

Abstract
How proteins specifically localize to the phospholipid monolayer surface of lipid droplets (LDs) is being unraveled. We review here the major known pathways of protein targeting to LDs and suggest a classification framework based on the localization origin for the protein. Class I proteins often have a membrane-embedded, hydrophobic ‘hairpin’ motif, and access LDs from the endoplasmic reticulum (ER) either during LD formation or after formation via ER-LD membrane bridges. Class II proteins access the LD surface from the cytosol and bind through amphipathic helices or other hydrophobic domains. Other proteins require lipid modifications or protein-protein interactions to bind to LDs. We summarize knowledge for targeting and removal of the different classes, and highlight areas needing investigation.
The Basics of LDs
LDs are important metabolic organelles in most cell types. The neutral lipids (e.g., triglycerides and sterol esters, see Glossary) stored in their cores provide a buffer for energy fluctuations and a reservoir for membrane lipid precursors. LDs also store other lipophilic molecules, such as fat-soluble vitamins. Both deficient and excessive storage of neutral lipids in LDs are associated with human diseases, including lipodystrophy, non-alcoholic fatty liver disease, atherosclerosis, and obesity/type 2 diabetes.
LDs are decorated by specific proteins, many of which mediate important metabolic functions. How proteins specifically target this organelle and how the LD protein composition is regulated are matters of active investigation. Numerous examples of disease-associated mutations in LD proteins highlight the importance of elucidating this biology for understanding physiology and disease.
In this review we focus on current knowledge of LD proteins and, specifically, on the mechanisms of their targeting and removal. Other aspects of LD cell biology can be found in several excellent recent comprehensive reviews [1–7].
LDs Are Dynamic Organelles
LDs are dynamic organelles, with their abundance coupled to the metabolic state of the cell. When fatty acids or sterols are in excess and drive neutral lipid synthesis, both the number and size of LDs increase. Conversely, LDs are consumed when energy or lipids for membranes are required. How the specific sizes and numbers of LDs are determined in particular cell types is unknown. It is also largely unknown how cells with fluctuating needs for lipid storage and mobilization regulate LD protein composition according to demand.
The number and size of individual LDs are influenced by the amount and type of phospholipids available to cover their surface [1]. The lipid composition of the surface monolayer is similar to the ER phospholipid composition [8,9]. When not enough phospholipids are available to densely cover the LD surface, the interfacial energy (known as surface tension) is high. To balance this energy cost, LDs fuse (or coalesce), thereby decreasing the overall surface, which then is covered more completely with phospholipids. LD coalescence is rarely observed under physiological conditions, but occurs for example in cells with a defect in phospholipid synthesis [10].
The distinct structure of LDs – a neutral lipid core bounded by a phospholipid monolayer-endows specific biophysical properties to this organelle (Box 1). Within a given cell, the amount of available LD surface for protein binding depends on the amount of cellular neutral lipids, which varies under different conditions. Dynamic changes in the limited surface area of LDs result in high susceptibility of protein binding to the influences of macromolecular crowding [11].
Box 1. Physical Properties of Lipid Droplets.
Because they have LDs, cells are emulsions. The hydrophobic neutral lipid core forms the dispersed phase, and the aqueous cytosol forms the continuous phase of the emulsion. Because the oil phase is much more viscous than the cytoplasm, LDs are harder to deform than are other membrane-bound organelles. To maintain the cellular emulsion in a metastable state instead of segregating into single oil and aqueous phases, surfactants are required. Surfactants reduce the energy cost of having molecules (e.g., TGs)atthe interface that do not interact with similar molecules in their phase. This energy cost is known as the surface tension. Particularly good surfactants for cellular LDs include amphiphilic glycerophospholipids such as phosphatidylcholine. By providing favorable interactions with the oil phase through their fatty acid side chains, and with the water phase through their polar headgroups, phosphatidylcholine and other phospholipids lower the surface tension of the LD-cytoplasm interface. In addition to surfactants, proteins at the LD surface may prevent the two oil phases from becoming sufficiently close to each other to favor coalescence.
LDs can expand or shrink, depending on metabolic conditions. New LDs can also be formed. Each of these processes affects the total LD surface area in a cell that is available for protein targeting. Because the LD surface is an interface between hydrophilic and hydrophobic phases, proteins that target LDs cannot have hydrophilic domains on both sides of the monolayer, as is commonly found for proteins inserted into bilayer membranes. Thus, most proteins that target LDs bind to the surface monolayer via hydrophobic interactions. This generally occurs via hydrophobic or amphipathic domains.
Cells Contain Distinct Populations of LDs
Different populations of LDs, based on size, protein, and lipid composition, have been identified in cells [12–14]. Based on their sizes and stage in the LD life cycle, LDs can be classified into two types. Initial LDs (iLDs) are formed from the ER, presumably through a budding process, and appear to range from 300 to 600 nm in diameter [12,15]. iLDs are thought to bud and detach from the ER in mammalian cells. In yeast, LDs generally appear to remain attached to the ER bilayer. A subset of iLDs can be converted into expanding LDs (eLDs) with distinct protein compositions, including triglyceride (TG) synthesis enzymes that mediate their expansion [12]. The Arf1/COPI machinery is required for this transition, likely by modifying the surfaces of LDs to enable the establishment of LD-ER membrane bridges that allow protein targeting from the ER [16] (discussed further below). In specific cell types, such as adipocytes, there are even larger LDs of several microns in diameter. Such LDs form from a combination of smaller LDs by the transfer of neutral lipids from one LD to another via a pore generated by proteins such as CIDEC/Fsp27 [17,18].
LDs Contain a Specific Set of Proteins
Proteomic experiments have identified proteins that copurify with LDs [19–27]. LD-associated proteins vary between cell and tissue types and typically number in the tens to hundreds. Common among them in mammalian cells are perilipins [28–31], which have regulatory functions, metabolic enzymes involved in TG synthesis and breakdown, and enzymes of phospholipid, retinol, and sterol ester metabolism [19,24,26,27].
Because mass spectrometry is so sensitive for identifying proteins, and because LDs are often in close contact with other cellular organelles such as the ER, mitochondria, or peroxisomes [32–35], many contaminants and peripherally associated proteins may copurify with LDs and thus may have been putatively identified as LD proteins. Measuring whether proteins are enriched in LD fractions of biochemical purifications through protein correlation profiling (PCP) can help to sort out contaminants from bona fide LD-associated proteins [24,26,36]. However, PCP does not distinguish proteins embedded in the LD monolayer from those enriching in copurifying membranes. Thus, immunogold labeling combined with electron microscopy remains the most reliable method for identifying proteins that localize to the LD surface monolayer (e.g., [12,37]).
A major question in LD biology is how proteins target LDs. In principle, this can be achieved either by proteins interacting with lipids of LDs or with other LD proteins. Because interactions between different LD proteins require an interaction of at least one of the partners specifically with the LD surface, we focus here primarily on the protein-lipid interactions of LD proteins.
In general, many proteins appear to access the LD surface by one of two major pathways – from the ER or from the cytosol. We designate proteins of the first pathway ‘class I’, and those from the second ‘class II’.
Targeting of Class I LD Proteins
Class I proteins have a dual localization in the ER and on LDs, and are found in the ER in the absence of LDs. They translocate from the ER to LDs either during iLD formation or after eLDs reconnect to the ER via membrane bridges (Figure 1). Class I proteins are embedded in the ER bilayer by hydrophobic sequences. Although the structure of these hydrophobic sequences is not known, they appear to lack ER-luminal domains, which enables them to embed into either the ER membrane or the LD monolayer.
