2012-100836. [DOI] [PubMed] [Google Scholar]
- 5.Calvo SE, Compton AG, Hershman SG, Lim SC, Lieber DS, Tucker EJ, Laskowski A, Garone C, Liu S, Jaffe DB, Christodoulou J, Fletcher JM, Bruno DL, Goldblatt J, Dimauro S, Thorburn DR, Mootha VK. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med. 2012;4118:118ra10. [DOI] [PMC free article] [PubMed]
- 6.Koene S, Rodenburg RJ, van der Knaap MS, Willemsen MA, Sperl W, Laugel V, Ostergaard E, Tarnopolsky M, Martin MA, Nesbitt V, Fletcher J, Edvardson S, Procaccio V, Slama A, van den Heuvel LP, Smeitink JA. Natural disease course and genotype-phenotype correlations in Complex I deficiency caused by nuclear gene defects: what we learned from 130 cases. J Inherit Metab Dis. 2012;355:737–747. doi: 10.1007/s10545-012-9492-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor RW, Pyle A, Griffin H, Blakely EL, Duff J, He L, Smertenko T, Alston CL, Neeve VC, Best A, Yarham JW, Kirschner J, Schara U, Talim B, Topaloglu H, Baric I, Holinski-Feder E, Abicht A, Czermin B, Kleinle S, Morris AA, Vassallo G, Gorman GS, Ramesh V, Turnbull DM, Santibanez-Koref M, McFarland R, Horvath R, Chinnery PF. Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA. 2014;3121:68–77. doi: 10.1001/jama.2014.7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kohda M, Tokuzawa Y, Kishita Y, Nyuzuki H, Moriyama Y, Mizuno Y, Hirata T, Yatsuka Y, Yamashita-Sugahara Y, Nakachi Y, Kato H, Okuda A, Tamaru S, Borna NN, Banshoya K, Aigaki T, Sato-Miyata Y, Ohnuma K, Suzuki T, Nagao A, Maehata H, Matsuda F, Higasa K, Nagasaki M, Yasuda J, Yamamoto M, Fushimi T, Shimura M, Kaiho-Ichimoto K, Harashima H, Yamazaki T, Mori M, Murayama K, Ohtake A, Okazaki Y. A Comprehensive genomic analysis reveals the genetic landscape of mitochondrial respiratory chain complex deficiencies. PLoS Genet. 2016;121:e1005679. doi: 10.1371/journal.pgen.1005679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neveling K, Feenstra I, Gilissen C, Hoefsloot LH, Kamsteeg EJ, Mensenkamp AR, Rodenburg RJ, Yntema HG, Spruijt L, Vermeer S, Rinne T, van Gassen KL, Bodmer D, Lugtenberg D, de Reuver R, Buijsman W, Derks RC, Wieskamp N, van den Heuvel B, Ligtenberg MJ, Kremer H, Koolen DA, van de Warrenburg BP, Cremers FP, Marcelis CL, Smeitink JA, Wortmann SB, van Zelst-Stams WA, Veltman JA, Brunner HG, Scheffer H, Nelen MR. A post hoc comparison of the utility of sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum Mutat. 2013;3412:1721–1726. doi: 10.1002/humu.22450. [DOI] [PubMed] [Google Scholar]
- 10.Wortmann SB, Koolen DA, Smeitink JA, van den Heuvel L, Rodenburg RJ. Whole exome sequencing of suspected mitochondrial patients in clinical practice. J Inherit Metab Dis. 2015;383:437–443. doi: 10.1007/s10545-015-9823-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Legati A, Reyes A, Nasca A, Invernizzi F, Lamantea E, Tiranti V, Garavaglia B, Lamperti C, Ardissone A, Moroni I, Robinson A, Ghezzi D, Zeviani M. New genes and pathomechanisms in mitochondrial disorders unraveled by NGS technologies. Biochim Biophys Acta. 2016;13:728. doi: 10.1016/j.bbabio.2016.02.022. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Cazorla A, De Lonlay P, Nassogne MC, Rustin P, Touati G, Saudubray JM. Long-term follow-up of neonatal mitochondrial cytopathies: a study of 57 patients. Pediatrics. 2005;1165:1170–1177. doi: 10.1542/peds.2004-2407. [DOI] [PubMed] [Google Scholar]