Figure 1. Mechanisms of Class I Protein Targeting to Lipid Droplets (LDs) from the Endoplasmic Reticulum (ER).

Class I LD proteins containing a hydrophobic hairpin motif are present in the ER in the absence of LDs. (A) Some class I proteins accumulate on nascent LDs in the ER and translocate to the LD surface during LD formation [15]. (B) After formation, other class I LD proteins target expanding LDs through membrane bridges from the ER [12]. (C) Electron micrograph showing ER-LD bridges in Drosophila S2 cells (image by Florian Wilfling, Morven Graham, and Xinran Liu). Abbreviations: ACSL3, acyl-CoA synthetase long-chain family member 3; eLD, expanding LD; GPAT4, glycerol-3-phosphate acyltransferase 4; LD, initial LD.
Many of the hydrophobic sequences that target class I proteins from the ER to LDs may form hairpin motifs. This topology has been clearly demonstrated for plant oleosins [38,39], but structural evidence for other proteins that might form this topology is lacking. Candidate proteins with sequences that may form hairpins include glycerol-3-phosphate acyltransferase 4 (GPAT4) and AUP1 [12,40], and possibly AGPAT3, acyl-CoA:diacylglycerol O-acyltransferase (DGAT2), and acyl-CoA synthetase long-chain family member 3 (ACSL3) [15,41,42] (Table 1 for examples). Hydrophobic hairpins consist of ∝-helical domains that form a V-shaped hydrophobic sequence entirely embedded in the membrane. Often, one or more proline residues at the midpoint of the hydrophobic sequences break the helix and might cause a kink, resulting in a hairpin conformation. With the hairpin topology, both ends of the helical domains face the cytosol when in the ER or on LDs (Figure 1).
Table 1.
Examples of Classes of LD Proteins and Targeting Pathways
| Class | Targeting Pathway | Binding Motif | Examples |
|---|---|---|---|
| I | From the ER | Hydrophobic hairpin/helix | ACSL3, GPAT4, DGAT2, AUP1, Ubxd8 |
| II | From the cytoplasm | Amphipathic and hydrophobic sequences | Perilipins, CCT∝, Cidea |
Although evidence for the hairpin structure of class I proteins is limited, some studies support and describe features of the putative hairpin topology. Mutation of the proline knot motif in some hairpin sequences, likely disrupting the hairpin topology, interferes with LD targeting [38,39]. Further characteristics ensuring correct orientation of the hairpin might be basic residues on the N-terminal side of the hairpin, which are required to promote correct targeting of hydrophobic hairpin proteins to LDs [43]. Alternatively, as suggested for ACSL3, an amphipathic helix located N-terminally of the hydrophobic hairpin might ensure proper orientation of the protein [41].
In addition to ER proteins with internal hairpins, other class I proteins have N-terminal hydrophobic sequences that are necessary and sufficient to target both the ER and LDs. The structure of these N-terminal LD targeting domains is not known. Some of them contain central proline residues, suggesting that they also have a hairpin structure. Examples of such proteins include ALDI, AAM-B, Cyb5r3, and 17β-HSD11 [44–50].
Some class I proteins target eLDs that are connected to the ER via membrane bridges (Figure 1). This pathway was discovered as the mechanism for targeting of Drosophila GPAT4 [12]. Based on the available data [32], it appears likely that the yeast TG synthesis enzyme Dga1 also relocates to LD via physical ER-LD bridges. The targeting of GPAT4 to eLDs was found to depend on activity of Arf1/COP-I proteins, which are also required for LD targeting of other proteins, including the TG lipase ATGL [16,51–53]. Recent work advanced our understanding of how the Arf1/COPI machinery regulates class I LD protein recruitment. An Arf1 guanine-nucleotide exchange factor (Arf1GEF) binds to LDs where it activates Arf1 locally to promote budding of nano-droplets from the surface to reduce phospholipid content [16,54–56]. This mechanism has been proposed to result in increased surface tension of LDs and to promote their connection to the ER via membrane bridges, which allows targeting of GPAT4 or potentially other class I proteins [16]. Conversely, inactivation of Arf1 by the Arf1 GTPase-activating factor (ArfGAP) Elmod2 on the LD surface results in decreased targeting of ATGL [57]. The small GTPase Rab18 has also been implicated in regulating ER-LD connections; however, its relationship to the Arf1/COPI machinery is unclear [58].
An important unresolved question for class I proteins is what drives their accumulation on LDs. For class I proteins that translocate from the ER to LDs via membrane bridges, it might be expected that these proteins would equilibrate between the ER and LD compartments. However, many proteins, such as GPAT4, accumulate on LD surfaces in vast excess over the ER pool [12] for unknown reasons.
Targeting of Class II LD Proteins from the Cytosol
Class II proteins are translated in the cytosol and bind directly to the LD surface (Figure 2). The characterized members of class II have distinct, and non-exclusive, targeting mechanisms. Most of the known class II proteins bind to LD surfaces through amphipathic helices or via multiple amphipathic and hydrophobic helices.
Figure 2. Mechanisms of Class II Protein Targeting Lipid Droplets (LDs) from the Cytosol.
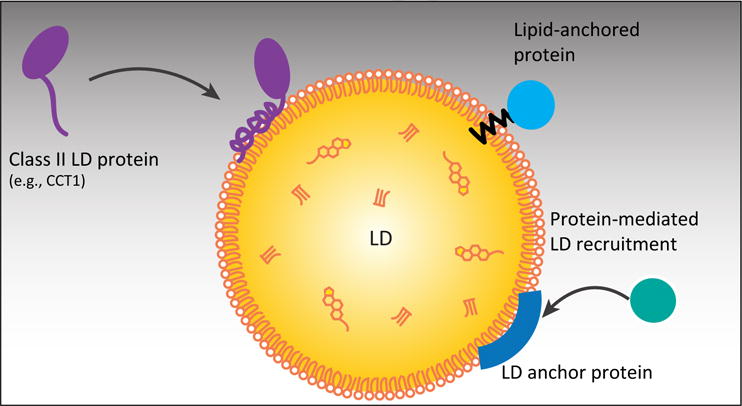
Class II proteins containing an amphipathic helix LD-binding motif or multiple amphipathic and hydrophobic domains target LDs probably from the cytosol. Some proteins bind to LDs with lipid anchors or by binding to other LD proteins; however, the mechanisms that regulate their targeting are not known.
Much investigation has led to a model for amphipathic helix binding to bilayer membranes. In this model, amphipathic targeting sequences are generally unfolded in solution but fold into a helix when binding a membrane surface [59,60]. When bound, the hydrophobic surface is embedded in the acyl-chain domain of the membrane, and the hydrophilic residues face the aqueous phase or interact with the polar head groups of the phospholipids [61]. The amphipathic helices might bind LD surfaces in a similar manner, but experimental evidence is lacking.
A well-studied example of a class II amphipathic helix protein is the CTP:phosphocholine cytidylyltransferase α isoform (CCT∝) [62]. CCT∝ is the rate-limiting enzyme of the Kennedy pathway responsible for the majority of phosphatidylcholine (PC) synthesis in most cells types [63,64]. CCT enzymes bind to the surface of expanding LDs in Drosophila cells as they become PC-deficient during growth. It is unknown what membrane properties exactly CCT senses as ‘PC deficiency’. The phosphatidylethanolamine (PE):PC ratio appears to be crucial [62], suggesting that CCT could be sensing higher surface tension on LD surfaces or lipid packing defects where the oil phase is insufficiently covered with PC. With oleate treatment of Drosophila cells, the binding of CCT∝ to LDs shifts much of the CCT∝ pool from the nucleus to the LD surface. CCToc becomes catalytically active upon binding, resulting in upregulation of PC synthesis [62,65–67]. CCT∝ functions as part of a homeostatic mechanism to maintain sufficient PC to coat eLD surfaces. Indeed, a mutation affecting binding of CCT∝ to LDs results in lipodystrophy, suggesting that LD binding is important for its metabolic function [68]. Aitchison et al. failed to observe CCT∝ binding to LDs in cultured murine adipocytes [69], perhaps owing to insufficient PC deficiency on LD surfaces in their system.
The binding of class II proteins to LD surfaces is likely influenced by factors other than lipid composition. For example, conical lipids that induce packing defects in membranes, such as diacylglycerol [70], promote CCToc binding to membranes [71], similarly to other amphipathic helices. In addition, CCT∝ binding to membranes is affected by phosphorylation of the protein, and this might modulate its binding to LDs [71,72]. The binding of CCT∝ and other amphipathic helix proteins to LDs is also sensitive to protein crowding at the LD surface [11]. For example, overexpression of some LD proteins can cause protein crowding at the surface and prevent binding by class II proteins. Another group of class II proteins targets LDs likely from the cytosol with multiple amphipathic and hydrophobic helical targeting domains (Figure 2). Hydrophobic helices, which are also found in transmembrane helices, differ from amphipathic helices in that all their residues tend to be hydrophobic. These include the perilipin/ADRP/TIP47 (PAT) proteins perilipins 1–5 and their orthologs [73,74] (Table 1). LD-binding of PAT proteins is mediated by a central 11-mer repeat-containing domain [75–77]. Similar 11-mer repeats also occur in a variety of other lipid-binding proteins, such as apolipoproteins and ∝-synuclein, and are thought to fold into amphipathic and hydrophobic 11/3 helices (three full turns every 11 residues) upon membrane binding [77–79]. In addition, perilipins 2 and 3 each contain a C-terminal four-helix bundle resembling the LDL receptor binding domain of apolipoprotein E, forming a lipid-binding hydrophobic cleft that likely contributes to LD targeting together with the central 11-mer repeat domain [77,80–82].
Although the binding of PAT proteins to LDs is less sensitive to the effects of protein crowding than amphipathic helix proteins [11] (discussed further below), there is evidence that competition of between perilipins or with other proteins influences protein targeting to LDs [11,82–84]. Mutations affecting perilipin expression, structure, and potentially their binding to LDs are associated with changes in TG metabolism and lipodystrophy in severe cases [85–88]. Future research might elucidate how each of the perilipins is bound to the LD and whether they form higher-order structural arrangements on the LD surface.
How class II proteins distinguish LD surfaces from other membranes, such as the ER bilayer, is unknown. Not all proteins with amphipathic helices, for example, appear to prefer binding to LDs. For some proteins with amphipathic ∝-helices that localize to bilayers, the mechanism for their binding to these membranes is understood. For example, proteins containing the amphipathic lipid packing defect sensor (ALPS) motif bind preferentially to highly curved bilayer membranes by inserting hydrophobic side chains into large phospholipid packing defects [89,90]. Why most of the these proteins appear to bind to bilayers rather than to LDs in cells is unclear.
Some class II proteins target from the cytoplasm to LDs by binding to other LD proteins (Figure 2). Hormone-sensitive lipase for example is recruited to LDs from the cytosol by perilipin 1 upon phosphorylation of both proteins [30,91]. Other examples are histones H2A, H2B, and H2Av, which are sequestered to LDs by the LD protein Jabba during Drosophila embryogenesis [92], and the Dengue virus capsid protein, which is potentially recruited to LDs by perilipin 3 [93].
The Mechanism for Targeting Some Proteins to LDs Remains Poorly Understood
Some proteins that clearly target LDs are so far difficult to classify. For example, how CGI-58, a protein that regulates lipolysis by ATGL [28], targets LDs is uncertain. Recent NMR studies revealed that an LD anchoring motif in the CGI-58 N-terminus depends on three tryptophan residues [48,49]. However, the topology of this hydrophobic N-terminal domain on LDs is unknown, as is whether CGI-58 targets via the class I or class II pathway.
Other proteins that are difficult to classify include those that use lipid modifications, such as palmitoylation, as lipid anchors to bind to the LD surface (Figure 2). For instance, the Arf-GAP ELMOD2 is palmitoylated [57], and aldehyde dehydrogenase ALDH3B2 is prenylated with a geranylgeranyl group [94]; both proteins require the lipid modification for LD targeting. Whether the lipid anchor motifs in ALDH3B2 or ELMOD2 are sufficient to target proteins specifically to LDs is unknown. Because similar motifs are found on other proteins that are not localized to LDs, other sequence elements are likely involved in targeting these proteins to LDs. Another example of a protein that utilizes a lipid anchor to target LDs is the small GTPase Rab18. Rab18 contains a mono-cysteine prenylation motif in its C-terminus, making it one of a few Rab family members that do not employ di-cysteine prenylation motifs [95].
Removal of Proteins from LDs
Because LDs dynamically cycle in size and abundance, depending on nutrient availability and cell proliferation, the LD proteome must be constantly adjusted to match metabolic requirements. In particular, the surface area of LDs decreases dramatically during LD consumption by lipolysis, begging the question of how proteins are removed from shrinking LDs.
Conceptually, removal of proteins from LDs must occur either by relocalization or by degradation. For example, class I and II proteins, in principle, could be relocalized to different locations in the cell (such as the ER and cytosol, respectively). Alternatively, proteins could be degraded at LD surfaces or by autophagy of LDs.
In autophagy, part of the cytoplasm, including some organelles, are engulfed by a new membrane-bound compartment (autophagosome) and delivered to lysosomes for degradation [96]. Autophagy may have a role in the degradation of LDs and specifically of neutral lipids [97–100]. Delivery of LDs to lysosomes via autophagy would allow lipids as well as proteins to be degraded in the lysosome by the action of lysosomal acidic lipase (LAL) and proteases, respectively, which play a role in TG and CE homeostasis in macrophages [101] (Figure 3). However, because autophagy in many cell types is most strongly induced under periods of prolonged fasting, it is unclear what role autophagy plays in LD turnover under normal metabolic fluctuations. Glucose deprivation for up to 24 h promotes interaction of LDs with mitochondria, and oxidation of fatty acids released from LDs, in a process that is dependent on AMPK and de-tyrosinated microtubules, but without inducing significant amounts of autophagy [102]. The fatty acids released from LDs can apparently be transferred directly from LDs to mitochondria–autophagy, on the other hand, might play a role in mobilizing lipids from other membranes for incorporation into LDs [103].
Figure 3. Mechanisms of Protein Removal from Lipid Droplets (LDs).