- 13.Honzik T, Tesarova M, Magner M, Mayr J, Jesina P, Vesela K, Wenchich L, Szentivanyi K, Hansikova H, Sperl W, Zeman J. Neonatal onset of mitochondrial disorders in 129 patients: clinical and laboratory characteristics and a new approach to diagnosis. J Inherit Metab Dis. 2012;355:749–759. doi: 10.1007/s10545-011-9440-3. [DOI] [PubMed] [Google Scholar]
- 14.Pronicka E, Piekutowska-Abramczuk D, Szymanska-Debinska T, Bielecka L, Kowalski P, Luczak S, Karkucinska-Wieckowska A, Migdal M, Kubalska J, Zimowski J, Jamroz E, Wierzba J, Sykut-Cegielska J, Pronicki M, Zaremba J, Krajewska-Walasek M. The natural history of SCO2 deficiency in 36 Polish children confirmed the genotype-phenotype correlation. Mitochondrion. 2013;136:810–816. doi: 10.1016/j.mito.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 15.Pronicka E, Weglewska-Jurkiewicz A, Taybert J, Pronicki M, Szymanska-Debinska T, Karkucinska-Wieckowska A, Jakobkiewicz-Banecka J, Kowalski P, Piekutowska-Abramczuk D, Pajdowska M, Socha P, Sykut-Cegielska J, Wegrzyn G. Post mortem identification of deoxyguanosine kinase (DGUOK) gene mutations combined with impaired glucose homeostasis and iron overload features in four infants with severe progressive liver failure. J Appl Genet. 2011;521:61–66. doi: 10.1007/s13353-010-0008-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pajdowska M, Gradowska W, Piekutowska-Abramczuk D, Baczyńska A, Iwanicka-Pronicka K, Sykut-Cegielska J. Urinary organic acid analysis by gas chromatography mass spectrometry (GC-MS) in the detection of mitochondrial disorders. Standardy Med Pediatria. 2012;94:552–561. [Google Scholar]
- 17.Morava E, van den Heuvel L, Hol F, de Vries MC, Hogeveen M, Rodenburg RJ, Smeitink JA. Mitochondrial disease criteria: diagnostic applications in children. Neurology. 2006;6710:1823–1826. doi: 10.1212/01.wnl.0000244435.27645.54. [DOI] [PubMed] [Google Scholar]
- 18.Ploski R, Pollak A, Muller S, Franaszczyk M, Michalak E, Kosinska J, Stawinski P, Spiewak M, Seggewiss H, Bilinska ZT. Does p. Q247X in TRIM63 cause human hypertrophic cardiomyopathy? Circ Res. 2014;1142:e2–e5. doi: 10.1161/CIRCRESAHA.114.302662. [DOI] [PubMed] [Google Scholar]
- 19.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;209:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;435:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;3816:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;291:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ciara E, Rokicki D, Halat P, Karkucinska-Wieckowska A, Piekutowska-Abramczuk D, Mayr J, Trubicka J, Szymanska-Debinska T, Pronicki M, Pajdowska M, Dudzinska M, Gizewska M, Krajewska-Walasek M, Ksiazyk J, Sperl W, Ploski R, Pronicka E. Difficulties in recognition of pyruvate dehydrogenase complex deficiency on the basis of clinical and biochemical features. The role of next-generation sequencing. Mol Genet Metab Rep. 2016;7:70–76. doi: 10.1016/j.ymgmr.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wortmann SB, Duran M, Anikster Y, Barth PG, Sperl W, Zschocke J, Morava E, Wevers RA. Inborn errors of metabolism with 3-methylglutaconic aciduria as discriminative feature: proper classification and nomenclature. J Inherit Metab Dis. 2013;366:923–928. doi: 10.1007/s10545-012-9580-0. [DOI] [PubMed] [Google Scholar]