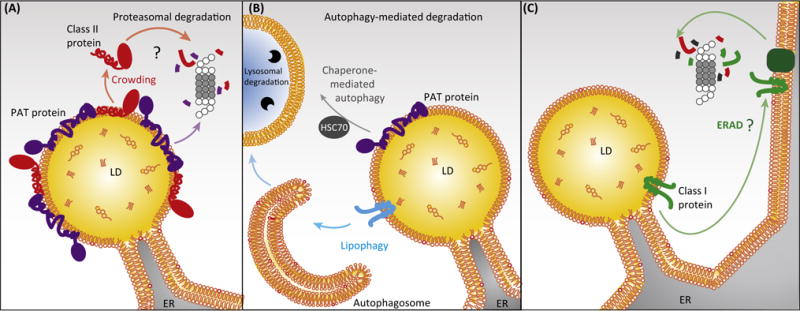
LD proteins are removed from LDs by relocalization or degradation. (A) Class II LD proteins are displaced from the LD surface by protein crowding during lipolysis [11] and could be degraded by the ubiquitin/proteasome system, which for instance degrades the class II protein perilipin 2 [116,117]. (B) Whole LDs or parts can undergo autophagy leading to protein as well as lipid degradation [120,121]. Perilipins 2 and 3 undergo chaperone-mediated autophagy during lipolysis [106]. (C) Mechanisms removing class I proteins from LDs are not known but might include relocalization back to the ER and ER-associated degradation (ERAD), or extraction and degradation directly from the LD surface by unknown factors. Abbreviations: ER, endoplasmic reticulum; PAT protein, member of the perilipin/adipose differentiation-related protein (ADRP)/tail-interacting protein of 47 kDa (TIP47) family; HSC70, heat shock cognate 70 kDa.
Turnover of Class I Proteins
It is unknown whether or how class I LD proteins are removed from LDs. Because class I proteins contain highly hydrophobic LD binding motifs likely interacting with the LD core, they probably require dedicated machinery to extract them from membranes. This could occur either after relocalization of the protein to the ER, where ER-associated degradation (ERAD) removes the protein, or by direct extraction and degradation at the LD.
Although the mechanisms remain to be elucidated, both the relocalization and turnover of class I LD proteins appear to be regulated by lipid-loading state. The class I LD proteins Ubxd8 and AAM-B apparently relocalize back to the ER when LDs are consumed [50] (Figure 3). In addition, binding to LDs results in the stabilization of many LD proteins, including class I proteins. PNPLA3, for example, shows a longer half-life in lipid-loaded cells or with proteasome inhibition [104]. Whether stabilization of LD proteins by TG/LDs is due to spatial segregation from the degradation machinery, stabilization of protein conformation, or through burying degrons in the hydrophobic environment of the LD is not known.
Some class I proteins might be degraded directly on the surface of LDs, as reported for oleosins and PAT proteins [105,106]. Several proteins involved in protein degradation, including AUP1, localize to LDs and could mediate such a process [107–110].
Overall, few data clearly define the route of degradation for any class I protein. More studies of the fate of individual class I proteins during lipolysis will be necessary to determine their mechanisms of removal and turnover.
Removal of Class II Proteins
Displacement by macromolecular crowding appears to be a major mechanism for removal of some class II proteins. In conditions that promote class II protein targeting to LDs, the coupling of amphipathic helix folding to surface binding for these proteins makes the binding reaction fairly stable [59,62]. When such proteins dissociate from LDs, however, this pathway is not simply reversed. Instead, proteins first fall off LDs and then unfold. In addition, concatenation of multiple binding helices or dimerization of many LD proteins likely makes binding essentially irreversible under steady-state conditions [11]. However, when LDs shrink during lipolysis, their surface compresses, and presumably the increased density of proteins at the surface results in macromolecular crowding of LD proteins [11]. This effectively increases the surface pressure of the LD surface monolayer, and proteins are displaced from the increasingly crowded surface to relieve the surface pressure. This occurs because crowding increases the likelihood of proteins colliding in a way where sufficient energy is transferred to displace a neighbor protein with weaker LD surface association [111]. At the same time, increased concentration of LD proteins on LD surfaces limits available binding sites, thus preventing the displaced proteins from re-binding to LDs. Whether there are actually changes in protein concentration on LD surfaces during LD shrinkage remains to be tested, although in vitro studies modeling shrinkage demonstrate slowing of diffusion, consistent with crowding [11]. The displacement of class II proteins, or other peripherally bound proteins, might be further regulated by post-translational modifications, such as phosphorylation, that could modify affinity for the LD surface. The surface-dependent displacement of class II proteins is similar to what has been found for exchangeable apolipoproteins on lipoprotein particles [112–115].
Chaperone-mediated autophagy is an alternative mechanism that may actively remove class II LD proteins with multiple amphipathic and hydrophobic helices (Figure 3). Evidence indicates that such a mechanism removes perilipins 2 and 3 from the LD surface during lipolysis, possibly allowing access of lipases to substrates [106]. ATGL association with the LD surface is prevented when HSC70-mediated lysosomal degradation of perilipin 2 and 3 is inhibited, resulting in decreased lipolysis and net TG accumulation. Therefore, it is possible that chaperone-mediated autophagy could be a mechanism to reduce crowding on the LD surface when lipolysis is actively stimulated. However, whether HSC70 acts directly on the LD surface to extract perilipins, and whether it is involved in the degradation of other LD proteins, is unknown. In addition, the proposed degradation of perilipin 2 by chaperone-mediated autophagy contradicts other studies showing that perilipin 2 is degraded by the ubiquitin/proteasome system [116,117]. How perilipin levels are adjusted to the abundance of TG remains to be determined.
Concluding Remarks
An understanding of protein targeting to LDs, and how this differs from targeting to other organelles, is emerging. We are beginning to understand the general mechanisms involved in protein targeting, such as the routes proteins take to the LD and what sequence motifs enable their localization. We know little, however, about how specificity is achieved or how proteins selectively accumulate at the organelle (see Outstanding Questions). Further examination of specific protein motifs will undoubtedly reveal sequence properties that determine this phenomenon. Specific lipid signatures at LD surfaces likely mediate the binding of particular proteins, as shown by a recent report that an amphipathic helix of CIDEA recognizes phosphatidic acid at LD surfaces [118].
Beyond elucidating targeting mechanisms for individual proteins, a major question is how the regulation of protein targeting affects the protein composition and cellular physiology of LDs. Many LD proteins are regulated by protein modifications, such as phosphorylation, depending on the metabolic state of the cell. Nonetheless, how these modifications affect overall protein composition is unknown. Mutations in LD proteins might also affect their binding to LDs and thus change the subcellular distribution of the protein. Mutations in CCT∝, for example, affect binding of the protein to LDs and result in lipodystrophy [68]. Moreover, owing to competition between LD proteins, changes in the abundance of a protein on LDs could change overall LD protein composition. For instance, a common polymorphism in PNPLA3 (I148 M) results in an apparent change in the protein composition of LDs and is a major determinant of hepatic lipid accumulation [119]. These examples highlight the need to determine whether changes in LD protein composition cause changes in physiology or result in disease.
Trends.
Tens to hundreds of proteins target to lipid droplets, where many carry out important metabolic functions.
Lipid droplets are unusual organelles with a neutral lipid core surrounded by a surface monolayer, presenting unique topological features for the targeting of specific proteins.
Most proteins that target lipid droplets do so either from the endoplasmic reticulum or from the cytosol, utilizing hydrophobic domains that interact with the monolayer and/or neutral lipid cores of lipid droplets.
Removal of proteins from lipid droplet surfaces is poorly understood but in some cases is due to macromolecular crowding on the shrinking LD surface.
Outstanding Questions.
What determines specificity for proteins targeting the LD monolayer versus bilayer membranes in cells?
What drives class I proteins from the ER to accumulate on LDs?
How are ER-LD membrane bridges maintained and regulated?
How are proteins containing hydrophobic hairpins removed from LDs?
How are LD proteins degraded?
Acknowledgments
The authors thank Gary Howard for editorial assistance. This work was supported by the Mathers foundation (T.C.W) and the National Institute of General Medical Sciences (NIGMS) (R01GM-097194, T.C.W; R01GM-099844, R.V.F.). T.C.W. is an HHMI investigator.
Glossary
- Acyl-CoA diacylglycerol O-acyltransferases (DGAT) 1 and 2
enzymes that catalyze triglyceride (TG) synthesis in mammals
- Acyl-CoA synthetase long-chain family member 3 (ACSL3)
a lipid droplet (LD)-targeted enzyme that catalyzes the synthesis of fatty acyl-CoAs. Accumulates on nascent LDs in the ER upon stimulation of TG synthesis
- Amphipathic helix
helices that have hydrophobic and hydrophilic residues on opposite sides allowing them to interact with membranes
- CTP: phosphocholine cytidylyltransferase α isoform (CCTα)
an enzyme that catalyzes the rate-limiting step of the de novo or Kennedy pathway of phosphatidylcholine synthesis. Contains a nuclear localization signal
- Expanding LDs (eLDs)
a population of LDs that are formed from iLDs; after acquiring iLDs they connect to the ER via membrane bridges and acquire TG synthesis enzymes from the ER. Owing to the localized expansion of lipids, eLDs can be several microns in size, depending on the cell type
- Glycerol-3-phosphate acyltransferase 4 (GPAT4)
one of several isoenzymes that catalyze the first step of glycerophospholipid synthesis and target to LDs via ER– LD membrane bridges
- Initial LDs (iLDs)
a population of LDs that are formed when neutral lipids are synthesized in the ER, presumably through a maturation and budding process. They are 200–600 nm in diameter. It is unknown whether iLDs are connected to the ER
- Macromolecular crowding
a phenomenon that alters the properties of molecules in a solution owing to high concentrations of macromolecules (for example, proteins), as found inside cells. On membranes, protein crowding results in stress from collisions between membrane-bound proteins. Stress is relieved either by bending of the membrane or by displacement of proteins from the membrane
- Neutral lipid
highly hydrophobic lipids that lack a charged group. They include triacylglycerols, sterol esters, ether lipids, retinyl esters, and waxes that are found in the cores of LDs. Storage forms of many membrane lipids in the cell
- Perilipins/PAT proteins
LD proteins, including perilipins 1 (perilipin), 2 (adipose differentiation-related protein, ADRP), 3 (tail-interacting protein of 47 kDa, TIP47), 4 (S3-12), and 5 (OXPAT), that contain an N-terminal (PAT) domain. Some perilipins regulate lipid storage by modulating lipase activity and possibly access to substrates
- Surface tensio
the interfacial energy existing between a hydrophobic and hydrophilic phase. Surface tension results from exposure of molecules of each phase to molecules of the other phase. Surfactants lower surface tension by interacting with molecules of both phases. High surface tension in LDs favors coalescence to minimize the surface tension. The inverse term of surface tension, often used for densely packed surfaces, is termed surface pressure
References
- 1.Thiam AR, et al. The biophysics and cell biology of lipid droplets. Nat Rev Mol Cell Biol. 2013;14:775–786. doi: 10.1038/nrm3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walther TC, Farese RV., Jr Lipid droplets and cellular lipid metabolism. Annu Rev Biochem. 2012;81:687–714. doi: 10.1146/annurev-biochem-061009-102430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilfling F, et al. Lipid droplet biogenesis. Curr Opin Cell Biol. 2014;29:39–45. doi: 10.1016/j.ceb.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Welte MA. Expanding roles for lipid droplets. Curr Biol. 2015;25:R470–R481. doi: 10.1016/j.cub.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gross DA, Silver DL. Cytosolic lipid droplets: from mechanisms of fat storage to disease. Crit Rev Biochem Mol Biol. 2014;49:304–326. doi: 10.3109/10409238.2014.931337. [DOI] [PubMed] [Google Scholar]
- 6.Hashemi HF, Goodman JM. The life cycle of lipid droplets. Curr Opin Cell Biol. 2015;33:119–124. doi: 10.1016/j.ceb.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barbosa AD, et al. Lipid droplet-organelle interactions: emerging roles in lipid metabolism. Curr Opin Cell Biol. 2015;35:91–97. doi: 10.1016/j.ceb.2015.04.017. [DOI] [PubMed] [Google Scholar]
- 8.Bartz R, et al. Lipidomics reveals that adiposomes store ether lipids and mediate phospholipid traffic. J Lipid Res. 2007;48:837–847. doi: 10.1194/jlr.M600413-JLR200. [DOI] [PubMed] [Google Scholar]
- 9.Tauchi-Sato K, et al. The surface of lipid droplets is a phospholipid monolayer with a unique fatty acid composition. J Biol Chem. 2002;277:44507–44512. doi: 10.1074/jbc.M207712200. [DOI] [PubMed] [Google Scholar]
- 10.Fei W, et al. A role for phosphatidic acid in the formation of ‘supersized’ lipid droplets. PLoS Genet. 2011;7:e1002201. doi: 10.1371/journal.pgen.1002201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kory N, et al. Protein crowding is a determinant of lipid droplet composition. Dev Cell. 2015;34:351–363. doi: 10.1016/j.devcel.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilfling F, et al. Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev Cell. 2013;24:384–399. doi: 10.1016/j.devcel.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolins NE, et al. S3-12, adipophilin, andTIP47 package lipid in adipocytes. J Biol Chem. 2005;280:19146–19155. doi: 10.1074/jbc.M500978200. [DOI] [PubMed] [Google Scholar]
- 14.Hsieh K, et al. Perilipin family members preferentially sequester to either triacylglycerol-specific or cholesteryl-ester-specific intracellular lipid storage droplets. J Cell Sci. 2012;125:4067–4076. doi: 10.1242/jcs.104943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kassan A, et al. Acyl-CoA synthetase 3 promotes lipid droplet biogenesis in ER microdomains. J Cell Biol. 2013;203:985–1001. doi: 10.1083/jcb.201305142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilfling F, et al. Arf1/COPI machinery acts directly on lipid droplets and enables their connection to the ER for protein targeting. Elife. 2014;3:0000000. doi: 10.7554/eLife.01607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gong J, et al. Fsp27 promotes lipid droplet growth by lipid exchange and transfer at lipid droplet contact sites. J Cell Biol. 2011;195:953–963. doi: 10.1083/jcb.201104142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jambunathan S, et al. FSP27 promotes lipid droplet clustering and then fusion to regulate triglyceride accumulation. PLoS ONE. 2011;6:e28614. doi: 10.1371/journal.pone.0028614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brasaemle DL, et al. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytes. J Biol Chem. 2004;279:46835–46842. doi: 10.1074/jbc.M409340200. [DOI] [PubMed] [Google Scholar]