- 25.Wortmann SB, Zietkiewicz S, Kousi M, Szklarczyk R, Haack TB, Gersting SW, Muntau AC, Rakovic A, Renkema GH, Rodenburg RJ, Strom TM, Meitinger T, Rubio-Gozalbo ME, Chrusciel E, Distelmaier F, Golzio C, Jansen JH, van Karnebeek C, Lillquist Y, Lucke T, Ounap K, Zordania R, Yaplito-Lee J, van Bokhoven H, Spelbrink JN, Vaz FM, Pras-Raves M, Ploski R, Pronicka E, Klein C, Willemsen MA, de Brouwer AP, Prokisch H, Katsanis N, Wevers RA. CLPB mutations cause 3-methylglutaconic aciduria, progressive brain atrophy, intellectual disability, congenital neutropenia, cataracts, movement disorder. Am J Hum Genet. 2015;962:245–257. doi: 10.1016/j.ajhg.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sofou K, Kollberg G, Holmstrom M, Davila M, Darin N, Gustafsson CM, Holme E, Oldfors A, Tulinius M, Asin-Cayuela J. Whole exome sequencing reveals mutations in NARS2 and PARS2, encoding the mitochondrial asparaginyl-tRNA synthetase and prolyl-tRNA synthetase, in patients with Alpers syndrome. Mol Genet Genom Med. 2015;31:59–68. doi: 10.1002/mgg3.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anttila V, Winsvold BS, Gormley P, Kurth T, Bettella F, McMahon G, Kallela M, Malik R, de Vries B, Terwindt G, Medland SE, Todt U, McArdle WL, Quaye L, Koiranen M, Ikram MA, Lehtimaki T, Stam AH, Ligthart L, Wedenoja J, Dunham I, Neale BM, Palta P, Hamalainen E, Schurks M, Rose LM, Buring JE, Ridker PM, Steinberg S, Stefansson H, Jakobsson F, Lawlor DA, Evans DM, Ring SM, Farkkila M, Artto V, Kaunisto MA, Freilinger T, Schoenen J, Frants RR, Pelzer N, Weller CM, Zielman R, Heath AC, Madden PA, Montgomery GW, Martin NG, Borck G, Gobel H, Heinze A, Heinze-Kuhn K, Williams FM, Hartikainen AL, Pouta A, van den Ende J, Uitterlinden AG, Hofman A, Amin N, Hottenga JJ, Vink JM, Heikkila K, Alexander M, Muller-Myhsok B, Schreiber S, Meitinger T, Wichmann HE, Aromaa A, Eriksson JG, Traynor BJ, Trabzuni D, Rossin E, Lage K, Jacobs SB, Gibbs JR, Birney E, Kaprio J, Penninx BW, Boomsma DI, van Duijn C, Raitakari O, Jarvelin MR, Zwart JA, Cherkas L, Strachan DP, Kubisch C, Ferrari MD, van den Maagdenberg AM, Dichgans M, Wessman M, Smith GD, Stefansson K, Daly MJ, Nyholt DR, Chasman DI, Palotie A. North American Brain Expression C, Consortium UKBE, International Headache Genetics C, Genome-wide meta-analysis identifies new susceptibility loci for migraine. Nat Genet. 2013;458:912–917. doi: 10.1038/ng.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spiegel R, Shaag A, Shalev S, Elpeleg O. Homozygous mutation in the APOA1BP is associated with a lethal infantile leukoencephalopathy. Neurogenetics. 2016. doi:10.1007/s10048-016-0483-3. [DOI] [PubMed]
- 29.Heide H, Bleier L, Steger M, Ackermann J, Drose S, Schwamb B, Zornig M, Reichert AS, Koch I, Wittig I, Brandt U. Complexome profiling identifies TMEM126B as a component of the mitochondrial complex I assembly complex. Cell Metab. 2012;164:538–549. doi: 10.1016/j.cmet.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 30.Andrews B, Carroll J, Ding S, Fearnley IM, Walker JE. Assembly factors for the membrane arm of human complex I. Proc Natl Acad Sci USA. 2013;11047:18934–18939. doi: 10.1073/pnas.1319247110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emahazion T, Brookes AJ. Mapping of the NDUFA2, NDUFA6, NDUFA7, NDUFB8, and NDUFS8 electron transport chain genes by intron based radiation hybrid mapping. Cytogenet Cell Genet. 1998;821–2:114. doi: 10.1159/000015081. [DOI] [PubMed] [Google Scholar]