- 20.Bouchoux J, et al. The proteome of cytosolic lipid droplets isolated from differentiated Caco-2/TC7 enterocytes reveals cell-specific characteristics. Biol Cell. 2011;103:499–517. doi: 10.1042/BC20110024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cermelli S, et al. The lipid-droplet proteome reveals that droplets are a protein-storage depot. Curr Biol. 2006;16:1783–1795. doi: 10.1016/j.cub.2006.07.062. [DOI] [PubMed] [Google Scholar]
- 22.D’Aquila T, et al. Characterization of the proteome of cytoplasmic lipid droplets in mouse enterocytes after a dietary fat challenge. PLoS ONE. 2015;10:e0126823. doi: 10.1371/journal.pone.0126823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ding Y, et al. Identification of the major functional proteins of prokaryotic lipid droplets. J Lipid Res. 2012;53:399–411. doi: 10.1194/jlr.M021899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krahmer N, et al. Protein correlation profiles identify lipid droplet proteins with high confidence. Mol Cell Proteomics. 2013;12:1115–1126. doi: 10.1074/mcp.M112.020230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang P, et al. Proteomic study and marker protein identification of Caenorhabditis elegans lipid droplets. Mol Cell Proteomics. 2012;11:317–328. doi: 10.1074/mcp.M111.016345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Currie E, et al. High confidence proteomic analysis of yeast LDs identifies additional droplet proteins and reveals connections to dolichol synthesis and sterol acetylation. J Lipid Res. 2014;55:1465–1477. doi: 10.1194/jlr.M050229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujimoto Y, et al. Identification of major proteins in the lipid droplet-enriched fraction isolated from the human hepatocyte cell line HuH7. Biochim Biophys Acta. 2004;1644:47–59. doi: 10.1016/j.bbamcr.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 28.Granneman JG, et al. Perilipin controls lipolysis by regulating the interactions of AB-hydrolase containing 5 (Abhd5) and adipose triglyceride lipase (Atgl) J Biol Chem. 2009;284:34538–34544. doi: 10.1074/jbc.M109.068478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Granneman JG, et al. Interactions of perilipin-5 (Plin5) with adipose triglyceride lipase. J Biol Chem. 2011;286:5126–5135. doi: 10.1074/jbc.M110.180711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sztalryd C, et al. Perilipin A is essential for the translocation of hormone-sensitive lipase during lipolytic activation of adipocytes. J Cell Biol. 2002;161:1103. doi: 10.1083/jcb.200210169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pollak NM, et al. The interplay of protein kinase A and perilipin 5 regulates cardiac lipolysis. J Biol Chem. 2015;290:1295–1306. doi: 10.1074/jbc.M114.604744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jacquier N, et al. Lipid droplets are functionally connected to the endoplasmic reticulum in Saccharomyces cerevisiae. J Cell Sci. 2011;124:2424–2437. doi: 10.1242/jcs.076836. [DOI] [PubMed] [Google Scholar]
- 33.Novikoff AB, et al. Organelle relationships in cultured 3T3-L1 preadipocytes. J Cell Biol. 1980;87:180–196. doi: 10.1083/jcb.87.1.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tarnopolsky MA, et al. Influence of endurance exercise training and sex on intramyocellular lipid and mitochondrial ultrastructure, substrate use, and mitochondrial enzyme activity. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1271–R12718. doi: 10.1152/ajpregu.00472.2006. [DOI] [PubMed] [Google Scholar]
- 35.Binns D, et al. An intimate collaboration between peroxisomes and lipid bodies. J Cell Biol. 2006;173:719–731. doi: 10.1083/jcb.200511125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andersen JS, et al. Proteomic characterization of the human centrosome by protein correlation profiling. Nature. 2003;426:570–574. doi: 10.1038/nature02166. [DOI] [PubMed] [Google Scholar]
- 37.Blanchette-Mackie EJ, et al. Perilipin is located on the surface layer of intracellular lipid droplets in adipocytes. J Lipid Res. 1995;36:1211–1226. [PubMed] [Google Scholar]
- 38.Abell BM, et al. Membrane protein topology of oleosin is constrained by its long hydrophobic domain. J Biol Chem. 2002;277:8602–8610. doi: 10.1074/jbc.M103712200. [DOI] [PubMed] [Google Scholar]
- 39.Abell BM, et al. Role of the proline knot motif in oleosin endoplasmic reticulum topology and oil body targeting. Plant Cell. 1997;9:1481–1493. doi: 10.1105/tpc.9.8.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stevanovic A, Thiele C. Monotopic topology is required for lipid droplet targeting of ancient ubiquitous protein 1. J Lipid Res. 2013;54:503–513. doi: 10.1194/jlr.M033852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Poppelreuther M, et al. The N-terminal region of acyl-CoA synthetase 3 is essential for both the localization on lipid droplets and the function in fatty acid uptake. J Lipid Res. 2012;53:888–900. doi: 10.1194/jlr.M024562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stone SJ, et al. Membrane topology and identification of key functional amino acid residues of murine acyl-CoA:diacylglycerol acyltransferase-2. J Biol Chem. 2006;281:40273–40282. doi: 10.1074/jbc.M607986200. [DOI] [PubMed] [Google Scholar]
- 43.Ingelmo-Torres M, et al. Hydrophobic and basic domains target proteins to lipid droplets. Traffic. 2009;10:1785–1801. doi: 10.1111/j.1600-0854.2009.00994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zehmer JK, et al. Identification of a novel N-terminal hydrophobic sequence that targets proteins to lipid droplets. J Cell Sci. 2008;12:1852–1860. doi: 10.1242/jcs.012013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Turró S, et al. Identification and characterization of associated with lipid droplet protein 1: a novel membrane-associated protein that resides on hepatic lipid droplets. Traffic. 2006;7:1254–1269. doi: 10.1111/j.1600-0854.2006.00465.x. [DOI] [PubMed] [Google Scholar]
- 46.Yokoi Y, et al. Regulated expression by PPARalpha and unique localization of 17beta-hydroxysteroid dehydrogenase type 11 protein in mouse intestine and liver. FEBS J. 2007;274:4837–4847. doi: 10.1111/j.1742-4658.2007.06005.x. [DOI] [PubMed] [Google Scholar]
- 47.Horiguchi Y, et al. Identification and characterization of the ER/lipid droplet-targeting sequence in 17beta-hydroxysteroid dehydrogenase type 11. Arch Biochem Biophys. 2008;479:121–130. doi: 10.1016/j.abb.2008.08.020. [DOI] [PubMed] [Google Scholar]
- 48.Gruber A, et al. The N-terminal region of comparative gene identification-58 (CGI-58) is important for lipid droplet binding and activation of adipose triglyceride lipase. J Biol Chem. 2008;285:12289–12298. doi: 10.1074/jbc.M109.064469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boeszoermenyi A, et al. Structure of a CGI-58 motif provides the molecular basis of lipid droplet anchoring. J Biol Chem. 2015;290:26361–26372. doi: 10.1074/jbc.M115.682203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zehmer JK, et al. Targeting sequences of UBXD8 and AAM-B reveal that the ER has a direct role in the emergence and regression of lipid droplets. J Cell Sci. 2009;122:3694–3702. doi: 10.1242/jcs.054700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soni KG, et al. Coatomer-dependent protein delivery to lipid droplets. J Cell Sci. 2009;122:1834–1841. doi: 10.1242/jcs.045849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beller M, et al. COPI complex is a regulator of lipid homeostasis. PLoS Biol. 2008;6:e292. doi: 10.1371/journal.pbio.0060292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakamura N, et al. Arf1-dependent PLD is localized to oleic acid-induced lipid droplets in NIH3T3 cells. Biochem Biophys Res Commun. 2005;335:117–123. doi: 10.1016/j.bbrc.2005.07.050. [DOI] [PubMed] [Google Scholar]