- 32.Wong LJ. Next generation molecular diagnosis of mitochondrial disorders. Mitochondrion. 2013;134:379–387. doi: 10.1016/j.mito.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 33.Moslemi AR, Darin N, Tulinius M, Oldfors A, Holme E. Two new mutations in the MTATP6 gene associated with Leigh syndrome. Neuropediatrics. 2005;365:314–318. doi: 10.1055/s-2005-872845. [DOI] [PubMed] [Google Scholar]
- 34.Castagna AE, Addis J, McInnes RR, Clarke JT, Ashby P, Blaser S, Robinson BH. Late onset Leigh syndrome and ataxia due to a T to C mutation at bp 9,185 of mitochondrial DNA. Am J Med Genet A. 2007;143A8:808–816. doi: 10.1002/ajmg.a.31637. [DOI] [PubMed] [Google Scholar]
- 35.Pitceathly RD, Murphy SM, Cottenie E, Chalasani A, Sweeney MG, Woodward C, Mudanohwo EE, Hargreaves I, Heales S, Land J, Holton JL, Houlden H, Blake J, Champion M, Flinter F, Robb SA, Page R, Rose M, Palace J, Crowe C, Longman C, Lunn MP, Rahman S, Reilly MM, Hanna MG. Genetic dysfunction of MT-ATP6 causes axonal Charcot-Marie-Tooth disease. Neurology. 2012;7911:1145–1154. doi: 10.1212/WNL.0b013e3182698d8d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Musumeci O, Andreu AL, Shanske S, Bresolin N, Comi GP, Rothstein R, Schon EA, DiMauro S. Intragenic inversion of mtDNA: a new type of pathogenic mutation in a patient with mitochondrial myopathy. Am J Hum Genet. 2000;666:1900–1904. doi: 10.1086/302927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blakely EL, Rennie KJ, Jones L, Elstner M, Chrzanowska-Lightowlers ZM, White CB, Shield JP, Pilz DT, Turnbull DM, Poulton J, Taylor RW. Sporadic intragenic inversion of the mitochondrial DNA MTND1 gene causing fatal infantile lactic acidosis. Pediatr Res. 2006;593:440–444. doi: 10.1203/01.pdr.0000198771.78290.c4. [DOI] [PubMed] [Google Scholar]
- 38.Valente L, Piga D, Lamantea E, Carrara F, Uziel G, Cudia P, Zani A, Farina L, Morandi L, Mora M, Spinazzola A, Zeviani M, Tiranti V. Identification of novel mutations in five patients with mitochondrial encephalomyopathy. Biochim Biophys Acta. 2009;17875:491–501. doi: 10.1016/j.bbabio.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 39.Shanske S, Coku J, Lu J, Ganesh J, Krishna S, Tanji K, Bonilla E, Naini AB, Hirano M, DiMauro S. The G13513A mutation in the ND5 gene of mitochondrial DNA as a common cause of MELAS or Leigh syndrome: evidence from 12 cases. Arch Neurol. 2008;653:368–372. doi: 10.1001/archneurol.2007.67. [DOI] [PubMed] [Google Scholar]
- 40.Zhadanov SI, Grechanina EY, Grechanina YB, Gusar VA, Fedoseeva NP, Lebon S, Munnich A, Schurr TG. Fatal manifestation of a de novo ND5 mutation: insights into the pathogenetic mechanisms of mtDNA ND5 gene defects. Mitochondrion. 2007;74:260–266. doi: 10.1016/j.mito.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 41.Swalwell H, Kirby DM, Blakely EL, Mitchell A, Salemi R, Sugiana C, Compton AG, Tucker EJ, Ke BX, Lamont PJ, Turnbull DM, McFarland R, Taylor RW, Thorburn DR. Respiratory chain complex I deficiency caused by mitochondrial DNA mutations. Eur J Hum Genet. 2011;197:769–775. doi: 10.1038/ejhg.2011.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taylor RW, Morris AA, Hutchinson M, Turnbull DM. Leigh disease associated with a novel mitochondrial DNA ND5 mutation. Eur J Hum Genet. 2002;102:141–144. doi: 10.1038/sj.ejhg.5200773. [DOI] [PubMed] [Google Scholar]