- 54.Guo Y, et al. Functional genomic screen reveals genes involved in lipid-droplet formation and utilization. Nature. 2008;453:657–661. doi: 10.1038/nature06928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bouvet S, et al. Targeting of the Arf-GEF GBF1 to lipid droplets and Golgi membranes. J Cell Sci. 2013;126:4794–4805. doi: 10.1242/jcs.134254. [DOI] [PubMed] [Google Scholar]
- 56.Thiam AR, et al. COPI buds 60-nm lipid droplets from reconstituted water-phospholipid-triacylglyceride interfaces, suggesting a tension clamp function. Proc Natl Acad Sci USA. 2013;110:13244–13249. doi: 10.1073/pnas.1307685110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Suzuki M, et al. ELMOD2 is anchored to lipid droplets by palmitoylation and regulates ATGL recruitment. Mol Biol Cell. 2015;26:2333–2342. doi: 10.1091/mbc.E14-11-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ozeki S, et al. Rab18 localizes to lipid droplets and induces their close apposition to the endoplasmic reticulum-derived membrane. J Cell Sci. 2005;118:2601–2611. doi: 10.1242/jcs.02401. [DOI] [PubMed] [Google Scholar]
- 59.Seelig J. Thermodynamics of lipid-peptide interactions. Biochim Biophys Acta. 2004;1666:40–50. doi: 10.1016/j.bbamem.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 60.Terzi E, et al. Interaction of Alzheimer beta-amyloid peptide(1–40) with lipid membranes. Biochemistry. 1997;36:14845–14852. doi: 10.1021/bi971843e. [DOI] [PubMed] [Google Scholar]
- 61.Hristova K, et al. An amphipathic alpha-helix at a membrane interface. A structural study using a novel X-ray diffraction method. J Mol Biol. 1999;290:99–117. doi: 10.1006/jmbi.1999.2840. [DOI] [PubMed] [Google Scholar]
- 62.Krahmer N, et al. Phosphatidylcholine synthesis for lipid droplet expansion is mediated by localized activation of CTP: phosphocholine cytidylyltransferase. Cell Metab. 2011;14:504–515. doi: 10.1016/j.cmet.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sundler R, Akesson B. Regulation of phospholipid biosynthesis in isolated rat hepatocytes. Effect of different substrates. J Biol Chem. 1975;250:3359–3367. [PubMed] [Google Scholar]
- 64.Kennedy EP, Weiss SB. The function of cytidine coenzymes in the biosynthesis of phospholipides. J Biol Chem. 1956;222:193–214. [PubMed] [Google Scholar]
- 65.Huang HK, et al. The membrane-binding domain of an amphitropic enzyme suppresses catalysis by contact with an amphipathic helix flanking its active site. J Mol Biol. 2013;425:1546–1564. doi: 10.1016/j.jmb.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 66.Lee J, et al. Structural basis for autoinhibition of CTP: phosphocholine cytidylyltransferase (CCT), the regulatory enzyme in phosphatidylcholine synthesis, by its membrane-binding amphipathic helix. J Biol Chem. 2014;289:1742–1755. doi: 10.1074/jbc.M113.526970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Friesen JA, et al. Enzymatic and cellular characterization of a catalytic fragment of CTP:phosphocholine cytidylyltransferase alpha. J Biol Chem. 1999;274:13384–13389. doi: 10.1074/jbc.274.19.13384. [DOI] [PubMed] [Google Scholar]
- 68.Payne F, et al. Mutations disrupting the Kennedy phosphatidylcholine pathway in humans with congenital lipodystrophy and fatty liver disease. Proc Natl Acad Sci USA. 2014;111:8901–8906. doi: 10.1073/pnas.1408523111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aitchison AJ, et al. Nuclear-localized CTP:phosphocholine cytidylyltransferase alpha regulates phosphatidylcholine synthesis required for lipid droplet biogenesis. Mol Biol Cell. 2015;26:2927–2938. doi: 10.1091/mbc.E15-03-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vamparys L, et al. Conical lipids in flat bilayers induce packing defects similar to that induced by positive curvature. Biophys J. 2013;104:585–593. doi: 10.1016/j.bpj.2012.11.3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arnold RS, et al. Binding of CTP:phosphocholine cytidylyltransferase to lipid vesicles: diacylglycerol and enzyme dephosphorylation increase the affinity for negatively charged membranes. Biochemistry. 1997;36:6149–6156. doi: 10.1021/bi970023z. [DOI] [PubMed] [Google Scholar]
- 72.Agassandian M, et al. Oxysterols inhibit phosphatidylcholine synthesis via ERK docking and phosphorylation of CTP: phosphocholine cytidylyltransferase. J Biol Chem. 2005;280:21577–21587. doi: 10.1074/jbc.M412409200. [DOI] [PubMed] [Google Scholar]
- 73.Arrese EL, et al. Function and structure of lipid storage droplet protein 1 studied in lipoprotein complexes. Arch Biochem Biophys. 2008;473:42–47. doi: 10.1016/j.abb.2008.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bickel PE, et al. PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim Biophys Acta. 2009;1791:419–440. doi: 10.1016/j.bbalip.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bulankina AV, et al. TIP47 functions in the biogenesis of lipid droplets. J Cell Biol. 2009;185:641–655. doi: 10.1083/jcb.200812042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Subramanian V, et al. Hydrophobic sequences target and anchor perilipin A to lipid droplets. J Lipid Res. 2004;45:1983–1991. doi: 10.1194/jlr.M400291-JLR200. [DOI] [PubMed] [Google Scholar]
- 77.Najt CP, et al. Structural and functional assessment of perilipin 2 lipid binding domain(s) Biochemistry. 2014;53:7051–7066. doi: 10.1021/bi500918m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bussell R, Jr, Eliezer D. A structural and functional role for 11-mer repeats in alpha-synuclein and other exchangeable lipid binding proteins. J Mol Biol. 2003;329:763–778. doi: 10.1016/s0022-2836(03)00520-5. [DOI] [PubMed] [Google Scholar]
- 79.Segrest JP, et al. The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J Lipid Res. 1992;33:141–166. [PubMed] [Google Scholar]
- 80.Chong BM, et al. The adipophilin C terminus is a self-folding membrane-binding domain that is important for milk lipid secretion. J Biol Chem. 2011;286:23254–23265. doi: 10.1074/jbc.M110.217091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hickenbottom SJ, et al. Structure of a lipid droplet protein; the PAT family member TIP47. Structure. 2004;12:1199–1207. doi: 10.1016/j.str.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 82.Ohsaki Y, et al. Recruitment of TIP47 to lipid droplets is controlled by the putative hydrophobic cleft. Biochem Biophys Res Commun. 2006;347:279–287. doi: 10.1016/j.bbrc.2006.06.074. [DOI] [PubMed] [Google Scholar]
- 83.Listenberger LL, et al. Adipocyte differentiation-related protein reduces the lipid droplet association of adipose triglyceride lipase and slows triacylglycerol turnover. J Lipid Res. 2007;48:2751–2761. doi: 10.1194/jlr.M700359-JLR200. [DOI] [PubMed] [Google Scholar]
- 84.Wolins NE, et al. A proposed model of fat packaging by exchangeable lipid droplet proteins. FEBS Lett. 2006;580:5484–5491. doi: 10.1016/j.febslet.2006.08.040. [DOI] [PubMed] [Google Scholar]
- 85.Magné J, et al. The minor allele of the missense polymorphism Ser251Pro in perilipin 2 (PLIN2) disrupts an ∝-helix, affects lipolysis, and is associated with reduced plasma triglyceride concentration in humans. FASEB J. 2013;27:3090–3099. doi: 10.1096/fj.13-228759. [DOI] [PubMed] [Google Scholar]
- 86.Gandotra S, et al. Perilipin deficiency and autosomal dominant partial lipodystrophy. N Engl J Med. 2011;364:740–748. doi: 10.1056/NEJMoa1007487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gandotra S, et al. Human frame shift mutations affecting the carboxyl terminus of perilipin increase lipolysis by failing to sequester the adipose triglyceride lipase (ATGL) coactivator AB-hydrolase-containing 5 (ABHD5) J Biol Chem. 2011;286:34998–35006. doi: 10.1074/jbc.M111.278853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kozusko K, et al. Clinical and molecular characterization of a novel PLIN1 frameshift mutation identified in patients with familial partial lipodystrophy. Diabetes. 2015;64:299–310. doi: 10.2337/db14-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Drin G, et al. A general amphipathic alpha-helical motif for sensing membrane curvature. Nat Struct Mol Biol. 2007;14:138–146. doi: 10.1038/nsmb1194. [DOI] [PubMed] [Google Scholar]
- 90.Bigay J, et al. ArfGAP1 responds to membrane curvature through the folding of a lipid packing sensor motif. EMBO J. 2005;24:2244–2253. doi: 10.1038/sj.emboj.7600714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Egan JJ, et al. Mechanism of hormone-stimulated lipolysis in adipocytes: translocation of hormone-sensitive lipase to the lipid storage droplet. Proc Natl Acad Sci USA. 1992;89:8537–8541. doi: 10.1073/pnas.89.18.8537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li Z, et al. Lipid droplets control the maternal histone supply of Drosophila embryos. Curr Biol. 2012;22:2104–2113. doi: 10.1016/j.cub.2012.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Carvalho FA, et al. Dengue virus capsid protein binding to hepatic lipid droplets (LD) is potassium ion dependent and is mediated by LD surface proteins. J Virol. 2012;86:2096–2108. doi: 10.1128/JVI.06796-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kitamura T, et al. Mouse aldehyde dehydrogenase ALDH3B2 is localized to lipid droplets via two C-terminal tryptophan residues and lipid modification. Biochem J. 2015;465:79–87. doi: 10.1042/BJ20140624. [DOI] [PubMed] [Google Scholar]
- 95.Leung KF, et al. Rab GTPases containing a CAAX motif are processed post-geranylgeranylation by proteolysis and methylation. J Biol Chem. 2007;282:1487–1497. doi: 10.1074/jbc.M605557200. [DOI] [PubMed] [Google Scholar]
- 96.Klionsky and Codogno (2013) Details to be added
- 97.Singh et al. (2009)a Details to be added
- 98.Singh et al. (2009)b Details to be added
- 99.van Zutphen et al. (2014) Details to be added
- 100.Yang et al. (2010)a Details to be added
- 101.Ouimet and Marcel (2012) Details to be added
- 102.Herms A, et al. AMPK activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation. Nat Commun. 2015;6:7176. doi: 10.1038/ncomms8176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rambold AS, et al. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell. 2015;32:678–692. doi: 10.1016/j.devcel.2015.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Huang Y, et al. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc Natl Acad Sci USA. 2010;107:7892–7897. doi: 10.1073/pnas.1003585107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Deruyffelaere C, et al. Ubiquitin-mediated proteasomal degradation of oleosins is involved in oil body mobilization during post-germinative seedling growth in Arabidopsis. Plant Cell Physiol. 2015;56:1374–1387. doi: 10.1093/pcp/pcv056. [DOI] [PubMed] [Google Scholar]
- 106.Kaushik S, Cuervo AM. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat Cell Biol. 2015;17:759–770. doi: 10.1038/ncb3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jo Y, et al. Ancient ubiquitous protein-1 mediates sterol-induced ubiquitination of 3-hydroxy-3-methylglutaryl CoA reductase in lipid droplet-associated endoplasmic reticulum membranes. Mol Biol Cell. 2013;24:169–183. doi: 10.1091/mbc.E12-07-0564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Klemm EJ, et al. Dual role of ancient ubiquitous protein 1 (AUP1) in lipid droplet accumulation and endoplasmic reticulum (ER) protein quality control. J Biol Chem. 2011;286:37602–37614. doi: 10.1074/jbc.M111.284794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Spandl J, et al. Ancient ubiquitous protein 1 (AUP1) localizes to lipid droplets and binds the E2 ubiquitin conjugase G2 (Ube2g2) via its G2 binding region. J Biol Chem. 2011;286:5599–5606. doi: 10.1074/jbc.M110.190785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Olzmann JA, et al. Spatial regulation of UBXD8 and p97/VCP controls ATGL-mediated lipid droplet turnover. Proc Natl Acad Sci USA. 2013;110:1345–1350. doi: 10.1073/pnas.1213738110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Derganc J, et al. Membrane bending: the power of protein imbalance. Trends Biochem Sci. 2013;38:576–584. doi: 10.1016/j.tibs.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 112.Mitsche MA, Small DM. Surface pressure-dependent conformation change of apolipoprotein-derived amphipathic alpha-helices. J Lipid Res. 2013;54:1578–1588. doi: 10.1194/jlr.M034462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang L, et al. Surface study of apoB1694-1880, a sequence that can anchor apoB to lipoproteins and make it nonexchangeable. J Lipid Res. 2009;50:1340–1352. doi: 10.1194/jlr.M900040-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ollila OH, et al. Interfacial tension and surface pressure of high density lipoprotein, low density lipoprotein, and related lipid droplets. Biophys J. 2012;103:1236–1244. doi: 10.1016/j.bpj.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dergunov AD. Local/bulk determinants of conformational stability of exchangeable apolipoproteins. Biochim Biophys Acta. 2011;1814:1169–1177. doi: 10.1016/j.bbapap.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 116.Masuda Y, et al. ADRP/adipophilin is degraded through the proteasome-dependent pathway during regression of lipid-storing cells. J Lipid Res. 2006;47:87–98. doi: 10.1194/jlr.M500170-JLR200. [DOI] [PubMed] [Google Scholar]
- 117.Xu G, et al. Post-translational regulation of adipose differentiation-related protein by the ubiquitin/proteasome pathway. J Biol Chem. 2005;280:42841–42847. doi: 10.1074/jbc.M506569200. [DOI] [PubMed] [Google Scholar]
- 118.Barneda D, et al. The brown adipocyte protein CIDEA promotes lipid droplet fusion via a phosphatidic acid-binding amphipathic helix. Elife. 2015;4:e07485. doi: 10.7554/eLife.07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Smagris E, et al. Pnpla3I148 M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology. 2015;61:108–118. doi: 10.1002/hep.27242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Singh R, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.van Zutphen T, et al. Lipid droplet autophagy in the yeast Saccharomyces cerevisiae. Mol Biol Cell. 2014;25:290–301. doi: 10.1091/mbc.E13-08-0448. [DOI] [PMC free article] [PubMed] [Google Scholar]