- 43.Hansen L, Tawamie H, Murakami Y, Mang Y, ur Rehman S, Buchert R, Schaffer S, Muhammad S, Bak M, Nothen MM, Bennett EP, Maeda Y, Aigner M, Reis A, Kinoshita T, Tommerup N, Baig SM, AbouJamra R. Hypomorphic mutations in PGAP2, encoding a GPI-anchor-remodeling protein, cause autosomal-recessive intellectual disability. Am J Hum Genet. 2013;924:575–583. doi: 10.1016/j.ajhg.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jezela-Stanek A, Ciara E, Piekutowska-Abramczuk D, Trubicka J, Jurkiewicz E, Rokicki D, Mierzewska H, Spychalska J, Uhrynowska M, Szwarc-Bronikowska M, Buda P, Said AR, Jamroz E, Rydzanicz M, Ploski R, Krajewska-Walasek M, Pronicka E. Congenital disorder of glycosylphosphatidylinositol (GPI)-anchor biosynthesis-The phenotype of two patients with novel mutations in the PIGN and PGAP2 genes. Eur J Paediatr Neurol. 2016;203:462–473. doi: 10.1016/j.ejpn.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 45.Haack TB, Danhauser K, Haberberger B, Hoser J, Strecker V, Boehm D, Uziel G, Lamantea E, Invernizzi F, Poulton J, Rolinski B, Iuso A, Biskup S, Schmidt T, Mewes HW, Wittig I, Meitinger T, Zeviani M, Prokisch H. Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat Genet. 2010;4212:1131–1134. doi: 10.1038/ng.706. [DOI] [PubMed] [Google Scholar]
- 46.Nouws J, Nijtmans L, Houten SM, van den Brand M, Huynen M, Venselaar H, Hoefs S, Gloerich J, Kronick J, Hutchin T, Willems P, Rodenburg R, Wanders R, van den Heuvel L, Smeitink J, Vogel RO. Acyl-CoA dehydrogenase 9 is required for the biogenesis of oxidative phosphorylation complex I. Cell Metab. 2010;123:283–294. doi: 10.1016/j.cmet.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 47.Steenweg ME, Ghezzi D, Haack T, Abbink TE, Martinelli D, Van berkel CG, Bley A, Diogo L, Grillo E, Te WaterNaude J, Strom TM, Bertini E, Prokisch H, Van derknaap MS, Zeviani M. Leukoencephalopathy with thalamus and brainstem involvement and high lactate ‘LTBL’ caused by EARS2 mutations. Brain. 2012;135(pt5):1387–1394. doi: 10.1093/brain/aws070. [DOI] [PubMed] [Google Scholar]
- 48.Wortmann SB, Vaz FM, Gardeitchik T, Vissers LE, Renkema GH, Schuurs-Hoeijmakers JH, Kulik W, Lammens M, Christin C, Kluijtmans LA, Rodenburg RJ, Nijtmans LG, Grunewald A, Klein C, Gerhold JM, Kozicz T, van Hasselt PM, Harakalova M, Kloosterman W, Baric I, Pronicka E, Ucar SK, Naess K, Singhal KK, Krumina Z, Gilissen C, van Bokhoven H, Veltman JA, Smeitink JA, Lefeber DJ, Spelbrink JN, Wevers RA, Morava E, de Brouwer AP. Mutations in the phospholipid remodeling gene SERAC1 impair mitochondrial function and intracellular cholesterol trafficking and cause dystonia and deafness. Nat Genet. 2012;447:797–802. doi: 10.1038/ng.2325. [DOI] [PubMed] [Google Scholar]
- 49.Kevelam SH, Bugiani M, Salomons GS, Feigenbaum A, Blaser S, Prasad C, Haberle J, Baric I, Bakker IM, Postma NL, Kanhai WA, Wolf NI, Abbink TE, Waisfisz Q, Heutink P, Van derknaap MS. Exome sequencing reveals mutated SLC19A3 in patients with an early-infantile, lethal encephalopathy. Brain. 2013;136(5):1534–1543. doi: 10.1093/brain/awt054. [DOI] [PubMed] [Google Scholar]
- 50.Zeng WQ, Al-Yamani E, Acierno JS, Jr, Slaugenhaupt S, Gillis T, MacDonald ME, Ozand PT, Gusella JF. Biotin-responsive basal ganglia disease maps to 2q36.3 and is due to mutations in SLC19A3. Am J Hum Genet. 2005;771:16–26. doi: 10.1086/431216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tucker EJ, Hershman SG, Kohrer C, Belcher-Timme CA, Patel J, Goldberger OA, Christodoulou J, Silberstein JM, McKenzie M, Ryan MT, Compton AG, Jaffe JD, Carr SA, Calvo SE, RajBhandary UL, Thorburn DR, Mootha VK. Mutations in MTFMT underlie a human disorder of formylation causing impaired mitochondrial translation. Cell Metab. 2011;143:428–434. doi: 10.1016/j.cmet.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wibom R, Lasorsa FM, Tohonen V, Barbaro M, Sterky FH, Kucinski T, Naess K, Jonsson M, Pierri CL, Palmieri F, Wedell A. AGC1 deficiency associated with global cerebral hypomyelination. N Engl J Med. 2009;3615:489–495. doi: 10.1056/NEJMoa0900591. [DOI] [PubMed] [Google Scholar]
- 53.Diodato D, Melchionda L, Haack TB, Dallabona C, Baruffini E, Donnini C, Granata T, Ragona F, Balestri P, Margollicci M, Lamantea E, Nasca A, Powell CA, Minczuk M, Strom TM, Meitinger T, Prokisch H, Lamperti C, Zeviani M, Ghezzi D. VARS2 and TARS2 mutations in patients with mitochondrial encephalomyopathies. Hum Mutat. 2014;358:983–989. doi: 10.1002/humu.22590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ghezzi D, Sevrioukova I, Invernizzi F, Lamperti C, Mora M, D’Adamo P, Novara F, Zuffardi O, Uziel G, Zeviani M. Severe X-linked mitochondrial encephalomyopathy associated with a mutation in apoptosis-inducing factor. Am J Hum Genet. 2010;864:639–649. doi: 10.1016/j.ajhg.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Edvardson S, Shaag A, Kolesnikova O, Gomori JM, Tarassov I, Einbinder T, Saada A, Elpeleg O. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet. 2007;814:857–862. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bourdon A, Minai L, Serre V, Jais JP, Sarzi E, Aubert S, Chretien D, de Lonlay P, Paquis-Flucklinger V, Arakawa H, Nakamura Y, Munnich A, Rotig A. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet. 2007;396:776–780. doi: 10.1038/ng2040. [DOI] [PubMed] [Google Scholar]
- 57.Ohba C, Okamoto N, Murakami Y, Suzuki Y, Tsurusaki Y, Nakashima M, Miyake N, Tanaka F, Kinoshita T, Matsumoto N, Saitsu H. PIGN mutations cause congenital anomalies, developmental delay, hypotonia, epilepsy, and progressive cerebellar atrophy. Neurogenetics. 2014;152:85–92. doi: 10.1007/s10048-013-0384-7. [DOI] [PubMed] [Google Scholar]
- 58.Lagier-Tourenne C, Tazir M, Lopez LC, Quinzii CM, Assoum M, Drouot N, Busso C, Makri S, Ali-Pacha L, Benhassine T, Anheim M, Lynch DR, Thibault C, Plewniak F, Bianchetti L, Tranchant C, Poch O, DiMauro S, Mandel JL, Barros MH, Hirano M, Koenig M. ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am J Hum Genet. 2008;823:661–672. doi: 10.1016/j.ajhg.2007.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mollet J, Delahodde A, Serre V, Chretien D, Schlemmer D, Lombes A, Boddaert N, Desguerre I, de Lonlay P, de Baulny HO, Munnich A, Rotig A. CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures. Am J Hum Genet. 2008;823:623–630. doi: 10.1016/j.ajhg.2007.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Saunders C, Smith L, Wibrand F, Ravn K, Bross P, Thiffault I, Christensen M, Atherton A, Farrow E, Miller N, Kingsmore SF, Ostergaard E. CLPB variants associated with autosomal-recessive mitochondrial disorder with cataract, neutropenia, epilepsy, and methylglutaconic aciduria. Am J Hum Genet. 2015;962:258–265. doi: 10.1016/j.ajhg.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gai X, Ghezzi D, Johnson MA, Biagosch CA, Shamseldin HE, Haack TB, Reyes A, Tsukikawa M, Sheldon CA, Srinivasan S, Gorza M, Kremer LS, Wieland T, Strom TM, Polyak E, Place E, Consugar M, Ostrovsky J, Vidoni S, Robinson AJ, Wong LJ, Sondheimer N, Salih MA, Al-Jishi E, Raab CP, Bean C, Furlan F, Parini R, Lamperti C, Mayr JA, Konstantopoulou V, Huemer M, Pierce EA, Meitinger T, Freisinger P, Sperl W, Prokisch H, Alkuraya FS, Falk MJ, Zeviani M. Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy. Am J Hum Genet. 2013;933:482–495. doi: 10.1016/j.ajhg.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bonnen PE, Yarham JW, Besse A, Wu P, Faqeih EA, Al-Asmari AM, Saleh MA, Eyaid W, Hadeel A, He L, Smith F, Yau S, Simcox EM, Miwa S, Donti T, Abu-Amero KK, Wong LJ, Craigen WJ, Graham BH, Scott KL, McFarland R, Taylor RW. Mutations in FBXL4 cause mitochondrial encephalopathy and a disorder of mitochondrial DNA maintenance. Am J Hum Genet. 2013;933:471–481. doi: 10.1016/j.ajhg.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kirby DM, Salemi R, Sugiana C, Ohtake A, Parry L, Bell KM, Kirk EP, Boneh A, Taylor RW, Dahl HH, Ryan MT, Thorburn DR. NDUFS6 mutations are a novel cause of lethal neonatal mitochondrial complex I deficiency. J Clin Invest. 2004;1146:837–845. doi: 10.1172/JCI20683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Spiegel R, Shaag A, Mandel H, Reich D, Penyakov M, Hujeirat Y, Saada A, Elpeleg O, Shalev SA. Mutated NDUFS6 is the cause of fatal neonatal lactic acidemia in Caucasus Jews. Eur J Hum Genet. 2009;179:1200–1203. doi: 10.1038/ejhg.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schuelke M, Smeitink J, Mariman E, Loeffen J, Plecko B, Trijbels F, Stockler-Ipsiroglu S, van den Heuvel L. Mutant NDUFV1 subunit of mitochondrial complex I causes leukodystrophy and myoclonic epilepsy. Nat Genet. 1999;213:260–261. doi: 10.1038/6772. [DOI] [PubMed] [Google Scholar]
- 66.Grad LI, Lemire BD. Mitochondrial complex I mutations in Caenorhabditis elegans produce cytochrome c oxidase deficiency, oxidative stress and vitamin-responsive lactic acidosis. Hum Mol Genet. 2004;133:303–314. doi: 10.1093/hmg/ddh027. [DOI] [PubMed] [Google Scholar]
- 67.Lebon S, Rodriguez D, Bridoux D, Zerrad A, Rotig A, Munnich A, Legrand A, Slama A. A novel mutation in the human complex I NDUFS7 subunit associated with Leigh syndrome. Mol Genet Metab. 2007;904:379–382. doi: 10.1016/j.ymgme.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 68.Davis CW, Hawkins BJ, Ramasamy S, Irrinki KM, Cameron BA, Islam K, Daswani VP, Doonan PJ, Manevich Y, Madesh M. Nitration of the mitochondrial complex I subunit NDUFB8 elicits RIP1- and RIP3-mediated necrosis. Free Radic Biol Med. 2010;482:306–317. doi: 10.1016/j.freeradbiomed.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Piekutowska-Abramczuk D, Popowska E, Pronicki M, Karczmarewicz E, Tylek-Lemanska D, Sykut-Cegielska J, Szymanska-Dembinska T, Bielecka L, Krajewska-Walasek M, Pronicka E. High prevalence of SURF1 c.845_846delCT mutation in Polish Leigh patients. Eur J Paediatr Neurol. 2009;132:146–153. doi: 10.1016/j.ejpn.2008.03.009. [DOI] [PubMed] [Google Scholar]